

Abstract
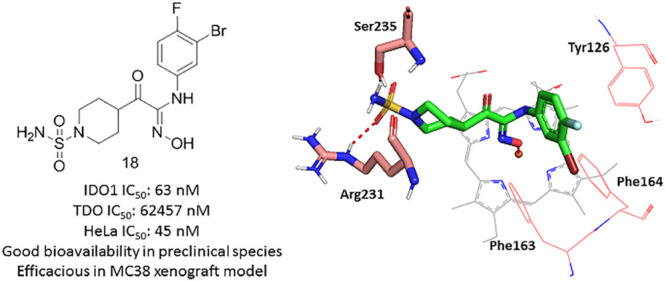
In this study, a series of novel hydroxyamidine derivatives were identified as potent and selective IDO1 inhibitors by structure-based drug design. Among them, compounds 13–15 and 18 exhibited favorable enzymatic and cellular activities. Compound 18 showed improved bioavailability in mouse, rat, and dog (F% = 44%, 58.8%, 102.1%, respectively). With reasonable in vivo pharmacokinetic properties, compound 18 was further evaluated in a transgenic MC38 xenograft mouse model. The combination of compound 18 with PD-1 monoclonal antibody showed a synergistic antitumor effect. These data indicated that compound 18 as a potential cancer immunotherapy agent should warrant further investigation.
Keywords: IDO1, cancer immunotherapy, hydroxyamidine derivatives, lead optimization, structure-based drug design, bioisostere
Over the past two decades, immunotherapy has been transformed into a mainstream treatment method with great potential for a variety of cancers.1 However, there are occasions where the immune system cannot effectively control the development of tumors due to immune tolerance. Indoleamine 2,3-dioxygenase 1 (IDO1), an immune regulatory enzyme, plays an important role in regulating the immune system through the control of the kynerenine pathway.2−4
IDO1 is a heme-containing monomeric enzyme that controls the rate-limiting step of catabolizing tryptophan to N-kynurenine along the kynurenine pathway, which is responsible for local immunosuppression.5−7 Plenty of studies indicate that the abnormal expression of IDO1 is related to tumor cells evading the immune system. IDO1 can oxidize and destroy tryptophan, which is an important amino acid for T cell activation. In this way, T cells lose the ability to kill tumors. In principle, blocking IDO1 can active T-cells and promote the immune system to kill cancer cells. Therefore, IDO1 is an attractive target for cancer immunotherapy.8
Epacadostat (INCB-24360) is developed as a selective IDO1 inhibitor. It has been used with checkpoint modulators for cancer treatment in clinical studies and showed an early sign of benefit in phase I/II trials.9−11 However, epacadostat in combination with pembrolizumab in the ECHO-301 phase III trial has failed to increase the overall and progression-free survival when compared to pembrolizumab alone.12 The disappointing phase III results have cooled down the research interest in the IDO1 inhibitors. However, IDO1 related therapy is still a promising field, as evidenced by multiple ongoing clinical trials from several companies.13,14 For example, an IDO1 inhibitor from Bristol-Myers Squibb, BMS-986205, is currently in an active phase 3 trial in Muscle Invasive Bladder Cancer (MIBC).15
The crystal structure of IDO1 in complex with epacadostat was published in 2018 with a PDB entry of 6E40,16 which opens the door for structure-based design of novel IDO1 inhibitors. In this crystal structure, epacadostat is positioned in the active pocket by three key contacts: (1) a π–π interaction between its substituted phenyl ring and the residue Tyr126; (2) a hydrogen bond formed by sulfamide and Arg231; (3) a dative bond formed between the N-hydroxylamidine oxygen and the heme iron (Figure 1). The furazan ring is often found in pyrotechnic compounds and propellants but is rarely used in medicines.17 Up to now, most medchem efforts have been focused on optimizing the hydroxyamidine motifs as well as the halogenated phenyl region.18−22 The sulfamide side chain and core modification especially the furazan ring replacement remain to be explored. So, herein, we describe our work that has led to the identification of novel IDO1 inhibitors by bioisosteric replacements of the furazan group, as well as the alternative groups to the sulfamide side chain.
Figure 1.
