

Abstract

A structure–activity relationship study unexpectedly showed that carbonothioates 4a and 4b, obtained by a unique alkaline hydrolysis of 2-alkylthio-oxazolines 3a and 3b, respectively, are a novel scaffold for indoleamine 2,3-dioxygenase 1 (IDO1) inhibitors. Derivatization of the carbonothioates enhanced inhibitory activity against IDO1 and cellular kynurenine production without cytotoxicity and led to the discovery of the related scaffolds carbonodithioates 5 and cyanocarbonimidodithioates 6 as IDO1 inhibitors. Incorporation of an OH group provided the most potent analogue 5i. UV–visible absorption spectroscopy of the Soret band, as well as docking and peptide mapping studies, suggested that these molecules bind to the heme in the active site of IDO1. Our unique IDO1 inhibitors are potential leads for future development.
Keywords: indoleamine 2,3-dioxygenase 1; inhibitor; kynurenine; carbonothioate; carbonodithioate; Soret band
Indoleamine 2,3-dioxygenase 1 (IDO1) is a cytosolic monomeric heme-containing dioxygenase that catalyzes oxidative cleavage of the C-2/C-3 bond of the indole ring of tryptophan (Trp) by addition of oxygen.1,2 This is the initial and rate-limiting step in the catabolism of the essential amino acid Trp to N-formylkynurenine, which is then rapidly converted to kynurenine via the kynurenine pathway. Kynurenine is further transformed into biologically active metabolites such as serotonin, excitotoxin quinolinic acid, N-methyl-d-aspartate (NMDA) receptor antagonist kynurenic acid, and nicotinamide adenine dinucleotide (NAD).3−5 Therefore, IDO1 is likely involved in a variety of physiological processes. IDO1 is expressed ubiquitously, but predominately in immune system cells, and is specifically induced by cytokines in dendritic cells and macrophages at the sites of inflammation.1 Constitutive IDO1 activity is also found at the maternal–fetal interface,6 and thus, IDO1 is thought to regulate acquired local and peripheral immune tolerance in normal and pathological scenarios. Indeed, IDO is overexpressed in a variety of immune-related diseases, including cancer,7 neurodegenerative disorders (e.g., Alzheimer’s disease),8 age-related cataract,9 and HIV encephalitis.10 IDO1 has been shown to play an important role in immune escape by tumors.1 The activation of Trp catabolism by IDO1 leads to the locoregional generation of kynurenine and depletion of Trp. This tumor microenvironment triggers several pathways that suppress effector T cell (Teff) proliferation and promotes differentiation into regulatory T cells (Treg).11 Also, kynurenine is a ligand of human aryl hydrocarbon receptor (AhR).12 AhR regulates the gene transcription of IDO113 and FoxP3, a key transcription factor that controls the differentiation of Treg.14,15 Therefore, the activation of AhR pathways leads to the differentiation and activation of Treg, the induction of anergy in Teff cells, and modulation of the function of antigen-presenting cells.12 Consequently, tumor cells evade the immune response and survive. This is consistent with the observation that increased expression of IDO1 in tumor cells is correlated with poor prognosis for survival in patients with serious ovarian and colorectal cancers.16,17
Increasing interest in IDO1 as a therapeutic target in cancer treatment has led to the identification of several IDO1 inhibitors (Figure 1). 1-Methyltryptophan is a conventional inhibitor with a weak Ki of 34 μM and has entered clinical development.18,19 The most advanced IDO1 inhibitor is epacadostat, which was studied in a phase 3 clinical trial.20 Linrodostat (BMS986205),21 navoximod (GDC-0919),22 PF-06840003,23 and MK-7162 (structure undisclosed)24 are currently in clinical trials. Although results to date are not satisfactory, research on IDO1 inhibitors remains active, including modifying advanced molecules (e.g., epacadostat)25,26 and expanding the chemical space via high-throughput screening campaigns or rational design.27
Figure 1.

Representative IDO1 inhibitors.
