

Abstract
Castration resistant prostate cancer (CRPC) remains androgen dependant despite castrate levels of circulating testosterone following androgen deprivation therapy, the first line of treatment for advanced metstatic prostate cancer. CRPC is characterized by alterations in the expression levels of steroidgenic enzymes that enable the tumour to derive potent androgens from circulating adrenal androgen precursors. Intratumoral androgen biosynthesis leads to the localized production of both canonical androgens such as 5α-dihydrotestosterone (DHT) as well as less well characterized 11-oxygenated androgens, which until recently have been overlooked in the context of CRPC. In this review we discuss the contribution of both canonical and 11-oxygenated androgen precursors to the intratumoral androgen pool in CRPC. We present evidence that CRPC remains androgen dependent and discuss the alterations in steroidogenic enzyme expression and how these affect the various pathways to intratumoral androgen biosynthesis. Finally we summarize the current treatment strategies for targeting adrenal derived androgen biosynthesis.
Keywords: prostate cancer, adrenal androgen precursors, 11β-hydroxyandrostenedione, 11-oxygenated androgens, 11-ketotestosterone
1. Introduction
The activation of the androgen receptor (AR) by endogenous androgens is essential for development and function of the normal prostate gland (1). In eugonadal men, the primary circulating androgen, testosterone (T), is produced by the Leydig cells of the testes. The androgen signal is amplified in peripheral target tissue, such as the prostate, by the steroid 5α-reductase (SRD5A) catalyzed conversion of T to the more potent androgen 5α-dihydrotestosterone (DHT) (2–6)
Huggins and Hodges were the first to demonstrate that prostate cancer (PCa) is a hormone dependent disease in 1941 (7). Androgen deprivation therapy (ADT) has since been adopted as the standard-of-care treatment for metastatic PCa (8). The aim of ADT is to reduce the circulating levels of T to castrate levels (50 ng/dL) and is accomplished by either surgical (orchiectomy) or chemical castration (LHRH agonists) (9–12). Despite the initial remission associated with ADT, the cancer inevitably returns in the vast majority of cases and is then termed castration-resistant prostate cancer (CRPC), which is uniformly fatal (13). Although originally considered androgen independent, it is now clear that CRPC remains androgen dependent and is driven, in part, by the intracrine activation of androgen precursors of adrenal origin. In this review, we present evidence that CRPC remains androgen dependent and discuss the intracrine pathways by which both canonical and 11-oxygenated adrenal androgen precursors are converted to active androgens capable of driving disease progression via activation of the AR. Moreover, we discuss how changes in steroidogenic enzyme expression profiles, coupled to the specificity of the relevant enzymes, regulate intracrine androgen metabolism. Finally, we provide an overview of current and potential treatment strategies targeting adrenal androgen precursor metabolism and action.
2. Evidence that Castration Resistant Prostate Cancer remains androgen dependent
While initially considered an androgen-independent or hormone-refractory cancer, it is now clear that CRPC remains androgen dependent as it is characterized by the reactivation of androgen signaling and increased serum prostatic-specific antigen (PSA) levels (14–17). Several mechanisms have been implicated in the reactivation of the AR-axis, and include the over expression of the AR, AR gene mutations, enhanced AR signal transduction through downstream regulatory molecules, co-factor recruitment alterations (increase expression of transcriptional co-activator proteins), cross-talk leading to the activation of the receptor independent of ligand binding, and AR splice alternatives (18–21). Importantly, many of these mechanisms are reliant on residual intratumoral androgens.
Although ADT achieves a 90–95% decrease in circulating levels of T, studies have consistently shown that a pool of potent androgens remain detectable in CRPC tumors and that this pool is sufficient for the activation of the AR (22). For example, the measurement of intratumoural levels of DHT by Sharifi and Auchus (22) demonstrated that DHT is present at approximately 1 nM (0.5 ng/g - 1.0 ng/g) in castrate patients, levels which have been suggested to be 50-fold higher than the concentration required for half maximal activation of the AR (2, 22). Similarly, Mohler et al demonstrated that DHT levels in locally recurrent tumors from castrated patients were only reduced to approximately 80% (0.4 ng/g) of the levels measured in benign prostatic hyperplasia (BPH) patients (23). The source of the residual intratumoral androgens has unanimously been attributed to the intratumoral conversion of adrenal androgen precursors to active androgens (24) as outlined in section 4 below. Two anti-androgen drugs have therefore been developed and approved for the treatment of CRPC. Abiraterone is a selective cytochrome P450 17A1 (steroid 17α-hydroxylase/17,20-lyase, CYP17A1) inhibitor which inhibits androgen biosynthesis by all tissues expressing CYP17A1 including the adrenal (25, 26). Enzalutamide is an AR antagonist, which prevents the activation of the AR by residual androgens by blocking the binding of androgens to the AR, inhibiting the translocation of activated AR to the nucleus and impairing the binding of activated AR to DNA (27, 28). Both abiraterone and enzalutamide have consistently been shown to increase the median survival of patients suffering from CRPC thereby confirming that residual androgens derived from adrenal precursors drive CRPC (29–38).
While initially intended for use following the development of CRPC, studies are now considering the use of antiandrogens together with castration for the treatment of PCa, again highlighting the contribution of adrenal androgen precursors. This strategy is termed “complete androgen blockade” or “combined androgen blockade” (CAB) and was first proposed in the 1980’s (39). In 1983 Labrie et al. demonstrated that ADT outcomes could be improved when LHRH agonist treatment was combined with the use of AR antagonists. This study demonstrated positive objective response in >95% of patients undergoing CAB as compared to 60–70% in patients who underwent chemical castration alone (17). A caveat to these early studies is that the LHRH agonists employed produced an initial flare reaction resulting in increased LH and testosterone for 1–3 weeks before providing functional antagonism due to downregulation of the LHRH receptors (40, 41). Some of the benefit of TAB was therefore in inhibiting the adverse consequences of the initial flare reaction (40). Nonetheless, studies by Geller et al demonstrated that residual intratumoral DHT levels following castration by orchiectomy, without the risk of flare reactions, could be further reduced by adrenal androgen blockade using ketoconazole, a non-specific CYP17A1 inhibitor (14–16, 42–44). Furthermore, recent studies have confirmed the findings of earlier CAB studies, though with the benefit of newer LHRH antagonists that do not induce the initial flare reaction. For example, Mostaghel et al demonstrated that intratumoral DHT levels could be reduced (0.92 ng/g to 0.03 ng/g) by treating with dutasteride and ketoconazole three months prior to prostatectomy in men suffering from localized PCa (45). Taplin et al demonstrated reductions of DHT levels (1.3 ng/g to 0.18 ng/g) as well as reduced levels of adrenal precursors androstenedione (A4) and dehydroepiandrosterone (DHEA) in prostate tissue in patients treated with a LHRH agonist and abiraterone three to six months prior to prostatectomy (46). Moreover, two recent clinical trials, STAMPEDE and LATTITUDE, have demonstrated similar promising results when comparing the addition of abiraterone to ADT alone for men with newly diagnosed metastatic prostate cancer (47–49).
Studies using xenograft models further support the role played by adrenal derived androgens in CRPC. Murine adrenal CYP17A1 expression has recently been demonstrated in intact and castrate mice (50), and has been positively correlated to orchiectomy (ORX) induced expression of the Luteinizing Hormone receptor (LH, Lhcgr) (51, 52). Thus, ORX, which increases circulating LH levels (53), leads to an auto-regulatory loop that induces CYP17A1 expression in murine adrenal and the production of sufficient adrenal androgen precursors to drive tumor growth (52). It should be noted that this regulatory loop may not be relevent in the case of the human adrenal due to only very low expression of LHCGR in the human adrenal cortex (54). However, unlike the murine adrenal the human adrenal expresses significant levels of CYP17A1 resulting in abundant androgen precursor production as described in section 3 below (55).
In line with the study described above, Huhtaniemi et al demonstrated that VCaP xenograft tumor growth in castrate mice was further inhibited by adrenalectomy (ADX) and was accompanied by decreases in serum T, DHT and PSA (52). Similarly, decreased intratumoral levels of T, 5α-dione and DHT were reported following ADX (52). Using C.B-17 SCID mice Mostaghel et al demonstrated that ADX suppressed tumor growth and androgen biosynthesis in two CRPC xenograft models and did so to a greater extend then previous studies with abiraterone (50). Furthermore, they measured detectible levels of the adrenal androgen precursors, DHEA and A4, as well as downstream metabolites, T, DHT and androsterone, in the adrenal glands of intact mice (50). Following castration the levels of A4, T and DHT were two orders of magnitude higher in the adrenal when compared to other organs which exhibit steroidogenic capacity, confirming the active androgen biosynthesis in the murine adrenal gland (50).
