

Abstract

Strong light-matter coupling provides a new strategy to manipulate the non-adiabatic dynamics of molecules by modifying potential energy surfaces. The vacuum field of nanocavities can couple strongly with the molecular degrees of freedom and form hybrid light-matter states, termed as polaritons or dressed states. The photochemistry of molecules possessing intrinsic conical intersections can be significantly altered by introducing cavity couplings to create new conical intersections or avoided crossings. Here, we explore the effects of optical cavities on the photo-induced hydrogen elimination reaction of pyrrole. Wave packet dynamics simulations have been performed on the two-state, two-mode model of pyrrole, combined with the cavity photon mode. Our results show how the optical cavities assist in controlling the photostability of pyrrole and influence the reaction mechanism by providing alternative dissociation pathways. The cavity effects have been found to be intensely dependent on the resonance frequency. We further demonstrate the importance of the vibrational cavity couplings and dipole-self interaction terms in describing the cavity-modified non-adiabatic dynamics.
Introduction
Photochemistry involves the interaction of matter with light and plays an important role in synthetic chemistry, biology, and material sciences.1−3 Photochemical reactions are crucial in processes such as photosynthesis,4 vision,5 and storage of solar energy,6 but they can also possess detrimental effects such as DNA damage7 and modification of the efficiency of solar cells.8 Therefore, photochemical reactions need to be either accelerated or suppressed depending on their applications. These manipulations can be achieved by chemical modifications and classical laser fields.9−12 In recent times, strong light-matter coupling introduced by the optical cavities has evolved as a new tool to control the photochemical processes.13−17 The light-matter interaction can be considered to be strong when the energy exchange rate between matter and the cavity mode outpaces all incoherent decay processes. This strong cavity coupling results in the formation of hybrid field-matter states called dressed states or polaritons and consequently modifies the energy landscape of the molecules involved in the chemical process.14−16,18 Tuning the resonance frequency of an optical nanocavity may in turn be used as a control knob for tuning the spectroscopic and dynamical properties of the molecular systems.19−31 Notable examples of their potential applications include modifying the branching ratio between multiple reaction pathways,32 controlling the photochemical reaction rates,23−25 and improving Raman signals.33 The field of polaritonic chemistry has also seen several theoretical developments including quantum optics and ab initio approaches to investigate and understand the cavity-modified dynamics.14,15,19,22,26,34−42
The cavity-modified photochemistry significantly differs from that of the bare molecule when entering a strong coupling regime. In the absence of a cavity, many of the photochemical reactions are governed by the conical intersections (CIs) through which the molecule can decay non-radiatively to a lower electronic state.43,44 The strong coupling enables mixing the cavity mode with the molecular degree of freedom and manipulates the potential energy surfaces (PESs) by creating additional CIs. These modified PESs correspond to the polaritonic states and determine the dynamics of the coupled cavity-molecular system.
The photochemistry of light-induced CIs is well explored27,45 and recent studies have started to investigate the phenomena of cavity-field induced CIs.13,14,46 Studies investigating the direct coupling of quantized light fields with the non-adiabatic dynamics of avoided crossings15 or intrinsic CIs16,47 are still limited. Previous studies have mainly considered polaritons formed by electronic states. In this work, we theoretically investigate the effect of the optical nanocavities on the non-adiabatic dynamics of the molecule possessing an intrinsic CI. Wave-packet dynamics calculations have been performed on the two-state, two-mode model of the pyrrole coupled to the cavity mode. In addition to the electronic–cavity interactions, we have also incorporated vibrational cavity couplings and dipole self-energy (DSE, which was found to be important in the ultrastrong cavity regime48) terms in the Hamiltonian used for the simulations. Our study involves exploring the influences of strong light-matter interaction on the photochemistry of pyrrole. The goal is to understand how an optical cavity controls the molecular photostability and affects the reaction mechanism. We have also attempted to assess the importance of vibrational cavity couplings and DSE terms in describing cavity-modified photochemistry.
Pyrrole and related compounds can be considered as interesting model systems for investigating the excited-state dynamics.49−56 The lowest-energy intense bands in the absorption spectrum of pyrrole appear around 6 eV.57,58 The assignment of these bands has become quite challenging due to the presence of a large number of electronic states in this energy range.59,60 Broad peaks in the UV absorption spectrum and small fluorescence quantum yields are also responsible for the ambiguity in assigning the spectral bands.59,60
Upon interacting with UV radiation, pyrrole undergoes dissociation along the NH bond. Based on previous theoretical studies, four lowest singlet excited states, 1A2(πσ*), 1B1(πσ*), 1B2(ππ*), and 1A1(ππ*), have been found to be involved in the photolysis reaction.55,61 Among these, ππ* states are the bright states, which correspond to the intense bands around 6 eV in the absorption spectrum. The molecule from the initially excited ππ* state relaxes to the πσ* state within an ultrafast time scale (≈20 fs).51 Because of the significant antibonding character of the σ* orbital, the latter state becomes repulsive in nature with respect to the NH bond.51 Consequently, the molecule dissociates and forms a conical intersection between the πσ* state and the ground state. In this paper, the photodissociation dynamics of pyrrole on the 1B1(πσ*), hereafter referred to as just πσ*, state has been considered for the study. Previous studies reveal NH stretching and out-of-plane bending modes as the tuning and the coupling mode coordinates for πσ*–S0 states, respectively.55 Thus, we here considered the two-state, two-mode model of pyrrole along with the cavity mode to unravel the influences of the strong light-matter interaction on the photo-dynamics of pyrrole. Nuclear wave-packet dynamics are used to simulate the coupled dynamics of electronic, vibrational, and photon mode by including all relevant cavity coupling terms—electronic, vibrational, and DSE—in the corresponding Hamiltonian. Since the primary objective here is to explore the cavity effects on the dynamics involving intrinsic CIs, we restricted ourselves to consider the ground and πσ* states, and their vibronic interactions to the other excited states have been ignored. It should be noted that these effects, which may influence the photodissociation dynamics of pyrrole, are not included in the current model.
