

Abstract
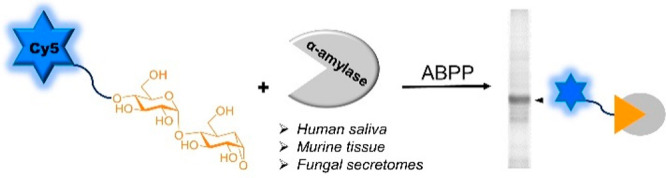
Amylases are key enzymes in the processing of starch in many kingdoms of life. They are important catalysts in industrial biotechnology where they are applied in, among others, food processing and the production of detergents. In man amylases are the first enzymes in the digestion of starch to glucose and arguably also the preferred target in therapeutic strategies aimed at the treatment of type 2 diabetes patients through down-tuning glucose assimilation. Efficient and sensitive assays that report selectively on retaining amylase activities irrespective of the nature and complexity of the biomaterial studied are of great value both in finding new and effective human amylase inhibitors and in the discovery of new microbial amylases with potentially advantageous features for biotechnological application. Activity-based protein profiling (ABPP) of retaining glycosidases is inherently suited for the development of such an assay format. We here report on the design and synthesis of 1,6-epi-cyclophellitol-based pseudodisaccharides equipped with a suite of reporter entities and their use in ABPP of retaining amylases from human saliva, murine tissue as well as secretomes from fungi grown on starch. The activity and efficiency of the inhibitors and probes are substantiated by extensive biochemical analysis, and the selectivity for amylases over related retaining endoglycosidases is validated by structural studies.
Introduction
Retaining α-amylases, belonging to the glycoside hydrolase (GH) 13 family (www.cazy.org),1 are starch-processing enzymes present in plants, animals, and microorganisms.2−4 They catalyze the hydrolysis of internal α-1,4-glycosidic linkages in starch and related polysaccharides through a two-step Koshland double displacement mechanism (Figure S2A). In this mechanism, the aglycon is protonated by the general acid/base residue, followed by SN2 nucleophilic attack by the catalytic nucleophile at the anomeric center to form a covalent enzyme–substrate intermediate with inversion of anomeric configuration. The covalent enzyme–substrate adduct is subsequently hydrolyzed to yield oligosaccharide products (for instance linear and α-1,6-branched malto-oligosaccharides) with net retention of α-anomeric configuration.5
Starch is the main product of major human and animal feedstock crops including wheat, rice, maize, tapioca, and potato. Traditionally, large-scale starch processing, an industry established over 100 years ago, relied on the degradation of starch by acid hydrolysis.6,7 Amylases have gained considerable traction as alternatives to conventional chemical starch hydrolysis, as they are more environmentally benign, are less energy intensive, and moreover may produce more defined oligosaccharide products from starch polymers.8,9 Today, a large number of α-amylases from microbial sources are commercially available and are widely used for the production of high glucose and fructose syrups.10,11 Another major application of α-amylases is found in the detergent industry, where they act at low temperatures and alkaline pH to remove tough starch stains from clothes and porcelains under mild conditions.12 α-Amylases are furthermore extensively employed in textile, food, paper, and biofuel industries. The α-amylases currently applied in industrial biotechnology originate from a limited number of fungal and bacterial strains, including Aspergillus oryzae and Bacillus licheniformis.11,13 The microbial world however may harbor many additional α-amylases including ones that differ functionally in terms of activity, substrate specificity, and resistance to (harsh) biotechnological conditions.
In addition to their potential in biotechnological applications, α-amylases have received increased interest in the past decades in the context of human health. In mammals, digestion of starch by salivary and pancreatic α-amylases leads to linear and branched malto-oligosaccharides. These are largely hydrolyzed to glucose by α-glucosidases located in the intestinal mucosa, which then enters the bloodstream by means of facilitated diffusion. Control of postprandial glucose levels, which has proven to be a key factor for the treatment of diabetes and obesity,14 might be achieved by activity modulation of starch-processing enzymes. Antidiabetic drugs currently used in the clinic include the α-glucosidase inhibitors,15,16 miglitol, voglibose, and acarbose, the last of which also inhibits human pancreatic α-amylase (HPA).17,18 However, their effectiveness is offset by undesirable side effects, including diarrhea, flatulence, and abdominal pain, secondary effects that arise predominantly from inhibition of intestinal α-glucosidases. Hence, selective inhibitors targeting exclusively HPA, the first enzyme in the digestion sequence, would be of great interest.19−24
Activity-based protein profiling (ABPP) has emerged as a powerful strategy to obtain qualitative and quantitative activity information on enzymes of interest in complex mixtures.25,26 ABPP is especially useful for retaining glycosidases that form a covalent enzyme–substrate intermediate during hydrolysis. It relies on the availability of suitable activity-based probes (ABPs) that are generally generated from covalent and irreversible inhibitors by equipping these with a reporter entity (a fluorophore or biotin) or a bio-orthogonal tag. By means of ABPP, enzyme activities can be rapidly identified and quantified (comparative ABPP) and the effect of inhibitors on target enzymes studied in cell extracts and living cells (competitive ABPP). Our work on retaining glycosidase ABPP is rooted in the known mechanism-based β-glucosidase inhibitor cyclophellitol, isolated from Phellinus sp.27 In the first instance we applied cyclophellitol-derived ABPs to the study of retaining exo-acting α- and β-glycosidases.28−32 More recently, we expanded the methodology toward retaining endoglycosidases in our work on xylobiose-configured ABPs to investigate endo-acting xylanases in Aspergillus secretomes.33,34 Here, we reveal the efficacy of retaining glycosidase ABPP by the design and chemical synthesis of a panel of maltobiose epi-cyclophellitol derived retaining α-amylase inhibitors and ABPs, the kinetic and structural analysis of their interactions with microbial α-amylases, and the application of these probes for rapid detection and identification of α-amylases in fungal secretomes, mouse tissue, and human saliva. Our work complements a recent24 study by Withers and colleagues, who reported a chemoenzymatic synthesis of maltobiose epi-cyclophellitol, to which we could match our chemically derived material and as well our structural work on α-amylase-inhibitor complexes. Our results moreover illustrate the potential of retaining α-amylase ABPP in the annotation of α-amylases in complex biological samples.
Results and Discussion
Design and Synthesis of α-Amylase Inhibitors and Activity-Based Probes
We have previously revealed that 1,6-epi-cyclophellitol and its aziridine and cyclic sulfate analogues (Figure 1A) are potent mechanism-based α-glucosidase inhibitors and that their tagged versions are effective activity-based retaining α-exoglucosidase probes.30,35 These monosaccharidic cyclitols however do not inhibit endo-acting α-amylases, which we argued is because of the absence of productive binding with the larger endo-glycosidase active sites (Figure S2C). In line with this consideration, Withers and co-workers subsequently reported that the maltose disaccharide mimetic α-1,4-glucopyranosyl epi-cyclophellitol (compound 1a, Figure 1B), which they prepared by enzymatic α-glucosylation of 1,6-epi-cyclophellitol, is an effective mechanism-based, covalent, and irreversible HPA inhibitor.24 Crystallographic analysis of an HPA–inhibitor complex points to the O4′ of the nonreducing end sugar, which may simply be alkylated to block potential exoglycosidase action as a promising site for orthogonal functionalization (as in 1b) to allow introduction of a reporter entity (a fluorophore or biotin, as in 1c and 1d). Alternatively and in line with our previous33 work on xylanase ABPs, we envisaged the design of maltose-configured cyclophellitol aziridines bearing the reporter tag on the aziridine nitrogen, either with or without an O4′ cap (compounds 2a–2c, Figure 1B). Finally and in line with our previous results on 1,6-epi-cyclophellitol cyclosulfate being a potent and selective retaining α-exoglucosidase inhibitor,30,35 we decided to produce the corresponding maltose cyclosulfate (compound 3a, Figure 1B) as well with the aim to investigate whether this Michaelis complex emulating inhibitor would also act on amylases.