Taken together, the studies presented above provide compelling evidence that CRPC remains androgen dependent following castration. The contribution of the adrenal to the intratumoral androgen pool and the pathways involved in intratumoral androgen activation are presented below.
3. The adrenal derived androgen precursor pool
The adult human adrenal produces adrenal androgen precursors, which include DHEA and its sulphate (DHEAS), 5-androstenediol (5-adiol) and its 3-sulphate (5-adiol3S), A4 and 11β-hydroxyandrostenedione (11OHA4) (56–62). The production of these androgen precursors is primarily from the zona reticularis, which develops during adrenarche between the ages of six and eight, and expresses the unique repertoire of steroidogenic enzymes required for their biosynthesis (63–65).
As with all de novo steroid biosynthesis, steroid production starts with the steroidogenic acute regulatory protein (StAR) mediated transfer of cholesterol from the outer mitochondrial membrane to the inner mitochondrial membrane. Here cholesterol is converted to pregnenolone, the precursor to all steroid hormones, by the activity of cytochrome P450 side-chain cleavage (CYP11A1). Pregnenolone is subsequently converted to 17α-hydroxypregnenolone by cytochrome P450 17α-hydroxylase/17,20-lyase (CYP17A1), which is localizes to the endoplasmic reticulum. The 17,20-lyase activity of CYP17A1 subsequently converts the C21 steroid 17α-hydroxylase to the C19 steroid DHEA (63, 64, 66, 67). This 17,20-lyase step is essential for the production of all androgen precursors and androgens and within the adrenal is specific to the zona reticularis due to the expression of cytochrome b5, which augments the 17,20-lyase activity of CYP17A1 (63, 64, 66, 67). While human CYP17A1 is able to 17α-hydroxylate the Δ4 steroid progesterone, the product 17α-hydroxyprogesterone is a poor substrate for the CYP17A1 catalyzed 17,20-lyase reaction (68, 69). As a result adrenal androgen precursor production follows the Δ5 pathway to DHEA and is aided by the reduced expression of 3β-hydroxysteroid dehydrogenase type 2 (HSD3B2) in the zona reticularis, thereby preventing the conversion of Δ5 to Δ4 steroids (66). While, DHEA can be released directly into circulation a large proportion of this steroid is first sulfonated by the activity of sulfotransferase 2A1 (SULT2A1) yielding DHEAS, the most abundant steroid in circulation (70). DHEA is also converted to A4 by the low levels of HSD3B2 expressed in the zona reticularis (71–73). Similarly, low levels of the aldo-keto reductase 1C3 (AKR1C3) can catalyze the conversion DHEA and A4 to 5-adiol and T, respectively. 5-adiol serves as a substrate for both SULT2A1 and HSD3B2, yielding 5-adiolS and T, respectively (74). It should however be noted that the levels of T produced by the adult adrenal are significantly lower than that produced by the Leydig cells of testes and that the primary C19 steroids produced by the adrenal are in the form of inactive androgen precursors.
Both A4 and T are readily 11β-hydroxylated by cytochrome P450 CYP11B1 and CYP11B2 enzymes (75) yielding 11OHA4 (76–78) and 11β-hydroxytestosterone (11OHT) (79), respectively. The levels of 11OHA4 produced are, however, significantly higher than that of 11OHT due to the differences in the availability of the precursors A4 and T (61, 77). While 11OHA4 is not a substrate for AKR1C3 (76, 80), both 11OHA4 and 11OHT can be converted to 11-ketoandrostenedione (11KA4) and 11-ketotestosterone (11KT), respectively by the low levels of 11β-hydroxysteroid dehydrogenase type 2 expressed in the adrenal (61, 76). 11KA4 can subsequently be converted to 11KT by AKR1C3 (61, 76, 80). However, adrenal output of 11KA4 and 11KT is low and as a result, it is thought that the circulating levels of these steroids are achieved by the peripheral conversion of 11OHA4 (61, 78, 81). 11OHA4 is likely converted to 11KA4 in mineralocorticoid target tissue, such as the kidney, expressing high levels of HSD11B2. The resulting circulating 11KA4 can subsequently be converted to 11KT by peripheral tissue such as adipose, which expressed AKR1C3 (80, 82). Importantly, circulating levels of 11KT (nM range) and 11OHT (sub nM) are similar in both men and women despite approximately 10-fold higher circulating levels of T in men (83–85). Moreover, the addition of exogenous T does not lead to an increase in the circulating levels of 11-oxygenated androgens (86). Together these findings confirm that 11KT and 11OHT are either derived directly from the adrenal or from the peripheral conversion of adrenal-derived 11OHA4 (78). This model is further supported by the observation of only trace amounts of 11-oxygenated steroids in patients diagnosed with 11β-hydroxylase deficiency and adrenal insufficiency (78). Interestingly, a recent study by Nanba et al have shown that unlike the canonical adrenal precursors, A4, DHEA(S) and potent androgen T, the circulating levels of 11-oxygenated androgens do not decrease with age in women when comparing premenopausal to postmenopausal women. These findings are attributed to the involution of the zona reticularis with age resulting in the zonal division of CYB5A and HSD3B2 expression becoming less distinct. The resulting overlap allows CYB5A to promote the 17,20-lyase activity of CYP17A1 (as in the zona reticularis), while the presence of HSD3B2 results in less DHEA and DHEAS being produced as DHEA is channeled to A4, which in turn serves as a substrate for 11β-hydroxylation by CYP11B1 yielding 11OHA4 (83). Similarly, 11-oxygenated androgen levels appear not to decrease with age in men when comparing a young group (20–60 years) to an older group (61–92 years) (87). Given that the occurrence of PCa is associated with age, these 11-oxygenated androgens may therefore make a significant contribution to the available androgen pool following castration as discussed in section 5.
4. Intratumoral conversion of adrenal derived precursors to DHT
4.1. Altered expression of steroidogenic enzymes in CRPC
A hallmark of CRPC is the altered expression of steroidogenic enzymes, which provide the tumor with the enzymatic machinery required for the biosynthesis of potent androgens from available adrenal androgen precursors (24, 52, 88–90).
Pfeiffer et al have demonstrated alterations in the expression levels of HSD17B2, HSD17B3, AKR1C3, SRD5A2, UGT2B15 and UGT2B17 in CRPC tissue (90). Mitsiades et al showed the upregulation of AKR1C3 and SRD5A1 in metastatic PCa when compared to normal prostate tissue or primary prostate carcinoma, while the expression of SRD5A2 was decreased (89). Similarly, when comparing CRPC tumors to primary prostate tumors, Montgomery et al reported increased HSD3B1, HSD3B2, CYP17A1, AKR1C3, HSD17B, UGT2B15 and UGT2B17 expression and decreased SRD5A2 expression (24). Stanbrough et al also measured increased HSD3B2, AKR1C3, SRD5A1, AKR1C2, AKR1C1, and UGT2B15 expression in CRPC tumors as compared to primary tumors (91).
A consistent change in the expression of 17β-hydroxysteroid dehydrogenases (HSD17Bs) suggests the utilization of adrenal androgens for intratumoral androgen production as discussed in section 5. Reductive 17β-hydroxysteroid dehydrogenases (AKR1C3 and HSD17B3) which activate androgens are upregulated (24, 88–90, 92, 93), while reduced expression of oxidative enzymes (HSD17B2 and HSD17B4) catalyzing the opposing inactivation reactions are reduced (88, 92, 94). In fact, the upregulation of AKR1C3 is the most frequent and striking of the changes observed and has been reported in PCa cell lines cultured in androgen deprived media (95), xenografts grown in castrate mice (95–97), soft tissue metastases (92) and in prostate tissue from several studies (80, 89, 90, 92, 95, 98–102). The upregulation of AKR1C3 has also been reported in enzalutamide-resistant PCa cells and xenograft tumors (97). Furthermore, overexpression of AKR1C3 in PCa cell lines confers resistance to abiraterone (96).
Although AKR1C3 expression is repressed by the activation of the AR in a negative feedback mechanism, this repression is overcome by the expression of the fusion protein TMPRSS2-ERG, a biomarker of advanced PCa (103). TMPRSS2-ERG is proposed to displace the AR from the AKR1C3 promoter thereby relieving the negative feedback. AKR1C3 expression in turns leads to an increase in the biosynthesis of potent androgens which further induces TMPRSS2-ERG expression leading to a positive feed-forward model for AKR1C3 expression (101). Interestingly, in addition to its function as an androgen activating enzyme AKR1C3 has also been shown to function as a coactivator of the AR suggesting a dual role for this key protein (104).