Theory and Model
Hamiltonian
The Hamiltonian describing the system of a molecule considered in an optical cavity comprises the molecular Hamiltonian ĤM, the Hamiltonian of the cavity mode ĤC, and the molecule–cavity interaction term ĤI(15,62,63)
| 1 |
where we assume that the dressed states can be expressed in field-free molecular electronic states.64 The molecular Hamiltonian includes two electronic states in a diabatic basis and the nuclear degrees of freedom q = (q1,q2)T
| 2 |
where mi are the reduced masses of the respective nuclear coordinates qi. The operators σ = |g⟩⟨e| and σ† = |e⟩⟨g| annihilate and create an excitation in the electronic subspace, respectively. The ground and first excited-state PESs are Vgg(q) and Vee(q), respectively, and Ŝge(q) is the diabatic coupling. The quantized cavity mode is described by
| 3 |
where â(†) is the annihilation (creation) operator of the cavity photon with mode frequency ωc. The coupling between the photon field and the molecule in the dipole approximation is given as
 |
4 |
where gij are the cavity couplings given by the vacuum Rabi frequency
| 5 |
and ⟨μ̂2⟩ij = ⟨i|μ̂2|j⟩ is the electronic
matrix of the
squared dipole operator, which describes the influence of the DSE
interaction.64 Here 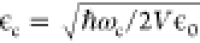 is the vacuum field amplitude of the cavity
mode with cavity mode volume V and μij are dipole functions for the permanent dipoles
(i = j) and transition dipole moments
(i ≠ j). The term gge is responsible for coupling the electronic
states, and the terms ggg and gee include the coupling of the vibrational levels
to the cavity.
is the vacuum field amplitude of the cavity
mode with cavity mode volume V and μij are dipole functions for the permanent dipoles
(i = j) and transition dipole moments
(i ≠ j). The term gge is responsible for coupling the electronic
states, and the terms ggg and gee include the coupling of the vibrational levels
to the cavity.
For a more convenient numerical treatment, we
use photon displacement coordinates,15,25 which are
obtained by expressing the corresponding ladder operators for the
cavity mode in terms of a pseudo spatial coordinate x̂ and a conjugate momentum  , where
, where
| 6 |
The Hamiltonian for the cavity mode then reads
| 7 |
and the light-matter coupling can be rewritten to read
 |
8 |
Note that in this form, the counter-rotating terms aσ and a†σ† from eq 4 are preserved.
In the present study, two nuclear coordinates corresponding to the NH bond are considered to be the active modes affecting the ground and the πσ* states of pyrrole (see below for more discussion). Consequently, the system under consideration becomes three-dimensional after adding the photon displacement coordinate x̂ as the third coordinate.
Potential Energy Surfaces
Based on the ab initio and quantum dynamics calculations, it is now well established that the πσ* state dynamics of pyrrole mainly involves NH stretching and out-of-plane bending modes.55,61 The former coordinate tunes the energy difference between the ground and the πσ* state, whereas the latter couples the two states. Consequently, these modes were referred to as the tuning and coupling modes, respectively. In the current work, we used Cartesian coordinate displacements for modeling the two-dimensional PESs. This choice simplifies the kinetic operator at the expense of grid points that are required to describe the nuclear wave packet. The tuning mode is considered to be the in-plane displacement of the hydrogen atom attached to the nitrogen atom, and the coupling mode is the vertical out-of-plane displacement (Figure 1a). These two coordinates are denoted as DIP (displacement in-plane) and DOP (displacement out-of-plane), respectively. Figure 1b shows the diabatic PESs of the ground and πσ* states as a function of DIP and DOP coordinates. It is observed that the two diabatic energy surfaces vary smoothly along the abovementioned displacement coordinates. There is a shallow well on the πσ* state from which the intersection between the ground and πσ* state around rNH = 2.1 Å is accessible by surmounting a small barrier.
Figure 1.

(a) Definition of tuning (DIP) and coupling (DOP) modes corresponding to S0–πσ* CI of pyrrole. (b) Two-dimensional diabatic PESs, highlighting the location of the curve crossing of ground (grey) and πσ* (red) states of pyrrole along DIP and DOP coordinates computed at the CASSCF(8,7)/aug-cc-pVDZ level of theory.
Our calculations predict the NH dissociation barrier on the πσ* state to be 0.19 eV. The vertical excitation energy from the ground state to the πσ* state is 4.9 eV. The computed dissociation limits for the S0 and πσ* states of pyrrole radical are 3.5 and 4.8 eV, respectively. These energetics are found to be lower than the values reported in the previous theoretical studies at more accurate levels of theory such as CASPT2 and MRCI.61 A maximum deviation of 0.8 eV has been observed in the case of vertical excitation energy. This underestimation of the energies with the CASSCF method is already pointed out by Domcke and co-workers in their theoretical study on the photochemistry of pyrrole.55 Furthermore, the energy ordering of the three states (S0, πσ* (1A2), and πσ* (1B1)) considered in our calculation and the location of the CI (rNH = 2.1 Å) are consistent with those in the earlier reports.54,55,61 Therefore, the computed PESs provide us a qualitative picture of the hydrogen detachment process in pyrrole.
Methods
Electronic Structure Methods
The reference geometry used to construct the PESs has been optimized at the DFT/B3LYP/aug-cc-pVDZ level of theory. Single point energies on the two-dimensional nuclear coordinate grid have been calculated using the 3-state-averaged CASSCF method along with the aug-cc-pVDZ basis set. The three states included in the state-averaging are S0, πσ* (1A2), and πσ* (1B1). A similar rigid scan methodology that does not consider the effect of the reaction coordinate displacement on the remaining molecular coordinates has also been applied successfully to describe the excited-state dynamics of pyrrole.55 The active space used for the CASSCF calculation consists of three π-orbitals, two π*-orbitals, and a pair of σ/σ*-orbitals corresponding to NH bond (Figure S1 of the Supporting Information). This active space can be denoted as CAS(8,7), which indicates the distribution of 8 electrons among 7 orbitals. All orbitals, including the core shells, were optimized in the CASSCF calculation. It is worth mentioning that the current active space yields small discontinuities around DOP ≈ −1.5 Å for DIP ≈ 0 to 1 Å (Figure 1b). However, this does not affect the dynamics because the wave packet has been observed to be never going beyond 1 Å along the DOP mode (more details in the results and discussion section). The other necessary electronic properties for the dynamics calculations are also obtained using the CASSCF method and are presented in the Supporting Information. These include diabatic couplings (Figure S2 of the Supporting Information), transition dipole moments (Figure S3 of the Supporting Information), and permanent dipole moments (Figures S4 and S5 of the Supporting Information). The diabatic PESs are used in time-dependent dynamics simulations. To obtain the diabatic states and the corresponding couplings, we employed the quasi-diabatization procedure of Simah et al. as implemented in MOLPRO-2019.65 The transformation matrix has been used to transform the remaining electronic properties such as permanent dipole moments and transition dipole moments from adiabatic to diabatic basis. All electronic structure calculations have been performed using the MOLPRO-2019 program package.66,67 The expectation value of the squared dipole operator ⟨μ̂2⟩ij(q) was obtained using the dipole moments. By inserting the resolution of identity, ⟨μ̂2⟩ij(q) takes the following form
| 9 |
Here, i, j, and k refer to the electronic eigenstates. A truncated subspace for the intermediate state k that includes the relevant electronic states involved in the dynamics has been used here.68 In the present case, the subset includes ground and excited electronic states of pyrrole55 and the sum in eq 9 then runs over g and e.