We previously reported IDO1 inhibitors that suppress cellular kynurenine production, namely, a derivative of the antihypertensive agent candesartan 1(28) and S-benzylisothiourea 2.29 In addition, our efforts to convert the isothiourea moiety of S-benzylisothiourea 2 to drug-like heterocycles30 provided oxazoline derivative 3a, which exhibits weak recombinant human IDO1 (rhIDO1) inhibition (40% @100 μM). During the course of this derivatization, we attempted alkaline hydrolysis of the 4-COOMe moiety of compound 3a. Contrary to our intention, carbonothioate 4a was obtained in 52% isolated yield. This ring cleavage of oxazoline is common in related compounds: for example, alkaline hydrolysis of 3b provided 4b in similar isolated yield. Carbonothioates 4a and 4b would be formed by the addition of OH– to the imidate of the oxazoline ring, followed by C–N bond cleavage (Scheme 1).
Scheme 1. Ring Cleavage of Oxazoline (3a, 3b) by Alkaline Hydrolysis.

The carbonothioate structure was confirmed using a different synthetic approach: condensation of Boc-Ser-OtBu and benzyl mercaptan with triphosgene, followed by deprotection with HCl, also provided carbonothioate 4b. The NMR and MS spectra of 4b synthesized by the two routes were identical (Scheme 2).
Scheme 2. Synthesis of Compound 4b for Structural Confirmation.

Reagents and conditions: (a) triphosgene, Et2O, RT; (b) PhCH2SH, Et3N, CH2Cl2, RT, 70% yield (2 steps); (c) 4 M HCl, EtOAc, RT, 99% yield.
We performed a preliminary evaluation of the carbonothioates 4a and 4b and, to our surprise, observed remarkable inhibitory activity against rhIDO1 (Table 1). While the inhibition of cell-based IDO1 (kynurenine production in A431 cells) was somewhat weaker than expected given the rhIDO1 inhibitory activity, these results prompted us to derivatize carbonothioate in an effort to enhance its inhibitory activity.
Table 1. Inhibitory Activity against rhIDO1 and Cellular Kynurenine Production by Carbonothioate (4a, 4b).

| cpd | R1 | rhIDO1 IC50 (μM) | KYNa % inhibition | cytotoxb IC50 (μM) |
|---|---|---|---|---|
| 4a | (CH2)2Ph | 0.94 | 44% @10 μM | >20 |
| 4b | H | 4.2 | 50% @10 μM | >20 |
Kynurenine production in A431 cells.
Cytotoxicity to A431 cells.
We initially synthesized the decarboxylated analogue 4c (ClogP 2.10) to increase its lipophilicity, given the extreme hydrophilicity of 4b (ClogP 0.0058). 2,4-Cl2 was also installed on the phenyl ring (4d) as the best substituent based on structure–activity relationship (SAR) studies of the precursor S-benzylisothiourea 2,29 since substituents on the phenyl ring clearly enhanced rhIDO1 inhibition (4a vs 4b). In addition, the sulfur isomer carbonodithioates (5a–5k) and cyanocarbonimidodithioates (6a–6b) with a modified right chain (R3 moiety) were prepared as derivatives of carbonothioate 4. The synthesis is summarized in Scheme 3. The carbonothioates 4c and 4d were synthesized by the condensation of N-Boc-ethanolamine and the corresponding benzyl mercaptan with N,N-carbonyldiimidazole (CDI), followed by deprotection with HCl. Carbonodithioates 5 were also synthesized by condensation of the corresponding thiol with CDI, followed by appropriate deprotection if necessary, similar to the synthesis of carbonothioates 4c and 4d. The synthesis of cyanocarbonimidodithioates 6 involved the introduction of R2PhCH2 and R3 by the corresponding alkyl halide into potassium N-cyanocarbamodithioic acid, which was obtained by reacting CS2 and cyanamide under basic conditions. The syn/anti isomers are tautomer.31
Scheme 3. Synthesis of Compounds 4c–4f, 5a–5k, 6a–6b.

Reagents and conditions: (a) CDI, CH2Cl2, RT; (b) R2-PhCH2SH, CH2Cl2, RT; (c) 4 M HCl in 1,4-dioxane, EtOAc, RT; (d) R3-SH, CH2Cl2, RT; (e) CS2, KOH, EtOH, RT; (f) R3-I, acetone, H2O, RT; (g) R2-PhCH2X (X = Br or Cl), EtOH, RT.
The biological activities of the synthesized compounds are listed in Table 2. Of the carbonothioates, the decarboxylated analogue 4c retained rhIDO1 inhibitory activity. Installation of 2,4-Cl2 (4d) remarkably enhanced rhIDO1 inhibition, as expected. Furthermore, 4d efficiently suppressed kynurenine production in A431 cells without cytotoxicity. The synthetic precursors (Boc-protected analogues 4e, 4f) showed the same trend in activity as the parent compounds (4c, 4d), indicating that 2,4-Cl2 enhances activity.