Another distinct switch in enzyme expression observed during the development of CRPC is the change in SRD5A isozyme expression. SRD5A2 is the dominant isoenzyme expressed in the healthy prostate. SRD5A1 expression is universally up-regulated in CRPC tumors (105–108), while SRD5A2 levels are down regulated (107, 109–112). Although they catalyze the same reaction, SRD5A1 and SRD5A2 demonstrate unique pH optima and substrate preferences (5, 113) and the switch in SRD5A isoenzyme expression therefore has a distinct effect of intratumoral androgen biosynthesis as discussed in section 4.2 below.
A limited number of studies have measured altered expression status of steroidogenic enzymes in metastatic lesions. Jernberg et al reported increased expression levels of SRD5A1, AKR1C2, AKR1C3 and HSD17B10 in bone metastases compared to non-malignant and/or malignant prostate tissue. The expression of CYP11A1, CYP17A1, HSD3B2, SRD5A2, and HSD17B6 mRNA levels were significantly lower in metastases. Moreover, a sub-group of metastases expressed very high levels of AKR1C3 and low levels of AR variants (AR-Vs), while high AR-V protein levels measured in metastases with low AKR1C3 levels suggesting different mechanisms of castration-resistance in individual bone metastases (100). Consistant with this finding, Mostaghel et more recently demonstrated several distinct staining profiles for the expression of steroidogenic enzymes in CRPC metastases. These included subsets of patients with AR and PSA negative tumours with low expression of steroidogenic enzymes; AR positive tumours with negative or positive PSA expression and low steroidogenic potential; and AR and PSA positive tumours with high steroidogenic potential due to the expression of HSD3B1 and HSD17B3/AKR1C3. Tumours in the latter group could be further subdivided based on potential for de novo steroidogenesis based on the expression of CYP17A1 (50).
4.2. The intratumoral biosynthesis of DHT from adrenal androgen precursors follows the 5α-dione pathway, which bypasses T
For many years, the intratumoral utilization of adrenal androgen precursors was assumed to follow the canonical pathway of androgen biosynthesis in which T is the central androgen. However, studies have revealed that this is not the case. Instead, A4 is preferentially 5α-reduced to 5α-androstanedione (5α-dione) by SRD5A1, with the resulting 5α-dione serving as a substrate for AKR1C3 and yielding DHT (114, 115). This pathway therefore entirely bypassed the production of T (116). The activity of the 5α-dione pathway is brought about by a change in the expression of SRD5A1 isozymes during the development of CRPC as discussed above in section 4.1. This switch in expression results in a change in the substrate preference and pathway followed as SRD5A1 catalyzes the 5α-reduction of A4 more efficiently than that of T (116). Coupled to this a recent study by Barnard et al has demonstrated that A4 is actually a poor substrate for AKR1C3 (80). Therefore, despite AKR1C3 being the most overexpressed steroidogenic enzyme in CRPC, the efficient SRD5A1 catalyzed 5α-reduction of A4 dominates and directs the flux via 5α-dione (116). The presence of the 5α-dione pathway has been confirmed by several studies (116–118). For example Chang et al. demonstrated that the flux from A4 to 5α-dione was more rapid than the conversion of A4 to T in all six of the PCa cell lines they tested. These results were confirmed in LAPC4 and LNCaP xenograft models as well as in freshly collected CRPC tumor biopsies (116). More recently, Dai et al collected prostate tissue from patients with clinically localized PCa. Tissue were cultured ex vivo and exposed to equimolar mixtures of A4 and T. Using stable isotopic tracing they demonstrated that this tissue reduced both T and A4, but that the reduction of A4 to 5α-dione was clearly favored in CRPC tissue (118).
4.3. DHEAS as an androgen precursor reservoir despite CYP17A1 inhibition
Given that DHEAS is the most abundant steroid in circulation and the only steroid to circulate in the μM range it is feasible that if inhibition of CYP17A1 is not complete, residual levels of DHEAS may remain in circulation and could serve as a reservoir for intracrine androgen production, thereby contributing to treatment resistance. Indeed, Taplin et al found that DHEAS and DHEA-glucuronide were only reduced to 10% of baseline levels following 24-week LHRH treatment combined with abiraterone (119). Similar results have been obtained by Tamae et al who conducted a Total Androgen Pathway Suppression Trial (TAPS) to investigate the effect of different treatment regimens on circulating androgen levels (120). Although patients who received a combination of goserelin, bicalutamide, dutasteride and ketoconazole showed an 85–90% decrease in T levels as well as a decrease in precursor androgen levels after 12 weeks of treatment, circulating DHEAS remained three orders of magnitude higher (50 μg/dL) than that of T (mean 10 ng/dL) (120). Similarly, Leuprolide treatment alone or combined with abiraterone and prednisone resulted in a 95–99% decrease in circulating T, with a concomitant decrease in DHEAS, DHEA and A4 in the group receiving the combination treatment (120). However, reduction of circulating DHEAS was incomplete (20 μg/dL) (120). This suggests that despite the best efforts to inhibit the activity of CYP17A1 a substantial reservoir of DHEAS remains, potentially providing a substrate for intratumoral androgen biosynthesis (121). However, unlike the majority of androgen precursors, DHEAS cannot passively diffuse across the cell membrane due to the presence of the negatively charge sulfate group and therefore requires the expression and activity of organic anion transporter polypeptides (OATPs) encoded by SLCO1A2, SLCO1B1, SLCO1B3, and SLCO2B1 (122, 123). Androgen deprivation has been shown to lead to the upregulation of OATPs and the enhanced uptake of steroid conjugates in PCa cell lines (123). Arakawa et al found that the mRNA expression levels of SLCO1A2 (4-fold), SLCO1B1 (2.5-fold) and SLCO1B2 (1.7-fold) were upregulated in both LNCaP and 22RV1 cells when grown in androgen depleted conditions (123). These increases were accompanied by concomitant increases in the cellular uptake of [3H]-DHEAS and cellular proliferation, while SLCO1A2 knockdown lead to a reduction in growth (123). Green et al found that PCa xenografts expressing both SLCO1B1- and SLCO2B1 showed and 3.9- and 1.9-fold increase in DHEAS accumulation and a 1.6- and 2.7-fold increase in DHEA levels respectively (124). Enhanced expression of OATPs as well as enhanced uptake of steroid has also been reported in prostate tumors (125–128). Hamada et al showed that OATP1B3 is overexpressed in PCa when compared to normal and BPH tissue (127). They also found that single-nucleotide polymorphisms (SNPs) in these transporters can affect patient outcome. Patients with the SLCO1B3 genotypes shown to more efficiently import T had a median 2-year shorter time to progression while on ADT (127). Overall survival was also improved to 8.5 years for patients with two of the mutant 334G/699 A allele, also known as the G allele when compared to the 6.4 years for patients carrying one or two copies of the wild-type SLCO1B3 334T/699G haplotypes or T allele (126). The latter exhibited higher maximal T uptake when compared to the wild-type allele. Sharifi et al have reported a significantly shorter time to the development of hormone-refractory PCa in patients with one or two copies of the T allele of SLCO1B3 undergoing ADT (126). Similarly a study by Yang et al identified three SNPs in SLCO2B1 that were associatied with time to progression for PCa patient receiving ADT (125). Collectively, these findings suggest that the change in the expression of SLCO transporters alter the response to ADT by regulating the availability of T and DHEAS for intracrine androgen biosynthesis.
Once inside a target cell, DHEAS needs to be deconjugated by steroid sulfatase (STS) before the resulting DHEA can be converted to DHT. STS expression has been reported in PCa cell lines (129, 130) as well as in localized PCas (131). While studies have demonstrated irreversible inhibition of STS, the use of STS inhibitors for the treatment of CRPC still need to be tested (121).
4.4. The HSD3B1(1245C) variant promotes the use of adrenal androgen precursors
HSD3B1 catalyzes the conversion of the adrenal precursor DHEA to A4, with A4 serving as the entry point to the 5α-dione pathway and therefore the expression of this enzyme regulates the amount of substrate available for intratumoral DHT biosynthesis (132). A germline SNP (1245C;N367T) has been identified in CRPC tumors with an allelic population frequency of between 15–35% reported in most cohorts (132–136). Population frequency appears to varies among ethnic groups with the highest frequency reported for Caucasians (62.7%) (136) and the lowest among Asian individuals (14.1%) (134).