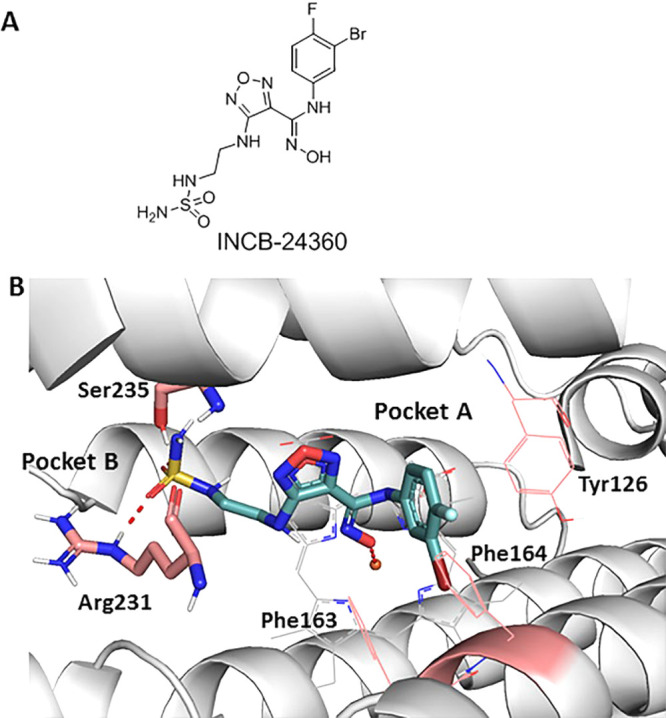
(A) Hydroxyamidine derivative as IDO1 inhibitor; (B) INCB-24360 (navy) binding mode in the crystal structure (PDB 6E40).
We designed a set of electron-withdrawing carbonyl groups to replace the furazan ring (compounds 1–6; the synthesis route of compounds 1–6 is shown in Schemes S1–S6), aiming to keep the acidity of the hydroxylamidine group. Subsequently, biological assays were employed to evaluate the biological activities of compounds 1–6, including enzymatic assays with purified recombinant human IDO1/TDO proteins and cellular IDO1 inhibition assays using HeLa cell lines. As shown in Table 1, compound 1 with thiazole substitution exhibited the best enzymatic activity (IDO1 IC50 = 51 nM) among those designs. It showed micromolar level activity in the HeLa cell line, which could be related to the membrane permeability of the compound. Compounds 2–4 with aromatic or heterocyclic aromatic substitutions also showed less potencies compared to epacadostat in enzymatic and cellular assays. Compound 5 with saturated six-member ring showed a modest enzymatic activity, and the best cellular activity among the designed compounds. As shown by the structure–activity relationship (SAR) data of compound 6, the carbonyl group of compound 5 was quite important for its enzymatic and cellular potencies.
Table 1. SAR of the Furazan Ring Replacement Groups with Charge and pKa Data.
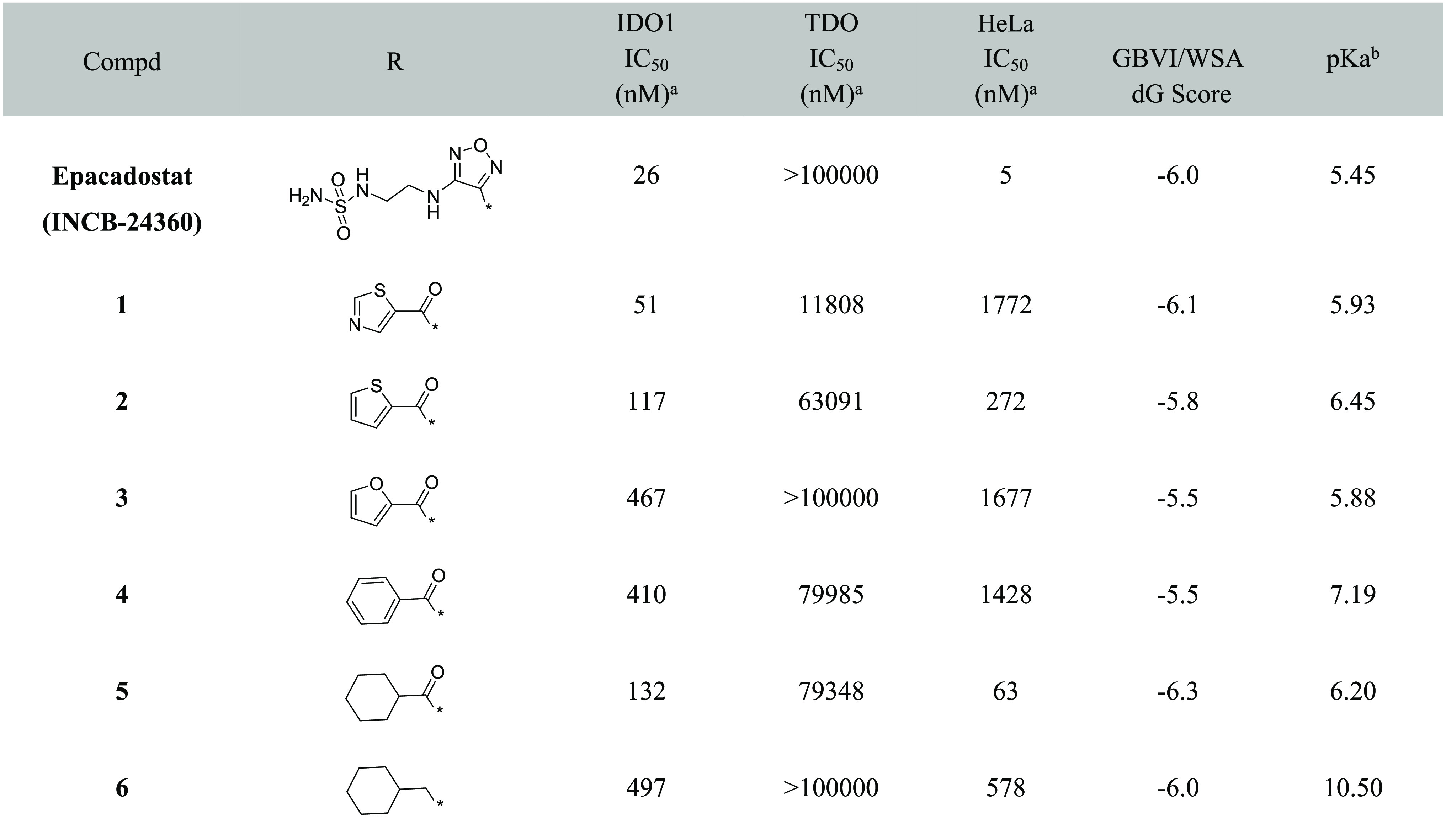
A molecular modeling study was carried out to further understand the SAR of these compounds. It was well-known that the interaction between the deprotonated oxygen and the heme iron is important for biological activity.18,19 So we hypothesized that the oxygen of the N-hydroxyamidine was in the deprotonated state and the heme iron was in its ferrous state (Fe2+). Compounds 1–6 were docked into the binding pocket using MOE.23,24 A commonly used docking score in MOE, GBVI/WSA dG28 alone, could not be used to distinguish actives from inactives (Table 1). Then, the pKa values25 of the heme interaction oxygens from each compounds were calculated; these are shown in Table 1. The calculated pKa values showed some correlation with the enzymatic activity. All the activities were similar to that of the reference compound except compound 3 which might be due to suboptimal binding reflected in the weaker docking score.