Figure 1.

Design of mechanism-based retaining α-glucosidase inhibitors and ABPs: (A) structures of retaining α-exoglucosidase inhibitors; (B) structures of retaining α-endoglucosidase inhibitors and ABPs, which are subject of the here-described studies.
We have recently reported the synthesis of xylobiose-cyclophellitols via the direct glycosylation of xylo-cyclophellitol (aziridine) acceptors.33 This route however appeared not feasible for the construction of compounds 1a–d since the epoxide functionality in partially protected 1,6-epi-cyclophellitols proved unstable toward stoichiometric amounts of Lewis acid under the conditions required to effect formation of α-1,4-glycosidic bonds using contemporary glycosylation conditions. Alternatively, cyclohexene 5 was found to readily undergo glycosylation with imidate donor 4a under N,N-dimethylformamide (DMF) and trifluoromethanesulfonic acid (TfOH) activating conditions, affording pseudodisaccharide 6a in good yield.36 Selective deprotection of the 4-methoxybenzyl group in 6a followed by epoxidation afforded an inseparable mixture of α/β-epoxides in a 1:3 ratio, which were silylated and separated by silica gel column chromatography to yield pure 8a and 9a. Desilylation and global debenzylation of 8a by Pearlman’s catalyst hydrogenation gave maltobiose epi-cyclophellitol 1a, of which all analytical and spectroscopical data proved to match those of the chemoenzymatically produced compound.24 The epoxide in 9a was converted to aziridine 14a by treatment with sodium azide followed by a Staudinger-type ring closure. Alkylation and global deprotection of 14a under Birch conditions afforded alkyl-aziridine 2a, which was equipped with a Cy5 fluorophore to produce ABP 2b (Scheme 1).
Scheme 1. Synthesis of the Inhibitors and ABPs Subject of the Here-Described Studies.
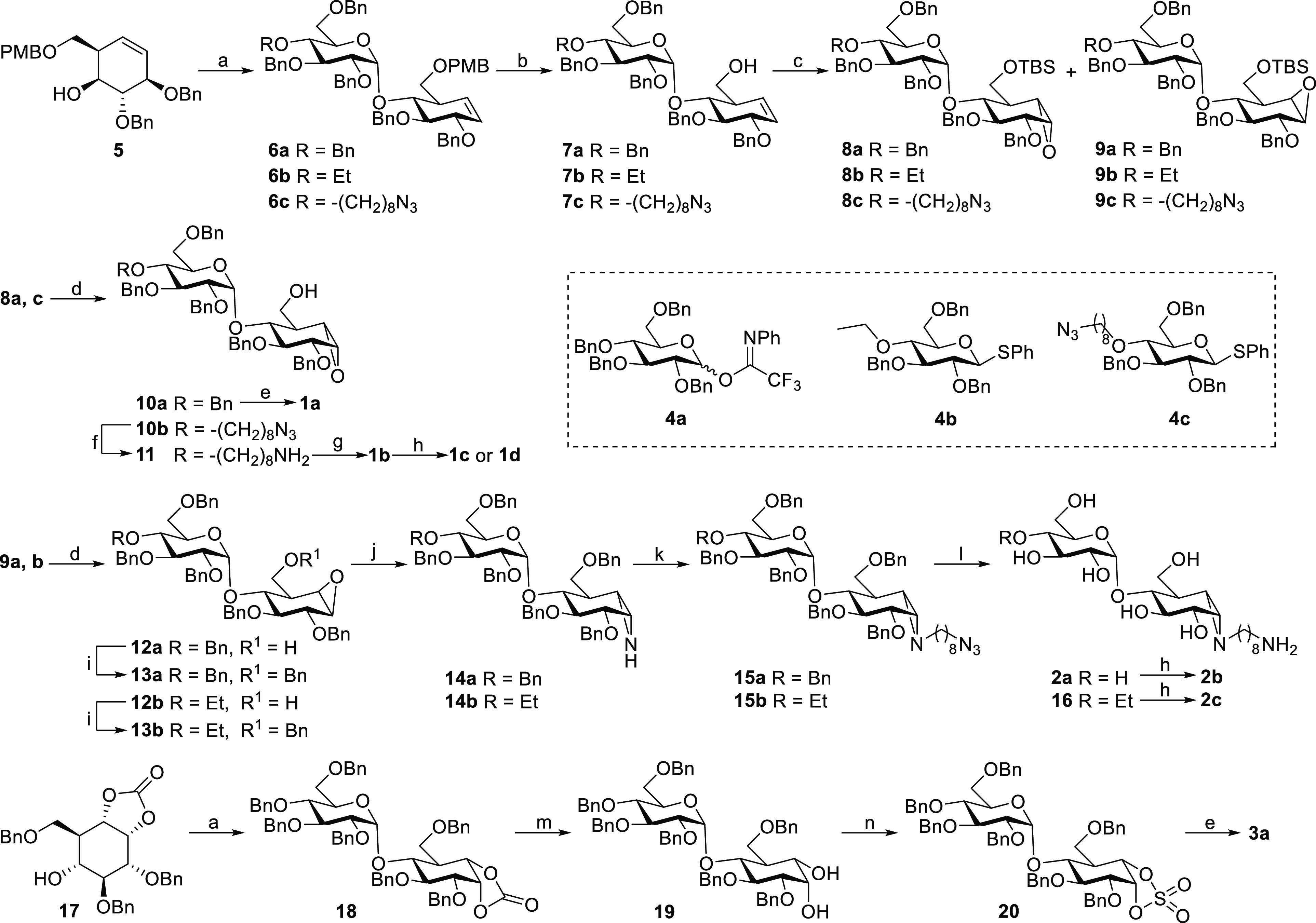
Reagents and conditions. (a) For 6a and 18: 4a, TfOH (1.5 equiv), DMF, DCM, 4 Å molecular sieves, −20 to 0 °C, 6a 96%, 18 57%. For 6b: 4b, NIS, TfOH (1.5 equiv), DMF, DCM, 4 Å molecular sieves, 0 °C, 88%. For 6c: 4c, NIS, TfOH (1.5 equiv), DMF, DCM, 4 Å molecular sieves, 0 °C, 87%. (b) DDQ, DCM/H2O (19/1), rt, 7a 80%, 7b 74%, 7c 71%; (c) (i) mCPBA, DCM, 0 °C to rt; (ii) TBSCl, DMAP, imidazole, DCM, rt, 8a 20%, 9a 58%, 8b 21%, 9b 65%, 8c 22%, 9c 63%; (d) TBAF, THF, rt, 10a 90%, 10b 87%, 12a 91%, 12b 88%; (e) Pd(OH)2/C, H2, MeOH/H2O/dioxane (2/1/2), rt, 1a 100%, 3a 89%; (f) polymer-bound PPh3, MeCN, H2O, 70 °C, 79%; (g) Pd(OH)2/C, H2, HOAc, tBuOH/H2O/dioxane (1/2/1), rt, 91%; (h) Cy5-OSu or biotin-OSu, DIPEA, DMF, rt, 1c 33%, 1d 16%, 2b 10%, 2c 17%; (i) BnBr, NaH, TBAI, DMF, 0 °C to rt, 13a 82%, 13b 75%; (j) (i) NaN3, LiClO4, DMF, 100 °C; (ii) polymer-bound PPh3, MeCN, 60 °C, 14a 48%, 14b 42%; (k) 8-azidooctyltrifluoromethanesulfonate, DIPEA, DCM, 0 °C to rt, 15a 90%, 15b 88%; (l) Na, NH3 (liq.), tBuOH, THF, −60 °C, 2a 79%, 16 94%; (m) NaOMe, DCM/MeOH (1/1), rt, 93%; (n) (i) SOCl2, TEA, DCM, 0 °C; (ii) RuCl3·3H2O, NaIO4, EtOAc/ACN/H2O (2/2/1), 0 °C, 58%.