The mutation does not affect the function of the enzyme but instead renders the it resistant to ubiquitination and degradation (132). The increase half-life of HSD3B1(1245C) variant therefore allows for increased DHT biosynthesis from DHEA (132). The expression of wild-type and HSD3B1 (1245C) variant is heterogeneous among PCa cell lines and tumors. LAPC-4 cells do not express the HSD3B1(1245C) variant and do not efficiently convert DHEA to A4 (132). Conversely, LNCaP have higher HSD3B1 protein expression levels due to the presence of the variant and efficiently convert DHEA to A4 (132).
The HSD3B1(1245C) variant has been shown to be a predictive biomarker for recurrence following ADT (133, 134, 136, 137). More rapid progression from the initiation of ADT to the development of CRPC has been observed in patients with aggressive PCa who inherited the HSD3B1(1245C) variant. It is thought that this is due to the variant increasing the CRPC tumors ability to make use of DHEA(S) for intratumoral androgen biosynthesis following ADT. It was therefore hypothesized that better clinical responses to pharmacological inhibition of adrenal androgen precursor biosynthesis could be achieved in these patients (138). Indeed, patients with the HSD3B1(1245C) variant have demonstrate improved clinical responses to ketoconazole, an antifungal azole that inhibits CYP17A1 among several other cytochrome P450s, when compared to those without the variant (138). An increase in the number of inherited HSD3B1 (1245C) variant alleles from 0 to 2 was associated with an increase in both the median duration of therapy (from 5.0 months to 12.3 months) and the median progression-free survival (from 5.4 months to 15.2 months) (138). Conversely, treatment with the steroidal CYP17A1 inhibitor, abiraterone, does not deliver the same beneficial outcomes in patients with the HSD3B1 (1245C) variant (139). This contradiction is brought about by the fact that the Δ5 structure of abiraterone is itself subject to metabolism by HSD3B1, yielding Δ4-abiraterone (D4A) (139). While D4A itself is a potent antiandrogen, its 5α-reduced metabolite 5α-abiraterone (5α-abi) is an AR agonist which works against the abiraterone mediated CYP17A1 blockade (discussed in section 6.1) (139). Alyamani et al have reported that the inheritance of 0, 1 and 2 copies of the HSD3B1(1245C) variant leads to a stepwise increase in the production of 5α-Abi, which counteract the benefits of CYP17A1 inhibition (140). A subsequent study has confirmed that the response to abiraterone can be predicted by the HSD3B1 (1245C) variant (141). It remains to be determined if combining of abiraterone with a SRD5A inhibitor to prevent the production of 5α-Abi will result in the same beneficial outcomes observed in patients treated with ketoconazole (138).
4.5. Look both ways: oxidative 17β-hydroxysteroid dehydrogenases regulate intratumoral androgen levels
The transition from benign prostate to CRPC is almost always accompanied by an increase in the expression of AKR1C3 as outlined in section 4.1 above (24, 88–90, 92, 93, 95–100, 102). The upregulation of AKR1C3 is believed to reflect the selection of tumors cells capable of efficiently producing DHT given that AKR1C3 is essential for the conversion of androgen precursors to active androgens irrespective of the pathway followed. A recent study by Barnard et al confirmed that both A4 and 5α-dione are in fact very poor substrates for AKR1C3 (80). Conversely, T and DHT were efficiently inactivated by the oxidative 17βHSD, HSD17B2 (80). The authors therefore made use of several in vitro models coupled to computational modeling to investigate the effect of different AKR1C3:HSD17B2 ratios on the activation of androgens (80). Their results revealed that the inactivation of the canonical androgens T and DHT overrides the activation of A4 and 5α-dione despite significantly increased AKR1C3:HSD17B2 expression ratios, suggesting that the loss of oxidative 17βHSD activity may be equally as important as the upregulation of AKR1C3 when considering the intratumoral androgen production (80). These results have been corroborated by two recent studies. First, Gao et al demonstrated that HSD17B2 expression is reduced as PCa progresses (142). The mechanisms involved in the functional silencing of HSD17B2 were shown to include DNA methylation and androgen stimulation, both of which decreased HSD17B2 expression. Furthermore, alternative splicing was shown to yield catalytic-deficient splice variants, the mRNA of which could bind to wild type HSD17B2 and promote its degradation (142).
Similar to HSD17B2, HSD17B4 catalyzes the oxidative reaction converting potent androgens to weaker androgens. Five splice variants of HSD17B4 with differing activity have been reported to date (143–147). Ko et al recently demonstrated that only HSD17B4 variant 2 functionally catalyzes the inactivation of DHT and that this variant is specifically suppressed during the develop of CRPC, while the expression of other HSD17B4 isoforms are increased (94). Collectively, these studies emphasize the importance of the oxidative 17βHSDs and suggest that the expression and activity of oxidative 17βHSD enzymes are equally as important as the overexpression of the reductive enzyme AKR1C3 when considering intratumoral androgen activation.
4.6. Androgen receptor splice variant 7
Although not an androgen dependent mechanism, the AR splice variant 7 (AR-V7) may be an important role player in a subset of CRPC cases (148–152). This splice variant of the AR lacks the ligand binding domain (LBD) resulting in a constitutively active ligand-independent truncated receptor (145, 153, 154). The expression levels of this splice variant has been shown to be increased relative to that of the full length AR (AR-FL) in abiraterone-resistant VCaP xenograft models following castration. Despite this induction, AR-V7 levels remained <1% relative to the AR-FL in both xenograft models and in clinical CRPC samples. However, these low levels have been proposed to allow for the maintenance of basal AR activity following androgen deprivation, thereby giving the tumour time to survive until more potent mechanisms of AR activation emerge (155). The presence of the AR-V7 splice variant therefore offers a resistance mechanism to standard antiandrogen treatment regimes, including enzalutamide and abiraterone. Indeed, the recent PROPHECY study confirmed that men with AR-V7 positive metastatic CRPC who were treated with enzalutamide or abiraterone had a shorter progression free survival and overall survival (156). Moreover, the detection of AR-V7 in treatment-naïve metastatic CRPC patients is more common in men with higher disease burden and signals a poor prognosis (157). Alternative agents targeting the AR splice variants therefore need to be developed to treat AR-V7 positive patients (157, 158). It should however be noted that the presence of AR-V7 is often accompanied by significant increases in the expression of AR-FL leading to the suggestion that both AR-V7 and AR-FL should be considered when predicting resistance against AR directed therapies (159). Indeed, AR amplification in CRPC bone metastases has been shown to be associated with increased expression of AR-V7, together leading to poor survival (162).
5. 11-oxygenated androgens are an additional source of intratumoral androgen
5.1. The 11OHA4 pathway
Although 11-oxygenated androgens have previously only been considered within the context of teleosts (a group of ray-finned fish), recent studies have shown that they are relevent in human physiology. The reader is referred to the following reviews for a detailed history of 11-oxygenated androgens and their role in human physiology: Swart and Storbeck. (77), Pretorius et al. (81), Turcu et al. (160, 161).
The abundant steroid 11OHA4 is produced by the human adrenal serves as a precursor for the production of the potent androgens 11KT and 11KDHT in peripheral tissue expressing the necessary enzymatic machinery, which include the prostate. The first step of this pathway is the conversion of 11OHA4 to 11KA4 by HSD11B2 as 11OHA4 is not a substrate for AKR1C3 or HSD17B3 (61, 76, 80). The conversion of 11OHA4 to 11KA4 by HSD11B2 is therefore an absolute requirement for the production of 11KT from 11OHA4. HSD11B2 is expressed in PCa cell lines (162, 163), prostate tissue (164, 165) and CRPC metastases (Mostaghel, unpublished data). Conversion of 11OHA4 to 11KA4 has been demonstrated in several PCa cell lines (76, 77, 166, 167). Barnard et al recently demonstrated that 11KA4 and its 5α-reduced metabolite, 11keto-5α-androstanedione (11K5α-dione) are excellent substrates for AKR1C3 (80). In fact, AKR1C3 catalyzed the conversion of 11KA4 and 11K-5α-dione 8- and 24-fold more efficiently than that of A4 or 5α-dione, respectively (80). Although the 5α-reduction of 11OHA4 and 11KA4 by SRD5A1 and SRD5A2 has been demonstrated, unpublished data from our group suggests that 11OHA4 is preferentially converted to 11KA4 by HSD11B2 followed by conversion to 11KT by AKR1C3. Moreover, while 11KT is efficiently 5α-reduced to 11KDHT by SRD5A2, 11KT is a poor substrate for SRD5A1, suggesting that the switch from SRD5A2 to SRD5A1 expression in CRPC may result in the accumulation of 11KT as the primary androgen.