Table 2. Inhibitory Activity against rhIDO1 and Cellular Kynurenine Production by Carbonothioates (4c–4f), Carbonodithioates (5a–5l), and Cyanocarbonimidodithioates (6a–6b).

| cpd | R2 | R3 | rhIDO1 IC50 (μM) | KYN productiona IC50 (μM) | cytotoxb IC50 (μM) |
|---|---|---|---|---|---|
| 4c | H | CH2CH2NH2 HCl | 3.3 | >10 | >10 |
| 4d | 2,4-Cl2 | CH2CH2NH2 HCl | 0.20 | 2.2 | >10 |
| 4e | H | CH2CH2NHBoc | 3.0 | >10 | >10 |
| 4f | 2,4-Cl2 | CH2CH2NHBoc | 0.59 | 1.4 | >10 |
| 5a | H | CH2CH2NH2 HCl | 5.3 | >10 | >10 |
| 5b | 2,4-Cl2 | CH2CH2NH2 HCl | 0.83 | 1.2 | >10 |
| 5c | H | CH2CH2NHBoc | >10 | >10 | >10 |
| 5d | 2,4-Cl2 | CH2CH2NHBoc | 1.6 | 1.1 | >10 |
| 5e | 2,4-Cl2 | CH2CH2OH | 0.31 | 0.39 | >10 |
| 5f | H | 3-HO-C6H4 | 0.60 | 2.6 | >10 |
| 5g | 2,4-Cl2 | 3-HO-C6H4 | 0.10 | 0.44 | >10 |
| 5h | H | 4-HO-C6H4 | 0.90 | 3.6 | >10 |
| 5i | 2,4-Cl2 | 4-HO-C6H4 | 0.10 | 0.40 | >10 |
| 5j | 2,4-Cl2 | 4-HOCH2–C6H4 | 0.12 | 0.27 | >10 |
| 5k | 2,4-Cl2 | 4-MeO-C6H4 | 0.20 | 0.27 | >10 |
| 5lc | >100 | >10 | >10 | ||
| 6a | H | Me | >10 | 6.1 | >10 |
| 6b | 2,4-Cl2 | Me | 0.90 | 3.7 | >10 |
Kynurenine production in A431 cells.
Cytotoxicity to A431 cells.
Purchased commercial product.
The carbonodithioates 5b and 5d also showed rhIDO1 inhibition and suppression of cellular kynurenine production without cytotoxicity. Their inhibitory activity against rhIDO1 were slightly weaker than that of the corresponding carbonothioates (4d, 4f), but their inhibitory activity against cellular kynurenine production was equal to or more potent than that of carbonothioate. The analogues without 2,4-Cl2 (5a, 5c) were weaker, similar to the corresponding carbonothioates (4c, 4e).
These results encouraged us to modify the right chain (R3). We introduced the OH group (5e–5j) since several known potent IDO1 inhibitors possess an OH moiety to interact with Ser23522 or Ser16732 in the active site of IDO1. Compound 5e (R3 = CH2CH2OH) showed potent inhibitory activity against both rhIDO1 and cellular kynurenine production, without cytotoxicity. The phenolic analogues 5g and 5i and the hydroxymethylphenyl analogue 5j were the most potent rhIDO1 inhibitors and strongly suppressed cellular kynurenine production, as we expected. The same SAR of the R2 moiety was also observed: 2,4-Cl2-benzyl analogues (5g, 5i) were much more potent than benzyl analogues (5f, 5h). In fact, the methylation of OH (5k) did not decrease potency, indicating that an OH/OMe moiety might act as a hydrogen bond acceptor. We simulated the interaction of 5i with rhIDO1 in a docking study (vide infra). Meanwhile, commercially available S,S’-dimethyl dithiocarbonate (5l) was completely inactive against both rhIDO1 and cellular kynurenine production, showing that the hydrophobic benzyl moiety is essential for rhIDO1 inhibition. Finally, cyanocarbonimidodithioate 6b demonstrated moderate inhibitory activity against rhIDO1 and cellular kynurenine production, and the beneficial effect of incorporating 2,4-Cl2 as R2 was also observed (vs 6a).