Our previous studies on endo-xylanases revealed that the xylobiose-cyclophellitol aziridine probe was susceptible to hydrolysis by exo-xylosidases present in Aspergillus secretomes.33 Thus, we hypothesized that “capping” the O4′ position of maltobiose epi-cyclophellitol azirines with an ethyl group would prevent cleavage by exo-acting α-glucosidases in complex biological samples. This was achieved by using O4 ethyl capped thiophenyl donor 4b for glycosylation with acceptor 5 under N-iodosuccinimide (NIS)/DMF/TfOH catalysis, giving 4′-ethyl pseudodisaccharide 6b in good yield. Following the same route as for 2a, 4′-ethyl capped maltobiose epi-cyclophellitol aziridine 16 could be readily obtained from intermediate 9b, which was equipped with a Cy5 fluorophore to produce ABP 2c. We constructed epoxide-based ABPs by using thiophenyl donor 4c, where the azidooctyl group was introduced in an early stage for later installation of reporter entities and also to prevent exo-glycosidase processing. Following the established route, epoxide 1b was obtained after desilylation, azide reduction, and debenzylation of 8c. Following coupling with a Cy5 fluorophore or biotin moiety, ABPs 1c and 1d were obtained, respectively.
The same strategy was also applied to construct maltobiose epi-cyclophellitol cyclosulfate 3a. Carbonate acceptor 17 was readily glycosylated with donor 4a under TfOH/DMF conditions, giving pseudodisaccharide 18 in moderate yield. Treatment of 18 with sodium methoxide gave diol 19. Generation of the cyclosulfite using thionyl chloride and subsequent oxidation gave perbenzylated cyclosulfate 20, the benzyl ethers of which were removed by Pearlman’s catalyst hydrogenation to afford inhibitor 3a.
Kinetic Studies for Time-Dependent Inhibition of Aspergillus oryzae α-Amylase by Maltobiose Inhibitors
To assess the potency of the synthesized maltobiose epi-cyclophellitols, we monitored the time-dependent inhibition of Aspergillus oryzae α-amylase (hereafter termed as Taka-amylase). Taka-amylase, one of the earliest amylases to be characterized and used commercially,37,38 is a retaining α-amylase belonging to the GH13 subfamily 1. This enzyme uses a pair of catalytic active site residues (Asp206 as a nucleophile and Glu230 as the acid/base) to cleave amylose chains and requires several subsites to be occupied to catalyze hydrolysis at meaningful rates.39
To assess time-dependent inhibition, the hydrolysis of 2-chloro-4-nitrophenyl maltotrioside (CNP-M3) by Taka-amylase was measured after preincubation with 2 mM of inhibitors 1a, 1b, 2a, and 3a. All four inhibitors displayed a time-dependent inhibition of Taka-amylase; however the rates of inhibition varied greatly (Figure 2). Both the epi-cyclophellitol epoxide 1a and aziridine 2a had slow rates of inactivation, with rate constants of 0.0061 ± 0.0002 min–1 and 0.005 ± 0.002 min–1, respectively. The rate constant for the inactivation by 1a is somewhat higher than was previously observed for its inhibition of HPA, determined to be 0.001 min–1 at a higher concentration of inhibitor (3.6 mM).24 It is somewhat surprising that the aziridine had a rate of inhibition slightly worse than that of the epoxide, as our previous work has demonstrated aziridine based inhibitors to be more potent for α-glucosidases.30 We speculate that N-alkylation of the aziridine may reduce the initial binding of inhibitor 2a, consequently reducing inactivation rates. Inhibition by the cyclosulfate inhibitor 3a was 4- to 5-fold faster (rate constant = 0.0250 ± 0.0009 min–1) when compared to 1a or 2a. This observation agrees well with our previous work, which showed that the monosaccharide epi-cyclophellitol cyclosulfate was indeed the most potent cyclophellitol type inhibitor of recombinant lysosomal acid α-glucosidase.35 Surprisingly, 4′-alkylamine 1b proved to be the most effective inhibitor tested, with an inhibition rate constant that was nearly 10 times that of the untagged 1a (rate constant = 0.057 ± 0.003 min–1). This suggests that initial binding of 1b is aided by interactions between the −3 subsite and the 4′-alkylamine present, which in turn increases the rate of inhibition. Previous kinetic analysis of Taka-amylase catalytic rates has shown dramatic rate increases for the hydrolysis of a maltotetrasaccharide over a maltotrioside,39 highlighting the importance of binding in the −3 position with a natural substrate.
Figure 2.

Time-dependent inactivation of Taka-amylase. Residual activity of Taka-amylase incubated with 2 mM of either 1a (◐), 1b (○), 2a (⊗), or 3a (●) was assayed over time and fit to an exponential decay curve.
Structural Analysis of Inhibitor–Enzyme Complexes
We next sought to use X-ray crystallography to further understand the binding of inhibitors 1a, 1b, 2a, and 3a to the active site of Taka-amylase. All four inhibitors formed covalent complexes with Taka-amylase, solved with resolution between 1.67 and 1.35 Å, and were covalently linked to the enzyme through the catalytic nucleophile Asp206 (Figure 3). The slow reaction rates necessitated prolonged incubation of the inhibitors with preformed crystals. The epi-cyclophellitol epoxides were soaked for 3 days. At this stage 1a reached 80% active site occupancy, while 1b had complete occupancy, reflective of its increased rate constant of inhibition when compared to 1a. After a week-long soak with 2a Taka-amylase crystals had approximately 70% inhibitor occupancy; thus soak length for this compound was further extended to 1 month after which complete occupancy was observed. The soak with 3a was also extended to 1 month to allow for complete active site occupancy.
Figure 3.

