

Abstract
The ability of cells to polarize and move toward external stimuli plays a crucial role in development, as well as in normal and pathological physiology. Migrating cells maintain dynamic complementary distributions of Ras activity and of the phospholipid phosphatidylinositol‐3,4‐bisphosphate (PI(3,4)P2). Here, we show that lagging‐edge component PI(3,4)P2 also localizes to retracting leading‐edge protrusions and nascent macropinosomes, even in the absence of phosphatidylinositol 3,4,5‐trisphosphate (PIP3). Once internalized, macropinosomes break up into smaller PI(3,4)P2‐enriched vesicles, which fuse with the plasma membrane at the rear of the cell. Subsequently, the phosphoinositide diffuses toward the front of the cell, where it is degraded. Computational modeling confirms that this cycle gives rise to stable back‐to‐front gradient. These results uncover a surprising “reverse‐fountain flow” of PI(3,4)P2 that regulates polarity.
Keywords: cell migration, excitability, membrane flow, neutrophils, refractory period
Subject Categories: Cell Adhesion, Polarity & Cytoskeleton; Membrane & Intracellular Transport
Macropinosome‐dependent delivery of the phospholipid PI(3,4)P2 to the trailing edge of the cell establishes its polarized distribution required for correct migratory behavior.
Introduction
Chemotaxis or directed cell migration is fundamental for a multitude of physiological and pathophysiological processes including organ formation during development, angiogenesis, wound healing, immune responses, and cancer metastasis (Sasaki et al, 2004, Devreotes et al, 2017; Stuelten et al, 2018, Li et al, 2020). In response to chemical stimulus, migrating cells display functionally distinct leading and lagging edges by relocating proteins or their activities selectively to the poles. Some events such as Ras and PI3K activation, or PIP3 accumulation are selectively activated or localized at the leading edge and are referred to as “front” events. Others, like Myosin II and PTEN distributions behave in a complementary fashion and are referred to as “back” events. These distributions of front and back molecules or activities are a crucial first step in establishing polarity and guiding cell migration, and are maintained even in the absence of a chemoattractant gradient (Parent et al, 1998; Weiner et al, 1999; Iijima & Devreotes, 2002; Janetopoulos et al, 2004; Parent, 2004; Andrew & Insall, 2007; Pal et al, 2019).
There exist different physiological situations such as macropinocytosis that parallel cell migration where such complementary distributions of molecules are conserved. Both macropinocytosis and migration require extensions of actin‐driven protrusions. During macropinocytosis in Dictyostelium ameba, the extending edges of the forming cups are decorated with front components, such as activated Ras and PIP3, whereas back components like PTEN, Myosin II, PI(3,4)P2, and GAP proteins localize to the base (Weiner et al, 1999; Luo et al, 2003; Dormann et al, 2004; Postma et al, 2004; Veltman et al, 2016; Kay et al, 2018; Li et al, 2018; Shellard et al, 2018). The conserved arrangement of these molecules in the macropinocytic cups and along the cell perimeter of migrating cells suggests a relationship between these two processes.
PI(3,4)P2 is reported to be important in both macropinocytosis and cell migration. In macropinocytosis, transient and sequential emergence of PI(3,4,5)P3 and PI(3,4)P2 is necessary (Dormann et al, 2004; Yoshida et al, 2009; Welliver & Swanson, 2012; Maekawa et al, 2014; Hawkins & Stephens, 2016; Kay et al, 2018). In migrating Dictyostelium cells and zebrafish neutrophils, PI(3,4)P2 is distributed in a back‐to‐front gradient (Lam et al, 2012; Li et al, 2018). There is a mutual inhibition between PI(3,4)P2 and Ras activities: When Ras is activated in the front, PI(3,4)P2 goes down; conversely, depletion of PI(3,4)P2 leads to elevated Ras activity. This mutual inhibitory feedback loop ensures that the back of the cell is silent, and the front is active. The resemblance between the back‐to‐front gradient across the cell and across the macropinocytic cups led us to hypothesize that PI(3,4)P2‐decorated macropinosomes could be a crucial component linking macropinocytosis and cell migration.
Here, we demonstrate that there is an ongoing flow of vesicular PI(3,4)P2 through the cell and a compensatory forward flow along the membrane, which establishes a back‐to‐front gradient of this phosphoinositide. Previous models have suggested that such vesicular and plasma membrane flows are important for cell migration, although the directions were reversed (Bretscher, 1996; Tanaka et al, 2017; O'Neill et al, 2018). Our findings show that PI(3,4)P2 is internalized on macropinosomes and transported on microtubules into the cytosol. These anterior macropinosomes break up into several smaller PI(3,4)P2‐enriched vesicles, which dock and fuse at the rear of the migrating cell. This surprising reverse‐fountain flow of PI(3,4)P2 and its role in establishing the back‐to‐front gradient may be essential for polarity in specific modes of cell migration.
Results
PI(3,4)P2 at the back diffuses toward the front
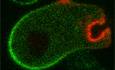
In migrating Dictyostelium ameba, PI(3,4)P2, observed using a tandem PH‐domain biosensor tPH‐CynA, is distributed to the back and is complementary to F‐actin and active Ras (Fig 1A and B). To investigate the dynamic distribution of PI(3,4)P2 at the back, we fused a green‐to‐red photoconvertible fluorescence Kikume Green‐Red (KikGR) with tPH‐CynA and expressed it in the cells. Using the Zeiss 780 bleaching mode with a 405 nm laser, photoconversion caused a decrease in green fluorescence and a simultaneous increase in red fluorescence. We photoconverted tPH‐CynA‐KiKGR in a small region of the membrane at the back of the cell and tracked the green and red channels. Photoconverted tPH‐CynA diffused symmetrically along the membrane toward the front by 50 s (Fig 1C and D, and Movie EV1). The signal decreased by less than 10% in the first 60 s (and less than 20% in the first 5 min) following photoconversion, indicating there is little exchange of membrane and cytosolic PI(3,4)P2 biosensor (Fig 1E). The lateral diffusion of the photoconverted tPH‐CynA on the membrane was found to be 2.05 μm2/s by comparing the experimental profiles with the simulated profiles from a stochastic simulation for a wide range of diffusion constants (see Materials and Methods, Appendix Fig S1A). At the side of the cell, the distribution of photoconverted PI(3,4)P2 biosensor spread asymmetrically, skewed toward the back of the cell and the total fluorescence decreased gradually during 145 s, the increase in the skewness was 52% in 60 s (Fig 1F and G, and Appendix Fig S1B). We reasoned that PI(3,4)P2 diffuses bi‐directionally on the membrane on the side of the cell, but it is degraded faster toward the front. Taken together, these observations suggest that there is a source of PI(3,4)P2 at the rear of the cell and that PI(3,4)P2 is degraded as it diffuses toward the front.
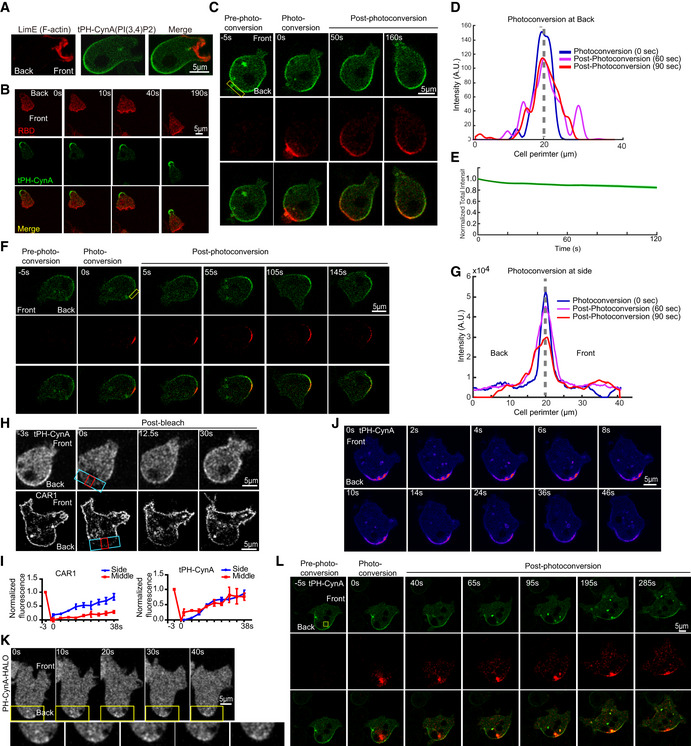
Figure 1. Small PI(3,4)P2 vesicles supply PI(3,4)P2 to the back of migrating Dictyostelium cells.
- Confocal images of growth‐stage wild‐type Dictyostelium AX3 cell co‐expressing tPH‐CynA‐KikGR (green) and an F‐actin biosensor LimE‐RFP (red). Front and back of the cell are shown.
- Time‐lapse confocal images of growth‐stage AX3 cell co‐expressing tPH‐CynA‐KikGR (green) and an activated Ras biosensor, RBD‐mCherry (red). Front and back of the cell are shown.
- Time‐lapse confocal images of tPH‐CynA‐KikGR expressing AX3 cells. Back region of cells was photoconverted from green fluorescence to red at t = 0 s. Front and back of the cell are shown, box showing the photoconverted region.
- Profiles of the converted red fluorescence at the back remain almost symmetric over time. The dotted line represents the axis of symmetry of the intensity profile of the photoconverted fluorophores at t = 0.
- Temporal profile of normalized intensity of total converted red fluorescence. Mean ± SEM is shown for n = 14 cells.
- Time‐lapse confocal images of tPH‐CynA‐KikGR expressing AX3 cells during photoconversion. Side region of cells was photoconverted at time 0. Green fluorescence was converted to red. Front and back of the cell are shown. Box shows the photoconverted region.
- Profiles of the converted red fluorescence at side become skewer (steeper near the front) over time. The dotted line represents the axis of symmetry of the intensity profile of the photoconverted fluorophores at t = 0.
- Confocal fluorescent images of tPH‐CynA‐KikGR (top) and CAR1‐mcherry (bottom) Dictyostelium AX3 cell showing the bleached area and the side (blue) and middle (red) boxes where the fluorescence recovery is monitored. Front and back of the cell are shown.
- The graphs depict the recovery of CAR1‐mcherry (left) and tPH‐CynA‐KikGR (right). Mean ± SEM is shown for n = 2 cells.
- Time‐lapse confocal images of tPH‐CynA‐KikGR expressing AX3 cells showing docking events at the back of a migrating Dictyostelium cell. Front and back of the cell are shown.