Based on the enzymatic and cellular data, as well as the calculated pKa, compound 5 was selected as the starting point for the next round SAR study. A series of cyclohexane substitutions were designed and synthesized (Table 2, compounds 7–10, Schemes S7–S10), none of them showed improved enzymatic potency. As inspired by our previous work,26 substituted piperidinyls were investigated (Table 2). Weak electron-withdrawing substitutions such as benzene (compound 11, Scheme S11) and benzaldehyde (compound 12,Scheme S12) showed similar potencies in IDO1 enzymatic assays as compound 5, with reduced cellular potencies. Interestingly, the phenylformamide substitution (compound 13, Scheme S13) showed 3-fold increase in the enzymatic potency compared to compound 5. Further substitution at the para-position with 1-methylpyrazol-4-yl (compound 14, Scheme S14) resulted in a 10-fold and a 4-fold increase in the enzymatic and cellular potencies, respectively. The benzylformamide substitution (compound 15, Scheme S15) and the sulfonyl substitution (compound 18, Scheme 1) showed similar potency levels in both enzymatic and cellular assays as compound 14.
Table 2. Structure–Activity Relationship Data of Cyclohexane and Piperidine Substituted Hydroxylamidine.

Values are expressed as the mean of at least two independent determinations.
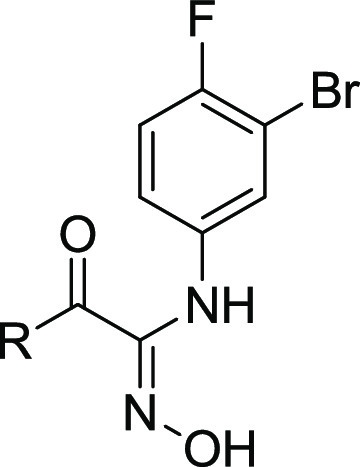
Scheme 1. Synthesis of Compound 18.

Reagents and conditions: (a) SeO2, 1,4-dioxane, 80 °C, 16 h; (b) NH2OH·HCl, K2CO3, CH3OH, rt, 2 h; (c) NCS, DMF, rt, 16 h; (d) 3-bromo-4-fluoroaniline, EtOH, rt, 3 h; (e) 4 M HCl in 1,4-dioxane, 2 h; (f) tert-butyl chlorosulfonylcarbamate, Et3N, DCM, 0 °C, 1 h; (g) 4 M HCl in 1,4-dioxane, MeOH, rt, 1 h.
To further analyze binding poses of the designed compounds, we also performed molecular modeling studies on compound 14 and 18. As shown in Figure 2, both compounds shared a similar binding pose in the pocket A. As a result, the deprotonated oxygen of the N-hydroxylamidine was positioned to bind the heme iron similarly as epacadostat. In the pocket B, compound 18 formed the aformentioned hydrogen bond with the residue Arg231 (Figure 2B). Interestingly, a significant cation−π interaction was formed at a distance of 2.47 Å between the Arg231 and the phenyl group in compound 14 (Figure 2A). Cation−π interactions were quite common in proteins, protein–ligands and protein–DNA complexes, and important for protein folding, molecular recognition and catalysis.29 Thus, it was reasonable to expect that the similar cation-π interactions in the pocket B could contribute to the improvement of binding potencies in compounds, 13–16.