Polariton Non-Adiabatic Dynamics
The excited-state dynamics of the pyrrole-cavity coupled system has been simulated by numerically solving the time-dependent Schrödinger equation with the Hamiltonian given in eq 1, which uses eqs 7 and 8 for ĤC and ĤI, respectively. Here, we consider the H atom Cartesian displacements in evaluating the molecular kinetic energy term in eq 2. This approximation is valid because of the much smaller H atom mass than that of the remaining molecular entity. The coupled nuclear-wave packet is propagated according to the Arnoldi propagation scheme69 on the PESs of pyrrole. These PESs are represented by a three-dimensional numerical grid with 256 points along DIP, 128 points along DOP, and 32 points for the photon displacement coordinate x̂. The initial condition of the system is prepared by vertically placing the ground electronic state with the lowest vibrational level on the πσ* state. This vertical excitation model, which has also been employed to investigate the photodissociation dynamics of pyrrole,55 can be justified as follows. According to Kasha’s rule, the initially populated ππ* state rapidly decays to the πσ* state after which the hydrogen-detachment process starts.70 Furthermore, the computed transition dipole moments suggest that the direct electronic excitation from the ground to the πσ* state should also be possible due to the nonzero dipole values along the symmetry breaking DOP mode (Figure S3 of the Supporting Information). Therefore, investigating the cavity-modified dynamics on the πσ* state is essential to understand the polaritonic effects on the photodissociation reaction of pyrrole. The vibrational ground state of the S0 potential is obtained by employing the imaginary time propagation method.71 Four different cavity mode frequencies, ωc = 0.28, 0.56, 1.56, and 3.54 eV, have been considered for the study (Figure 2). The nuclear wave packet has been evolved for 726 fs with a time step of 48 as using our in-house quantum dynamics code (QDng). The perfect matched layer (PML) has been placed adjacent to the edges of the DIP and DOP coordinates, as the absorbing boundary conditions.72
Figure 2.

One-dimensional potential energy curves of ground (black) and πσ* (red) states of pyrrole along NH in-plane displacement computed at CASSCF/aug-cc-pVDZ level of theory. Vertical arrows with green color represent the cavity mode frequencies ωc, considered in the current study.
The two-dimensional PES is separated by a radial dividing surface located slightly above the surface crossing region (Figure S6 of the Supporting Information). The areas before and after the dividing surface of the PES are designated as the bonding and dissociative regions, respectively. In other words, the molecule reaching the region beyond the intersection in the vicinity of DIP = 1 Å has been considered dissociative. The population probability in the bonding region can be obtained by the expectation value of the elliptical step function with the time-dependent nuclear wave function.
| 10 |
where q1 and q2 refer to the DIP and DOP coordinates, respectively. The elliptical step function f(q1, q2) is defined as
 |
11 |
The dynamics results discussed in the main article are obtained by considering the cavity field along the y-component of the molecular dipole moments. The other components (x and z) are found to be very small, and the corresponding dynamics are very similar to that of the bare molecule (Figures S7 and S8 of the Supporting Information).
Results and Discussion
To investigate the effects of the cavity coupling on the photolysis reaction of pyrrole, wave packet dynamics calculations were performed. Here we have assumed a vertical excitation from the ground electronic state, starting with a product wave function comprising the respective cavity, electronic, and vibrational subsystems: Ψ(t = 0) = |0⟩⊗|πσ*⟩⊗|vX = 0⟩. The quantized cavity field was included in the Hamiltonian (eq 1) used for the simulations. Figure 2 displays the 1-D potential energy profile of ground and πσ* states as a function of NH in-plane displacement (rDOP= 0). There exist a conical intersection (at rN–H ≈ 2.1 Å) between the ground and πσ* states along the DIP coordinate and is accessible for the excited-state molecule by surmounting a small barrier (Figure 2). As a result, pyrrole undergoes hydrogen-dissociation reaction upon photo-excitation. The branching ratio between the ground and excited-state dissociative channels was found to be dependent on the preparation of the initial state.55 The study by Domcke and co-workers considers different initial states with different vibrational levels of the ground electronic state and suggests that the vibrational excitation of NH modes (DIP and DOP) influences the branching ratio between different dissociation channels.55 The present work aims to understand the influences of the cavity coupling on the photo-induced hydrogen-elimination reaction of pyrrole.
The impact of the light-matter interaction on modifying the dynamics not only depends on the strength of the cavity coupling but also on the region of the PES in which the cavity is in resonance with the electronic states. We have considered different cavity mode frequencies ωc that range from coupling near the Franck–Condon (FC) region to coupling in the CI vicinity (Figure 2). The molecular-cavity couplings corresponding to electronic and vibrational polaritons, and DSE terms are included in the Hamiltonian (eq 8) to describe the photon–matter interaction. The photonic-molecular PESs are represented by a three-dimensional numerical grid, including the two molecular degrees of freedom and the photon field coordinate.
We have analyzed the time evolution of the overall population probabilities (Pgbr + Pe) of the molecule in the bonding region, which will be referred to as the survival probabilities hereafter. Figure 3a–d shows the variation of these probabilities by increasing the cavity vacuum field strength ϵc at different cavity frequencies ωc. The black curve in each plot represents the survival probability for the bare molecule (ϵc = 0.00 GV/m), which decays with a dissociation rate of 0.02 fs–1 and fully dissociates within 200 fs. This scenario changes markedly when the cavity field is added to the Hamiltonian. The observed influence is distinct for some cavity parameters (Figure 3a–d) and will be discussed for each cavity resonance frequency separately in the following. The excited-state lifetimes of the molecule in the bonding region are obtained by exponential fitting of the corresponding survival probability decay curves and presented in Figure 4.
Figure 3.

Time evolution of the survival probabilities upon excitation to the πσ* state of pyrrole under the influence of different cavity field strengths (ϵc) with cavity frequency (ωc), 0.28 eV (a), 0.54 eV (b), 1.36 eV (c), and 3.54 eV (d).
Figure 4.

Lifetimes of pyrrole in the bonding region following vertical excitation as a function of the cavity field strength εc. The inset figure is to show the lifetime reduction for the smallest cavity frequency ωc = 0.28 eV.
At the smallest cavity frequency ωc = 0.28 eV, the cavity couples 0.1 Å before the wave packet reaches the CI. The two lowest cavity field strengths (ϵc = 0.78 and 1.96 GV/m) show only minimal effects on the time evolution of the survival probability (Figure 3a). For the two higher field strengths (ϵc = 3.92 and 5.87 GV/m), the dynamics is significantly altered and the photodissociation reaction is accelerated, reducing the lifetime from ≈50 fs (bare molecule) to ≈10 fs (Figure 4). Consequently, the bonding region’s population diminishes to zero much more quickly than the other cavity field strengths and resonance frequencies.The expectation value for the square of the DIP and DOP coordinates with the full polaritonic wave function, ⟨qi2⟩, is computed using time-dependent wave functions. This yields the spread of the wave packet in DIP and DOP modes as a function of time (Figures S9 and S10 of the Supporting Information). Upon increasing the cavity field strength at 0.28 eV resonance frequency, the spread of DOP coordinate on the excited state increases from ≈0.1 Å (bare molecule) to ≈0.8 Å for ϵc = 5.87 GV/m (Figure S10a of the Supporting Information), which suggests the involvement of out-of-plane mode for the dissociation. Unlike the field-free molecule case, the hydrogen detachment process here proceeds along both DIP and DOP coordinates, and multiple pathways are possible.