- Time‐lapse confocal images of PH‐CynA‐HALO expressing AX3 cells showing high PH‐CynA vesicles accumulation at the back of a migrating Dictyostelium cell. Front and back of the cell are shown.
- Time‐lapse confocal images of tPH‐CynA‐KikGR (green) expressing AX3 cells. Back cortical region of cell was photoconverted at time 0. Green fluorescence was converted to red. Front and back of the cell is shown, box showing the photoconverted region.
Source data are available online for this figure.
Small PI(3,4)P2 vesicles supply PI(3,4)P2 to the back of migrating Dictyostelium cells
To determine the nature of the source of PI(3,4)P2 at the back, we conducted fluorescence recovery after photobleaching (FRAP). As a control we used chemoattractant receptor cAR1, which is uniformly distributed on the plasma membrane of cells. We photobleached a small portion of the periphery at the back of the cell and carefully monitored the fluorescence recovery. As expected, for cAR1, the recovery proceeds in a vectoral fashion from the adjacent unbleached areas of the membrane, since the receptor diffuses in the plane of the membrane. In contrast, for tPH‐CynA there was an even fluorescence recovery in the middle and boundaries of the bleached zone (Fig 1H and I, and Appendix Fig S1C and D). Since the biosensor stably marks PI(3,4)P2 (Fig 1E), this shows that there is a cytosolic source of PI(3,4)P2.
We next carefully examined the dynamic behavior of PI(3,4)P2 at the back using two similar biosensors with different affinities and tags, tPH‐CynA‐KikGR and PH‐CynA‐HALO. With both biosensors, we observed small PI(3,4)P2‐containing vesicles docking in the back region (Fig 1J and K, and Movie EV2). To confirm this result, we photoconverted the intracellular pool of small vesicles close to the membrane at the back of the cell. We found that PI(3,4)P2‐containing small intracellular vesicles moved close to the membrane, and appeared to dock with it, whereupon PI(3,4)P2 diffused away quickly (Fig 1L). We propose that small vesicles fuse with membrane and diffuse fast, but we allow the possibility that vesicles dock at the back and then slide along the membrane to the front.
PI(3,4)P2 trails F‐actin and PIP3 on the leading‐edge macropinosomes
To understand the origins of these vesicles, we sought the first appearance of enriched PI(3,4)P2. There was a low amount of PI(3,4)P2 on the membrane at the front of the cells, which disappeared from newly formed actin‐rich protrusions. PI(3,4)P2 then appeared de novo as the leading‐edge protrusions retracted or evolved into nascent macropinosomes (Li et al, 2018). We directly compared the localization of PI(3,4)P2 with newly polymerized F‐actin. Time‐lapse two‐color images showed that F‐actin appeared on the tips of forming macropinosomes in the regions where PI(3,4)P2 had disappeared, then gradually dissipated as the structures closed. Simultaneously, PI(3,4)P2 appeared and remained on the fully formed macropinosomes. The changes in intensity and distribution of these two biosensors on the forming macropinosomes were shown (Fig 2A and Movie EV3). Quantification of relative intensities of F‐actin and PI(3,4)P2 on macropinosomes at different time points showed that PI(3,4)P2 lags F‐actin and lasts longer (Fig 2C and Appendix Fig S2).
Figure 2. PI(3,4)P2 trails F‐actin and PIP3 on the leading‐edge macropinosomes.
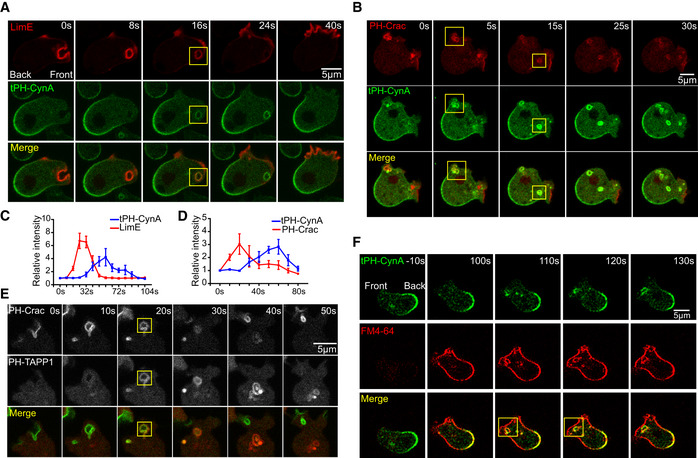
- Time‐lapse confocal images of Dictyostelium AX3 cell co‐expressing tPH‐CynA‐KikGR (green) and LimE‐RFP (red). Front and back of the cell are shown. Box shows the vesicle.
- Time‐lapse confocal images of Dictyostelium AX3 cell co‐expressing tPH‐CynA‐GFP (green) and PH‐Crac‐RFP (red). Box shows the vesicle.
- Relative intensities of tPH‐CynA‐KikGR on macropinosomes and LimE‐RFP on macropinosomes in experiments in (A). Mean ± SEM is shown for n = 5 cells.
- Relative intensities of tPH‐CynA‐KikGR on macropinosomes and PH‐Crac‐RFP on macropinosomes in experiment in Fig 3B. Mean ± SEM is shown for n = 3 cells.
- Time‐lapse confocal images of AX3 cell co‐expressing PH‐Crac‐GFP (green) and another established PI(3,4)P2 biosensor tPH‐TAPP1‐RFP (red). Box shows the vesicle.
- Time‐lapse confocal images of tPH‐CynA‐KikGR (green) expressing wild‐type AX3 cells. FM4‐64 (red) was added at time 0 during live cell imaging. Front and back of the cell are shown. Box shows the vesicle.
Source data are available online for this figure.
Next, we compared the dynamic localization of PI(3,4)P2 with PIP3, detected by the biosensor PH‐Crac. PIP3 resembles the pattern of F‐actin, increasing and then decreasing during the formation of macropinosomes. Again, dynamic distributions of tPH‐CynA showed a complementary pattern only appearing on the forming macropinosomes (Fig 2B and D). This distribution of PI(3,4)P2 was further demonstrated by another established biosensor PH‐TAPP1, which localized to macropinosomes only as PIP3 is subsiding (Fig 2E). These data showed that lagging‐edge component PI(3,4)P2 localized to retracting protrusions and nascent macropinosomes in a distinct pattern compared with F‐actin and PIP3.
To determine the prevalence of PI(3,4)P2‐enriched vesicles in membrane trafficking, we applied the lipophilic dye, FM4‐64. It fluoresces only when it is incorporated in the plasma membrane and, once internalized, it becomes trapped in intracellular vesicles (Kriebel et al, 2008). Thus, FM4‐64 labeling is a useful tool to visualize the membranes of pre‐macropinosomes. When FM4‐64 was added during a live cell imaging sequence, we observed increasing plasma membrane labeling within 100 s. By 110 s, the signal internalizes to label intracellular compartments or macropinosomes, which were labeled by PI(3,4)P2. Taken together, PI(3,4)P2 is detectable on majority of the dye‐labeled macropinosomes at the leading edge of the cells (Fig 2F and Movie EV4).
PI(3,4)P2 appears in absence of PIP3
We next assessed the relative contribution of PIP3 degradation to PI(3,4)P2 levels on the vesicles and plasma membrane with PI3K inhibitors and in PI3K1 − 2 − cells. In wild‐type cells, we investigated PI(3,4)P2 dynamics before and after addition of PI3K inhibitor, wortmannin. Surprisingly, PI(3,4)P2 remained on the plasma membrane at the back of the cell after wortmannin treatment while PIP3 was depleted (Fig 3A). Kymograph showed that over time PIP3 disappeared and line scan quantification demonstrated that dynamic distribution of PI(3,4)P2 is similar before and after PI3K inhibition (Fig 3B and C, and Appendix Fig S3). To verify this result, we first treated cells with latrunculin A to eliminate the cell shape changes and then treated cells with the well‐established PI3K inhibitor LY2924002. Again, PI(3,4)P2 remained on the membrane, and, in fact, its level was even higher (Fig 3D and E). To confirm this observation, we treated PI3K1 − 2 − cells with latrunculin A and found that even though there is no detectable PIP3 on the membrane, PI(3,4)P2 levels are nearly maintained (Fig 3F and G). Furthermore, PI3K1 − 2 − cells expressing tPH‐CynA‐KiKGR and LimE‐RFP showed that localization of PI(3,4)P2 on macropinosomes was also maintained (Fig 3H and I, and Movie EV5). Taken together, these observations suggest that PI(3,4)P2 dynamics can be regulated in the absence of PIP3, and this regulation is conserved in diverse cell types.
Figure 3. PI(3,4)P2 appears in absence of PIP3.
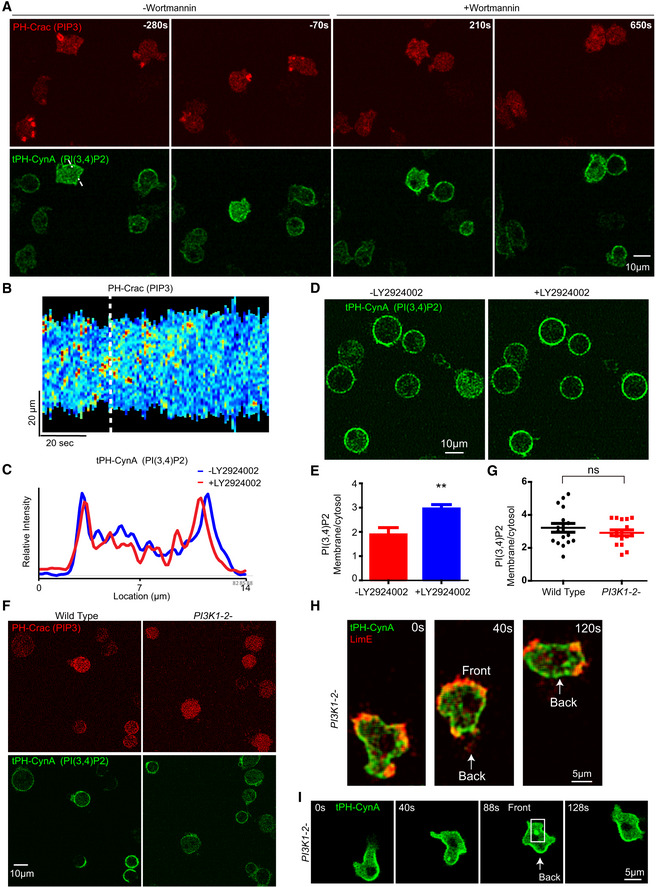
- Time‐lapse confocal images of wild‐type AX3 cells expressing tPH‐CynA‐KikGR (green) and PH‐crac (red) before and after PI3K inhibition by 5 µM Wortmannin.