Covalent inhibition of Taka-amylase with glycosylated cyclophellitols. Crystal structures are shown between Taka-amylase and each of 1a (A), 1b (B), 3a (C), and 2a (D). Electron density (2Fo – Fc) is shown for both the ligand and the catalytic nucleophile (Asp206) as a blue mesh contoured at 1.5σ (1a = 0.59 e–/Å3, 1b = 0.61 e–/Å3, 2a = 0.46 e–/Å3, 3a = 0.54 e–/Å3). The polypeptide is shown in cartoon form with active site residues shown as sticks. Apparent hydrogen bonding interactions are shown as dotted black lines. Active-site residue Arg204 is omitted for clarity.
The crystal structure of 1a bound to the active site nucleophile (Asp206) showed the cyclitol moiety to adopt a 4C1 chair conformation, which is also observed for 1b and 3a. This conformation matches that observed for the −1 sugar by Caner et al.24 for the binding of enzymatically synthesized 1a to HPA and the conformation of an in situ generated 5-fluoro-idose trisaccharide, also to HPA.23 Although a detailed computational analysis of the conformational itineraries for a GH13 α-amylase has yet to be done, a detailed analysis of a homologous GH13 amylosucrase from Neisseria polysaccharea has been published.40 This analysis proposes a 4C1 → [3H4]⧧ → 4C1 trajectory for the glycosylation step, consistent with the observed conformation of the inhibitor bound in the −1 position. The nucleophile, Asp206, in the 1a complex structure is present in two conformations, one minor unbound conformation which aligns with the uncomplexed form (PDB code 6YQ7) and the other rotated approximately 120° from the uncomplexed orientation that we ascribe to the conformation present in the covalently inhibited enzyme (Figure 3, panel A, PDB code 6YQ9). This second conformation is also seen for the 1b complex which is at full occupancy. Bound 1a also forms nine direct hydrogen bonds with Taka-amylase (with His122, Arg204, Thr207, His296, and Asp297 in the −1 subsite and Trp83, Asp340, and Arg344 in the −2 subsite), replicating those seen in the structure of a complex between Taka-amylase and acarbose (PDB code 7TAA), with the exception of the hydrogen bond between the 6-OH of 1a and Thr207, and the lack of an H-bond with the catalytic nucleophile that is observed for acarbose.41
The structure of 4′-alkylamine epi-cyclophellitol 1b bound in the active site (Figure 3, panel B, PDB code 6YQC) maintains an identical hydrogen bonding network to that seen in the complex with 1a, the only difference being the complete occupancy of the inhibitor, resulting in the absence of an unbound Asp206 conformer and the presence of an alkyl tail. The 4′ tail of 1b can only be modeled to the sixth carbon (away from the maltose core) indicating that the terminal amine may form multiple interactions in the −3 subsite. The portion of the alkyl tail that could be modeled with confidence is appropriately positioned to form a C–H/π hydrogen bond through the second carbon which is 3.9 Å away from Tyr75. This interaction may in part be responsible for better binding of 1b to Taka-amylase and the resulting increased rate of inhibition.
The structure of the epi-cyclophellitol cyclosulfate 3a complex has many similarities with complexes of the epoxide inhibitors 1a and 1b (Figure 3, panel C, PDB code 6YQB). The cyclitol is covalently attached to Asp206, again rotated approximately 120° from the unreacted enzyme, and is in a 4C1 conformation. The −2 sugar is positioned exactly as for 1a, however the presence of an exocyclic sulfur at the 6 position has resulted in Thr207 being positioned within hydrogen bonding distance to the O6 that forms the sulfate ester. The sulfate group is also positioned within hydrogen bonding distance to the acid base residue, Glu230; however this residue appears in two different conformations in the structure.
Binding of 2a to Taka-amylase is strikingly dissimilar to the other three inhibitor complexes (Figure 3, panel D, PDB code 6YQA). The inhibitor is again covalently bound to Asp206; however the nucleophile is rotated nearly 180° when compared to the other three complexes such that the nonbonded δO is positioned above the “β” face of the bound cyclitol ring rather than below it. The hydrogen bonds between the inhibitor and the enzyme −1 subsite are also altered, with the cyclitol methoxy alcohol rotated to form a hydrogen bond with Thr207 (instead of His122), and the C6 nitrogen forms a hydrogen bond with the acid/base residue (Glu230). Furthermore, the reacted cyclitol, rather than 4C1, is in an unprecedented E3 conformation, the emergence of which we attribute to the presence of the alkyl-chain. We have observed that cyclophellitol aziridines with N-alkylation are generally well accepted in both exo-acting enzymes as these often have open + subsites28−32 and endo-acting enzymes that target β-linkages,33 which typically contain long, tunnel, or groove-like active sites. The active site of Taka-amylase, conversely, is a V-shaped surface depression with a severe angle between the −1 and +1 sugar binding sites; this in turn constrains the conformation of the aziridine alkylation and the conformation of the cyclitol ring.
In Vitro Labeling of Recombinant Human Saliva α-Amylase with ABPs 1c, 2b, and 2c
With the fluorescent ABPs 1c, 2b, and 2c in hand, we first investigated their labeling efficiency and specificity toward recombinant human saliva α-amylase (type XIII-A, HSA). Kinetic analysis for inactivation of Taka-amylase revealed the parent inhibitors 1a, 1b, and 2a as slow enzyme binders. Thus, recombinant α-amylase was incubated with ABPs 1c, 2b, or 2c at two quite high concentrations (100 μM or 10 μM, 1 h, 37 °C, pH 7). As shown in Figure 4A, HSA was effectively labeled by epoxide-based ABP 1c, while the labeling potency of aziridine-based ABPs 2b and 2c was dramatically reduced, and only very weak bands were observed even at 100 μM probe concentrations. In accordance with that of kinetic studies, the epoxide inhibitor 1b is much more potent than aziridine inhibitor 2a. This observation is supported by the X-ray structure analysis of inhibitor–enzyme complexes, which indicates that interactions between the −3 subsite in α-amylase active sites and the O4′ substituent may contribute to the initial binding of 1b and the resulting increased inhibition potency while the alkylation of aziridine warhead in 2a is not preferred during enzyme binding.
Figure 4.

(A) Fluorescent labeling of recombinant human saliva α-amylase (type XIII-A) with ABPs 1c, 2b, and 2c at pH 7 after 1 h incubation at 37 °C. (B) Fluorescent labeling of concentrated human saliva, mouse pancreas, and salivary gland lysates (55 μg total protein) with different concentrations of ABP 1c (pH 7.0, 37 °C). (C) pH-dependent labeling with ABP 1c in complex biological samples. The optimal pH is approximately 5.0 in these three samples. (D) Detection limit of α-amylase in complex biological samples labeled with ABP 1c (pH 5.0, 37 °C). (E) Time-dependent labeling of α-amylase in complex biological samples with ABP 1c (pH 5.0, 37 °C). (F) Competitive ABPP on amylases in extracts from human saliva (HS), mouse pancreas (MP), and mouse salivary gland (MSG), offset against Western blot detection of amylase abundance using an anti-amylase antibody.