Figure 2.
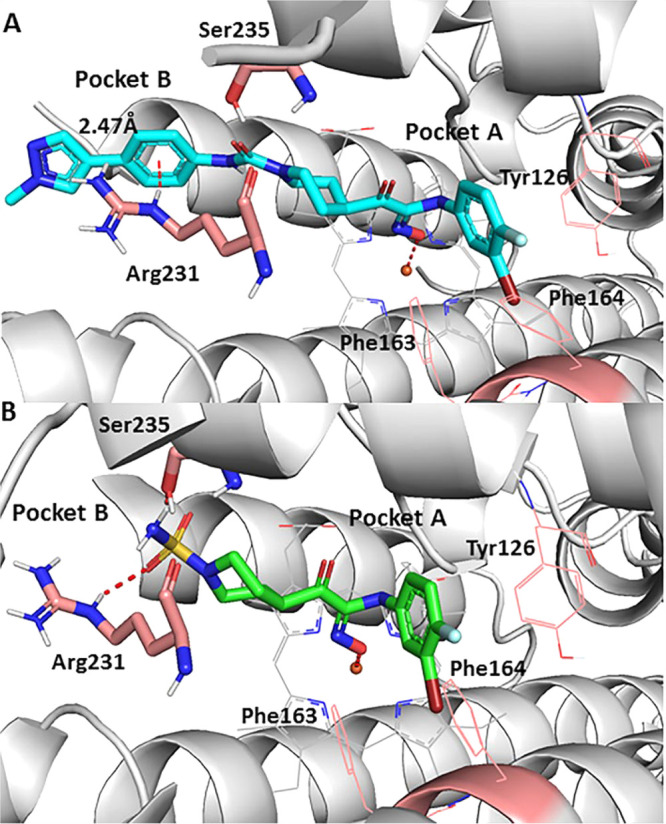
Molecular docking of active compounds binding to the IDO1 active site (PDB code: 6E40). (A) Compound 14 (cyan); (B) compound 18 (purple).
To further evaluate the ADMET properties of compounds 13–15 and 18, we profiled them in CYP and hERG inhibition assays. As shown in Table S2, all the compounds except 15 had clean CYP and hERG profiles. In addition to compound 18, we selected the most potent compound 14 for further in vivo studies among the compounds forming cation−π interactions. Two animal models (rat and dog) were used to evaluate the pharmacokinetics of those two compounds. As listed in Table 3, compound 14 showed poor oral pharmacokinetics in dog, while compound 18 had a better profile and good oral exposure in both species.
Table 3. Oral Pharmacokinetic Profiles of Compound 14 and 18.
| rat
PK@3mg/kg |
dog
PK@2mg/kg |
|||||
|---|---|---|---|---|---|---|
| compd | Cmax (ng/mL) | AUC (ng/mL·h) | t1/2 (h) | Cmax (ng/mL) | AUC (ng/mL·h) | t1/2 (h) |
| 14 | 173 | 1034 | 5.08 | 140 | 255 | 1.10 |
| 18 | 179 | 1527 | 6.14 | 742 | 3633 | 3.04 |
The synthesis route and in vitro and in vivo Profile for compound 18 are shown in Scheme 1 and Table 4. In vitro data indicated that compound 18 was a highly potent and selective IDO1 inhibitor with clean CYP and hERG profiles. Its pharmacokinetic profiles in animal models (mouse, rat, and dog) demonstrated an increased oral exposure and bioavailability from mouse, rat, to dog (F = 44%, 58%, and 102.1%, respectively). Meanwhile, compared with epacadostat, compound 18 exhibited a superior pharmacokinetic profile in a nonrodent species (dog) with lower clearance and better bioavailability. Compound 18 had the potential to show better pharmacokinetic profile in humans than epacadostat.
Table 4. Profiling of Compound 18.