- Kymographs of PH‐Crac intensity on the perimeter of one cell undergoing random migration in (A). White dotted line shows addition of Wortmannin.
- Intensity plot across the white dotted line in tPH‐CynA‐KikGR expressing cell before and after Wortmannin treatment in (A).
- Confocal images of tPH‐CynA‐KikGR expressing wild‐type Dictyostelium AX3 cells before (left) and after (right) LY2924002 treatment.
- Ratio of membrane to cytosol intensity of tPH‐CynA‐KikGR in wild‐type Dictyostelium AX3 cells before and after LY2924002 treatment, t‐test was carried out by GraphPad Prism, **P ≤ 0.01 vs. LY2924002 group (mean ± SEM, n = 7).
- Confocal images of tPH‐CynA‐KikGR (green) and PH‐Crac (red) expressing wild‐type Dictyostelium AX3 cells (left) and PI3K1 − 2 − Dictyostelium cells (right) treated with 5 µM latrunculin A.
- Ratio of membrane to cytosol intensity of tPH‐CynA‐KikGR in wild‐type Ax3 and PI3K1 − 2 − cells. There was no statistically significant difference in the biosensor on the membrane between the two groups (mean ± SEM, n = 17).
- Confocal images of tPH‐CynA‐KikGR (green) and LimE (labeling F‐actin) expressing PI3K1 − 2 − cells. Front and back of the cell are shown and white arrow showing the back localization.
- Confocal images of tPH‐CynA‐KikGR (green) expressing PI3K1 − 2 − cells, white box showing the vesicle, and white arrow showing the back localization.
Source data are available online for this figure.
PI(3,4)P2 macropinosomes are transported along microtubules and break apart
To determine the relationship between PI(3,4)P2‐enriched vesicles and the microtubule network, we fixed cells expressing the PI(3,4)P2 biosensor, tPH‐CynA‐KikGR, stained them with an anti‐α‐tubulin antibody, and performed z‐stack confocal microscopy. In untreated wild‐type AX3 cells, the microtubular network was intact and PI(3,4)P2‐enriched vesicles appeared to be associated with microtubules (Fig 4A and Movie EV6).
Figure 4. Macropinocytic vesicle processing is necessary for PI(3,4)P2 enrichment at the back of migrating cells.
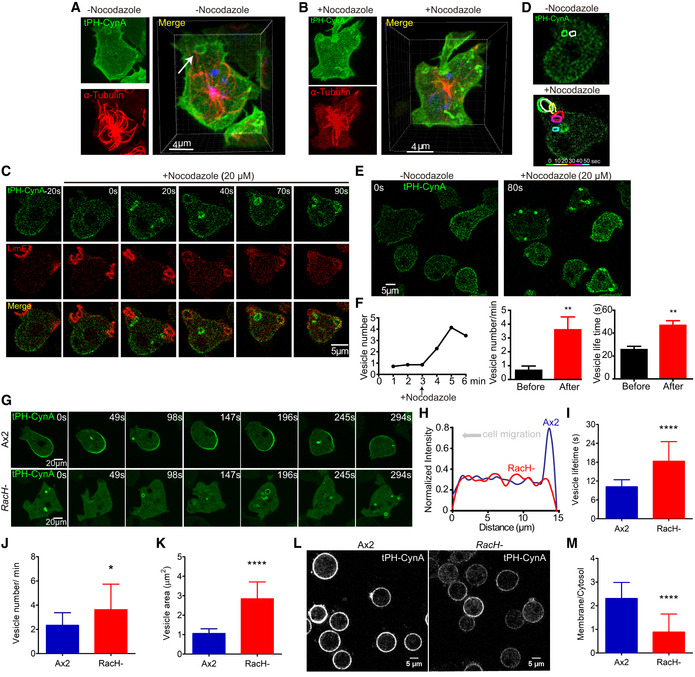
-
AConfocal images showing localization of PI(3,4)P2 (tPH‐CynA‐KiKGR; green) and α‐tubulin (red) in fixed wild‐type Dictyostelium AX3 cells in 3D. Arrow shows a vesicle attaching to the α‐tubulin labeled microtubules.
-
BConfocal images showing localization of PI(3,4)P2 (green) and α‐tubulin (red) in fixed AX3 cells after Nocodazole treatment in 3D.
-
CTime‐lapse confocal images showing dynamic localization of PI(3,4)P2 (green) and F‐actin (red) in wild‐type Dictyostelium cells. Nocodazole was added at time 0 during live cell imaging.
-
DColor‐coded tracing of vesicle at 10‐sec intervals from experiment in (C).
-
ETime‐lapse confocal images showing dynamic localization of PI(3,4)P2. Nocodazole was added at time 0 during the live cell imaging.
-
FQuantification of vesicle number, vesicle number/min, and vesicle lifetime (s) from experiment in E. t‐Test was carried out by GraphPad Prism, **P ≤ 0.01 vs. After Nocodazole group; mean ± SEM (n = 10).
-
GTime‐lapse confocal images of individual cells of growth‐stage wild‐type Dictyostelium AX2 or RacH‐ strain highlighting tPH‐CynA‐KikGR‐coated macropinocytic vesicles and back‐to‐front membrane gradient. Images were captured every 7 s.
-
HIntensity plot across wild‐type Dictyostelium AX2 or RacH‐ strain in image “147 s” shown in (G).
-
I–K(I) Lifetime, (J) number, or (K) area of tPH‐CynA‐KikGR‐coated macropinocytic vesicles in wild‐type AX2 and RacH‐ cells. t‐Test was carried out by GraphPad Prism, *P ≤ 0.05, ****P ≤ 0.0001 vs. AX2 group; mean ± SEM (n = 20).
-
LRepresentative confocal images of tPH‐CynA‐KikGR in wild‐type Dictyostelium AX2 or RacH‐ cells treated with 5 μM latrunculin A. Scale bar: 5 μm.
-
MRatio of membrane to cytosol intensity of tPH‐CynA‐KikGR in wild‐type Dictyostelium AX2 and RacH‐ cells. t‐Test was carried out by GraphPad Prism, ****P ≤ 0.0001 vs. AX2 group; mean ± SEM (n = 33).
Source data are available online for this figure.
Treatment of the wild‐type AX3 cells with 20 µM Nocodazole for 5 min caused a substantial disassembly of the microtubule network. Simultaneously, more PI(3,4)P2‐enriched vesicles were seen, which were no longer associated with the microtubules and were clustered around the closure of the macropinocytic cups (Fig 4B). The LimE channel demonstrated that macropinocytic cups continued to form, but the increase in the number of vesicles in the tPH‐CynA channel was due to a slower disappearance (Fig 4C and D). The macropinosomes were trapped along the membrane and were unable to be internalized in 50 s (Fig 4E and Movie EV7). The number of vesicles viewed per minute, and the vesicle lifetime increased upon Nocodazole treatment (Fig 4F). Disassembly of microtubule network with Nocodazole in another wild‐type strain, AX2, also resulted in more PI(3,4)P2‐enriched vesicles near the membrane (Appendix Fig S4).
We investigated whether inhibition of processing of PI(3,4)P2‐enriched macropinocytic vesicles by genetic perturbation would abrogate its accumulation at the back of a migrating Dictyostelium cell. To test this hypothesis, we used a RacH‐ Dictyostelium strain whose vesicle trafficking is disrupted due to defective acidification of early endosomes. This impairs macropinocytosis and chemotaxis (Somesh et al, 2006; Balest et al, 2011) As shown in Fig 4G, whereas a back‐to‐front gradient in PI(3,4)P2 membrane association was apparent in wild‐type Dictyostelium AX2 cells, in the RacH‐ cells “back” regions were weaker and there were more cytosolic PI(3,4)P2‐enriched vesicles. Line scans of pixel intensity across wild‐type or mutant cells confirmed greater PI(3,4)P2 accumulation at the back of wild‐type cells (Fig 4H).
Next, we quantified the lifetimes of the PI(3,4)P2‐enriched vesicles, their numbers per unit of time, surface areas, and membrane‐cytosol ratio. In RacH‐ cells, the vesicles were almost twice as long‐lived as those in wild‐type AX2 cells (Fig 4I). The vesicles in the mutant were also 40% greater in number and 63% larger (Fig 4J and K). Moreover, the plasma membrane‐to‐cytosol ratio of PI(3,4)P2 was 2.30 ± 0.67 in wild‐type AX2 cells but 0.88 ± 0.76 in RacH‐ cells (Fig 4L and M). Assuming that the degradation rate of PI(3,4)P2 is the same in the mutant and wild‐type, this result suggests that at least 50% of the PI(3,4)P2 on the membrane depends on vesicular trafficking.
Under live‐cell 3D super‐resolution imaging, we further found the macropinosomes are transported into the cytosol where they appear to break apart into smaller “satellite” vesicles and disappear (Fig 5A and Movie EV8). In Nocodazole treated or RacH‐ cells, macropinosomes remained on the perimeter and were not processed into the smaller vesicles. Taken together, these results suggest that PI(3,4)P2‐enriched macropinosomes are transported along microtubules into the cytosol where they are likely to be processed into smaller vesicles and disperse.
Figure 5. A link between PI(3,4)P2 on front macropinosomes and the back‐to‐front gradient of PI(3,4)P2.
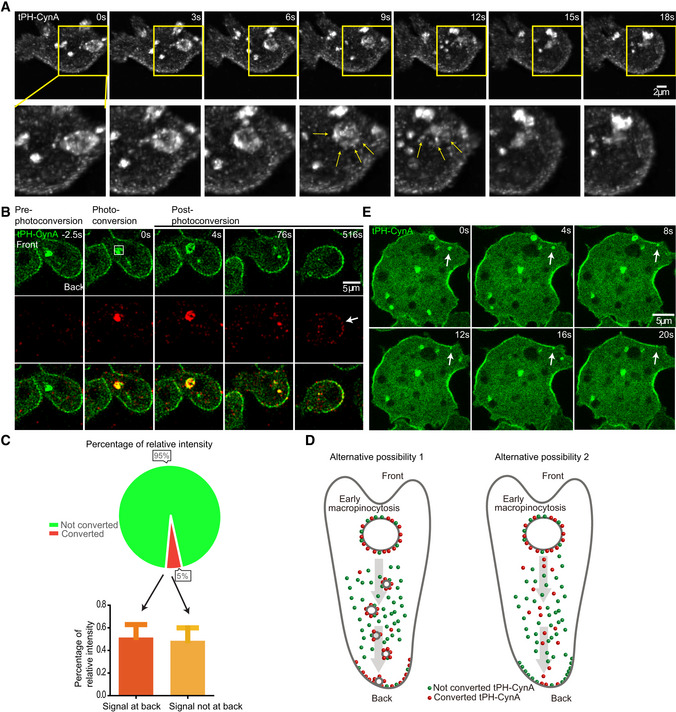
- Time‐lapse confocal images of tPH‐CynA‐KikGR expressing Dictyostelium AX3 cells. Below are zoomed in images from images above. Yellow arrows point to small satellite vesicles.