Activity-Based Protein Profiling of Retaining α-Amylases in Complex Biological Samples
As the next research objective, we attempted to visualize α-amylases in concentrated human saliva and lysates from mouse pancreas and salivary gland with ABP 1c. As shown in Figure 4B, ABP 1c labeled a distinct band at ∼57 kDa, corresponding to the molecular weight of HSA.42 The same band was also observed in mouse pancreas lysates where significant labeling appeared at 25 μM (2 h, 37 °C, pH 7), while at higher probe concentrations background fluorescence was observed. Interestingly, labeling in mouse salivary gland lysates with 1c showed two clear bands, not only the one at ∼57 kDa but also a slightly stronger band at ∼70 kDa. Next, the pH dependence of labeling was determined (Figure 4C). ABP 1c exhibits a similar pH-dependent activity toward α-amylase (at the time unknown ∼57 kDa band) in these three biological samples, and the optimal pH for labeling is determined at 5.0, different from the reported neutral pH optimum in HPA21 and HSA42 activity. Of note, in mouse salivary gland lysates, the band at ∼70 kDa only appeared following labeling at pH ranging from 6.0 to 8.0 with optimal labeling observed at pH 7.5. Additionally, the efficiency and the rate of labeling of α-amylase were further investigated under optimal pH 5.0 (Figure 4D and Figure 4E). In these three samples, α-amylase (the ∼57 kDa band) was selectively labeled by 1c in a concentration- and time-dependent manner, consistent with the irreversible inhibition mechanism one would expect for the maltose cyclophellitols.
Having established that amylase activities could be visualized in extracts from human saliva, murine pancreas, and murine salivary glands, competitive ABPP (cABPP) experiments were performed to see whether these activities could be negated by inclusion of a nontagged inhibitor. Preincubation of biological samples with inhibitor 1d followed by labeling with Cy5 ABP 1c resulted in a decrease of fluorescent signals similar to denaturation of the samples with 2% SDS (Figure 4F). After fluorescent imaging, proteins were transferred to a polyvinylidene fluoride (PVDF) membrane for Western blot analysis with an anti-amylase antibody. The observed fluorescent bands and chemiluminescence bands match well where no inhibitor is included, with the chemiluminescence bands remaining equal in intensity where the ABP signal decreases in an inhibitor-dependent fashion, confirming that ABP 1c labeled amylase. cABPP assays were also performed with inhibitors 1a and 1b in human saliva (Figure S5). Compared with 1d, the inhibition potency of these two was reduced and only minor competition was observed with “untagged” 1a at the highest concentration applied (100 μM), further indicating that alkylation of the O4′ position of the epoxide-based inhibitor may improve its inhibition potency.
ABPP of Fungal Secretomes
The probes developed herein can also be applied to monitor the production of amylases by biotechnologically relevant fungi. Secretomes from the saprophytic ascomycete Aspergillus nidulans grown on various starches have previously been shown to contain a complex mixture of amylases, glucosidases, and oxidative enzymes.43 We performed ABPP of secretomes from a time course of A. nidulans strain FGSC A4 growth in minimal media supplemented with 1% wheat starch as a carbon source. Secretome samples were taken daily over a 5-day period, and the fluorescent Cy5 ABP 1c was incubated with these secretome samples to reveal a complex pattern of fluorescent bands that could first be detected starting by day 3. The intensity of amylase bands increased after day 3 with a maximum intensity at day 5 (Figure 5C).
Figure 5.

(A) Fluorescent labeling of day 5 A. nidulans secretome with ABP 1c at pH 3–8 after incubation for 1 h at 37 °C. (B) Temperature dependent labeling of day 5 A. nidulans secretome with ABP 1c after incubation at pH 5.0 for 1 h at 25–55 °C. (C) Fluorescent labeling of day 1–5 A. nidulans secretomes with ABP 1c at pH 5.0 after incubation for 1 h at 37 °C. (D) Competitive ABPP of day 5 A. nidulans secretome with acarbose. The secretome was preincubated with acarbose at varying concentrations (30 min, pH 5.0, 37 °C) prior to labeling with ABP 1c (67.5 μM, 1 h, pH 5.0, 37 °C). (E) Competitive ABPP of day 5 A. nidulans secretome with 3a. The secretome was preincubated with 3a at varying concentrations (30 min, pH 5.0, 37 °C) prior to labeling with ABP 1c (67.5 μM, 1 h, pH 5.0, 37 °C).
Conventional industrial starch hydrolysis relies on enzymes that are both thermostable and acid tolerant.44,45 Both of these metrics can be measured directly from the secretome using ABPP, capturing these parameters in the native enzyme environment. Labeling of the day 5 secretome in the presence of McIlvaine buffers at pH between 3.0 and 8.0 revealed pH dependent labeling patterns (Figure 5A). The most intense band (observed at ∼66 kDa) was labeled between pH 4.0 and pH 7.0, while the bands appearing at ∼52 and ∼48 kDa were constrained to labeling between 4.0 and 5.5 and 5.0–7.0, respectively. We next assessed the temperature-dependent labeling of the day 5 A. nidulans secretome at pH 5.0 and at temperatures between 25 and 55 °C (Figure 5B). Within this range the bands at ∼66, ∼52, and ∼48 kDa all showed temperature dependent labeling, with labeling efficiency decreased above 37 °C, presumably due to denaturation of the corresponding proteins at this temperature. Conversely the bands at ∼58 kDa and ∼44 kDa remained present at the highest temperature tested.
We next used cABPP to show the sensitivity of enzyme labeling to both competitive and covalent inhibitors (Figure 5D,E). As mentioned above, cABPP allows for the identification of active enzyme inhibitors, without the need for purification and even if the identity of the active enzyme is unknown.46,47 Preincubation of secretomes for 30 min with either acarbose or 3a prior to labeling with ABP 1c resulted in decreased labeling of specific bands in a concentration dependent manner. Enzyme bands at ∼66, ∼58, ∼52, and ∼48 kDa were all inhibited by either acarbose or 3a, while the band at 44 kDa appeared to be insensitive to these α-amylase inhibitors.
Identification of ABP Labeled Bands
To further determine the specificity of the developed ABPs, we have analyzed the ABP-labeled proteins in human saliva sample. For this purpose, we incubated human saliva sample with biotinylated ABP 1d, a no probe control (DMSO only), or a competitor-inhibited control (first treatment with 1b). ABP 1d-labeled proteins were enriched by using magnetic streptavidin beads. After on-bead tryptic digestion, the resulting peptides were analyzed by LC–MS/MS and matched against the human UniProt database, using the Mascot search engine as previously described.48 As expected, AMY1A (P04745) was clearly abundant in the ABP 1d pulldown experiment (Table S2). Other proteins with high scores were background proteins such as enolase standard and keratin contaminations. Pretreatment of the saliva sample with inhibitor 1b caused a significant loss of AMY1A peptide abundance, and almost no AMY1A was found in the untreated control. These results demonstrate AMY1A in human saliva sample can be selectively labeled and identified via biotinylated ABP 1d.