| assay | 18 | INCB-24360 (epacadostat) |
|---|---|---|
| enzymatic IDO1 IC50 (nM) | 63 | 26 |
| enzymatic TDO IC50 (nM) | 62 457 | >10 000 |
| cellular HeLa IC50 (nM) | 45 | 5.3 |
| CYP inhibition (1A2, 2C9, 2C19, 2D6, 3A4) | >10uM | >10uM |
| hERG | >30 uM | >30 uM |
| PPB (rat/dog/human) | 92.4%/96.0%/96.4% | 97.0%/97.5%/98.5% |
| liver microsome stability (rat/human) T1/2 (min) | 93.4/348.7 | 73.1/293 |
| mouse PK@3mg/kg | ||
| Cmax (ng/mL) | 127 | 253 |
| AUC (ng/mL·h) | 358 | 1016 |
| t1/2 (h) | 1.21 | 2.41 |
| Cl (μL/min/mg) | 61.2 | 49.2 |
| bioavailability (F%) | 44% | 44% |
| rat PK@3mg/kg | ||
| Cmax (ng/mL) | 179 | 133 |
| AUC (ng/mL·h) | 1527 | 807 |
| t1/2 (h) | 6.14 | 2.73 |
| Cl (μL/min/mg) | 16.7 | 29 |
| bioavailability (F%) | 58.8% | 55% |
| dog PK@2mg/kg | ||
| Cmax (ng/mL) | 742 | 245 |
| AUC (ng/mL·h) | 3633 | 676 |
| t1/2 (h) | 3.04 | 5.03 |
| Cl (μL/min/mg) | 9.3 | 36.6 |
| bioavailability (F%) | 102.1% | 50% |
To further understand the mechanism of action, the in vivo pharmacodynamics (PD) study of compound 18 was carried out in a mouse model. After administrated orally to C57 mice (300 mg/kg single dose), compound 18 was able to reduce the level of kynurenine down to 87.8% at 2 h after dosing (Figure 3). The concentration change of kynurenine was proportional to the exposure level of the compound.
Figure 3.
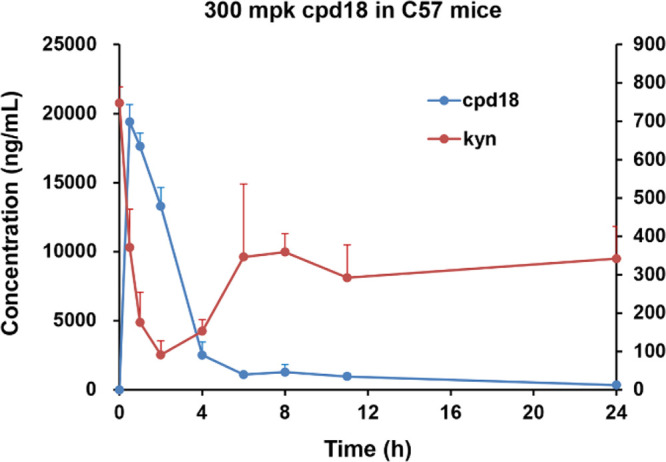
Kynurenine reduction of oral administration of compound 18 in the C57 mouse model.
The antitumor effects of compound 18 in combination with PD-1 antibody were further evaluated using the MC38 tumor growth inhibition model in hPD-1 transgenic mice. Due to the lower exposure seen in mice, higher doses of compound 18 were used to match the exposure level of epacadostat. As shown in Figure 4, Oral treatment of compound 18 combined with PD-1 antibody (PD-1, 3 mg/kg, ip, qod ×8; compound 18, 300 or 600 mg/kg, po, bid ×14) showed good dose-dependent tumor growth inhibition (150 mg/kg, TGI = 60.3%; 300 mg/kg, TGI = 71.7%; 600 mg/kg, TGI = 86.8%). The PD-1 combo treatment groups with 300 or 600 mg/kg compound 18 showed better antitumor efficacy compared to either PD-1 alone (3 mg/kg, ip, qod ×8, TGI = 57.3%) or the combination usage of epacadostat and PD-1 (PD-1, 3 mg/kg, ip, qod ×8; epacadostat, 100 mg/kg, po bid ×14, TGI = 66.9%). No body weight losses were observed in all the treatment groups.
Figure 4.
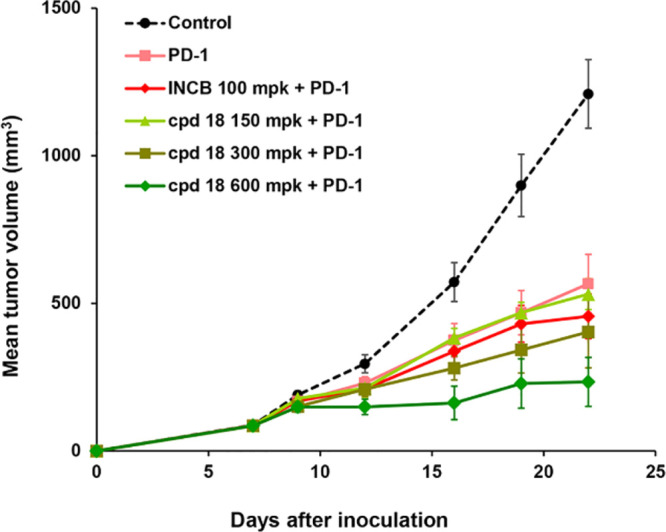
Efficacy study of compound 18 in combination with PD-1 monoclonal antibody in the MC38 xenograft model in hPD-1 transgenic mice.