- Time‐lapse confocal images of tPH‐CynA‐KikGR expressing Dictyostelium AX3 cells. The macropinosome at the front of the cell was photoconverted at time 0. White arrow points to PI(3,4)P2 localization at the back of the membrane. Cell front and back are shown, box showing the photoconverted region.
- Pie chart quantification of percentage of relative intensity of total green fluorescence (not converted) and converted red fluorescence (converted), showing 4.97% of total fluorescence was converted to red (top). Bottom: 4.97% of total fluorescence was converted to red, and around 50% of converted red fluorescence end up at the back of membrane (red), less than 50% of the converted red fluorescence not at back (orange). n = 7.
- Schematic representation of two potential working alternative possibilities. Red dots show photoactivated tPH‐CynA, and green dots show not converted tPH‐CynA. Left shows that if the biosensor remains associated with the vesicles, a larger proportion of the red signal will appear at the back. Right shows that if the biosensor disassociates from the vesicles, a much smaller amount of the red signal will appear at the back.
- Time‐lapse confocal images of tPH‐CynA‐KikGR in giant Dictyostelium AX3 cells. Arrows point to vesicles.
A link between PI(3,4)P2 on front macropinosomes and the back‐to‐front gradient of PI(3,4)P2
Our observations on the “satellite” vesicles suggest these might be the same as the docking vesicles at the back (Fig 1J–L). To gain insight into the connection between PI(3,4)P2 on the front macropinosomes and the back‐to‐front gradient of plasma membrane, we photoconverted the PI(3,4)P2 biosensor, tPH‐CynA‐KiKGR, on late‐macropinosomes at the leading edge of the cells and tracked the signal. Figure 5B shows an example of photoconversion of a front macropinosome which eventually resulted in increased signal at the back of the cell. Another example is shown in Appendix Fig S5 where the cell changes direction after photoconversion and the delivery to the back is quicker. We quantified the relative percentage of green and red signal upon photoconversion. An average 4.97% of green fluorescence was converted to red while 51.43% of the generated red fluorescence ended up at the back, indicating that vesicles labeled with PI(3,4)P2 biosensor travel all the way to the back of membrane before the biosensor dissociates (Fig 5C). The data argue against the alternative possibility that the biosensor dissociates from the macropinosomes and rebinds to smaller vesicles which go to the back (Fig 5D).
Observation of docking and fusing events in single cells are limited by the small cell boundary. Electrofused “giant” cells provide a more accessible system for characterizing these dynamics. Giant cells have multiple “front” regions, which include F‐actin, ruffles, and macropinosomes, and “back” regions which lack these events (Gerisch et al, 2013; Gerhardt et al, 2014). In these cells, we observed large and small PI(3,4)P2 vesicles arising in multiple front regions and others docking at the nearby back regions. This is consistent with docking activities observed in single cells (Fig 5E).
We developed a stochastic mathematical model to explore the effect of different vesicle fusion parameters on the creation and maintenance of a stable back region in a cell (see Materials and Methods). We found that for creation of a stable back, the directed insertion of the satellite vesicles into the regions of highest PI(3,4)P2 levels on the membrane was an important condition (Appendix Fig S6A). Other important parameters were the mean arrival time of the PI(3,4)P2 vesicles, and the rate of PI(3,4)P2 diffusion and decay (Fig 6A and B). Increase in the mean arrival time resulted discontinuity in the back profile unless the decay rate was reduced appropriately (Appendix Fig S6B). Figure 6A and B shows the results of increasing the mean arrival time of the PI(3,4)P2 vesicles and decreasing the PI(3,4)P2 decay rates. These changes created a broader back without causing a large change in the overall amount (the change was less than 10%). A broader distribution of PI(3,4)P2 at the back would imply a larger fraction of the cell is inactive, which would make the cell more polarized. Consistently, our experimental results showed that the decay rate in more polarized cells was indeed fourfold lower than that in less polarized cells, and the extent of cell membrane covered by PI(3,4)P2 is smaller in less polarized cells compared with more polarized cells. The mean relative width of the back regions and decay rate obtained from the simulated kymographs was consistent with experimental results (Fig 6C and D).
Figure 6. A link between PI(3,4)P2 on front macropinosomes and the back‐to‐front gradient of PI(3,4)P2.
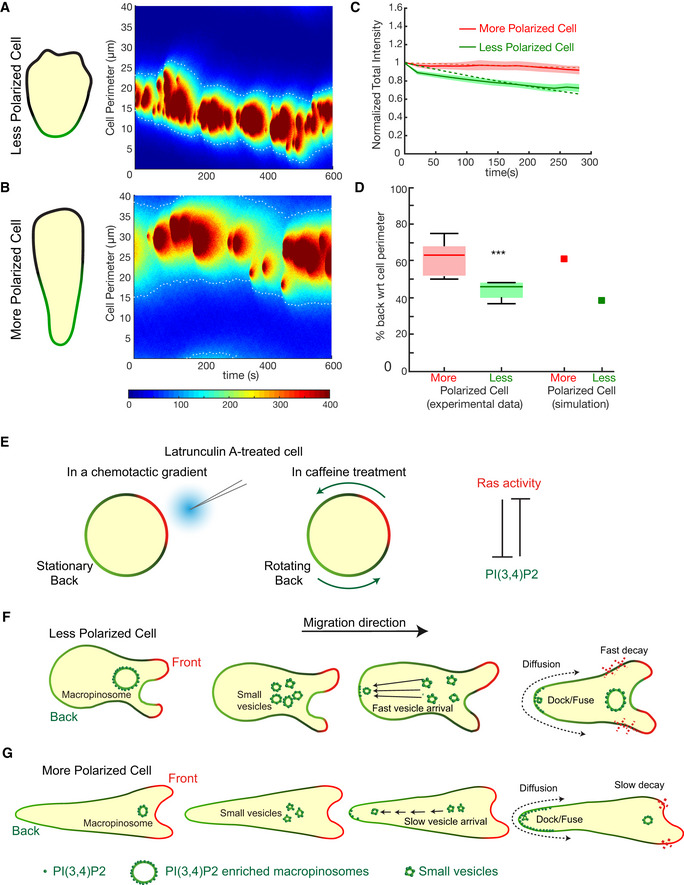
- Simulated kymograph of a less polarized cell with a narrow PI(3,4)P2 enriched region. The white dashed lines correspond to the periphery of the back regions which spanned on average 30% of the cell perimeter.
- Simulated kymograph of a more polarized cell with broader PI(3,4)P2 enriched region compared (A). Slower vesicle arrival time and reduced decay rate for PI(3,4)P2 was assumed in comparison with parameters used for the simulation in (A).
- Total intensity profiles (Mean ± SEM) of tPH‐CynA‐KikGR on the membrane over time show different decay rates for less polarized (green, n = 5) and more polarized (red, n = 8) Dictyostelium cells. The dashed lines show the respective single exponential fit to the data. Mean half time for vegetative cells = 7.5 min, for developed cells = 55 min (from single exponential fit to the data).
- The boxplot shows the relative width of the back regions in less (green) and more (red) polarized Dictyostelium cells. The square markers of respective colors show the mean relative width of the back regions obtained from the simulated kymographs in (A) and (B). ***P < 0.001 (n = 5). Mann–Whitney–Wilcoxon test was performed. The plots are standard box plots showing medians (central bands) along with the respective first and third quartiles.
- Schematic representation of our previous study demonstrating the mutual inhibition between Ras activity and PI(3,4)P2 which establishes polarity even in immobilized latrunculin A‐treated cells in a chemotactic gradient (with stationary back) or in caffeine treatment (with rotating back). In Dictyostelium, caffeine is used to inhibit the synthesis of the chemoattractant cAMP.
- Schematic representation of the working model in less polarized Dictyostelium cells. PI(3,4)P2 is internalized on macropinosomes and transported into the cytosol. These anterior macropinosomes break up into smaller PI(3,4)P2 vesicles, which dock and fuse at the rear of the cell.
- Schematic representation of the working model in more polarized Dictyostelium cells. Slow vesicle arrival of the PI(3,4)P2 vesicles and slower decay rates of PI(3,4)P2 created a broader back.
To access how much the vesicular trafficking contributes to the overall production of PI(3,4)P2, we measured turnover before and after latrunculin A treatment. Latrunculin A acutely blocks macropinosome formation. After latrunculin A treatment, there was a 30% decrease in tPH‐CynA on the membrane and a photoconversion experiment showed that the degradation rate of PI(3,4)P2 decreased threefold (Appendix Fig S7 and Fig 1E). Together these data suggest that the vesicular trafficking is responsible for more than 50% of the levels of PI(3,4)P2, which is consistent with the estimate made above when vesicular trafficking is blocked by RacH‐ mutant cells (Fig 4L and M).
Dynamics of PI(3,4)P2 in human neutrophils
As PI(3,4)P2 is distributed to the back of migrating Dictyostelium, complementary to F‐actin and active Ras (Fig 1A and B), we sought to investigate its spatio‐temporal distribution in migrating human leukemia neutrophil‐like HL‐60 cells. In Dictyostelium cells, the two biosensors for PI(3,4)P2, tPH‐CynA and PH‐TAPP1, display slightly different patterns, despite the fact that they are identical on lipid strips and have preference to PI(3,4)P2. Li and Edwards et al have previously established in Dictyostelium that tPH‐CynA and PH‐TAPP1 colocalize to the base of macropinocytic cups and back of the cell membrane (Li et al, 2018; Goulden et al, 2019). However, while tPH‐CynA labeled both structures strongly, the labeling of the back by PH‐TAPP1 was relatively weaker (Appendix Fig S8A–C). In fixed cells with depleted cytosolic signal, the presence of PH‐TAPP1 as a sharp band on the membrane at the back was detectable and colocalized with tPH‐CynA (Appendix Fig S8D).