In the case of ωc = 0.54 eV, the time evolution of the survival probability curves is similar at all cavity field strengths, except for 3.92 GV/m where the probability increases slightly (Figure 3b). The out-of-plane dissociation becomes significant with this frequency too, although the contribution is only ≈30% to that of the previous cavity frequency of 0.28 eV (Figure S10b of the Supporting Information). The lifetime is maximized for a field strength of ϵc = 3.92 GV/m with ≈300 fs and drops when the field strength is further increased (Figure 4).
The nature of the decay curves changes again with the next higher cavity resonance frequency 1.56 eV, but in contrast to 0.28 eV, we observe a significant increase in the lifetime (Figure 3c) for higher field strengths. The photoreaction is suppressed drastically for stronger cavity fields (ϵc = 3.92 and 5.87 GV/m), which leads to a huge rise in the survival probability and increases the lifetime to ≈1500 fs (Figure 4). At the end of the simulation time scale (726 fs), more than 50% of the wave packet remains in the bonding region due to the massive suppression of the hydrogen-elimination reaction at ϵc = 5.87 GV/m. By further increasing the resonance frequency ωc to 3.54 eV, the influence of the cavity mode on the dynamics is decreased (Figure 3d). Analyzing the population dynamics at the abovementioned cavity frequencies (Figure 3a–d), it becomes clear that there is an optimum resonance that allows for maximizing and minimizing excited-state lifetimes, respectively.
To gain more insight into the effects of the cavity on the photolysis of pyrrole, we have evaluated the influence of the light-matter coupling terms such as gii (vibrational cavity couplings) and ⟨μ̂2⟩ij (DSE interactions) for the description of the dynamics. For this purpose, two more sets of wave packet dynamics calculations were performed, where we have dropped coupling terms from the calculation. The corresponding reduced Hamiltonians, Ĥred2 and Ĥred1, have the following form
| 12 |
| 13 |
Figure 5a–d displays the corresponding survival probabilities of these two calculations along with those of the full Hamiltonian for the strongest cavity field ϵc = 5.87 GV/m. Excluding both, the DSE and the coupling to the vibrational motion, leaves the dynamics nearly unaffected, when compared to the field-free case (cyan curve in Figure 5a–d). This highlights the importance of the vibrational cavity couplings, especially at higher field strengths. On the other hand, ignoring only the DSE interaction terms, ⟨μ2⟩ij, results in the survival probability to remain significant at all resonance frequencies (blue curve in Figure 5a–d), overestimating the stabilizing effect of the cavity field. Interestingly, these cavity terms were also observed to be playing a role in the cavity-modified dynamics of pyrrole for other cavity field strengths, except ϵc = 0.78 GV/m (Figures S11, S12, and S13 of the Supporting Information). This emphasizes the necessity to consider the cavity coupling to the vibrational motion and the DSE when investigating the cavity-induced modifications on the dynamics of the molecule.
Figure 5.

Time evolution of the survival probabilities upon excitation to the πσ* state of pyrrole under the influence of the cavity field strength, ϵc = 5.87 GV/m, with different resonance frequencies, ωc. Full Hamiltonian represents the Hamiltonian in eq 8, whereas Ĥred1 and Ĥred2 refer to the reduced Hamiltonians. DSE term, ⟨μ2⟩ij, is excluded in the former reduced Hamiltonian, and both DSE and gii terms are excluded in the latter.
The results discussed above suggest that the hydrogen-dissociation dynamics on the πσ* state of pyrrole coupled to an optical cavity are largely determined by the light-matter coupling terms gii and ⟨μ̂2⟩ii which depend on the molecular permanent dipole moments. Since these two terms appear in the diagonal part of the electronic Hamiltonian, their magnitudes corresponding to the different electronic states must be considerably different to affect the overall dynamics. Thus, there should be a significant jump in the permanent dipole moments going from the ground electronic state to the πσ* state of pyrrole. The excited-state dipole moments of pyrrole are indeed higher than those of the ground state, which can be attributed to the diffused/Rydberg-type nature of the σ* orbital involved in the electronic transition (Figures S1, S4, and S5 of the Supporting Information). In order to discern the factors controlling the dissimilar dynamics (Figures 3a–d and 5a–d), one-dimensional cuts of the polaritonic PESs are obtained by diagonalizing the Hamiltonian in the basis of the Fock states (Figure 6a–c). We would like to emphasize here that the displayed polaritonic curves are along only the DIP coordinate and are obtained within rotating wave approximation where the counter-rotating terms in eq 4 are ignored. On the other hand, the simulations were performed on a three-dimensional surface (DIP, DOP, and x̂), and no further approximations were made to the Hamiltonian. Thus, the polaritionic curves shown here are meant to deliver a qualitative understanding of the mechanisms, especially for the higher field strengths.
Figure 6.

One dimensional cuts of polaritonic PESs for highest cavity field strength ϵc = 5.87 GV/m. Full Hamiltonian represents the Hamiltonian in eq 8, whereas Ĥred1 and Ĥred2 refer to the reduced Hamiltonians. DSE term, ⟨μ2⟩ij, is excluded in the former Hamiltonian and both DSE and gii terms are excluded in the latter.
Based on the analysis of the polaritonic curves at different cavity frequencies, two quantities have been found to be significantly affected. One is the dissociation barrier (ΔEbar) on the excited-state PES and the other is the energy gap (ΔEgap) between the upper and lower polaritons at the new avoided crossing formed by the hybrid field-matter states. These energetics are compiled in Table 1. In the full Hamiltonian case, the dissociation barrier gradually increases going from ωc = 0.28 to 3.54 eV (Figure 6a). At the two lowest resonance frequencies (0.28 and 0.54 eV), the curves exhibit negligible dissociation barriers with a moderate energy gap. Though the two curves possess similar ΔEbar and ΔEgap, they mainly differ by the energy of the excited-state minimum, around 6 eV for the former and 4.5 eV for the latter. In the field-free case, the FC is around 1 eV above the CI. The higher energy difference with respect to the CI causes the wave packet to reach the crossing region with higher kinetic energy. Combined with the minuscule barrier, this explains the short lifetimes for ωc = 0.28 eV (Figure 4). Furthermore, the paltry dissociation barrier makes the excited-state minimum very flat and allows the wave packet to evolve in all directions of the surface. As a result, multiple dissociation paths, along DIP or DOP or any combination of them, become accessible. The presence of these competing pathways rationalizes the increased DOP contributions for the dissociation process at the two lowest resonance frequencies (Figure S10 of the Supporting Information). This process is also responsible for the sudden drop in the lifetimes observed at the strongest cavity field for ωc = 0.54 eV (blue curve in (Figure 4). An increase in the cavity field strength leads to an increased Rabi splitting between the polaritonic curves, separating the upper polariton states, thus stabilizing it.14 The final field strength (ϵc = 5.87 GV/m) however reduces the lifetime by allowing the molecule to dissociate via another competing pathway. For ωc = 1.56 eV, the barrier and energy gap become quite significant. Consequently, molecular photostability increases greatly. The energy gap again decreases at the highest resonance frequency 3.54 eV. Here, the polaritonic states (see Figure 6a) are not well separated, and a vertical excitation leads to a partial population of the lower polariton state, thus reducing the stabilizing effect that has been observed for ωc = 1.56 eV.