On the basis of the known content of A. nidulans secretome,43 we tentatively assigned the most prominent ∼66 kDa band labeled with 1c as the mature form of α-amylase AmyB (ANIA_03402); see Table S4 for list of mature α-amylase molecular masses. The two minor bands at ∼52 and ∼48 kDa were also tentatively assigned as AmyA (ANIA_03388) and AmyF (ANIA_0201) α-amylases, respectively. As the wheat starch grown secretome of A. nidulans only contains three amylases,43 the final two minor bands could not be confidently assigned. We however speculate that these are proteins that are highly abundant in the secretome.
To identify the labeled enzymes in the A. nidulans secretome, we also performed an activity-based protein pulldown. The day 5 secretome was treated with either the biotinylated ABP 1d or an equivalent volume of DMSO, and then a pulldown was performed using streptavidin beads, followed by an on bead tryptic digest and LC–MS/MS analysis. All three amylases from the A. nidulans secretome (AmyA, AmyF, and AmyB) were detected at elevated levels in the probe 1d-treated samples relative to the negative control and total secretome, confirming our initial assignment of the major and two minor bands observed by in-gel fluorescence (Table S3). We also detected ChiB, an endochitanase, and AgdB, an α-glucosidase, both of which are highly abundant in the secretome (Table S3). The molecular weights of the detected amylases match well with the labeled bands seen from ABPP with probe 1c. Furthermore, these results suggest the identities of the minor bands seen at 44 and 58 kDa may belong respectively to CbiB and AgdB, the former, which is proteolytically processed from its 108 kDa form to a heterodimer with the catalytic protomer, having a 58 kDa molecular weight.49
Conclusion
We have synthesized a panel of maltobiose-configured epi-cyclophellitol derivatives as mechanism-based covalent inhibitors and ABPs for retaining α-amylases. Kinetic studies of the inhibition of Taka-amylase revealed that the cyclosulfate warhead (3a) is the most potent, compared with its epoxide and aziridine counterparts (1a and 2a). As well, alkylation of O4′ of the nonreducing end sugar in 1a led to compound 1b that inhibits α-amylase up to 10-fold more potently than the parent compound. Compound conformations can strongly influence the inhibitory potencies and selectivities of cyclophellitol analogues. Our X-ray studies have shown that both epoxide and cyclosulfate-based inhibitors bind to the active site nucleophilic residue of Taka-amylase in a 4C1 chair conformation, while the structure of aziridine 2a complex was found in an unprecedented E3 conformation, likely due to the presence of the aziridine alkyl-chain, which might account for its lower potency. The inability of fluorescent ABPs 2b and 2c to modify recombinant human saliva α-amylase further indicated that substituted aziridines are not accepted by the tested amylases. In contrast, epoxide-based ABP 1c selectively labels α-amylases in human saliva and mice tissue in a concentration- and time-dependent manner, with optimum labeling at pH 5.0. In addition, it can also effectively detect α-amylases in fungal secretomes and allow easy readout of pH and temperature dependence and susceptibility to inhibitors. Looking ahead, we speculate that installation of reporter entities at O4′ of the nonreducing end sugar of cyclosulfate-based inhibitor 3a would yield more potent ABPs based on kinetic and crystallographic studies. In conclusion, our maltobiose epi-cyclophellitol ABPs allow activity-based profiling of α-amylases in complex biological samples and may find use in the discovery of new and effective human amylase inhibitors that may have potential as therapeutic reagents for the treatment of type 2 diabetes and in the discovery of new microbial amylases with potentially advantageous properties for biotechnological application.
Acknowledgments
We thank the China Scholarship Council (CSC, Ph.D. Grant to Y.C.), the Royal Society (Ken Murray Research Professorship to G.J.D.), and the Biotechnology and Biological Sciences Research Council (BBSRC) (Grant BB/R001162/1 to G.J.D. and Z.A.). We also thank the Novo Nordisk Foundation Post-doc grant within the Biotechnology-Based Synthesis and Production Research program (Grant NNF17OC0025642). We thank the Diamond Light Source for access to beamlines i04 and i03 (Proposal Numbers mx-18598-21 and mx-18598-25) that contributed to the results presented here and Johan Turkenburg and Sam Hart for coordinating data collection.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications Web site at DOI: (PDF) (XLSX) The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c13059.
Author Contributions
Y.C. and Z.A. contributed equally to this paper.
The authors declare no competing financial interest.
Supplementary Material
References
- Lombard V.; Golaconda Ramulu H.; Drula E.; Coutinho P. M.; Henrissat B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42 (D1), D490–D495. 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang N.; Stebbins G. L.; Rodriguez R. L. Classification and evolution of α-amylase genes in plants. Proc. Natl. Acad. Sci. U. S. A. 1992, 89, 7526–7530. 10.1073/pnas.89.16.7526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brayer G. D.; Luo Y.; Withers S. G. The structure of human pancreatic a-amylase at 1.8 Å resolution and comparisons with related enzymes. Protein Sci. 1995, 4, 1730–1742. 10.1002/pro.5560040908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta R.; Gigras P.; Mohapatra H.; Goswami V. K.; Chauhan B. Microbial α-amylases: a biotechnological perspective. Process Biochem. 2003, 38 (11), 1599–1616. 10.1016/S0032-9592(03)00053-0. [DOI] [Google Scholar]
- Zechel D. L.; Withers S. G. Glycosidase mechanisms: Anatomy of a finely tuned catalyst. Acc. Chem. Res. 2000, 33 (1), 11–18. 10.1021/ar970172+. [DOI] [PubMed] [Google Scholar]
- Nakazawa Y.; Wang Y. J. Acid hydrolysis of native and annealed starches and branch-structure of their Naegeli dextrins. Carbohydr. Res. 2003, 338 (24), 2871–2882. 10.1016/j.carres.2003.09.005. [DOI] [PubMed] [Google Scholar]
- Wang S.; Copeland L. Effect of acid hydrolysis on starch structure and functionality: a review. Crit. Rev. Food Sci. Nutr. 2015, 55 (8), 1081–1097. 10.1080/10408398.2012.684551. [DOI] [PubMed] [Google Scholar]