In the course of these derivatization studies, we initially speculated that compounds 4–6 inhibited rhIDO1 by forming a covalent bond, given their electrophilic structures, and thus, we conducted conventional peptide mapping analysis. Trypsin digestion of the rhIDO1–4a complex and analysis by LCMS/MS allowed mapping of the inhibitor reactive residue. Contrary to our speculation, compound 4a did not covalently bind to the enzyme since no change was observed by MS (data not shown). To investigate the mode of inhibition of our IDO1 inhibitors, we conducted a heme binding study using UV–visible absorption spectroscopy. Heme is essential for IDO1 enzymatic activity, comprises a porphyrin ring with an iron atom at the center, and has characteristic light absorption at around λ = 400 nm (Soret band). The optical properties of the heme group are extremely sensitive to changes in the polarity of the heme environment upon ligand/substrate binding, which changes the spectral properties of the heme.33 Ligand binding typically red-shifts the Soret band λmax. We measured the UV–visible spectra of rhIDO1 with/without rhIDO1 inhibitors (Figure 2). In the absence of inhibitor, the Soret band maximum was at 404 nm, consistent with the literature.34 The addition of compound 5b or 5i clearly red-shifted the Soret band from 404 nm to >420 nm, whereas no apparent shift was observed following the addition of inactive carbonodithioate analogue 5l. These results indicate that our IDO1 inhibitors almost certainly bind to the heme of IDO1 in the active site.
Figure 2.
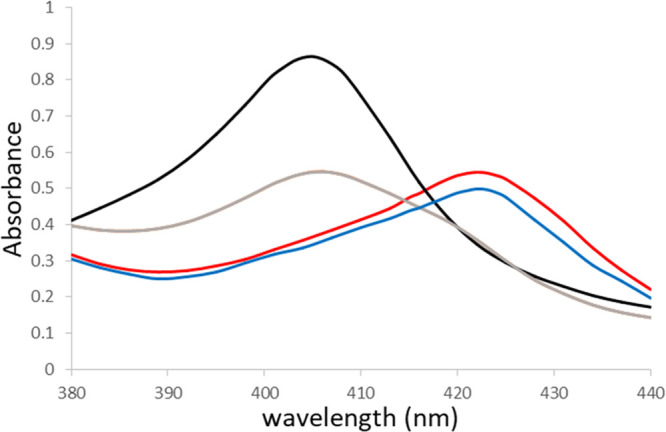
UV spectra of ferric IDO1 with vehicle (black) and compounds 5b (red), 5i (blue), and inactive 5l (gray).
Finally, we conducted a docking study of the most potent analogue 5i with IDO1 using the FRED program (OpenEye, Inc.) and the crystal structure of IDO1/inhibitor (PDB: 6R63) as the template. As shown in Figure 3, 5i coordinates to the heme iron via an oxygen atom. An O-based coordination bond is also observed in the crystal structure of IDO1 complexed with epacadostat.35 The 2,4-Cl2-benzyl group inserts deep into the vertical region of the heme called “pocket A” formed by the hydrophobic residues Phe163, Leu234, Phe164, Val130, and Tyr126. The 4-Cl atom sits next to Cys129, suggesting the formation of a Cl···S halogen bond, similar to other IDO1 inhibitors containing a halogen atom.35,36 This interaction would significantly improve rhIDO1 inhibition by 2,4-Cl2-benzyl analogues. Compound 5i contains a 4-OHPh moiety, with the OH hydrogen bonding with the side chain of Arg231, rather than our intended interaction with Ser235. This interaction is reconciled by the methylation (5k) of OH, thereby retaining potent rhIDO1 inhibition (vide supra). This interaction with Arg231 is also observed in the crystal structure of epacadostat-IDO135 and would contribute to the increased potency of our compounds (such as 5i) against IDO1.
Figure 3.