Whereas tPH‐CynA did not express in HL‐60 cells, the distribution of PH‐TAPP1 was similar in HL‐60 cells and Dictyostelium. We used cPHX3‐TAPP1‐GFP, recently developed in the Hammond lab, as the biosensor for PI(3,4)P2 in HL‐60 cells. We generated a stable HL‐60 cell line expressing both RFP‐LifeAct (another F‐actin biosensor in human cells) and cPHX3TAPP1‐GFP (Goulden et al, 2019). Under live‐cell imaging, PI(3,4)P2 accumulated on the membrane, in a broadband at the front and a thin faint line at the back of migrating neutrophils (Fig EV1A–C). We further examined the distribution of PI(3,4)P2 in fixed HL‐60 cells which have depleted cytosolic signal. Under these conditions, the presence of PI(3,4)P2 as a sharp band on the membrane at the back is clear (Fig EV1D and E). Consistently, PI(3,4)P2 was found at the leading and trailing edges in zebrafish neutrophils (Lam et al, 2012).
Figure EV1. Dynamic localization of PI(3,4)P2 in neutrophils.
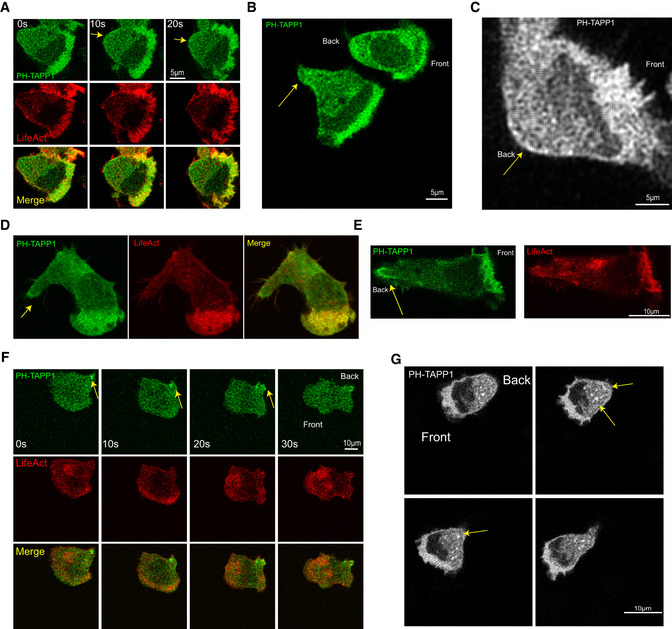
-
ATime‐lapse confocal images of human leukemia neutrophil‐like HL‐60 cell line stably co‐expressing PI(3,4)P2 biosensor, trimers of the isolated cPH (residues 169–329 in human TAPP1) (cPHx3TAPP1‐GFP), and RFP‐LifeAct (labeling F‐actin) showing front and back localization of PI(3,4)P2. Yellow arrow points to PI(3,4)P2 localization at the back of the membrane. Front and back of the cell are shown.
-
B, CConfocal images of HL‐60 cell line stably co‐expressing PI(3,4)P2 biosensor cPHx3TAPP1‐GFP and RFP‐LifeAct. Yellow arrow points to PI(3,4)P2 localization at the back of the membrane. Front and back of the cell are shown.
-
DConfocal images showing front and back localization of PI(3,4)P2 in fixed HL‐60 cells. Yellow arrow points to PI(3,4)P2 localization at the back of the membrane.
-
EA maximum intensity projection of Z‐stack of confocal images showing front and back localization of PI(3,4)P2 in fixed HL‐60 cells. Yellow arrow points to PI(3,4)P2 localization at the back of the membrane
-
FTime‐lapse confocal images of HL‐60 cell co‐expressing cPHx3TAPP1‐GFP and RFP‐LifeAct showing PI(3,4)P2 vesicles at the back of cells. Yellow arrow points to PI(3,4)P2 vesicle localization at the back of the HL‐60 cell.
-
GTime‐lapse confocal images of HL‐60 cell expressing cPHx3TAPP1‐GFP showing PI(3,4)P2 vesicles at the back of cells. Yellow arrow points to some PI(3,4)P2 vesicles at the back of the HL‐60 cell. Front and back of the cell are shown.
Next, we looked for PI(3,4)P2‐enriched vesicles using the cPHX3TAPP1‐GFP biosensor in HL‐60 cells. We observed an intracellular pool of PI(3,4)P2‐enriched vesicles clustered toward the back of the neutrophils (Fig EV1F and G). Some of these vesicles appeared very close to the membrane at the back and then disappear, suggesting that they might be fusing at the back of the membrane.
Discussion
Our studies can be summarized in the model shown in Fig 6E–G. Our previous study demonstrated the mutual inhibition between Ras activity and PI(3,4)P2 establishes polarity even in immobilized latrunculin A‐treated cells in the presence of a chemoattractant gradient or caffeine (Fig 6E; Arai et al, 2010; Wang et al, 2013; Li et al, 2018). We now propose an additional mechanism involving a specific vesicle recycling path which sharpens the back‐to‐front gradient in migrating Dictyostelium cells. PI(3,4)P2 disappears from protrusions at the leading edges of macropinocytic cups, and then accumulates on the macropinosomes at the end of the internalization process. The macropinosomes are processed into smaller satellite PI(3,4)P2‐tagged vesicles which then dock at the back. The PI(3,4)P2 molecules incorporated at the back diffuse along the membrane toward the front, where they are degraded (Fig 6F). In more polarized cells, owing to a slower arrival time of PI(3,4)P2 vesicles and a slower decay rate of PI(3,4)P2 on the membrane, the PI(3,4)P2 signal at the back broadens and extends further to the front (Fig 6G). Our results suggest that a similar mechanism could exist in polarized migrating mammalian cells.
Excitable network hypothesis and the formation of macropinocytic cups
This model is consistent with the excitable network hypothesis, which has been proposed to explain the behaviors of migrating cells (Li et al, 2020). According to this hypothesis, propagating waves have an active region where front molecules are high and back molecules are low. This region is followed by a refractory region, where front molecules are very low and back components strongly accumulate (Xiong et al, 2010; Huang et al, 2013; Tang et al, 2014; Li et al, 2018). Here, our findings suggest macropinosomes are formed by a spreading wave whose trailing edge forms the base of the macropinocytic cups. This suggests that the base of the macropinocytic cups would be in a refractory state, which is heavily decorated by the back molecule PI(3,4)P2. If PI(3,4)P2 is delivered to the back of the cell as we propose, it would shut off this region and further polarize the cell.
Many other signaling molecules such as PTEN, Myosin II, IQGAP1 and 2, cortexillin I and II, RasGAP2 (RG2) and RapGAP3 (RG3) localize to the back of the cells (Ramalingam et al, 2015; Li et al, 2018, 2020). Among those molecules, PTEN, RG2, RG3, and Myosin II are also reported to localize to the base of the cups or retracting protrusions. In addition, RG2 and RG3 bind to PI(3,4)P2 in vitro. Our results raise the possibility that the cells might be bringing the refractory state carrying many of those molecules from front to back.
Regulation of vesicle recycling controls polarity
In the reverse‐fountain flow model, we were proposing that vesicular trafficking plays an important role in polarity. Yet, the vesicle recycling mechanism we described is more obvious in less polarized cells. How can we explain this apparent discrepancy? Highly polarized cells are generally characterized by their typical elongated morphology, which is a result of having a larger proportion of the cell with elevated backmarkers. In fact, our simulations showed that the relatively longer arrival time of the vesicles and the slower decay rate increased the back region. Subsequently, experiment measurements did show a slower decay rate in highly polarized cells. Thus, both experimental data and simulation results are consistent in supporting the reverse‐fountain flow model (Fig 6 and Appendix Fig S6).
PI(3,4)P2 is a distinct signaling component during cell migration
Our observations are consistent with an emerging view that PI(3,4)P2 is a signaling molecule on its own right, regulating cellular events (Li & Marshall, 2015). Numerous studies have shown PI(3,4)P2 localize to the leading/lagging edge during cell migration, early endosomes, and lysosomes during clathrin‐mediated endocytosis, and back of macropinocytic cups and early macropinosomes. This PI(3,4)P2 localization dynamics regulate all of these processes and there appears to be a consistent pattern of PI(3,4)P2 localization involved in protrusion formation during cell migration and other aforementioned cellular events (Zhou et al, 1998; Hasegawa et al, 2011; Posor et al, 2013; Maekawa et al, 2014; Valenzuela‐Iglesias et al, 2015; Wang et al, 2018; Li et al, 2020).
In some studies of cell motility and cytoskeletal events, PI(3,4)P2 acts as a negative regulator (Lam et al, 2012; Ghosh et al, 2018). Huttenlocher’s group reported that Src‐homology 2‐containing inositol 5′ phosphatase (SHIP) limits the motility of neutrophils and their recruitment to wounds in live zebrafish (Yoo et al, 2010). Observations by Wu showed that PI(3,4)P2 defines the refractory period of the oscillation in cortical waves in mast cells (Xiong et al, 2016; Yang et al, 2017). Our lab reported that PI(3,4)P2 negatively regulates cell motility by inhibiting Ras activity, even in the absence of PIP3.
Our results depart from the canonical view that PI(3,4)P2 is only a byproduct of PIP3 hydrolysis (Czech, 2000). We demonstrated in mammalian neutrophils and in Dictyostelium cells that the level of PI(3,4)P2 was maintained on the plasma membrane under PI3K inhibition (Fig 3A–E). Additionally, in PI3K1 − 2 − Dictyostelium cells, PI(3,4)P2 still accumulated on the membrane at the back of the cells and the macropinosomes (Fig 3F–I). This suggests that a fraction of PI(3,4)P2 must come from a source other than PIP3 and there is regulation of enzymes that generate PI(3,4)P2 from PI3P or PI4P. As there are multiple phosphoinositide kinases in Dictyostelium that have not been characterized, it is possible that one of these corresponds to a novel PI3K.
Endocytic trafficking of PI(3,4)P2 as a universal means of establishing polarity
Our current findings reveal that the PI(3,4)P2 decorated macropinosomes and its connection with PI(3,4)P2 gradient at the rear of the plasma membrane in regulating polarity during cell migration. Consistently, there are reports suggesting recycling of PI(3,4)P2‐tagged vesicles and other endocytic events play an important role in polarity of epithelial cells. A study by the Bryant group recently described the function of PI(3,4)P2 in apical domain morphogenesis in MDCK cysts. Apical PI(3,4)P2 is supplied by the endosomal pool, rather than conversion from basolateral PIP3 by SHIP1 and is a determinant of apical membrane identity (Roman‐Fernandez et al, 2018). These observations reinforce the parallels between basolateral and apical surfaces of epithelial cells and front and back of migrating cells (Martin‐Belmonte et al, 2007; Nelson, 2009; Bryant et al, 2014). Another parallel could be the establishment of polarity during cytokinesis, where PI(3,4)P2 is found elevated in the cleavage furrow (Li et al, 2020). The potential role of vesicular trafficking in cytokinesis has not been investigated.