Table 1. Excited-State Dissociation Barriers, ΔEbar (eV), Energy Gap between Upper and Lower Polaritonic States ΔEgap (eV) at the Avoided Crossing for Three Different Hamiltonians.
| full
Hamiltonian |
Ĥred1 (no ⟨μ2⟩ij) |
Ĥred2 (no gii + ⟨μ2⟩ij) |
||||
|---|---|---|---|---|---|---|
| cavity freq (eV) | ΔEbar | ΔEgap | ΔEbar | ΔEgap | ΔEbar | ΔEgap |
| ωc = 0.28 | 0.05 | 0.24 | 0.60 | 0.20 | 0.19 | 0.02 |
| ωc = 0.54 | 0.06 | 0.28 | 0.60 | 0.35 | 0.19 | 0.09 |
| ωc = 1.56 | 0.28 | 0.40 | 0.60 | 0.42 | 0.19 | 0.35 |
| ωc = 3.54 | 0.46 | 0.13 | 0.56 | 0.33 | 0.00 | 0.64 |
We now turn to discuss the dynamics of the reduced Hamiltonians, Ĥred1 and Ĥred2, using the corresponding polaritonic curves (Figure 6b,c). Upon excluding the self-interaction term from the Hamiltonian, the curves exhibit high barriers with significant energy gaps at all four frequencies (Table 1). Furthermore, the FC-minimum on the excited state is energetically similar to the avoided crossing, which is unlike the full Hamiltonian case resulting in a wave packet that reaches the crossing region with less kinetic energy. Accordingly, the dissociation process becomes less efficient, and the survival probability remains large. With Ĥred2, on the other hand, the barriers are similar to the bare molecule for all frequencies except for ωc = 3.54 eV, which exhibits no significant barrier. The minimal energy gaps at 0.28 and 0.54 eV are consistent with the corresponding dynamics that are identical to those of the molecule without the cavity field. The higher energy gaps for the next two frequencies do not explain the observed dynamics. However, the energy difference between the FC-minimum and the avoided crossing for ωc = 1.56 eV and the absence of the barrier for ωc = 3.54 eV may lead to an efficient dissociation process.
The role of the collective light-matter couplings on the photodissociation dynamics of pyrrole has not been considered in the present work. In a recent study by Pérez-Sánchez and Yuen-Zhou on polariton-assisted down-conversion of photons, collective coupling effects have been shown to be less significant.73 The smaller collective effects were due to the cavity resonating the molecules far away from the FC region. As a result, the coupling of the cavity with the initially excited single molecule is possible only after sufficient progress of the reaction, while the other molecules at the FC region remain off-resonant. The resonating frequencies employed in the present work are also not very close to the FC region, so similar collective coupling effects can be expected. However, an extensive study with a detailed account of such collective cavity couplings is necessary to ascertain their effects on the photolysis of pyrrole.
Additionally, the leakages of cavity photons may also play a role in altering the dynamics of the polariton states. More recently, the influence of photon loss on the excited-state dynamics has been addressed by several authors:74−77 certain reaction channels, such as dissociation, may actually be suppressed by short photon life times. Along with these effects, other molecular modes and the vibronic interactions of πσ* with the remaining excited states could also be important in describing the photodissociation dynamics of pyrrole. A detailed investigation by considering all these effects is beyond the scope of this paper and deferred to future work.
Conclusions
In summary, the optical cavity can either accelerate or inhibit the photodissociation reaction of pyrrole depending on their frequency and coupling strength. The longest lifetime (1.5 ps) has been observed for a cavity frequency of 1.56 eV (at ϵc = 5.87 GV/m), which we found to be an optimum for stabilizing the photodissociation dynamics of pyrrole. The shortest lifetime that has been observed is ≈10 fs (at ωc = 0.28 eV with ϵc = 5.87 GV/m), whereas the value is ≈50 fs for the field-free molecule (Figure 4). Subsequently, the hydrogen elimination reaction proceeds along an alternative pathway involving DOP mode in addition to the in-plane dissociation. Therefore, the cavity allows the molecule to dissociate along multiple reaction pathways. These modifications are mainly due to the cavity coupling with the vibrational modes and self-polarization terms (Figure 5a–d). The rapid local changes of the permanent dipoles in the vicinity of the CI and their difference between the two electronic states are responsible for the stark difference in the simulations. They further add to the mixing between electronic and vibrational degrees of freedom in the vicinity of the CI. A second factor that has been found to be important is the change of the PES in the FC region; varying the resonance frequency of the cavity affects barriers and the relative energy shift with respect to the crossing region. For an intrinsic CI, the electronic and vibrational degrees are already heavily mixed, thus making the inclusion of the full dipole matrix into light-matter couplings essential. While this may be expected, the strong influence of the self-polarization terms has so far mainly been considered in the context of vibrational strong coupling in the ground state.78 This highlights the importance of these couplings and their consideration for studying the cavity-modified photochemistry of molecules with intrinsic CIs.
Acknowledgments
This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (grant agreement no. 852286). Support from the Swedish Research Council (grant no. VR 2018-05346) is acknowledged.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpca.0c09252.