In summary, we developed a series of novel hydroxyamidine based IDO1 inhibitors using the structure-based drug design approach. Among these derivatives, compounds 14 and 18 showed favorable enzymatic and cellular activities against IDO1, which indicated that the carbonyl group could be used as a bioisostere replacement for the furazan ring in drug design. As compound 14 showed a poor dog PK, further in vivo studies were focused on compound 18. In the transgenic MC38 xenograft model, compound 18 was orally efficacious in combination with PD-1 monoclonal antibody and showed a synergistic antitumor effect. Together with the increased bioavailability from rodent to larger nonrodent animals, these in vivo PD and efficacy studies demonstrated that compound 18 warrant further investigation as a potential add-on cancer immunotherapy agent to PD-1 antibody.
Acknowledgments
We thank Wang Jingfang for proof reading the manuscript and Chen Yiqian, Jiang Hongjian, Qin Qiang, Xu Anchao, Zhang Baolei, Yuan Jijun, Wang Dan, Mao Yuchang, Zhu Ying, Feng Jun, Wang Qian, Yang Changyong, Fan Yuanmin, and Liu Xiaolan for their contributions to the project.
Glossary
Abbreviations
- IDO1
Indoleamine 2, 3-dioxygenase 1
- TDO1
tryptophan 2,3-dioxygenase 1
- MOE
Molecular Operating Environment
- PD-1
programmed death 1
- DMHH
N,O-dimethylhydroxylamine hydrochloride
- DMAP
4-dimethylaminopyridine
- DCM
dichloromethane
- NCS
N-chlorosuccinimide
- EDC
3-(ethyliminomethylideneamino)-N,N-dimethylpropan-1-amine
- THF
tetrahydrofuran
- DMF
dimethylformamide
- SAR
structure–activity relationship
- CYP
cytochrome p450 enzyme
- hERG
human ether-a-go-go-related gene
- PPB
plasma protein bonding
- PK
pharmacokinetic
- po
orally
- ip
intraperitoneally
- bid
twice daily
- qod
every other day
- TGI
tumor growth inhibition.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00443.
Biological assays, pharmacokinetic assays, in vivo efficacy study, experimental procedures, and analytical data for compound 18 (PDF)
Author Present Address
H.L.: Room 102, No. 51, 560 Heqing Rd, Minhang District, Shanghai, China.
Author Present Address
W.T.: Building 37, Lane 1000, Zhangheng Road, Pudong New District, Shanghai, China.
Author Present Address
Y.Z.: BeiGene (Beijing) Co., Ltd., Beijing, China.
Author Contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Khalil D. N.; Smith E. L.; Brentjens R. J.; Wolchok J. D. The Future of Cancer Treatment: Immunomodulation, CARs and Combination Immunotherapy. Nat. Rev. Clin. Oncol. 2016, 13, 273–290. 10.1038/nrclinonc.2016.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munn D. H.; Mellor A. L. Indoleamine 2,3 Dioxygenase and Metabolic Control of Immune Responses. Trends Immunol. 2013, 34, 137–143. 10.1016/j.it.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung A. W. S.; Terentis A. C.; King N. J. C.; Thomas S. R. Role of Indoleamine 2,3-Dioxygenase in Health and Disease. Clin. Sci. 2015, 129, 601–672. 10.1042/CS20140392. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Wang B.; Liu J.; Sun P.; Liu H. An Overview on the Methods of Determining the Activity of Indoleamine 2, 3-Dioxygenase 1. J. Drug Target 2019, 27, 724–731. 10.1080/1061186X.2018.1523416. [DOI] [PubMed] [Google Scholar]
- KHK2455 (IDO Inhibitor) Plus Avelumab in Adult Subjects With Advanced Bladder Cancer. ClinicalTrials.gov, 2019. 10.31525/ct1-nct03915405. [DOI]
- Takikawa O.; Yoshida R.; Kido R.; Hayaishi O. Tryptophan Degradation in Mice Initiated by Indoleamine 2,3-Dioxygenase. J. Biol. Chem. 1986, 261, 3648–3653. 10.1016/S0021-9258(17)35696-X. [DOI] [PubMed] [Google Scholar]