5.2. 11KT and 11KDHT are bona fide androgens
Several studies have confirmed that 11KT and 11KDHT are androgens capable of activating the human AR at physiologically relevant concentrations (61, 76, 168–170). Rege et al (61), Yazawa et al (169), Storbeck et al (76) and Campana et al (170) demonstrated that 11KT activates the AR in a similar manner to T. Storbeck et al also found that 11KDHT elicits a comparable tranactivation response to DHT via the AR at a concentration of 1 nM. Pretorius et al subsequently demonstrated that 11KT and 11KDHT bind to the AR with affinities comparable to that of T and DHT (168). Moreover, using dose response assays they demonstrated that 11KT and 11KDHT are as potent and efficacious as T and DHT, respectively (168). Pretorius et al, went on to show that both 11KT and 11KDHT could up-regulate the expression of AR-regulated genes, KLK3, TMPRSS2 and FKBP5 in LNCaP and VCaP cells, and induce cell growth in both cell lines (168). Subsequent, proteomic analysis of VCaP cells treated with 11KDHT and 11KT confirmed an androgenic response (168).
A study by Gent et al recently confirmed that LNCaP cells convert the precursor 11OHA4 to the potent androgen 11KT, resulting in elevated levels of PSA (167). Furthermore, they demonstrated that inhibition of HSD11B2 activity prevented the production of 11KT and the concomitant increase in PSA (167).
5.3. Active 11-oxygenated androgens have the potential to accumulate in CRPC tissue.
A4 has for many years been considered as the major substrate for AKR1C3. However, as mentioned above (section 4.5) Barnard et al have recently demonstrated that 11-oxygenated androgen precursors such as 11KT are the preferred substrates (80). Using several in vitro models as well as a computational model this study demonstrated that increased AKR1C3:HSD17B2 ratios significantly favors the production of the potent 11-oxygenated androgen, 11KT, while the activity of the oxidative enzyme HSD17B2 prevents the production of T or DHT despite elevated AKR1C3:HSD17B2 ratios (80). Their validated computational model predicts that higher steady state concentrations of active 11-oxygenated androgens can be achieved when even low levels of oxidative 17βHSDs are present and that this effect is independent of substrate concentration (80).
There is also increasing evidence that 11-oxygenated androgens are resistant to inactivation. Inactivation of active androgens is achieved by metabolism by 3α-hydroxysteroid dehydrogenases and/or glucuronidation reactions carried out by uridine 5’-diphospho-glucuronosyltransferase (UGT) enzymes. Pretorius et al demonstrated that the inactivation of 11KT and 11KDHT by LNCaP and VCaP PCa cells occurred at a significantly lower rate than that of T and DHT, respectively, but did not investigate the mechanism of inactivation (168). However, subsequent studies have revealed that 11KT and 11KDHT are not efficiently glucuronidated by UGT enzymes (166).
Taken together, the increased and robust activation of 11-oxygenated androgens by AKR1C3 coupled to a lower rate of glucuronidation suggests that 11-oxygenated androgens have the potential to accumulate at higher levels than canonical androgens in CRPC tissue. Indeed, a recent study measured higher levels of 11-oxygenated androgens than canonical androgens in two PCa tissue samples (166). Further studies measuring a complete androgen profile by mass spectrometry are therefore required as it is increasingly clear that current measurements of residual T and DHT may severely underestimate the pool of residual androgens available to activate the AR in CRPC.
6. Targeting intratumoral androgen biosynthesis
6.1. CYP17A1 inhibitors
From the evidence presented above it is clear that enzymes catalyzing the biosynthesis or subsequent activation of adrenal androgen precursors are logical drug targets for the treatment of CRPC. CYP17A1 remains a highly viable target for inhibiting the production of adrenal androgen precursors given that the CYP17A1 catalysed 17,20-lyase reaction which is essential for the biosynthesis of all C19 steroids.
6.1.1. Abiraterone
Abiraterone was the first specific CYP17A1 inhibitor approved for the treatment of CRPC. Abiraterone acts by binding tightly to the active pocket of CYP17A1 and inhibiting both the 17α-hydroxylase and 17,20-lyase reactions. As a result, abiraterone treatment is combined with the administration of glucocorticoids in order to prevent mineralocorticoid excess associated with the inhibition of cortisol biosynthesis (36, 171–173). Phase I (36), II (31, 33) and III (34, 37) clinical studies with abiraterone have shown that the majority of patients respond well to abiraterone with primary outcomes including decreases in PSA levels (31, 33, 34, 36, 37, 171–173) along with a overall increase in patient survival compared to the placebo group (34, 37). Circulating levels of T, downstream androgens and estradiol were all suppressed with cocominant increases in steroids upstream of CYP17A1 (36).
Acquired resistance to abiraterone has however subsequently been reported (174) and could result from a variety of mechanisms including AR overexpression (175), AR splice variants (143, 145, 176) and AR point mutations. The AR point mutations T878A and L702H lead to AR activation by progesterone and pregnenolone both of which accumulate during abiraterone treatment (177). A recent study has shown that pregnenolone and progesterone induce cell growth in both castration‐naïve and CRPC cell lines via both the wild type and mutant AR and that this effect could be inhibited by the addition of selective AR antagonist (178). A similar effect has also been observed with prednisone which is often co-administered with abiraterone (179, 180). Furthermore, abiraterone is subject to metabolism by the steroidogenic machinery, yielding both antiandrogenic and androgenic metabolites with both beneficial and detrimental activities. The Δ5, 3β-hydroxyl-structure of abiraterone makes it susceptible to conversion by the HSD3B isoenzymes yielding D4A, which is detected in the serum of CRPC patients treated with abiraterone (181). Li et al have demonstrated that D4A maintains the ability to inhibit the activity of CYP17A1, but that it also inhibits both HSD3B1 and HSD3B2, isozymes vital to the production of the delta-4, 3-keto moiety shared by all active androgens (181). D4A was also shown to inhibit the SRD5A isozymes, thereby preventing the production of the potent 5α-reduced androgens, DHT and 11KDHT (181). Moreover, D4A was shown to bind to and antagonize both the wild type and T877A mutant AR with half-maximal inhibitory concentration (IC50) values comparable to that of enzalutamide. D4A therefore targets multiple sites downstream of CYP17A1, thus leading to a more comprehensive enzymatic blockade than abiraterone itself. However, the delta-4, 3-keto moiety of D4A makes it a target for irreversible 5α- or 5β-reduction by SRD5A and AKR1D1, respectively. The resulting 5α-abiraterone (5α-Abi) and 5β-abiraterone (5β-Abi) are subject to 3-keto-reduction reversibly yielding the respective 3α-hydroxy and 3β-hydroxy congeners. Metabolism of D4A therefore yields a total of three 5α-reduced and three 5β-reduced metabolites all of which are detectable in the serum of patients treated with abiraterone (182). While metabolism of D4A might attenuate CYP17A1 inhibition, the 5α-reduced product (5α-Abi) has been shown to act as an agonist of the AR, thus working against the beneficial effects of abiraterone and D4A (182). Moreover, the long-term propagation of PCa cells in the presence of abiraterone or D4A has been shown to result in the upregulation of SRD5A1 expression, thus suggesting that the 5α-reduction of D4A may serve as a drug resistance mechanism. This is supported by clinical findings showing that SRD5A is one of the most upregulated steroidogenic enzymes observed during resistance to abiraterone (183), as well as shortened progression-free survival (defined as tumor size <1000mm) in CRPC xenograft models treated with 5α-Abi compared to controls (183). The metabolism of abiraterone can, however, be fine-tuned as to yield optimal anti-androgen outcomes as demonstrated by Li et al who showed that the co-administration of abiraterone and dutasteride substantially decreases the mean concentration of 5α-Abi, while increasing that of D4A (182). It is therefore clear from the example of abiraterone that the metabolism of steroidal drugs and the activities of the resulting metabolites need to be taken into account when developing new treatments. Moreover, with this knowledge at hand the metabolism and outcomes can be manipulated in order to ensure optimal outcomes.