- Kirk O.; Borchert T. V.; Fuglsang C. C. Industrial enzyme applications. Curr. Opin. Biotechnol. 2002, 13 (4), 345–351. 10.1016/S0958-1669(02)00328-2. [DOI] [PubMed] [Google Scholar]
- van der Maarel M. J. E. C.; van der Veen B.; Uitdehaag J. C. M.; Leemhuis H.; Dijkhuizen L Properties and applications of starch-converting enzymes of the α-amylase family. J. Biotechnol. 2002, 94, 137–155. 10.1016/S0168-1656(01)00407-2. [DOI] [PubMed] [Google Scholar]
- Souza P. M. d.; Magalhães P. d. O. e. Application of microbial α-amylase in industry – a review. Braz. J. Microbiol. 2010, 41, 850–861. 10.1590/S1517-83822010000400004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundarram A.; Murthy T. P. K. α-Amylase production and applications: a review. J. Appl. Environ. Microbiol. 2014, 2, 166–175. 10.12691/jaem-2-4-10. [DOI] [Google Scholar]
- Hmidet N.; El-Hadj Ali N.; Haddar A.; Kanoun S.; Alya S.-K.; Nasri M. Alkaline proteases and thermostable α-amylase co-produced by Bacillus licheniformis NH1: Characterization and potential application as detergent additive. Biochem. Eng. J. 2009, 47 (1–3), 71–79. 10.1016/j.bej.2009.07.005. [DOI] [Google Scholar]
- Rana N.; Walia A.; Gaur A. α-Amylases from microbial sources and its potential applications in various industries. Natl. Acad. Sci. Lett. 2013, 36 (1), 9–17. 10.1007/s40009-012-0104-0. [DOI] [Google Scholar]
- Mooradian A. D.; Thurman J. E. Drug therapy of postprandial hyperglycaemia. Drugs 1999, 57 (1), 19–29. 10.2165/00003495-199957010-00003. [DOI] [PubMed] [Google Scholar]
- Scott L. J.; Spencer C. M. Miglitol: a review of its therapeutic potential in type 2 diabetes mellitus. Drugs 2000, 59, 521–549. 10.2165/00003495-200059030-00012. [DOI] [PubMed] [Google Scholar]
- Chiasson J.-L.; Josse R. G.; Gomis R.; Hanefeld M.; Karasik A.; Laakso M. Acarbose for prevention of type 2 diabetes mellitus: the STOP-NIDDM randomised trial. Lancet 2002, 359 (9323), 2072–2077. 10.1016/S0140-6736(02)08905-5. [DOI] [PubMed] [Google Scholar]
- Li C.; Begum A.; Numao S.; Park K. H.; Withers S. G.; Brayer G. D. Acarbose rearrangement mechanism implied by the kinetic and structural analysis of human pancreatic α-amylase in complex with analogues and their elongated counterparts. Biochemistry 2005, 44, 3347–3357. 10.1021/bi048334e. [DOI] [PubMed] [Google Scholar]
- Nahoum V.; Roux G.; Anton V.; RougÉ P.; Puigserver A.; Bischoff H.; Henrissat B.; Payan F. Crystal structures of human pancreatic α-amylase in complex with carbohydrate and proteinaceous inhibitors. Biochem. J. 2000, 346 (1), 210–208. [PMC free article] [PubMed] [Google Scholar]
- Tarling C. A.; Woods K.; Zhang R.; Brastianos H. C.; Brayer G. D.; Andersen R. J.; Withers S. G. The search for novel human pancreatic alpha-amylase inhibitors: high-throughput screening of terrestrial and marine natural product extracts. ChemBioChem 2008, 9 (3), 433–438. 10.1002/cbic.200700470. [DOI] [PubMed] [Google Scholar]
- Williams L. K.; Zhang X.; Caner S.; Tysoe C.; Nguyen N. T.; Wicki J.; Williams D. E.; Coleman J.; McNeill J. H.; Yuen V.; Andersen R. J.; Withers S. G.; Brayer G. D. The amylase inhibitor montbretin A reveals a new glycosidase inhibition motif. Nat. Chem. Biol. 2015, 11 (9), 691–696. 10.1038/nchembio.1865. [DOI] [PubMed] [Google Scholar]
- Braun C.; Brayer G. D.; Withers S. G. Mechanism-based inhibition of yeast α-glucosidase and human pancreatic α-amylase by a new class of inhibitors. J. Biol. Chem. 1995, 270, 26778–26781. 10.1074/jbc.270.45.26778. [DOI] [PubMed] [Google Scholar]
- Zhang R.; McCarter J. D.; Braun C.; Yeung W.; Brayer G. D.; Withers S. G. Synthesis and testing of 2-deoxy-2,2-dihaloglycosides as mechanism-based inhibitors of α-Glycosidases. J. Org. Chem. 2008, 73, 3070–3077. 10.1021/jo702565q. [DOI] [PubMed] [Google Scholar]
- Zhang R.; Li C.; Williams L. K.; Rempel B. P.; Brayer G. D.; Withers S. G. Directed “in situ” inhibitor elongation as a strategy to structurally characterize the covalent glycosyl-enzyme intermediate of human pancreatic alpha-amylase. Biochemistry 2009, 48 (45), 10752–10764. 10.1021/bi901400p. [DOI] [PubMed] [Google Scholar]
- Caner S.; Zhang X.; Jiang J.; Chen H. M.; Nguyen N. T.; Overkleeft H.; Brayer G. D.; Withers S. G. Glucosyl epi-cyclophellitol allows mechanism-based inactivation and structural analysis of human pancreatic alpha-amylase. FEBS Lett. 2016, 590 (8), 1143–1151. 10.1002/1873-3468.12143. [DOI] [PubMed] [Google Scholar]
- Cravatt B. F.; Wright A. T.; Kozarich J. W. Activity-based protein profiling: from enzyme chemistry to proteomic chemistry. Annu. Rev. Biochem. 2008, 77, 383–414. 10.1146/annurev.biochem.75.101304.124125. [DOI] [PubMed] [Google Scholar]
- Willems L. I.; Overkleeft H. S.; van Kasteren S. I. Current developments in activity-based protein profiling. Bioconjugate Chem. 2014, 25 (7), 1181–1191. 10.1021/bc500208y. [DOI] [PubMed] [Google Scholar]
- Atsumi S.; Umezawa K.; Iinuma H.; Naganawa H.; Nakamura H.; Iitaka Y.; Takeuchi T. Production, isolation and structure determination of a novel beta-glucosidase inhibitor, cyclophellitol, from Phellinus sp. J. Antibiot. 1990, 43 (1), 49–53. 10.7164/antibiotics.43.49. [DOI] [PubMed] [Google Scholar]
- Jiang J.; Kallemeijn W. W.; Wright D. W.; van den Nieuwendijk A.; Rohde V. C.; Folch E. C.; van den Elst H.; Florea B. I.; Scheij S.; Donker-Koopman W. E.; Verhoek M.; Li N.; Schurmann M.; Mink D.; Boot R. G.; Codée J. D. C.; van der Marel G. A.; Davies G. J.; Aerts J.; Overkleeft H. S. In vitro and in vivo comparative and competitive activity-based protein profiling of GH29 alpha-l-fucosidases. Chem. Sci. 2015, 6 (5), 2782–2789. 10.1039/C4SC03739A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willems L. I.; Beenakker T. J.; Murray B.; Scheij S.; Kallemeijn W. W.; Boot R. G.; Verhoek M.; Donker-Koopman W. E.; Ferraz M. J.; van Rijssel E. R.; Florea B. I.; Codée J. D.; van der Marel G. A.; Aerts J. M.; Overkleeft H. S. Potent and selective activity-based probes for GH27 human retaining alpha-galactosidases. J. Am. Chem. Soc. 2014, 136 (33), 11622–11625. 10.1021/ja507040n. [DOI] [PubMed] [Google Scholar]