- Platten M.; Nollen E. A. A.; Röhrig U. F.; Fallarino F.; Opitz C. A. Tryptophan Metabolism as a Common Therapeutic Target in Cancer, Neurodegeneration and Beyond. Nat. Rev. Drug Discovery 2019, 18, 379–401. 10.1038/s41573-019-0016-5. [DOI] [PubMed] [Google Scholar]
- Gostner J. M.; Becker K.; Überall F.; Fuchs D. The Potential of Targeting Indoleamine 2,3-Dioxygenase for Cancer Treatment. Expert Opin. Ther. Targets 2015, 19, 605–615. 10.1517/14728222.2014.995092. [DOI] [PubMed] [Google Scholar]
- Beatty G. L.; O’Dwyer P. J.; Clark J.; Shi J. G.; Bowman K. J.; Scherle P. A.; Newton R. C.; Schaub R.; Maleski J.; Leopold L.; Gajewski T. F. First-in-Human Phase I Study of the Oral Inhibitor of Indoleamine 2,3-Dioxygenase-1 Epacadostat (INCB024360) in Patients with Advanced Solid Malignancies. Clin. Cancer Res. 2017, 23, 3269–3276. 10.1158/1078-0432.CCR-16-2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristeleit R.; Davidenko I.; Shirinkin V.; El-Khouly F.; Bondarenko I.; Goodheart M. J.; Gorbunova V.; Penning C. A.; Shi J. G.; Liu X.; Newton R. C.; Zhao Y.; Maleski J.; Leopold L.; Schilder R. J. A Randomised, Open-Label, Phase 2 Study of the IDO1 Inhibitor Epacadostat (INCB024360) versus Tamoxifen as Therapy for Biochemically Recurrent (CA-125 Relapse)–Only Epithelial Ovarian Cancer, Primary Peritoneal Carcinoma, or Fallopian Tube Cancer. Gynecol. Oncol. 2017, 146, 484–490. 10.1016/j.ygyno.2017.07.005. [DOI] [PubMed] [Google Scholar]
- Komiya T.; Huang C. H. Updates in the Clinical Development of Epacadostat and Other Indoleamine 2,3-Dioxygenase 1 Inhibitors (IDO1) for Human Cancers. Front. Oncol. 2018, 8, 423. 10.3389/fonc.2018.00423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long G. V.; Dummer R.; Hamid O.; Gajewski T. F.; Caglevic C.; Dalle S.; Arance A.; Carlino M. S.; Grob J.-J.; Kim T. M.; Demidov L.; Robert C.; Larkin J.; Anderson J. R.; Maleski J.; Jones M.; Diede S. J.; Mitchell T. C. Epacadostat plus Pembrolizumab versus Placebo plus Pembrolizumab in Patients with Unresectable or Metastatic Melanoma (ECHO-301/KEYNOTE-252): A Phase 3, Randomised, Double-Blind Study. Lancet Oncol. 2019, 20, 1083–1097. 10.1016/S1470-2045(19)30274-8. [DOI] [PubMed] [Google Scholar]
- Neoadjuvant Celecoxib in Newly Diagnosed Patients With Endometrial Carcinoma. ClinicalTrials.gov, 2019. 10.31525/ct1-nct03896113. [DOI]
- Luke J. J.; Tabernero J.; Joshua A.; Desai J.; Varga A. I.; Moreno V.; Gomez-Roca C. A.; Markman B.; De Braud F. G.; Patel S. P.; Carlino M. S.; Siu L. L.; Curigliano G.; Liu Z.; Ishii Y.; Wind-Rotolo M.; Basciano P. A.; Azrilevich A.; Gelmon K. A. BMS-986205, an Indoleamine 2, 3-Dioxygenase 1 Inhibitor (IDO1), in Combination with Nivolumab (Nivo): Updated Safety across All Tumor Cohorts and Efficacy in Advanced Bladder Cancer (AdvBC). J. Clin. Oncol. 2019, 37, 358. 10.1200/JCO.2019.37.7_suppl.358. [DOI] [Google Scholar]
- Sonpavde G.; Necchi A.; Gupta S.; Steinberg G. D.; Gschwend J. E.; Van Der Heijden M. S.; Garzon N.; Elegbe A.; Raybold B.; Liaw D.; Rutstein M.; Galsky M. D. A Phase 3 Randomized Study of Neoadjuvant Chemotherapy (NAC) Alone or in Combination with Nivolumab (NIVO) ± BMS-986205 in Cisplatin-Eligible Muscle Invasive Bladder Cancer (MIBC). J. Clin. Oncol. 2019, 37, TPS4587. 10.1200/JCO.2019.37.15_suppl.TPS4587. [DOI] [Google Scholar]
- Luo S.; Xu K.; Xiang S.; Chen J.; Chen C.; Guo C.; Tong Y.; Tong L. High-Resolution Structures of Inhibitor Complexes of Human Indoleamine 2,3-Dioxygenase 1 in a New Crystal Form. Acta Crystallogr., Sect. F: Struct. Biol. Commun. 2018, 74, 717–724. 10.1107/S2053230X18012955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor R. D.; MacCoss M.; Lawson A. D. G. Rings in Drugs. J. Med. Chem. 2014, 57, 5845–5859. 10.1021/jm4017625. [DOI] [PubMed] [Google Scholar]