6.1.2. Galeterone
Galeterone (formerly TOK-001 or VN/124–1) is a steroidal multi-targeted inhibitor. Galeterone preferentially inhibits the 17,20-lyase activity of CYP17A1, induces the degredation of the AR and antagonizes the AR (184–186). In vitro studies have shown that galeterone is effective in PCa cell lines which expresses AR mutants (184) as well as in AR-negative PCa cell lines through a mechanism involving phosphorylation of eukaryotic Initiation Factor 2 (eIF2α), an integral component of the eukaryotic mRNA translation complex (187). Like abiraterone, the steroidal structure of galeterone makes is susceptible to metabolism by HSD3B to form Δ4-galeterone (D4G), as well as subsequent metabolism by SRD5A and 3αHSD enzymes yielding 3-keto-5α-galeterone (5αG), 3α-OH-5α-galeterone, and 3β-OH-5α-galeterone (188). D4G has been shown to bind to both mutant (LNCaP cells) and wild type AR (LAPC4 cells) with greater affinity than galeterone, with both inhibiting AR regulated gene expression in a manner comparable to that of D4A (188). Moreover, D4G demonstrated similar tumor suppressing activity in a CRPC xenograft model when compared to galeterone (188). While the 5α-reduced metabolite, 5αG, has demonstrated weak agonist activity via the AR, this is significantly less that that of 5α-Abi (188).
Phase I and II Androgen Receptor Modulation Optimized for Response (ARMOR) clinical trials have shown significant anti-tumor activity in both non-metastatic treatment-naïve CRPC and metastatic treatment-naïve CRPC patients treated with galeterone alone (189, 190), while additional clinical studies have shown that combining abiraterone and galeterone treatment is highly effective (191). A phase 3 trial, ARMOR 3-SV, set out to investigate combined galeterone and enzalutamide treatment in patients with treatment-naïve metastatic CRPC and who are positive for AR-V7 (due to galeterone’s ability to induce the degredation of the AR). This study was discontinued, however, as it was concluded that the trial would most likely not meet the primary endpoint of improved progression-free survival (192). At time of writing, the drug is therefore not being further advanced into clinical development.
6.1.3. Seviteronel
Seviteronel (INO-464) is an orally bioavailable nonsteroidal CYP17A1 inhibitor, which inhibits the 17,20-lyase activity with up to 10-fold selectivity over the 17α-hydroxylase activity (193). Morover, Seviteronel functions as an antagonist of both the wild-type and mutated forms of the AR (194). In vitro studies have shown that seviteronel inhibits cell growth in several CRPC cell lines (194–196). Phase I clinical trials have shown a significant decline in PSA levels of between ≥30% and ≥50% depending on the doses administered (174). There is currently an ongoing Phase II trial investigating the effects of oral seviteronel in CRPC patients previously treated with enzalutamide.
6.2. SRD5A inhibitors
Finasteride and dutasteride are the two most common SRD5A inhibitors and were first designed and approved for the treatment of BPH (197–200). Finasteride is a selective SRD5A1 inhibitor, while dutasteride inhibits both SRD5A1 and SRD5A2. Given the antiandrogenic nature of these inhibitors both have been used in the prevention and treatment of PCa. Both the Prostate Cancer Prevention Trial, which employed finasteride, and the Dutasteride of Prostate Cancer Events (REDUCE) trials demonstrated a modest reduction in the risk of developing PCa compared to placebo groups, however, treatment groups demonstrated an increased risk of developing high-grade cancer (201–204). However, a subsequent study has contradicted these findings (205). Dutasteride has since been used in combination with other treatments to aid in achieving total androgen blockage. For example Mostaghel et al. combined dutasteride with ketoconazole as discussed in section 2. Morover, by combining dutasteride with enzalutamide and leuprolide treatment, Montgomery et al demonstrated pathologic complete response rates similar to that achieved with abiraterone (206). Recently, the co-administration of dutasteride with abiraterone has been shown to to be a way to fine-tune abiraterone metabolism, preventing the production of the unfavourable metabolite 5α-Abi, as discussed in section 6.1.1 above (182).
6.3. CYP11B1 inhibitors
The CYP11B1 inhibitor metyrapone is already in use for the treatment of hypercortisolism associated with Cushing’s syndrome (207, 208). The increased evidence that 11-oxygenated androgens contribute to the androgen pool in CRPC brings CYP11B1 into focus as a potential drug target for CRPC. Metyrapone could therefore potentially be used in concert with CYP17A1 inhibitors. Alternatively a drug with dual CYP17A1 and CYP11B1 inhibitory properties could be of interest. Such a compound (CFG920) has previously been developed (207, 208) and made it to phase I clinical trials, however, the study was terminated, although not due to safety reasons.
6.4. AKR1C3 inhibitors
AKR1C3 is an essential enzyme for the activation of all androgen precursors irrespective of the pathway followed. The expression of AKR1C3 is consistently upregulated in CRPC as discussed in section 4.1. Furthermore, studies in PCa cell lines and xenograft models have demonstrated that elevated AKR1C3 levels confer resistance to both abiraterone and enzalutamide while inhibition or knockdown restores drug sensitivity as discussed in section 6.2 (96). While AKR1C3 is therefore an attractive drug target which could inhibit intracrine androgen biosynthesis and potentially increase sensitivity to existing drugs, the enzyme shares >86% sequence identity with AKR1C1 and AKR1C2, thereby complicating the development of target-specific drugs (209). Furthermore, the most striking preclinical proof-of-principle studies of AKR1C3 inhibition performed to date have made use of the nonsteroidal anti-inflammatory drug, indomethacin, which is also an inhibitor of cyclooxygenase 1 and 2 (210, 211). Despite the significant challenge posed by developing a specific AKR1C3 inhibitor, several studies have reported promising lead compounds with improved selectivities (212–214). Moreover, the development of specific AKR1C3 inhibitors with dual AR antagonist activity is also being explored (215). The potential clinical benefit of AKR1C3 inhibitors remains to be determined following further preclinical optimization of current compounds.
7. Conclusion
The development and progression of CRPC remains dependent on the activation of the AR by intratumoral androgens derived from the circulating androgen precursers of adrenal origin. These precursors include both canonical and 11-oxygenated androgen precursors which are converted to the potent androgens DHT and 11KT by pathways which share common steroidogenic enzymes. Indeed primary PCa and CRPC tumors are characterized by alterations in the expression levels of these steroidogenic enzymes favouring the production of potent androgens which bind and activate the AR. While the metabolism of canonical androgens has been well characterized, the recent identification of the 11-oxygenated androgens as role players in CRPC has revealed that the extent of the intratumoral androgen pool remaning following ADT has been underestimated to date and further studies are needed in order to obtain the full picture. Furthermore, these 11-oxygenated androgens need to be taken into account when evaluating treatment targets and drug efficacy. Nonetheless, steroidogenic enzymes remain key targets for the development of new drugs. However, understanding of intratumoral and systemic steroid metabolising enzymes is vital when developing drugs based on steroidal structures given that these compounds may be subject to metabolism themselves. Current strategies in drug development focus on combination therapies, inhibiting one or more steroidogenic enzymes in combination with the AR. Treatments may not however be a one-size-fits-all as the specific steroidogenic enzyme repertoire, which include splice varients and SNPs, have been shown to significantly influence treatment outcomes and can be predictive of the most appropriate treatment strategy. A comprehensive understanding of steroid metabolizing enzymes and pathways therefore remains a prerequisite for our growing understanding of CRPC and the development of novel precision treatments.
Figure 1.

Schematic overview of adrenal androgen biosynthesis. Arrows are labelled with the catalyzing enzyme and isoform where appropriate. Potent androgens are shown in grey. Essential accessory proteins are also indicated: adrenodoxin (ADX); adrenodoxin reductase (ADXR); cytochrome b5 (b5); cytochrome P450 oxidoreductase (POR); hexose-6-phosphate dehydrogenase (H6PDH); PAPS synthase 2 (PAPSS2); steroidogenic acute regulatory protein (StAR).
Figure 2.