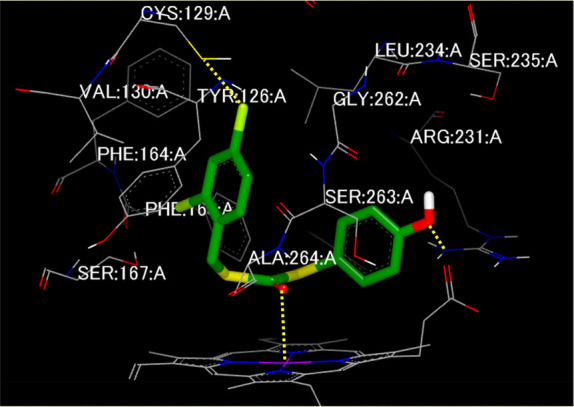
Plausible docking mode for 5i (green) in the active site of IDO1 using the crystal structure of IDO1 with the inhibitor (PDB: 6R63) generated by FRED (Openeye, Inc.). Interactions between IDO1 and 5i are shown with yellow dotted lines.
In conclusion, we found that alkaline hydrolysis of 2-alkylthio-oxazoline (3a, 3b) provided the carbonothioates 4a and 4b, and these molecules unexpectedly inhibited rhIDO1. Derivatization enhanced inhibitory activity against rhIDO1 and cellular kynurenine production without cytotoxicity and led to carbonodithioates 5 and cyanocarbonimidodithioates 6 as novel series of rhIDO1 inhibitors. Furthermore, the incorporation of an OH group in the R3 moiety enhanced activity. Peptide mapping, UV–visible absorption spectroscopy, and docking studies showed that these molecules bind to the heme in the active site of IDO1 and do not covalently bind to the enzyme. Additionally, the incorporation of OH in 5i may allow hydrogen bond formation with Arg231, increasing IDO1 inhibition. The unique IDO1 inhibitors described here are potential leads for future development.
Acknowledgments
The authors thank Ms. Shiori Toyama and Mr. Shun-suke Fukasawa for technical assistance in compound analysis. The authors thank Prof. Yasutada Imamura and Dr. Yongchol Shin (Kogakuin University) for their kind technical advice on UV–visible absorption spectroscopy. The authors also thank Prof. Takaaki Miyaji (Okayama University) for his technical advice on peptide mapping study. The authors thank OpenEye Scientific Software, Inc. (Santa Fe, NM) for providing a free academic license for molecular modeling software.
Glossary
Abbreviations
- IDO1
indoleamine 2,3-dioxygenase 1
- Trp
tryptophan
- Teff
effector T cell
- Treg
regulatory T cells
- AhR
aryl hydrocarbon receptor
- rhIDO1
recombinant human indoleamine 2,3-dioxygenase 1
- SAR
structure activity relationship
- CDI
N,N-carbonyldiimidazole
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00527.
Experimental sections and characterization of the synthesized compounds (PDF)
Author Present Address
∥ H.M.: Present address: Drug Discovery Initiative, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan.
Author Contributions
The manuscript was written through contributions of all authors.
This study was supported by a Japan Society for the Promotion of Science (JSPS) Grant-in-Aid for Scientific Research (C) (grant number JP25460149 to Kenji Matsuno) and by a grant from the Strategic Research Foundation Grant-aided Project for Private Universities (grant number S1411005 to Yasutada Imamura and Kenji Matsuno) from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
The authors declare no competing financial interest.
Supplementary Material
References
- Takikawa O. Biochemical and medical aspects of the indoleamine 2,3-dioxygenase-initiated L-tryptophan metabolism. Biochem. Biophys. Res. Commun. 2005, 338, 12–19. 10.1016/j.bbrc.2005.09.032. [DOI] [PubMed] [Google Scholar]
- Munn D. H.; Mellor A. L. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J. Clin. Invest. 2007, 117, 1147–1154. 10.1172/JCI31178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sono M.; Roach M. P.; Coulter E. D.; Dawson J. H. Heme-containing oxygenases. Chem. Rev. 1996, 96, 2841–2888. 10.1021/cr9500500. [DOI] [PubMed] [Google Scholar]