Molecular orbitals involved in the active space, diabatic couplings, transition dipole moments, permanent dipole moments correspond to ground and πσ* states, bonding and dissociative regions, survival probability plots corresponding to the dynamics where x- and z-components of dipole moments are considered, the expectation value for the spread of DIP and DOP coordinates, and comparison of survival probabilities obtained using full and reduced Hamiltonians (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Turro N. J.Modern Molecular Photochemistry; University Science Books: Sausalito, 1991. [Google Scholar]
- Balzani V.; Ceroni P.; Juris A.. Photochemistry and Photophysics: Concepts, Research, Applications; John Wiley & Sons, 2014. [Google Scholar]
- Alarcos N.; Cohen B.; Ziółek M.; Douhal A. Photochemistry and Photophysics in Silica-Based Materials: Ultrafast and Single Molecule Spectroscopy Observation. Chem. Rev. 2017, 117, 13639–13720. 10.1021/acs.chemrev.7b00422. [DOI] [PubMed] [Google Scholar]
- Emerson R.; Arnold W. The Photochemical Reaction in Photosynthesis. J. Gen. Physiol. 1932, 16, 191–205. 10.1085/jgp.16.2.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polli D.; Altoè P.; Weingart O.; Spillane K. M.; Manzoni C.; Brida D.; Tomasello G.; Orlandi G.; Kukura P.; Mathies R. A.; Garavelli M.; Cerullo G. Conical intersection dynamics of the primary photoisomerization event in vision. Nature 2010, 467, 440–443. 10.1038/nature09346. [DOI] [PubMed] [Google Scholar]
- Kucharski T. J.; Tian Y.; Akbulatov S.; Boulatov R. Chemical solutions for the closed-cycle storage of solar energy. Energy Environ. Sci. 2011, 4, 4449–4472. 10.1039/c1ee01861b. [DOI] [Google Scholar]
- Sinha R. P.; Häder D.-P. UV-induced DNA damage and repair: a review. Photochem. Photobiol. Sci. 2002, 1, 225–236. 10.1039/b201230h. [DOI] [PubMed] [Google Scholar]
- Zietz B.; Gabrielsson E.; Johansson V.; El-Zohry A. M.; Sun L.; Kloo L. Photoisomerization of the cyanoacrylic acid acceptor group - a potential problem for organic dyes in solar cells. Phys. Chem. Chem. Phys. 2014, 16, 2251–2255. 10.1039/c3cp54048k. [DOI] [PubMed] [Google Scholar]
- Warren W. S.; Rabitz H.; Dahleh M. Coherent Control of Quantum Dynamics: The Dream Is Alive. Science 1993, 259, 1581–1589. 10.1126/science.259.5101.1581. [DOI] [PubMed] [Google Scholar]
- Rabitz H.; de Vivie-Riedle R.; Motzkus M.; Kompa K. Whither the Future of Controlling Quantum Phenomena?. Science 2000, 288, 824–828. 10.1126/science.288.5467.824. [DOI] [PubMed] [Google Scholar]
- Brif C.; Chakrabarti R.; Rabitz H. Control of quantum phenomena: past, present and future. New J. Phys. 2010, 12, 075008. 10.1088/1367-2630/12/7/075008. [DOI] [Google Scholar]
- Hoff P. v. d.; Thallmair S.; Kowalewski M.; Siemering R.; Vivie-Riedle R. d. Optimal control theory - closing the gap between theory and experiment. Phys. Chem. Chem. Phys. 2012, 14, 14460. 10.1039/c2cp41838j. [DOI] [PubMed] [Google Scholar]
- Ulusoy I. S.; Gomez J. A.; Vendrell O. Modifying the Nonradiative Decay Dynamics through Conical Intersections via Collective Coupling to a Cavity Mode. J. Phys. Chem. A 2019, 123, 8832–8844. 10.1021/acs.jpca.9b07404. [DOI] [PubMed] [Google Scholar]
- Kowalewski M.; Bennett K.; Mukamel S. Non-adiabatic dynamics of molecules in optical cavities. J. Chem. Phys. 2016, 144, 054309. 10.1063/1.4941053. [DOI] [PubMed] [Google Scholar]
- Kowalewski M.; Bennett K.; Mukamel S. Cavity Femtochemistry: Manipulating Nonadiabatic Dynamics at Avoided Crossings. J. Phys. Chem. Lett. 2016, 7, 2050–2054. 10.1021/acs.jpclett.6b00864. [DOI] [PubMed] [Google Scholar]
- Gu B.; Mukamel S. Manipulating nonadiabatic conical intersection dynamics by optical cavities. Chem. Sci. 2020, 11, 1290–1298. 10.1039/c9sc04992d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vendrell O. Collective Jahn-Teller Interactions through Light-Matter Coupling in a Cavity. Phys. Rev. Lett. 2018, 121, 253001. 10.1103/physrevlett.121.253001. [DOI] [PubMed] [Google Scholar]
- Groenhof G.; Climent C.; Feist J.; Morozov D.; Toppari J. J. Tracking Polariton Relaxation with Multiscale Molecular Dynamics Simulations. J. Phys. Chem. Lett. 2019, 10, 5476–5483. 10.1021/acs.jpclett.9b02192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feist J.; Galego J.; Garcia-Vidal F. J. Polaritonic Chemistry with Organic Molecules. ACS Photonics 2018, 5, 205–216. 10.1021/acsphotonics.7b00680. [DOI] [Google Scholar]
- Galego J.; Garcia-Vidal F. J.; Feist J. Many-Molecule Reaction Triggered by a Single Photon in Polaritonic Chemistry. Phys. Rev. Lett. 2017, 119, 136001. 10.1103/physrevlett.119.136001. [DOI] [PubMed] [Google Scholar]
- Galego J.; Garcia-Vidal F. J.; Feist J. Suppressing photochemical reactions with quantized light fields. Nat. Commun. 2016, 7, 13841. 10.1038/ncomms13841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galego J.; Garcia-Vidal F. J.; Feist J. Cavity-Induced Modifications of Molecular Structure in the Strong-Coupling Regime. Phys. Rev. X 2015, 5, 041022. 10.1103/physrevx.5.041022. [DOI] [Google Scholar]
- Herrera F.; Spano F. C. Cavity-Controlled Chemistry in Molecular Ensembles. Phys. Rev. Lett. 2016, 116, 238301. 10.1103/physrevlett.116.238301. [DOI] [PubMed] [Google Scholar]
- Semenov A.; Nitzan A. Electron transfer in confined electromagnetic fields. J. Chem. Phys. 2019, 150, 174122. 10.1063/1.5095940. [DOI] [PubMed] [Google Scholar]
- Schäfer C.; Ruggenthaler M.; Appel H.; Rubio A. Modification of excitation and charge transfer in cavity quantum-electrodynamical chemistry. Proc. Natl. Acad. Sci. U.S.A. 2019, 116, 4883–4892. 10.1073/pnas.1814178116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett K.; Kowalewski M.; Mukamel S. Novel photochemistry of molecular polaritons in optical cavities. Faraday Discuss. 2016, 194, 259–282. 10.1039/c6fd00095a. [DOI] [PubMed] [Google Scholar]