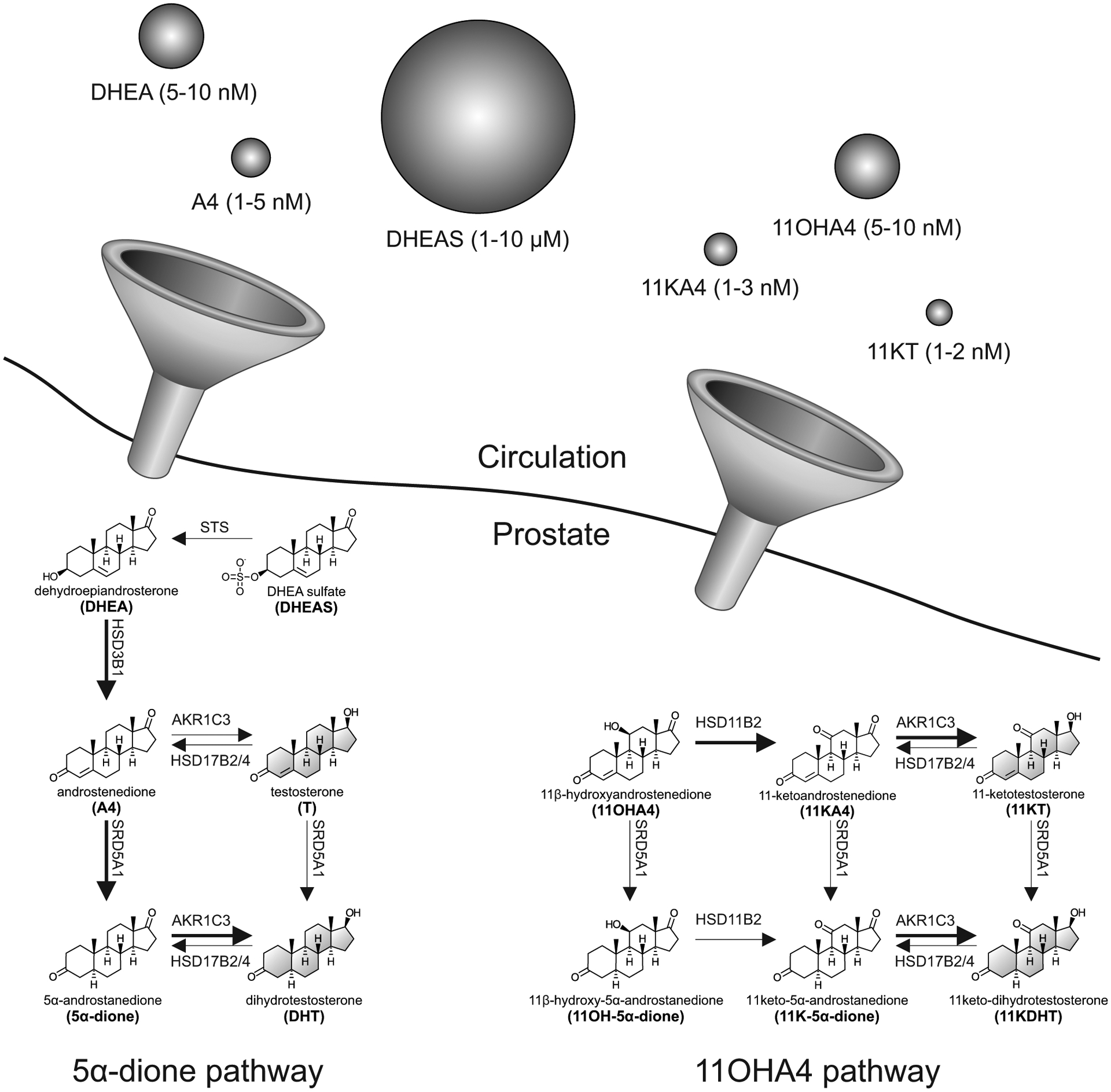
Schematic overview of the intratumoral pathways converting circulating adenal androgen precursors to potent androgens. The relative circulating concentrations of adrenal androgens are indicated by the size of spheres (not to scale). Arrows are labelled with the catalyzing enzyme and isoform where appropriate with preferred reactions in the 5α-dione and 11OHA4 pathways shown with bold arrows. Potent androgens are shown in grey.
Acknowlegements
We would like to thank Trish Storbeck for the preparation of the figures. This work is based on the research supported in part by the National Research Foundation of South Africa (Grant Number 98886 to KS), the Medical Research Council of South Africa (KS), the Cancer Association of South Africa (KS), the NIH Pacific Northwest Prostate Cancer SPORE (P50 CA97186 to EAM), the NIH (P01 CA163227 to EAM and R01GM086596 to RJA) and the DOD (W81XWH-15-1-0150 and W81XWH-12-1-0208 to EAM).
Abbreviations:
- 5αG
3-keto-5α-galeterone
- 5-Adiol
androstenediol
- 5-adiol3S
androstenediol-3-sulphate
- 11K-5α-dione
11keto-5α-androstanedione
- 11KA4
11-ketoandrostenedione
- 11KDHT
11-keto-5α-dihydrotestosterone
- 11KT
11-ketotestosterone
- 11OHA4
11β-hydroxyandrostenedione
- 11OHT
11β-hydroxytestosterone
- A4
androstenedione
- ACTH
adrenocorticotropic hormone
- ADT
androgen deprivation therapy
- ADX
adrenalectomy
- AKR1C3
aldo-keto reductase 1C3
- AR
androgen receptor
- BPH
benign prostatic hyperplasia
- CAB
combined androgen blockade
- CRPC
castration resistant prostate cancer
- CYP11B1
cytochrome P450 11β-hydroxylase
- CYP17A1
cytochrome P450 17α-hydroxylase/17,20-lyase
- D4A
Δ4-abiraterone
- D4G
Δ4-galeterone
- DHEA
dehydroepiandrosterone
- DHEAS
dehydroepiandrosterone sulphate
- DHT
5α-dihydrotestosterone
- eIF2α
eukaryotic Initiation Factor 2
- HSD3B2
3β-hydroxysteroid dehydrogenase type 2
- HSD11B11
11β-hydroxysteroid dehydrogenase type 1
- HSD17B2
17β-hydroxysteroid dehydrogenase type 2
- HSD17B4
17β-hydroxysteroid dehydrogenase type 4
- LH
luteinizing Hormone
- OATP
organic anion transporter polypeptides
- ORX
orchiectomy
- PSA
prostatic-specific antigen
- SNP
single-nucleotide polymorphisms
- SRD5A
steroid 5α-reductase
- STS
steroid sulfatase
- StAR
steroidogenic acute regulatory protein
- SULT2A1
sulfotransferase family 2A member 1
- T
testosterone
- TAPS
total androgen pathway suppression
- TMPRSS22
transmembrane protease serine 2
- TN
treatment-naïve
- UGT
uridine 5’-diphospho-glucuronosyltransferase
References
- 1.Heinlein CA, and Chang C (2004) Androgen receptor in prostate cancer. Endocr. Rev 25, 276–308 [DOI] [PubMed] [Google Scholar]
- 2.Deslypere JP, Young M, Wilson JD, and McPhaul MJ (1992) Testosterone and 5 alpha-dihydrotestosterone interact differently with the androgen receptor to enhance transcription of the MMTV-CAT reporter gene. Mol. Cell. Endocrinol 88, 15–22 [DOI] [PubMed] [Google Scholar]
- 3.Luu-The V, Bélanger A, and Labrie F (2008) Androgen biosynthetic pathways in the human prostate. Best Pract. Res. Clin. Endocrinol. Metab 22, 207–221 [DOI] [PubMed] [Google Scholar]
- 4.Penning TM, Jin Y, Rizner TL, and Bauman DR (2008) Pre-receptor regulation of the androgen receptor. Mol. Cell. Endocrinol 281, 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Russell DW, and Wilson JD (1994) Steroid 5α-Reductase: Two genes/two enzymes. Annu. Rev. Biochem 63, 25–61 [DOI] [PubMed] [Google Scholar]
- 6.Scher HI, and Sawyers CL (2005) Biology of progressive, castration-resistant prostate cancer: directed therapies targeting the androgen-receptor signaling axis. J. Clin. Oncol 23, 8253–8261 [DOI] [PubMed] [Google Scholar]
- 7.Huggins C, and Hodges CV (1941) Studies on Prostatic Cancer. I. The Effect of Castration, of Estrogen and of Androgen Injection on Serum Phosphatases in Metastatic Carcinoma of the Prostate. Cancer Res. 1, 293 LP – 297 [DOI] [PubMed] [Google Scholar]
- 8.Shafi AA, Yen AE, and Weigel NL (2013) Androgen receptors in hormone-dependent and castration-resistant prostate cancer. Pharmacol. Ther 140, 223–38 [DOI] [PubMed] [Google Scholar]
- 9.Heidenreich A, Bastian PJ, Bellmunt J, Bolla M, Joniau S, van der Kwast T, Mason M, Matveev V, Wiegel T, Zattoni F, and Mottet N (2014) EAU guidelines on prostate cancer. Part II: Treatment of advanced, relapsing, and castration-resistant prostate cancer. Eur. Urol 65, 467–479 [DOI] [PubMed] [Google Scholar]
- 10.Huggins C, and Hodges CV (1972) Studies on prostatic cancer. I. The effect of castration, of estrogen and androgen injection on serum phosphatases in metastatic carcinoma of the prostate. CA. Cancer J. Clin 22, 232–240 [DOI] [PubMed] [Google Scholar]
- 11.Ozyigit G, Hurmuz P, Yuce D, and Akyol F (2019) Prognostic significance of castrate testosterone levels for patients with intermediate and high risk prostate cancer. World J. Clin. Oncol 10, 283–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sharifi R, Bruskewitz RC, Gittleman MC, Graham SDJ, Hudson PB, and Stein B (1996) Leuprolide acetate 22.5 mg 12-week depot formulation in the treatment of patients with advanced prostate cancer. Clin. Ther 18, 647–657 [DOI] [PubMed] [Google Scholar]
- 13.Penning TM (2010) New frontiers in androgen biosynthesis and metabolism. Curr. Opin. Endocrinol. Diabetes. Obes 17, 233–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Geller J, Albert J, Yen SS, Geller S, and Loza D (1981) Medical castration of males with megestrol acetate and small doses of diethylstilbestrol. J. Clin. Endocrinol. Metab 52, 576–580 [DOI] [PubMed] [Google Scholar]
- 15.Geller J, Albert J, Loza D, Geller S, Stoeltzing W, and de la Vega D (1978) DHT concentrations in human prostate cancer tissue. J. Clin. Endocrinol. Metab 46, 440–444 [DOI] [PubMed] [Google Scholar]
- 16.Geller J, and Albert J (1987) Effects of castration compared with total androgen blockade on tissue dihydrotestosterone (DHT) concentration in benign prostatic hyperplasia (BPH). Urol. Res 15, 151–153 [DOI] [PubMed] [Google Scholar]