- Botting N. P. Chemistry and neurochemistry of the kynurenine pathway of tryptophan metabolism. Chem. Soc. Rev. 1995, 24, 401–412. 10.1039/cs9952400401. [DOI] [Google Scholar]
- Stone T. W.; Darlington L. G. Endogenous kynurenines as targets for drug discovery and development. Nat. Rev. Drug Discovery 2002, 1, 609–620. 10.1038/nrd870. [DOI] [PubMed] [Google Scholar]
- Munn D. H.; Zhou M.; Attwood J. T.; Bondarev I.; Conway S. J.; Marshall B.; Brown C.; Mellor A. L. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science 1998, 281, 1191–1193. 10.1126/science.281.5380.1191. [DOI] [PubMed] [Google Scholar]
- Uyttenhove C.; Pilotte L.; Theate I.; Stroobant V.; Colau D.; Parmentier N.; Boon T.; van den Eynde B. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat. Med. 2003, 9, 1269–1274. 10.1038/nm934. [DOI] [PubMed] [Google Scholar]
- Guillemin G. J.; Brew B. J.; Noonan C. E.; Takikawa O.; Cullen K. M. Indoleamine 2,3 dioxygenase and quinolinic acid immunoreactivity in Alzheimer’s disease hippocampus. Neuropathol. Appl. Neurobiol. 2005, 31, 395–404. 10.1111/j.1365-2990.2005.00655.x. [DOI] [PubMed] [Google Scholar]
- Fujigaki H.; Saito K. Inhibition of increased indoleamine 2,3-dioxygenase activity exacerbates neuronal cell death in various CNS disorders. Int. Congr. Ser. 2007, 1304, 314–323. 10.1016/j.ics.2007.07.050. [DOI] [Google Scholar]
- Potula R.; Poluektova L.; Knipe B.; Chrastil J.; Heilman D.; Dou H.; Takikawa O.; Munn D. H.; Gendelman H. E.; Persidsky Y. Inhibition of indoleamine 2,3-dioxygenase (IDO) enhances elimination of virus-infected macrophages in an animal model of HIV-1 encephalitis. Blood 2005, 106, 2382–2390. 10.1182/blood-2005-04-1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munn D. H.; Shafizadeh E.; Attwood J. T.; Bondarev I.; Pashine A.; Mellor A. L. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J. Exp. Med. 1999, 189, 1363–1372. 10.1084/jem.189.9.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opitz C. A.; Litzenburger U. M.; Sahm F.; Ott M.; Tritschler I.; Trump S.; Schumacher T.; Jestaedt L.; Schrenk D.; Weller M.; Jugold M.; Guillemin G. J.; Miller C. L.; Lutz C.; Radlwimmer B.; Lehmann I.; von Deimling A.; Wick W.; Platten M. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 2011, 478, 197–203. 10.1038/nature10491. [DOI] [PubMed] [Google Scholar]
- Nguyen N. T.; Kimura A.; Nakahama T.; Chinen I.; Masuda K.; Nohara K.; Fujii-Kuriyama Y.; Kishimoto T. Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 19961–19966. 10.1073/pnas.1014465107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mezrich J. D.; Fechner J. H.; Zhang X.; Johnson B. P.; Burlingham W. J.; Bradfield C. A. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J. Immunol. 2010, 185, 3190–3198. 10.4049/jimmunol.0903670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spits H.; Mjösberg J. The aryl hydrocarbon receptor: a sentinel safeguarding the survival of immune cells in the gut. Immunity 2012, 36, 5–7. 10.1016/j.immuni.2012.01.004. [DOI] [PubMed] [Google Scholar]