- Jiang J.; Kuo C. L.; Wu L.; Franke C.; Kallemeijn W. W.; Florea B. I.; van Meel E.; van der Marel G. A.; Codée J. D.; Boot R. G.; Davies G. J.; Overkleeft H. S.; Aerts J. M. Detection of active mammalian GH31 alpha-glucosidases in health and disease using in-class, broad-spectrum activity-based probes. ACS Cent. Sci. 2016, 2 (5), 351–358. 10.1021/acscentsci.6b00057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L.; Jiang J.; Jin Y.; Kallemeijn W. W.; Kuo C.-L.; Artola M.; Dai W.; van Elk C.; van Eijk M.; van der Marel G. A.; Codée J. D. C.; Florea B. I.; Aerts J. M. F. G.; Overkleeft H. S.; Davies G. J. Activity-based probes for functional interrogation of retaining β-glucuronidases. Nat. Chem. Biol. 2017, 13, 867–873. 10.1038/nchembio.2395. [DOI] [PubMed] [Google Scholar]
- McGregor N. G. S.; Artola M.; Nin-Hill A.; Linzel D.; Haon M.; Reijngoud J.; Ram A.; Rosso M. N.; van der Marel G. A.; Codée J. D. C.; van Wezel G. P.; Berrin J. G.; Rovira C.; Overkleeft H. S.; Davies G. J. Rational Design of Mechanism-based inhibitors and activity-based probes for the identification of retaining alpha-L-arabinofuranosidases. J. Am. Chem. Soc. 2020, 142 (10), 4648–4662. 10.1021/jacs.9b11351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröder S. P.; de Boer C.; McGregor N. G. S.; Rowland R. J.; Moroz O.; Blagova E.; Reijngoud J.; Arentshorst M.; Osborn D.; Morant M. D.; Abbate E.; Stringer M. A.; Krogh K.; Raich L.; Rovira C.; Berrin J. G.; van Wezel G. P.; Ram A. F. J.; Florea B. I.; van der Marel G. A.; Codée J. D. C.; Wilson K. S.; Wu L.; Davies G. J.; Overkleeft H. S. Dynamic and functional profiling of xylan-degrading enzymes in Aspergillus secretomes using activity-based probes. ACS Cent. Sci. 2019, 5 (6), 1067–1078. 10.1021/acscentsci.9b00221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Boer C.; McGregor N. G. S.; Peterse E.; Schröder S. P.; Florea B. I.; Jiang J.; Reijngoud J.; Ram A. F. J.; van Wezel G. P.; van der Marel G. A.; Codée J. D. C.; Overkleeft H. S.; Davies G. J. Glycosylated cyclophellitol-derived activity-based probes and inhibitors for cellulases. RSC Chemical Biology 2020, 1 (3), 148–155. 10.1039/D0CB00045K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artola M.; Wu L.; Ferraz M. J.; Kuo C. L.; Raich L.; Breen I. Z.; Offen W. A.; Codée J. D. C.; van der Marel G. A.; Rovira C.; Aerts J.; Davies G. J.; Overkleeft H. S. 1,6-Cyclophellitol cyclosulfates: A new class of irreversible glycosidase inhibitor. ACS Cent. Sci. 2017, 3 (7), 784––793. 10.1021/acscentsci.7b00214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Overkleeft H. S.; van der Marel G. A.; Codée J. D. C. Reagent controlled stereoselective synthesis of alpha-glucans. J. Am. Chem. Soc. 2018, 140 (13), 4632–4638. 10.1021/jacs.8b00669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman H. C.; Kendall E. C.; Clark E. D. Studies on Amylases. I. An examination of methods for the determination of diastatic power. J. Am. Chem. Soc. 1910, 32 (9), 1073–1086. 10.1021/ja01927a010. [DOI] [Google Scholar]
- Takamine J. Diastatic substances from fungus growths. J. Soc. Chem. Ind. 1898, 17 (2), 118–120. [Google Scholar]
- Kato S.; Shimizu-Ibuka A.; Mura K.; Takeuchi A.; Tokue C.; Arai S. Molecular cloning and characterization of an α-amylase from Pichia burtonii15-1. Biosci., Biotechnol., Biochem. 2007, 71 (12), 3007–3013. 10.1271/bbb.70407. [DOI] [PubMed] [Google Scholar]
- Alonso-Gil S.; Coines J.; André I.; Rovira C. Conformational itinerary of sucrose during hydrolysis by retaining amylosucrase. Front. Chem. 2019, 7, 269. 10.3389/fchem.2019.00269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brzozowski A. M.; Davies G. J. Structure of the Aspergillus oryzae α-amylase complexed with the inhibitor acarbose at 2.0 Å resolution. Biochemistry 1997, 36 (36), 10837–10845. 10.1021/bi970539i. [DOI] [PubMed] [Google Scholar]
- Fisher S. Z.; Govindasamy L.; Tu C.; Agbandje-McKenna M.; Silverman D. N.; Rajaniemi H. J.; McKenna R. Structure of human salivary alpha-amylase crystallized in a C-centered monoclinic space group. Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun. 2006, 62 (2), 88–93. 10.1107/S1744309105042491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nekiunaite L.; Arntzen M. O.; Svensson B.; Vaaje-Kolstad G.; Abou Hachem M. Lytic polysaccharide monooxygenases and other oxidative enzymes are abundantly secreted by Aspergillus nidulans grown on different starches. Biotechnol. Biofuels 2016, 9 (1), 187–202. 10.1186/s13068-016-0604-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parashar D.; Satyanarayana T. An insight into ameliorating production, catalytic efficiency, thermostability and starch saccharification of acid-stable alpha-amylases from acidophiles. Front. Bioeng. Biotechnol. 2018, 6, 125. 10.3389/fbioe.2018.00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabb W. D.; Mitchinson C. Enzymes involved in theprocessing of starch to sugars. Trends Biotechnol. 1997, 15, 349–352. 10.1016/S0167-7799(97)01082-2. [DOI] [Google Scholar]
- Wu L.; Armstrong Z.; Schröder S. P.; de Boer C.; Artola M.; Aerts J. M.; Overkleeft H. S.; Davies G. J. An overview of activity-based probes for glycosidases. Curr. Opin. Chem. Biol. 2019, 53, 25–36. 10.1016/j.cbpa.2019.05.030. [DOI] [PubMed] [Google Scholar]
- Armstrong Z.; Kuo C. L.; Lahav D.; Liu B.; Johnson R.; Beenakker T. J. M.; de Boer C.; Wong C. S.; van Rijssel E. R.; Debets M. F.; Florea B. I.; Hissink C.; Boot R. G.; Geurink P. P.; Ovaa H.; van der Stelt M.; van der Marel G. M.; Codée J. D. C.; Aerts J.; Wu L.; Overkleeft H. S.; Davies G. J. Manno-epi-cyclophellitols enable activity-based protein profiling of human alpha-mannosidases and discovery of new Golgi mannosidase II inhibitors. J. Am. Chem. Soc. 2020, 142 (30), 13021–13029. 10.1021/jacs.0c03880. [DOI] [PubMed] [Google Scholar]
- Kuharev J.; Navarro P.; Distler U.; Jahn O.; Tenzer S. In-depth evaluation of software tools for data-independent acquisition-based label-free quantification. Proteomics 2015, 15 (18), 3140–3151. 10.1002/pmic.201400396. [DOI] [PubMed] [Google Scholar]
- Kato N.; Suyama S.; Shirokane M.; Kato M.; Kobayashi T.; Tsukagoshi N. Novel alpha-glucosidase from Aspergillus nidulans with strong transglycosylation activity. Appl. Environ. Microbiol. 2002, 68 (3), 1250–1256. 10.1128/AEM.68.3.1250-1256.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.