- Szidarovszky T.; Halász G. J.; Császár A. G.; Cederbaum L. S.; Vibók Á. Conical intersections induced by quantum light: field-dressed spectra from the weak to the ultrastrong coupling regimes. J. Phys. Chem. Lett. 2018, 9, 6215. 10.1021/acs.jpclett.8b02609. [DOI] [PubMed] [Google Scholar]
- Hertzog M.; Wang M.; Mony J.; Börjesson K. Strong light-matter interactions: a new direction within chemistry. Chem. Soc. Rev. 2019, 48, 937–961. 10.1039/c8cs00193f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Martínez L. A.; Du M.; Ribeiro R. F.; Kéna-Cohen S.; Yuen-Zhou J. Polariton-Assisted Singlet Fission in Acene Aggregates. J. Phys. Chem. Lett. 2018, 9, 1951–1957. 10.1021/acs.jpclett.8b00008. [DOI] [PubMed] [Google Scholar]
- Du M.; Martínez-Martínez L. A.; Ribeiro R. F.; Hu Z.; Menon V. M.; Yuen-Zhou J. Theory for polariton-assisted remote energy transfer. Chem. Sci. 2018, 9, 6659–6669. 10.1039/c8sc00171e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Ballestero C.; Feist J.; Moreno E.; Garcia-Vidal F. J. Harvesting excitons through plasmonic strong coupling. Phys. Rev. B: Condens. Matter Mater. Phys. 2015, 92, 121402. 10.1103/physrevb.92.121402. [DOI] [Google Scholar]
- Thomas A.; Lethuillier-Karl L.; Nagarajan K.; Vergauwe R. M. A.; George J.; Chervy T.; Shalabney A.; Devaux E.; Genet C.; Moran J.; et al. Tilting a ground-state reactivity landscape by vibrational strong coupling. Science 2019, 363, 615–619. 10.1126/science.aau7742. [DOI] [PubMed] [Google Scholar]
- Shalabney A.; George J.; Hiura H.; Hutchison J. A.; Genet C.; Hellwig P.; Ebbesen T. W. Enhanced Raman Scattering from Vibro-Polariton Hybrid States. Angew. Chem. 2015, 54, 7971–7975. 10.1002/anie.201502979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera F.; Owrutsky J. Molecular polaritons for controlling chemistry with quantum optics. J. Chem. Phys. 2020, 152, 100902. 10.1063/1.5136320. [DOI] [PubMed] [Google Scholar]
- Flick J.; Narang P. Cavity-Correlated Electron-Nuclear Dynamics from First Principles. Phys. Rev. Lett. 2018, 121, 113002. 10.1103/physrevlett.121.113002. [DOI] [PubMed] [Google Scholar]
- Flick J.; Rivera N.; Narang P. Strong light-matter coupling in quantum chemistry and quantum photonics. Nanophotonics 2018, 7, 1479–1501. 10.1515/nanoph-2018-0067. [DOI] [Google Scholar]
- Flick J.; Ruggenthaler M.; Appel H.; Rubio A. Atoms and molecules in cavities, from weak to strong coupling in quantum-electrodynamics (QED) chemistry. Proc. Natl. Acad. Sci. U.S.A. 2017, 114, 3026–3034. 10.1073/pnas.1615509114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mordovina U.; Bungey C.; Appel H.; Knowles P. J.; Rubio A.; Manby F. R. Polaritonic coupled-cluster theory. Phys. Rev. Res. 2020, 2, 023262. 10.1103/physrevresearch.2.023262. [DOI] [Google Scholar]
- Buchholz F.; Theophilou I.; Nielsen S. E. B.; Ruggenthaler M.; Rubio A. Reduced Density-Matrix Approach to Strong Matter-Photon Interaction. ACS Photonics 2019, 6, 2694–2711. 10.1021/acsphotonics.9b00648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva R. E. F.; Pino J. d.; García-Vidal F. J.; Feist J. Polaritonic molecular clock for all-optical ultrafast imaging of wavepacket dynamics without probe pulses. Nat. Commun. 2020, 11, 1423. 10.1038/s41467-020-15196-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vendrell O. Coherent dynamics in cavity femtochemistry: Application of the multi-configuration time-dependent Hartree method. Chem. Phys. 2018, 509, 55–65. 10.1016/j.chemphys.2018.02.008. [DOI] [Google Scholar]; , high-dimensional quantum dynamics (on the occasion of the 70th birthday of Hans-Dieter Meyer)
- Ribeiro R. F.; Martínez-Martínez L. A.; Du M.; Campos-Gonzalez-Angulo J.; Yuen-Zhou J. Polariton chemistry: controlling molecular dynamics with optical cavities. Chem. Sci. 2018, 9, 6325–6339. 10.1039/c8sc01043a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robb M. A.; Bernardi F.; Olivucci M. Conical intersections as a mechanistic feature of organic photochemistry. Pure Appl. Chem. 1995, 67, 783–789. 10.1351/pac199567050783. [DOI] [Google Scholar]
- Bernardi F.; Olivucci M.; Robb M. A. Potential energy surface crossings in organic photochemistry. Chem. Soc. Rev. 1996, 25, 321–328. 10.1039/cs9962500321. [DOI] [Google Scholar]
- Csehi A.; Halász G. J.; Cederbaum L. S.; Vibók Á. Competition between Light-Induced and Intrinsic Nonadiabatic Phenomena in Diatomics. J. Phys. Chem. Lett. 2017, 8, 1624–1630. 10.1021/acs.jpclett.7b00413. [DOI] [PubMed] [Google Scholar]
- Csehi A.; Kowalewski M.; Halász G. J.; Vibók Á. Ultrafast dynamics in the vicinity of quantum light-induced conical intersections. New J. Phys. 2019, 21, 093040. 10.1088/1367-2630/ab3fcc. [DOI] [Google Scholar]
- Fregoni J.; Granucci G.; Coccia E.; Persico M.; Corni S. Manipulating azobenzene photoisomerization through strong light–molecule coupling. Nat. Commun. 2018, 9, 4688. 10.1038/s41467-018-06971-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rokaj V.; Welakuh D. M.; Ruggenthaler M.; Rubio A. Light–matter interaction in the long-wavelength limit: no ground-state without dipole self-energy. J. Phys. B: At., Mol. Opt. Phys. 2018, 51, 034005. 10.1088/1361-6455/aa9c99. [DOI] [Google Scholar]
- Blank D. A.; North S. W.; Lee Y. T. The ultraviolet photodissociation dynamics of pyrrole. Chem. Phys. 1994, 187, 35–47. 10.1016/0301-0104(94)00230-4. [DOI] [Google Scholar]
- Trofimov A. B.; Zaitseva I. L.; Moskovskaya T. E.; Vitkovskaya N. M. Theoretical investigation of photoelectron spectra of furan, pyrrole, thiophene, and selenole. Chem. Heterocycl. Compd. 2008, 44, 1101–1112. 10.1007/s10593-008-0159-5. [DOI] [Google Scholar]
- Sobolewski A. L.; Domcke W.; Dedonder-Lardeux C.; Jouvet C. Excited-state hydrogen detachment and hydrogen transfer driven by repulsive 1πσ* states: A new paradigm for nonradiative decay in aromatic biomolecules. Phys. Chem. Chem. Phys. 2002, 4, 1093–1100. 10.1039/b110941n. [DOI] [Google Scholar]