- Okamoto A.; Nikaido T.; Ochiai K.; Takakura S.; Saito M.; Aoki Y.; Ishii N.; Yanaihara N.; Yamada K.; Takikawa O.; Kawaguchi R.; Isonishi S.; Tanaka T.; Urashima M. Indoleamine 2,3-dioxygenase serves as a marker of poor prognosis in gene expression profiles of serous ovarian cancer cells. Clin. Cancer Res. 2005, 11, 6030–6039. 10.1158/1078-0432.CCR-04-2671. [DOI] [PubMed] [Google Scholar]
- Brandacher G.; Perathoner A.; Ladurner R.; Schneeberger S.; Obrist P.; Winkler C.; Werner E. R; Werner-Felmayer G.; Weiss H. G; Göbel G.; Margreiter R.; Königsrainer A.; Fuchs D.; Amberger A. Prognostic value of indoleamine 2,3-dioxygenase expression in colorectal cancer: effect on tumor-infiltrating T cells. Clin. Cancer Res. 2006, 12, 1144–1151. 10.1158/1078-0432.CCR-05-1966. [DOI] [PubMed] [Google Scholar]
- Cady S. G.; Sono M. 1-Methyl-DL-tryptophan, beta-(3-benzofuranyl)-DL-alanine (the oxygen analog of tryptophan), and beta-[3-benzo(b)thienyl]-DL-alanine (the sulfur analog of tryptophan) are competitive inhibitors for indoleamine 2,3-dioxygenase. Arch. Biochem. Biophys. 1991, 291, 326–333. 10.1016/0003-9861(91)90142-6. [DOI] [PubMed] [Google Scholar]
- Muller A. J.; Scherle P. A. Targeting the mechanisms of tumoral immune tolerance with small-molecule inhibitors. Nat. Rev. Cancer 2006, 6, 613–625. 10.1038/nrc1929. [DOI] [PubMed] [Google Scholar]
- Yue E. W.; Sparks R.; Polam P.; Modi D.; Douty B.; Wayland B.; Glass B.; Takvorian A.; Glenn J.; Zhu W.; Bower M.; Liu X-d.; Leffet L.; Wang Q.; Bowman K. J.; Hansbury M. J.; Wei M.; Li Y-l.; Wynn R.; Burn T. C.; Koblish H. K.; Fridman J. S.; Emm T.; Scherle P. A.; Metcalf B.; Combs A. P. INCB24360 (Epacadostat), a highly potent and selective indoleamine-2,3-dioxygenase 1 (IDO1) inhibitor for immuno-oncology. ACS Med. Chem. Lett. 2017, 8, 486–491. 10.1021/acsmedchemlett.6b00391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham K. N.; Yeh S. R. Mapping the binding trajectory of a suicide inhibitor in human indoleamine 2,3-dioxygenase 1. J. Am. Chem. Soc. 2018, 140, 14538–14541. 10.1021/jacs.8b07994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S.; Waldo J. P.; Jaipuri F. A.; Marcinowicz A.; Van Allen C.; Adams J.; Kesharwani T.; Zhang X-x.; Metz R.; Oh A. J.; Harris S. F.; Mautino M. R. Discovery of clinical candidate (1R,4r)-4-((R)-2-((S)-6-fluoro-5H-imidazo[5,1-a]isoindol-5-yl)-1-hydroxyethyl)cyclohexan-1-ol (Navoximod), a potent and selective inhibitor of indoleamine 2,3-dioxygenase 1. J. Med. Chem. 2019, 62, 6705–6733. 10.1021/acs.jmedchem.9b00662. [DOI] [PubMed] [Google Scholar]
- Crosignani S.; Bingham P.; Bottemanne P.; Cannelle H.; Cauwenberghs S.; Cordonnier M.; Dalvie D.; Deroose F.; Feng J. L.; Gomes B.; Greasley S.; Kaiser S. E.; Kraus M.; Négrerie M.; Maegley K.; Miller N.; Murray B. W.; Schneider M.; Soloweij J.; Stewart A. E.; Tumang J.; Torti V. R.; Van Den Eynde B.; Wythes M. discovery of a novel and selective indoleamine 2,3-dioxygenase (IDO-1) inhibitor 3-(5-fluoro-1H-indol-3-yl)pyrrolidine-2,5-dione (EOS200271/PF-06840003) and its characterization as a potential clinical candidate. J. Med. Chem. 2017, 60, 9617–9629. 10.1021/acs.jmedchem.7b00974. [DOI] [PubMed] [Google Scholar]
- Study of MK-7162 in combination with pembrolizumab (MK-3475) in adult participants with advanced solid tumors (MK-7162-002). ClinicalTrials.gov identifier: NCT03364049.