Reverse‐fountain flow model of PI(3,4)P2
During migration, the additional membrane required to enable the expending protrusions could come from unfolding of invaginations of the membrane or from vesicular trafficking.
Multiple different studies have suggested that cells migrate following a fountain flow model: Membrane precursor vesicles fuse with the anterior cell membrane at the protrusions, both the dorsal and ventral membranes flow toward the rear, and membrane is internalized at the rear (Lee et al, 1990; Tanaka et al, 2017). In support of the fountain flow models, it has been shown that blocking of vesicular trafficking is required for movement and that particles sticking on the outside of the cells as well as photobleached membrane patches flow from the front to the back.
In our reverse‐fountain flow model, PI(3,4)P2‐enriched vesicles are taken from macropinosomes at the anterior protrusion and eventually fuse to the membrane at the rear. Furthermore, plasma membrane PI(3,4)P2 at the rear diffuses toward the front. While this reverse‐fountain flow model of PI(3,4)P2 sheds light on the role of this phospholipid in regulating polarity during cell migration, further study is needed to determine the direction of flow of other membrane components. One study showed that photoactivation of cAR1 did move toward the front, which support the reverse‐fountain flow model (Traynor & Kay, 2007). In addition, vesicles containing adenylyl cyclase fuse with the back of Dictyostelium cells and vesicles carrying growth factor receptor fuse at the back of cells (Kriebel et al, 2008; Zoncu et al, 2009). A recent study by Moreau et al (2019) showed that migrating immature dendritic cells form macropinosomes at their leading edge which traverse the cytoplasm and ultimately release their fluid content at the back. This agrees with our model. Membrane folding, fountain flow model, or reverse‐fountain flow models may operate in specific cells during different migratory modes.
Materials and Methods
Reagents and Tools Table
| Reagent/Resource | Reference or Source | Identifier or catalog number |
|---|---|---|
| Experimental models | ||
| Chemically Competent Escherichia coli | Thermo Fisher | C404010 |
| Wild‐type Dictyostelium discoideum cells of AX3 | Devreotes Lab | N/A |
| Wild‐type Dictyostelium discoideum cells of AX2 | Devreotes Lab | N/A |
| Neutrophil‐like HL‐60 cells | Orion Weiner lab | N/A |
| RacH‐ strain | Miho Iijima lab | N/A |
| Recombinant DNA | ||
| LimEΔcoil‐RFP | Devreotes Lab | N/A |
| PH‐Crac‐RFP | Devreotes Lab | N/A |
| tPH‐CynA‐KikGR | Devreotes Lab | N/A |
| cPH‐TAPP1‐RFP | Devreotes Lab | N/A |
| cPHx3TAPP1‐GFP in pHR_SFFV | This study | N/A |
| PHCynA‐HALO | This study | N/A |
| CAR1‐KikGR | Masahiro Ueda lab | N/A |
| pHR_SFFV | Addgene | Cat # 79121 |
| RFP‐LifeAct | Desiderio Lab | N/A |
| Antibodies | ||
| Mouse anti α‐Tubulin monoclonal antibody | Sigma‐Aldrich | Cat #T9026 |
| Oligonucleotides and other sequence‐based reagents | ||
| PCR primers of HALO‐tagged PHCynA | This study | In Materials and Methods |
| Chemicals, enzymes, and other reagents | ||
| RPMI 1640 Medium, HEPES | Thermo Fisher Scientific | # 22400089 |
| Heat‐inactivated fetal bovine serum | Gibco | # 16140071 |
| Penicillin–Streptomycin | Sigma | # P0781‐100ML |
| Fibronectin | Millipore Sigma | # F2006 |
| Eight‐well coverslip chamber slide | Lab‐Tek, Thermo Scientific. | #155411 |
| FM4‐64 dye | Thermo Fisher Scientific | # T3166 |
| CELLSTAR T25 Flask | Greiner | # 82051‐074 |
| DMSO | Thermo Fisher Scientific | #D128‐1 |
| Polybrene | Sigma | # TR‐1003‐G |
| Hygromycin B | Thermo Fisher Scientific | # 10687010 |
| HEPES | Thermo Fisher Scientific | # 22400089 |
| Lipofectamine® 3000 | Invitrogen | #L3000008 |
| Software | ||
| Fiji | https://imagej.net/Fiji/Downloads | |
| MATLAB | https://www.mathworks.com/products/matlab.html | |
| GraphPad Prism 5.0. | https://www.graphpad.com/ | |
| Other | ||
| LSM780‐FCS/Quasar confocal microscope | Zeiss | N/A |
Methods and Protocols
Cell culture and differentiation
Wild‐type Dictyostelium discoideum cells of AX3 and AX2 strains were used in this study. These strains are established lab cell lines because they can form fruiting bodies upon starvation and their morphology and behavior can be easily examined during chemotactic assays. RacH‐ strain, generated in AX2 background, was provided by Dr. Miho Iijima (JHU) and generated as described previously (Somesh et al, 2006). All of these strains were cultured axenically in HL5 medium at 22°C, either in suspension for obtaining high cell densities for cell differentiation or on tissue culture dishes for cell line maintenance. Cells expressing LimEΔcoil‐RFP, PH‐Crac‐RFP, cPHx3‐TAPP1‐GFP, tPH‐CynA‐KikGR, PHCynA‐HALO, or CAR1‐KikGR constructs were generated by electroporation of the appropriate plasmids in wild‐type or mutant cells. Transformed cells were selected and maintained in HL5 medium containing 20 μg/ml G418, 50 μg/ml hygromycin B, or both. For microscopy, cells were plated in DB buffer to reduce HL5 medium‐associated autofluorescence and photosensitivity of growing cells. This enabled prolonged and frequent imaging of cells.
Neutrophil‐like HL‐60 cells, used in this study, were procured from Dr. Orion Weiner (UCSF). Cells were grown in RPMI 1640 medium containing HEPES (Thermo Fisher Scientific # 22400089) and heat‐inactivated fetal bovine serum (10% v/v; Gibco). Cultures were maintained at a density of 0.1–1.0 million cells/ml at 37°C/5% CO2. Differentiated HL‐60s were obtained by adding 1.5% (vol/vol) DMSO (Sigma‐Aldrich) to actively growing cells (at a density of 0.3 million cells/ml) followed by incubation for an additional 5–6 days (Millius & Weiner, 2009).
Human embryonic kidney 293 (HEK293T) cells were used to generate lentivirus to perform lentivirus transduction in HL‐60 cells. HEK293T cells were grown in DMEM (MediaTek) containing 10% (vol/vol) heat‐inactivated fetal bovine serum and maintained at 37°C/5% CO2.
Plasmid construction
cPHx3TAPP1‐GFP and pPamCherry1‐C1‐TAPP1‐cPHx3 were provided by Gerald R.V. Hammond (Pitt) and Brady D. Goulden (Goulden et al, 2019). It was subsequently cloned into pDM358 vector or SFFV vector. RFP‐LifeAct was obtained from Stephen Desiderio Lab (JHU) and was subsequently cloned into SFFV vector (Addgene Cat # 79121). LimEΔcoil‐RFP, cPH‐TAPP1‐RFP, PH‐Crac‐RFP, and tPH‐CynA‐KikGR constructs were created in previous studies (Li et al, 2018). C‐terminal HALO‐tagged PHCynA construct was generated by cloning the PHCynA ORF from tPH‐CynA‐KikGRk‐KF2 construct using the primers 5′ Fwd; TATAAGATCTAAAAAAATGAAATCCTCAAATGGTGTCGGTTGTAC and 3′ Rev; TATAACTAGTTGAAATTGGTTTTGGCGGAAGAGGAG into BglII/SpeI sites of pHK12neo plasmid.
Lentivirus transduction of HL‐60 cells
HEK293T cells were seeded into six‐well plates and grown until 80% confluent. For each well, 5 μg cPHx3TAPP1‐GFP in SFFV vector, 1.25 μg VSVG, and 3.5 μg cytomegalovirus 8.91 vector were mixed and prepared for transfection using Lipofectamine® 3000 following manufacturer’s instructions. After transfection, cells were grown for an additional 1–3 days, after which virus‐containing supernatants were harvested. 50–200 μl virus was mixed with 1 million HL‐60 cells in growth medium supplemented with polybrene (8 μg/ml), and incubated overnight. HL‐60 cells expressing cPHx3TAPP1‐GFP or RFP‐LifeAct were selected by FACS after 5 days.
Microscopy
Growth‐phase Dictyostelium or differentiated HL‐60 cells were placed on uncoated or fibronectin‐coated eight‐well coverslip chamber slide (Lab‐Tek, Thermo Scientific, Nunc). Confocal imaging was performed with the help of a Zeiss LSM780 single‐point laser‐scanning microscope. Time‐lapse images were obtained with DIC, GFP, or RFP illumination at 3‐ to 25‐s intervals for 20–180 min.
Confocal microscopy with FM4‐64. Dictyostelium cells were pretreated with 0.1 M sorbitol in PB to reduce formation of contractile vacuoles, which tend to get extensively labeled with FM4‐64 dye (Millius & Weiner, 2010). 2 μM FM4‐64 was added to cells during the live cell imaging.
Confocal microscopy with Photoconversion. Cells expressing tPH‐CynA‐KikGR were visualized using a 488 nm Argon laser and a 40× objective. For photoconversion of KikGR, a 405 nm diode laser was used with 1 × 5 iterations. Furthermore, a region of interest was moved to the center of the viewing field, and the field diaphragm was closed to its smallest opening to make a photoconvertible spot using a 541‐nm laser for red fluorescence excitation. Imaging of both photoconverted and unconverted KikGR was done using 488‐nm Ag laser for green fluorescence and 541‐nm laser for red fluorescence in multi‐track mode.
Fluorescence recovery after photobleaching (FRAP). To minimize the contribution of diffusion from above and below the bleach box, the box was expanded to include a portion of the top and bottom membranes. A section of the cell periphery in zoom was exposed to two iterations of saturating 488‐nm laser light causing a partial bleach.
PHCynA‐HALO labeling and imaging using HaloTag® TMR Ligand. PH‐CynA‐Halo expressing Dictyostelium cells were harvested, washed in DB, and incubated with 3 µM TMR‐conjugated HaloTag ligand (Promega) for 30 min at RT. Excess stain was washed away using DB, and cells were imaged using the Zeiss LSM780 confocal microscope.