- Wei J.; Kuczmann A.; Riedel J.; Renth F.; Temps F. Photofragment velocity map imaging of H atom elimination in the first excited state of pyrrole. Phys. Chem. Chem. Phys. 2003, 5, 315–320. 10.1039/b208132f. [DOI] [Google Scholar]
- Wei J.; Riedel J.; Kuczmann A.; Renth F.; Temps F. Photodissociation dynamics of pyrrole: Evidence for mode specific dynamics from conical intersections. Faraday Discuss. 2004, 127, 267–282. 10.1039/b403191c. [DOI] [PubMed] [Google Scholar]
- Grygoryeva K.; Rakovský J.; Vinklárek I. S.; Votava O.; Fárník M.; Poterya V. Vibrationally mediated photodissociation dynamics of pyrrole. AIP Adv. 2019, 9, 035151. 10.1063/1.5091974. [DOI] [Google Scholar]
- Vallet V.; Lan Z.; Mahapatra S.; Sobolewski A. L.; Domcke W. Photochemistry of pyrrole: Time-dependent quantum wave-packet description of the dynamics at the π1σ*-S0 conical intersections. J. Chem. Phys. 2005, 123, 144307. 10.1063/1.2049250. [DOI] [PubMed] [Google Scholar]
- Ashfold M. N. R.; King G. A.; Murdock D.; Nix M. G. D.; Oliver T. A. A.; Sage A. G. πσ* excited states in molecular photochemistry. Phys. Chem. Chem. Phys. 2010, 12, 1218–1238. 10.1039/b921706a. [DOI] [PubMed] [Google Scholar]
- Mullen P. A.; Orloff M. K. Ultraviolet Absorption Spectrum of Pyrrole Vapor Including the Observation of Low-Energy Transitions in the Far Ultraviolet. J. Chem. Phys. 1969, 51, 2276–2278. 10.1063/1.1672329. [DOI] [Google Scholar]
- Cooper C. D.; Williamson A. D.; Miller J. C.; Compton R. N. Resonantly enhanced multiphoton ionization of pyrrole, N-methyl pyrrole, and furan. J. Chem. Phys. 1980, 73, 1527–1537. 10.1063/1.440358. [DOI] [Google Scholar]
- Palmer M. H.; Walker I. C.; Guest M. F. The electronic states of pyrrole studied by optical (VUV) absorption, near-threshold electron energy-loss (EEL) spectroscopy and ab initio multi-reference configuration interaction calculations. Chem. Phys. 1998, 238, 179–199. 10.1016/s0301-0104(98)00285-7. [DOI] [Google Scholar]
- Wan J.; Meller J.; Hada M.; Ehara M.; Nakatsuji H. Electronic excitation spectra of furan and pyrrole: Revisited by the symmetry adapted cluster-configuration interaction method. J. Chem. Phys. 2000, 113, 7853–7866. 10.1063/1.1316034. [DOI] [Google Scholar]
- Celani P.; Werner H.-J. Analytical energy gradients for internally contracted second-order multireference perturbation theory. J. Chem. Phys. 2003, 119, 5044–5057. 10.1063/1.1597672. [DOI] [PubMed] [Google Scholar]
- Jaynes E. T.; Cummings F. W. Comparison of quantum and semiclassical radiation theories with application to the beam maser. Proc. IEEE 1963, 51, 89–109. 10.1109/proc.1963.1664. [DOI] [Google Scholar]
- Schleich W. P.Quantum Optics in Phase Space; John Wiley & Sons, Ltd, 2001; Chapter 4, pp 99–151. [Google Scholar]
- Flick J.; Appel H.; Ruggenthaler M.; Rubio A. Cavity Born-Oppenheimer Approximation for Correlated Electron-Nuclear-Photon Systems. J. Chem. Theory Comput. 2017, 13, 1616–1625. 10.1021/acs.jctc.6b01126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simah D.; Hartke B.; Werner H.-J. Photodissociation dynamics of H2S on new coupled ab initio potential energy surfaces. J. Chem. Phys. 1999, 111, 4523–4534. 10.1063/1.479214. [DOI] [Google Scholar]
- Werner H.-J.; Knowles P. J.; Knizia G.; Manby F. R.; Schütz M. Molpro: a general-purpose quantum chemistry program package. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2012, 2, 242–253. 10.1002/wcms.82. [DOI] [Google Scholar]
- Werner H.-J.; Knowles P. J.; Knizia G.; Manby F. R.; Schütz M.; Celani O.; Györffy W.; Kats D.; Korona T.; Lindh R., et al. MOLPRO, version 2019.2, a Package of ab Initio Programs, 2019.
- Mandal A.; Krauss T. D.; Huo P. Polariton-Mediated Electron Transfer via Cavity Quantum Electrodynamics. J. Phys. Chem. B 2020, 124, 6321–6340. 10.1021/acs.jpcb.0c03227. [DOI] [PubMed] [Google Scholar]
- Smyth E. S.; Parker J. S.; Taylor K. T. Numerical integration of the time-dependent Schrödinger equation for laser-driven helium. Comput. Phys. Commun. 1998, 114, 1–14. 10.1016/s0010-4655(98)00083-6. [DOI] [Google Scholar]
- Kasha M. Characterization of electronic transitions in complex molecules. Discuss. Faraday Soc. 1950, 9, 14–19. 10.1039/df9500900014. [DOI] [Google Scholar]
- Kosloff R.; Tal-Ezer H. A direct relaxation method for calculating eigenfunctions and eigenvalues of the schrödinger equation on a grid. Chem. Phys. Lett. 1986, 127, 223–230. 10.1016/0009-2614(86)80262-7. [DOI] [Google Scholar]
- Nissen A.; Karlsson H. O.; Kreiss G. A perfectly matched layer applied to a reactive scattering problem. J. Chem. Phys. 2010, 133, 054306. 10.1063/1.3458888. [DOI] [PubMed] [Google Scholar]
- Pérez-Sánchez J. B.; Yuen-Zhou J. Polariton Assisted Down-Conversion of Photons via Nonadiabatic Molecular Dynamics: A Molecular Dynamical Casimir Effect. J. Phys., Lett. 2020, 11, 152–159. 10.1021/acs.jpclett.9b02870. [DOI] [PubMed] [Google Scholar]
- Davidsson E.; Kowalewski M. Simulating Photodissociation Reactions in Bad Cavities with the Lindblad Equation. J. Chem. Phys. 2020, 153, 234304. 10.1063/5.0033773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulusoy I. S.; Vendrell O. Dynamics and spectroscopy of molecular ensembles in a lossy microcavity. J. Chem. Phys. 2020, 153, 044108. 10.1063/5.0011556. [DOI] [PubMed] [Google Scholar]
- Antoniou P.; Suchanek F.; Varner J. F.; Foley J. J. Role of Cavity Losses on Nonadiabatic Couplings and Dynamics in Polaritonic Chemistry. J. Phys. Chem. Lett. 2020, 11, 9063–9069. 10.1021/acs.jpclett.0c02406. [DOI] [PubMed] [Google Scholar]
- Felicetti S.; Fregoni J.; Schnappinger T.; Reiter S.; de Vivie-Riedle R.; Feist J. Photoprotecting Uracil by Coupling with Lossy Nanocavities. J. Phys. Chem. Lett. 2020, 11, 8810–8818. 10.1021/acs.jpclett.0c02236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galego J.; Climent C.; Garcia-Vidal F. J.; Feist J.. Cavity Casimir-Polder Forces and Their Effects in Ground-State Chemical Reactivity. Phys. Rev. X 2019, 9. 10.1103/physrevx.9.021057 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.