- Yue E. W.; Sparks R.; Polam P.; Modi D.; Douty B.; Wayland B.; Glass B.; Takvorian A.; Glenn J.; Zhu W.; Bower M.; Liu X.; Leffet L.; Wang Q.; Bowman K. J.; Hansbury M. J.; Wei M.; Li Y.; Wynn R.; Burn T. C.; Koblish H. K.; Fridman J. S.; Emm T.; Scherle P. A.; Metcalf B.; Combs A. P. INCB24360 (Epacadostat), a Highly Potent and Selective Indoleamine-2,3-Dioxygenase 1 (IDO1) Inhibitor for Immuno-Oncology. ACS Med. Chem. Lett. 2017, 8, 486–491. 10.1021/acsmedchemlett.6b00391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue E. W.; Douty B.; Wayland B.; Bower M.; Liu X.; Leffet L.; Wang Q.; Bowman K. J.; Hansbury M. J.; Liu C.; Wei M.; Li Y.; Wynn R.; Burn T. C.; Koblish H. K.; Fridman J. S.; Metcalf B.; Scherle P. A.; Combs A. P. Discovery of Potent Competitive Inhibitors of Indoleamine 2,3-Dioxygenase with in Vivo Pharmacodynamic Activity and Efficacy in a Mouse Melanoma Model. J. Med. Chem. 2009, 52, 7364–7367. 10.1021/jm900518f. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Liang X.; Dong W.; Fang Y.; Lv J.; Zhang T.; Fiskesund R.; Xie J.; Liu J.; Yin X.; Jin X.; Chen D.; Tang K.; Ma J.; Zhang H.; Yu J.; Yan J.; Liang H.; Mo S.; Cheng F.; Zhou Y.; Zhang H.; Wang J.; Li J.; Chen Y.; Cui B.; Hu Z.-W.; Cao X.; Xiao-Feng Qin F.; Huang B. Tumor-Repopulating Cells Induce PD-1 Expression in CD8+ T Cells by Transferring Kynurenine and AhR Activation. Cancer Cell 2018, 33, 480–494.e7. 10.1016/j.ccell.2018.02.005. [DOI] [PubMed] [Google Scholar]
- Liu C.; Nan Y.; Xia Z.; Gu K.; Chen C.; Dong X.; Ju D.; Zhao W. Discovery of Novel Hydroxyamidine Derivatives as Indoleamine 2,3-Dioxygenase 1 Inhibitors with in Vivo Anti-Tumor Efficacy. Bioorg. Med. Chem. Lett. 2020, 30, 127038. 10.1016/j.bmcl.2020.127038. [DOI] [PubMed] [Google Scholar]
- Weng T.; Qiu X.; Wang J.; Li Z.; Bian J. Recent Discovery of Indoleamine-2,3-Dioxygenase 1 Inhibitors Targeting Cancer Immunotherapy. Eur. J. Med. Chem. 2018, 143, 656–669. 10.1016/j.ejmech.2017.11.088. [DOI] [PubMed] [Google Scholar]
- Vilar S.; Cozza G.; Moro S. Medicinal Chemistry and the Molecular Operating Environment (MOE): Application of QSAR and Molecular Docking to Drug Discovery. Curr. Top. Med. Chem. 2008, 8, 1555–1572. 10.2174/156802608786786624. [DOI] [PubMed] [Google Scholar]
- Muegge I.; Bergner A.; Kriegl J. M. Computer-Aided Drug Design at Boehringer Ingelheim. J. Comput.-Aided Mol. Des. 2017, 31, 275–285. 10.1007/s10822-016-9975-3. [DOI] [PubMed] [Google Scholar]
- Calculator plugins were used for structure property prediction and calculation: Calculator Plugins, Marvin 15.1.5, 2015; ChemAxon. http://www.chemaxon.com.
- Tu W.; Yang F.; Xu G.; Chi J.; Liu Z.; Peng W.; Hu B.; Zhang L.; Wan H.; Yu N.; Jin F.; Hu Q.; Zhang L.; He F.; Tao W. Discovery of Imidazoisoindole Derivatives as Highly Potent and Orally Active Indoleamine-2,3-Dioxygenase Inhibitors. ACS Med. Chem. Lett. 2019, 10, 949–953. 10.1021/acsmedchemlett.9b00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cumming J. G.; Davis A. M.; Muresan S.; Haeberlein M.; Chen H. Chemical Predictive Modelling to Improve Compound Quality. Nat. Rev. Drug Discovery 2013, 12, 948–962. 10.1038/nrd4128. [DOI] [PubMed] [Google Scholar]
- Waring M. J.; Arrowsmith J.; Leach A. R.; Leeson P. D.; Mandrell S.; Owen R. M.; Pairaudeau G.; Pennie W. D.; Pickett S. D.; Wang J.; Wallace O.; Weir A. An Analysis of the Attrition of Drug Candidates from Four Major Pharmaceutical Companies. Nat. Rev. Drug Discovery 2015, 14, 475–486. 10.1038/nrd4609. [DOI] [PubMed] [Google Scholar]
- Kumar K.; Woo S. M.; Siu T.; Cortopassi W. A.; Duarte F.; Paton R. S. Cation−π Interactions in Protein–Ligand Binding: Theory and Data-Mining Reveal Different Roles for Lysine and Arginine. Chem. Sci. 2018, 9, 2655–2665. 10.1039/C7SC04905F. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.