Cell fixation. Growth‐stage Dictyostelium cells were allowed to attach to cover‐glass for 30 min to 2 h in an eight‐well chamber slide. Cells were incubated with freshly made fixative (2% buffered paraformaldehyde, 0.25% glutaraldehyde, and 0.1% Triton X‐100 in HL5) for 10 min at room temperature, and subsequently quenched in 1 mg/ml sodium borohydride for 3 min. Cells were then washed twice with TBS (supplemented with 0.25% BSA and 0.05% Triton X‐100) and visualized using the Zeiss LSM780 confocal microscope. In each experiment, images from different samples were taken consecutively using identical settings.
Cell fusion by electroporation
Growth‐phase tPH‐CynA‐KikGR expressing Dictyostelium cells were grown in suspension, harvested, washed, and resuspended in SB (17 mM Soerensen buffer containing 15 mM KH2PO4 and 2 mM Na2HPO4, pH 6.0) at a density of 15 million cells/ml. 10 ml of cells was rolled gently for 20–45 min. 800 μl of rolled cells was transferred to a 4‐mm‐gap electroporation cuvette. Electroporation was carried out with the following settings: 1,000 V, 3 μF for once, and 1,000 V, 1 μF for three times, with 1–2 s between pulses. 50 μl of cells was then transferred to the center of a well in an 8‐well chamber and was allowed to adhere for 10 min. 450 μl of SB (containing 2 mM CaCl2 and 2 mM MgCl2) was added to the well and was pipetted gently to resuspend the cells evenly. Unadhered cells were removed by washing with 450 μl of fresh SB, after which the attached cells were incubated for 1 h for recovery before imaging (Miao et al, 2019).
Computational study
In this present study, we modeled the cell membrane as a 1D spatial domain of length 40 μm with periodic boundary condition. The arrival of the satellite vesicles on the membrane was modeled as a discrete Poisson process with a variable mean arrival time—(i) for the vegetative cell: = 10 s, (b) for the developed cell: = 40 s. We assumed two separate pools of PI(3,4)P2 molecules: PI(3,4)P2 on the docked vesicle (U) and on the membrane (V). For the purpose of modeling, we simplified the process of vesicle fusion as follows:
At a randomly chosen time instant ta (ta ~ chosen with an exponential distribution with mean arrival time λ), n(n ~ Normal distribution) molecules of U per unit length (i.e. n × r total molecules where r is the radius of the vesicle) appeared on the membrane centered around a docking site, xd. The docking site, xd, is randomly selected sampled either from a probability distribution with the cumulative density function, where is the value of V at spatial index k (directed insertion case) or from a discrete uniform distribution (random insertion case).
During the docking time interval, , U remained unchanged.
After , U converted to V with a linear conversion rate expression . V diffuses freely on the membrane with diffusion constant, .
V is also subjected to a degradation with reaction rate expression where
The first part of the decay function denotes a spatially independent basal decay rate, and the second part represents the effect of spatially variable decay rate as observed experimentally (the skewed distribution of the photoconverted red fluorescent profile validated this assumption; see Fig 1D and G).
The stochastic simulations of the model were performed using URDME (Drawert et al, 2012) approach using custom code written in MATLAB 2019b (MathWorks, Natick, MA, USA). The simulation domain was considered as a 1D domain of length 40 μm with spatial grid resolution of 0.1 μm. The completely unpolarized cell membrane ((U,V) = (0,0)) was taken as the initial condition. The code and the simulation data for Fig 6 A,B are in Code EV1.
The nominal values of the parameters along with the respective standard deviations are listed in Appendix Table S1.
For the estimation of the diffusion constant in Fig 1D (see Appendix Fig S1A), a simple diffusion model was simulated using the URDME as explained above using the converted red fluorescence profile at 0 s as the initial condition. The optimum diffusion coefficient was obtained by minimizing the following cost function:
where = {0, 60s, 90s}.
Quantification and statistical analysis
Image analysis
All images were processed using Fiji (like ImageJ) (National Institutes of Health) and MATLAB. The mean intensity of the membrane portion and cytosol portion was measured, and the ratio was calculated using ImageJ as described previously (Li et al, 2018). Cells were segmented by a multistep process using commands from the Image Processing Toolbox in MATLAB. Colors were assigned linearly in the same fashion across all kymographs, with blue indicating the lowest intensity and red the highest intensity. Biosensor patches were defined regions of fluorescent intensity that are twofold over background.
Statistical analysis
Mean values and standard deviations/standard error of the mean were calculated and used to assess differences between two groups by GraphPad Prism. P‐values ≤ 0.05 were considered statistically significant, throughout this study. Images presented in figures are representative of more than three independent experiments.
Author contributions
XL performed experiments in Figs 1, 2, 3, 4, 5, 6 and Fig EV1, and Appendix Figs S1–S3, S5, S7, and S8; DSP performed experiments in Fig 4G–M, Appendix Fig S4 and generated constructs/strains for Fig 1K; DB and PAI conducted computational simulations and quantification in Fig 6A–D, Appendix Figs S1 and S6. XL, DSP, DB, PAI, and PND analyzed the data and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflicts of interest.
Supporting information
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Movie EV6
Movie EV7
Movie EV8
Code EV1
Source Data for Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Acknowledgements
The authors would like to thank all members of the Devreotes and Iglesias labs for helpful suggestions for this work. We thank the Johns Hopkins University Microscope Facility for confocal microscopy. We thank Dr. Carolyn Machamer, Dr. Geraldine Seydoux, Dr. Erin Goley, Dr. Shigeki Watanabe, and Dr. Doug Robinson for helpful suggestions. We thank Jane Borleis for providing PI3K1 − 2 − cells. We thank Dr. Masahiro Ueda lab for PTEN‐Halo and CAR1‐KikGR constructs. We thank Dr. Stephen Desiderio Lab for RFP‐LifeAct construct. We thank Dr. Gerald R.V. Hammond and Brady D. Goulden for providing cPHx3TAPP1 construct. We thank Dr. Miho Iijima for generating and providing RacH‐ cells. We thank Dr. Orion Weiner lab for providing HL‐60 cell line. This work was supported by This work was supported by National Institutes of Health grant R35 GM118177 (to P.N.D.), AFOSR MURI FA95501610052, DARPA HR0011‐16‐C‐0139, as well as NIH Grant S10 OD016374 (to S. Kuo of the JHU Microscope Facility).
The EMBO Journal (2021) 40: e105094.
Contributor Information
Xiaoguang Li, Email: xli82@jhu.edu.
Peter N Devreotes, Email: pnd@jhmi.edu.
Data availability
This study includes no data deposited in external repositories.
References
- Andrew N, Insall RH (2007) Chemotaxis in shallow gradients is mediated independently of PtdIns 3‐kinase by biased choices between random protrusions. Nat Cell Biol 9: 193–200 [DOI] [PubMed] [Google Scholar]
- Arai Y, Shibata T, Matsuoka S, Sato MJ, Yanagida T, Ueda M (2010) Self‐organization of the phosphatidylinositol lipids signaling system for random cell migration. Proc Natl Acad Sci USA 107: 12399–12404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balest A, Peracino B, Bozzaro S (2011) Legionella pneumophila infection is enhanced in a RacH‐null mutant of Dictyostelium . Commun Integr Biol 4: 194–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretscher MS (1996) Getting membrane flow and the cytoskeleton to cooperate in moving cells. Cell 87: 601–606 [DOI] [PubMed] [Google Scholar]
- Bryant DM, Roignot J, Datta A, Overeem AW, Kim M, Yu W, Peng X, Eastburn DJ, Ewald AJ, Werb Z et al (2014) A molecular switch for the orientation of epithelial cell polarization. Dev Cell 31: 171–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czech MP (2000) PIP2 and PIP3: complex roles at the cell surface. Cell 100: 603–606 [DOI] [PubMed] [Google Scholar]
- Devreotes PN, Bhattacharya S, Edwards M, Iglesias PA, Lampert T, Miao Y (2017) Excitable signal transduction networks in directed cell migration. Annu Rev Cell Dev Biol 33: 103–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dormann D, Weijer G, Dowler S, Weijer CJ (2004) In vivo analysis of 3‐phosphoinositide dynamics during Dictyostelium phagocytosis and chemotaxis. J Cell Sci 117: 6497–6509 [DOI] [PubMed] [Google Scholar]
- Drawert B, Engblom S, Hellander A (2012) URDME: a modular framework for stochastic simulation of reaction‐transport processes in complex geometries. BMC Syst Biol 6: 76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhardt M, Ecke M, Walz M, Stengl A, Beta C, Gerisch G (2014) Actin and PIP3 waves in giant cells reveal the inherent length scale of an excited state. J Cell Sci 127: 4507–4517 [DOI] [PubMed] [Google Scholar]
- Gerisch G, Ecke M, Neujahr R, Prassler J, Stengl A, Hoffmann M, Schwarz US, Neumann E (2013) Membrane and actin reorganization in electropulse‐induced cell fusion. J Cell Sci 126: 2069–2078 [DOI] [PubMed] [Google Scholar]
- Ghosh S, Scozzaro S, Ramos AR, Delcambre S, Chevalier C, Krejci P, Erneux C (2018) Inhibition of SHIP2 activity inhibits cell migration and could prevent metastasis in breast cancer cells. J Cell Sci 131: jcs216408 [DOI] [PubMed] [Google Scholar]
- Goulden BD, Pacheco J, Dull A, Zewe JP, Deiters A, Hammond GRV (2019) A high‐avidity biosensor reveals plasma membrane PI(3,4)P2 is predominantly a class I PI3K signaling product. J Cell Biol 218: 1066–1079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa J, Tokuda E, Tenno T, Tsujita K, Sawai H, Hiroaki H, Takenawa T, Itoh T (2011) SH3YL1 regulates dorsal ruffle formation by a novel phosphoinositide‐binding domain. J Cell Biol 193: 901–916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins PT, Stephens LR (2016) Emerging evidence of signalling roles for PI(3,4)P2 in Class I and II PI3K‐regulated pathways. Biochem Soc Trans 44: 307–314 [DOI] [PubMed] [Google Scholar]