- Zhang H-j.; Liu K.; Pu Q-l.; Achab A.; Ardolino M. J.; Cheng M-g.; Deng Y-q.; Doty A. C.; Ferguson H.; Fradera X.; Knemeyer I.; Kurukulasuriya R.; Lam Y-h.; Lesburg C. A.; Martinot T. A.; McGowan M. A.; Miller J. R.; Otte K.; Biju P. J.; Sciammetta N.; Solban N.; Yu W-s.; Zhou H.; Wang X.; Bennett D. J.; Han Y-x. Discovery of Amino-cyclobutarene-derived indoleamine-2,3-dioxygenase 1 (IDO1) inhibitors for cancer immunotherapy. ACS Med. Chem. Lett. 2019, 10, 1530–1536. 10.1021/acsmedchemlett.9b00344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steeneck C.; Kinzel O.; Anderhub S.; Hornberger M.; Pinto S.; Morschhaeuser B.; Braun F.; Kleymann G.; Hoffmann T. Discovery of hydroxyamidine based inhibitors of IDO1 for cancer immunotherapy with reduced potential for glucuronidation. ACS Med. Chem. Lett. 2020, 11, 179–187. 10.1021/acsmedchemlett.9b00572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serafini M.; Torre E.; Aprile S.; Grosso E. D.; Gesù A.; Griglio A.; Colombo G.; Travelli C.; Paiella S.; Adamo A.; Orecchini E.; Coletti A.; Pallotta M. T.; Ugel S.; Massarotti A.; Pirali T.; Fallarini S. Discovery of highly potent benzimidazole derivatives as indoleamine 2,3-dioxygenase-1 (IDO1) inhibitors: from structure-based virtual screening to in vivo pharmacodynamic activity. J. Med. Chem. 2020, 63, 3047–3065. 10.1021/acs.jmedchem.9b01809. [DOI] [PubMed] [Google Scholar]
- Matsuno K.; Yamazaki H.; Isaka Y.; Takai K.; Unno Y.; Ogo N.; Ishikawa Y.; Fujii S.; Takikawa O.; Asai A. Novel candesartan derivatives as indoleamine-2,3-dioxygenase inhibitors. MedChemComm 2012, 3, 475–479. 10.1039/c2md00278g. [DOI] [Google Scholar]
- Matsuno K.; Takai K.; Isaka Y.; Unno Y.; Sato M.; Takikawa O.; Asai A. S-Benzylisothiourea derivatives as small-molecule inhibitors of indoleamine-2,3-dioxygenase. Bioorg. Med. Chem. Lett. 2010, 20, 5126–5129. 10.1016/j.bmcl.2010.07.025. [DOI] [PubMed] [Google Scholar]
- Fukuda M.; Sasaki T.; Hashimoto T.; Miyachi H.; Waki M.; Asai A.; Takikawa O.; Ohno O.; Matsuno K. Cyclic analogue of S-benzylisothiourea that suppresses kynurenine production without inhibiting indoleamine 2,3-dioxygenase activity. Bioorg. Med. Chem. Lett. 2018, 28, 2846–2849. 10.1016/j.bmcl.2018.07.034. [DOI] [PubMed] [Google Scholar]
- Walek W.; Preiß A.; Dietzel S. D-NMR-Untersuchungen zur E/Z-Isomerie in unsymmetrisch substituierten Cyanimidodithiokohlensäureestern. Z. Chem. 1978, 18, 144–145. 10.1002/zfch.19780180412. [DOI] [Google Scholar]
- Brant M. G.; Goodwin-Tindall J.; Stover K. R.; Stafford P. M.; Wu F.; Meek A. R.; Schiavini P.; Wohnig S.; Weaver D. F. Identification of Potent indoleamine 2,3-dioxygenase 1 (IDO1) inhibitors based on a phenylimidazole scaffold. ACS Med. Chem. Lett. 2018, 9, 131–136. 10.1021/acsmedchemlett.7b00488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenkman J. B.; Sligar S. G.; Cinti D. L. Substrate interaction with cytochrome P-450. Pharmacol. Ther. 1981, 12, 43–71. 10.1016/0163-7258(81)90075-9. [DOI] [PubMed] [Google Scholar]
- Röhrig U. F.; Majjigapu S. R.; Grosdidier A.; Bron S.; Stroobant V.; Pilotte L.; Colau D.; Vogel P.; Van den Eynde B. J.; Zoete V.; Michielin O. Rational design of 4-aryl-1,2,3-triazoles for indoleamine 2,3-dioxygenase 1 inhibition. J. Med. Chem. 2012, 55, 5270–5290. 10.1021/jm300260v. [DOI] [PubMed] [Google Scholar]
- Lewis-Ballester A.; Pham K. N.; Batabyal D.; Karkashon S.; Bonanno J. B.; Poulos T. L.; Yeh S.-R. Structural insights into substrate and inhibitor binding sites in human indoleamine 2,3-dioxygenase 1. Nat. Commun. 2017, 8, 1693. 10.1038/s41467-017-01725-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Röhrig U. F.; Majjigapu S. R.; Vogel P.; Zoete V.; Michielin O. Challenges in the discovery of indoleamine 2,3-dioxygenase 1 (IDO1) inhibitors. J. Med. Chem. 2015, 58, 9421–9437. 10.1021/acs.jmedchem.5b00326. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.