- Huang CH, Tang M, Shi C, Iglesias PA, Devreotes PN (2013) An excitable signal integrator couples to an idling cytoskeletal oscillator to drive cell migration. Nat Cell Biol 15: 1307–1316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iijima M, Devreotes P (2002) Tumor suppressor PTEN mediates sensing of chemoattractant gradients. Cell 109: 599–610 [DOI] [PubMed] [Google Scholar]
- Janetopoulos C, Ma L, Devreotes PN, Iglesias PA (2004) Chemoattractant‐induced phosphatidylinositol 3,4,5‐trisphosphate accumulation is spatially amplified and adapts, independent of the actin cytoskeleton. Proc Natl Acad Sci USA 101: 8951–8956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay RR, Williams TD, Paschke P (2018) Amplification of PIP3 signalling by macropinocytic cups. Biochem J 475: 643–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriebel PW, Barr VA, Rericha EC, Zhang G, Parent CA (2008) Collective cell migration requires vesicular trafficking for chemoattractant delivery at the trailing edge. J Cell Biol 183: 949–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam PY, Yoo SK, Green JM, Huttenlocher A (2012) The SH2‐domain‐containing inositol 5‐phosphatase (SHIP) limits the motility of neutrophils and their recruitment to wounds in zebrafish. J Cell Sci 125: 4973–4978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Gustafsson M, Magnusson KE, Jacobson K (1990) The direction of membrane lipid flow in locomoting polymorphonuclear leukocytes. Science 247: 1229–1233 [DOI] [PubMed] [Google Scholar]
- Li H, Marshall AJ (2015) Phosphatidylinositol (3,4) bisphosphate‐specific phosphatases and effector proteins: a distinct branch of PI3K signaling. Cell Signal 27: 1789–1798 [DOI] [PubMed] [Google Scholar]
- Li X, Edwards M, Swaney KF, Singh N, Bhattacharya S, Borleis J, Long Y, Iglesias PA, Chen J, Devreotes PN (2018) Mutually inhibitory Ras‐PI(3,4)P2 feedback loops mediate cell migration. Proc Natl Acad Sci USA 115: E9125–E9134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Miao Y, Pal DS, Devreotes PN (2020) Excitable networks controlling cell migration during development and disease. Semin Cell Dev Biol 100: 133–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo HR, Huang YE, Chen JC, Saiardi A, Iijima M, Ye K, Huang Y, Nagata E, Devreotes P, Snyder SH (2003) Inositol pyrophosphates mediate chemotaxis in Dictyostelium via pleckstrin homology domain‐PtdIns(3,4,5)P3 interactions. Cell 114: 559–572 [DOI] [PubMed] [Google Scholar]
- Maekawa M, Terasaka S, Mochizuki Y, Kawai K, Ikeda Y, Araki N, Skolnik EY, Taguchi T, Arai H (2014) Sequential breakdown of 3‐phosphorylated phosphoinositides is essential for the completion of macropinocytosis. Proc Natl Acad Sci USA 111: E978–E987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin‐Belmonte F, Gassama A, Datta A, Yu W, Rescher U, Gerke V, Mostov K (2007) PTEN‐mediated apical segregation of phosphoinositides controls epithelial morphogenesis through Cdc42. Cell 128: 383–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao Y, Bhattacharya S, Banerjee T, Abubaker‐Sharif B, Long Y, Inoue T, Iglesias PA, Devreotes PN (2019) Wave patterns organize cellular protrusions and control cortical dynamics. Mol Syst Biol 15: e8585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millius A, Weiner OD (2009) Chemotaxis in neutrophil‐like HL‐60 cells. Methods Mol Biol 571: 167–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millius A, Weiner OD (2010) Manipulation of neutrophil‐like HL‐60 cells for the study of directed cell migration. Methods Mol Biol 591: 147–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau HD, Blanch‐Mercader C, Attia R, Maurin M, Alraies Z, Sanseau D, Malbec O, Delgado MG, Bousso P, Joanny JF et al (2019) Macropinocytosis overcomes directional bias in dendritic cells due to hydraulic resistance and facilitates space exploration. Dev Cell 49: 171–188.e5 [DOI] [PubMed] [Google Scholar]
- Nelson WJ (2009) Remodeling epithelial cell organization: transitions between front‐rear and apical‐basal polarity. Cold Spring Harb Perspect Biol 1: a000513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill PR, Castillo‐Badillo JA, Meshik X, Kalyanaraman V, Melgarejo K, Gautam N (2018) Membrane flow drives an adhesion‐independent amoeboid cell migration mode. Dev Cell 46: 9–22.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal DS, Li X, Banerjee T, Miao Y, Devreotes PN (2019) The excitable signal transduction networks: movers and shapers of eukaryotic cell migration. Int J Dev Biol 63: 407–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent CA, Blacklock BJ, Froehlich WM, Murphy DB, Devreotes PN (1998) G protein signaling events are activated at the leading edge of chemotactic cells. Cell 95: 81–91 [DOI] [PubMed] [Google Scholar]
- Parent CA (2004) Making all the right moves: chemotaxis in neutrophils and Dictyostelium . Curr Opin Cell Biol 16: 4–13 [DOI] [PubMed] [Google Scholar]
- Posor Y, Eichhorn‐Gruenig M, Puchkov D, Schoneberg J, Ullrich A, Lampe A, Muller R, Zarbakhsh S, Gulluni F, Hirsch E et al (2013) Spatiotemporal control of endocytosis by phosphatidylinositol‐3,4‐bisphosphate. Nature 499: 233–237 [DOI] [PubMed] [Google Scholar]
- Postma M, Roelofs J, Goedhart J, Loovers HM, Visser AJ, Van Haastert PJ (2004) Sensitization of Dictyostelium chemotaxis by phosphoinositide‐3‐kinase‐mediated self‐organizing signalling patches. J Cell Sci 117: 2925–2935 [DOI] [PubMed] [Google Scholar]
- Ramalingam N, Franke C, Jaschinski E, Winterhoff M, Lu Y, Bruhmann S, Junemann A, Meier H, Noegel AA, Weber I et al (2015) A resilient formin‐derived cortical actin meshwork in the rear drives actomyosin‐based motility in 2D confinement. Nat Commun 6: 8496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman‐Fernandez A, Roignot J, Sandilands E, Nacke M, Mansour MA, McGarry L, Shanks E, Mostov KE, Bryant DM (2018) The phospholipid PI(3,4)P2 is an apical identity determinant. Nat Commun 9: 5041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki AT, Chun C, Takeda K, Firtel RA (2004) Localized Ras signaling at the leading edge regulates PI3K, cell polarity, and directional cell movement. J Cell Biol 167: 505–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shellard A, Szabo A, Trepat X, Mayor R (2018) Supracellular contraction at the rear of neural crest cell groups drives collective chemotaxis. Science 362: 339–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somesh BP, Neffgen C, Iijima M, Devreotes P, Rivero F (2006) Dictyostelium RacH regulates endocytic vesicular trafficking and is required for localization of vacuolin. Traffic 7: 1194–1212 [DOI] [PubMed] [Google Scholar]
- Stuelten CH, Parent CA, Montell DJ (2018) Cell motility in cancer invasion and metastasis: insights from simple model organisms. Nat Rev Cancer 18: 296–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M, Kikuchi T, Uno H, Okita K, Kitanishi‐Yumura T, Yumura S (2017) Turnover and flow of the cell membrane for cell migration. Sci Rep 7: 12970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang M, Wang M, Shi C, Iglesias PA, Devreotes PN, Huang CH (2014) Evolutionarily conserved coupling of adaptive and excitable networks mediates eukaryotic chemotaxis. Nat Commun 5: 5175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynor D, Kay RR (2007) Possible roles of the endocytic cycle in cell motility. J Cell Sci 120: 2318–2327 [DOI] [PubMed] [Google Scholar]
- Valenzuela‐Iglesias A, Sharma VP, Beaty BT, Ding Z, Gutierrez‐Millan LE, Roy P, Condeelis JS, Bravo‐Cordero JJ (2015) Profilin1 regulates invadopodium maturation in human breast cancer cells. Eur J Cell Biol 94: 78–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veltman DM, Williams TD, Bloomfield G, Chen BC, Betzig E, Insall RH, Kay RR (2016) A plasma membrane template for macropinocytic cups. Elife 5: e20085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Senoo H, Sesaki H, Iijima M (2013) Rho GTPases orient directional sensing in chemotaxis. Proc Natl Acad Sci USA 110: E4723–E4732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Lo WT, Vujicic Zagar A, Gulluni F, Lehmann M, Scapozza L, Haucke V, Vadas O (2018) Autoregulation of class II alpha PI3K activity by its lipid‐binding PX‐C2 domain module. Mol Cell 71: 343–351.e4 [DOI] [PubMed] [Google Scholar]
- Weiner OD, Servant G, Welch MD, Mitchison TJ, Sedat JW, Bourne HR (1999) Spatial control of actin polymerization during neutrophil chemotaxis. Nat Cell Biol 1: 75–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welliver TP, Swanson JA (2012) A growth factor signaling cascade confined to circular ruffles in macrophages. Biol Open 1: 754–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y, Huang CH, Iglesias PA, Devreotes PN (2010) Cells navigate with a local‐excitation, global‐inhibition‐biased excitable network. Proc Natl Acad Sci USA 107: 17079–17086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong D, Xiao S, Guo S, Lin Q, Nakatsu F, Wu M (2016) Frequency and amplitude control of cortical oscillations by phosphoinositide waves. Nat Chem Biol 12: 159–166 [DOI] [PubMed] [Google Scholar]
- Yang Y, Xiong D, Pipathsouk A, Weiner OD, Wu M (2017) Clathrin assembly defines the onset and geometry of cortical patterning. Dev Cell 43: 507–521.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo SK, Deng Q, Cavnar PJ, Wu YI, Hahn KM, Huttenlocher A (2010) Differential regulation of protrusion and polarity by PI3K during neutrophil motility in live zebrafish. Dev Cell 18: 226–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida S, Hoppe AD, Araki N, Swanson JA (2009) Sequential signaling in plasma‐membrane domains during macropinosome formation in macrophages. J Cell Sci 122: 3250–3261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou K, Pandol S, Bokoch G, Traynor‐Kaplan AE (1998) Disruption of Dictyostelium PI3K genes reduces [32P]phosphatidylinositol 3,4 bisphosphate and [32P]phosphatidylinositol trisphosphate levels, alters F‐actin distribution and impairs pinocytosis. J Cell Sci 111(Pt 2): 283–294 [DOI] [PubMed] [Google Scholar]
- Zoncu R, Perera RM, Balkin DM, Pirruccello M, Toomre D, De Camilli P (2009) A phosphoinositide switch controls the maturation and signaling properties of APPL endosomes. Cell 136: 1110–1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Movie EV6
Movie EV7
Movie EV8
Code EV1
Source Data for Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Data Availability Statement
This study includes no data deposited in external repositories.