- 17.Labrie F, Dupont A, Belanger A, Lacoursiere Y, Raynaud JP, Husson JM, Gareau J, Fazekas AT, Sandow J, and Monfette G (1983) New approach in the treatment of prostate cancer: complete instead of partial withdrawal of androgens. Prostate. 4, 579–594 [DOI] [PubMed] [Google Scholar]
- 18.Yuan X, and Balk SP (2009) Mechanisms Mediating Androgen Receptor Reactivation After Castration. Urol Oncol. 27, 36–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taplin ME, Bubley GJ, Shuster TD, Frantz ME, Spooner AE, Ogata GK, Keer HN, and Balk SP (1995) Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. N. Engl. J. Med 332, 1393–1398 [DOI] [PubMed] [Google Scholar]
- 20.Knudsen KE, and Penning TM (2010) Partners in crime: deregulation of AR activity and androgen synthesis in prostate cancer. Trends Endocrinol. Metab 21, 315–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Taplin M-E, Rajeshkumar B, Halabi S, Werner CP, Woda BA, Picus J, Stadler W, Hayes DF, Kantoff PW, Vogelzang NJ, and Small EJ (2003) Androgen receptor mutations in androgen-independent prostate cancer: Cancer and Leukemia Group B Study 9663. J. Clin. Oncol 21, 2673–2678 [DOI] [PubMed] [Google Scholar]
- 22.Sharifi N, and Auchus RJ (2012) Steroid biosynthesis and prostate cancer. Steroids. 77, 719–726 [DOI] [PubMed] [Google Scholar]
- 23.Mohler JL, Gregory CW, Ford OH 3rd, Kim D, Weaver CM, Petrusz P, Wilson EM, and French FS (2004) The androgen axis in recurrent prostate cancer. Clin. Cancer Res 10, 440–448 [DOI] [PubMed] [Google Scholar]
- 24.Montgomery RB, Mostaghel EA, Vessella R, Hess DL, Kalhorn TF, Higano CS, True LD, and Nelson PS (2008) Maintenance of Intratumoral Androgens in Metastatic Prostate Cancer : A Mechanism for Castration-Resistant Tumor Growth. 10.1158/0008-5472.CAN-08-0249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Potter GA, Barrie SE, Jarman M, and Rowlands MG (1995) Novel steroidal inhibitors of human cytochrome P45017 alpha (17 alpha-hydroxylase-C17,20-lyase): potential agents for the treatment of prostatic cancer. J. Med. Chem 38, 2463–2471 [DOI] [PubMed] [Google Scholar]
- 26.Barrie SE, Potter GA, Goddard PM, Haynes BP, Dowsett M, and Jarman M (1994) Pharmacology of novel steroidal inhibitors of cytochrome P450(17) alpha (17 alpha-hydroxylase/C17–20 lyase). J. Steroid Biochem. Mol. Biol 50, 267–273 [DOI] [PubMed] [Google Scholar]
- 27.Watson PA, Chen YF, Balbas MD, Wongvipat J, Socci ND, Viale A, Kim K, and Sawyers CL (2010) Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc. Natl. Acad. Sci . 107, 16759–16765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, Wongvipat J, Smith-Jones PM, Yoo D, Kwon A, Wasielewska T, Welsbie D, Chen CD, Higano CS, Beer TM, Hung DT, Scher HI, Jung ME, and Sawyers CL (2009) Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 324, 787–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ryan CJ, Smith MR, Fizazi K, Saad F, Mulders PFA, Sternberg CN, Miller K, Logothetis CJ, Shore ND, Small EJ, Carles J, Flaig TW, Taplin M-E, Higano CS, de Souza P, de Bono JS, Griffin TW, De Porre P, Yu MK, Park YC, Li J, Kheoh T, Naini V, Molina A, and Rathkopf DE (2015) Abiraterone acetate plus prednisone versus placebo plus prednisone in chemotherapy-naive men with metastatic castration-resistant prostate cancer (COU-AA-302): final overall survival analysis of a randomised, double-blind, placebo-controlled phase 3 study. Lancet. Oncol 16, 152–160 [DOI] [PubMed] [Google Scholar]
- 30.O’Donnell A, Judson I, Dowsett M, Raynaud F, Dearnaley D, Mason M, Harland S, Robbins A, Halbert G, Nutley B, and Jarman M (2004) Hormonal impact of the 17alpha-hydroxylase/C(17,20)-lyase inhibitor abiraterone acetate (CB7630) in patients with prostate cancer. Br. J. Cancer 90, 2317–2325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Danila DC, Morris MJ, de Bono JS, Ryan CJ, Denmeade SR, Smith MR, Taplin M-E, Bubley GJ, Kheoh T, Haqq C, Molina A, Anand A, Koscuiszka M, Larson SM, Schwartz LH, Fleisher M, and Scher HI (2010) Phase II multicenter study of abiraterone acetate plus prednisone therapy in patients with docetaxel-treated castration-resistant prostate cancer. J. Clin. Oncol 28, 1496–1501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beer TM, Armstrong AJ, Rathkopf DE, Loriot Y, Sternberg CN, Higano CS, Iversen P, Bhattacharya S, Carles J, Chowdhury S, Davis ID, de Bono JS, Evans CP, Fizazi K, Joshua AM, Kim C-S, Kimura G, Mainwaring P, Mansbach H, Miller K, Noonberg SB, Perabo F, Phung D, Saad F, Scher HI, Taplin M-E, Venner PM, and Tombal B (2014) Enzalutamide in metastatic prostate cancer before chemotherapy. N. Engl. J. Med 371, 424–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ryan CJ, Shah S, Efstathiou E, Smith MR, Taplin M-E, Bubley GJ, Logothetis CJ, Kheoh T, Kilian C, Haqq CM, Molina A, and Small EJ (2011) Phase II study of abiraterone acetate in chemotherapy-naive metastatic castration-resistant prostate cancer displaying bone flare discordant with serologic response. Clin. Cancer Res 17, 4854–4861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ryan CJ, Smith MR, de Bono JS, Molina A, Logothetis CJ, de Souza P, Fizazi K, Mainwaring P, Piulats JM, Ng S, Carles J, Mulders PFA, Basch E, Small EJ, Saad F, Schrijvers D, Van Poppel H, Mukherjee SD, Suttmann H, Gerritsen WR, Flaig TW, George DJ, Yu EY, Efstathiou E, Pantuck A, Winquist E, Higano CS, Taplin M-E, Park Y, Kheoh T, Griffin T, Scher HI, and Rathkopf DE (2013) Abiraterone in metastatic prostate cancer without previous chemotherapy. N. Engl. J. Med 368, 138–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim W, Zhang L, Wilton JH, Fetterly G, Mohler JL, Weinberg V, Morse A, Szmulewitz RZ, Friedlander TW, Fong L, Lin AM, Harzstark AL, Molina A, Small EJ, and Ryan CJ (2014) Sequential use of the androgen synthesis inhibitors ketoconazole and abiraterone acetate in castration-resistant prostate cancer and the predictive value of circulating androgens. Clin. Cancer Res 20, 6269–6276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Attard G, Reid AHM, Yap TA, Raynaud F, Dowsett M, Settatree S, Barrett M, Parker C, Martins V, Folkerd E, Clark J, Cooper CS, Kaye SB, Dearnaley D, Lee G, and de Bono JS (2008) Phase I clinical trial of a selective inhibitor of CYP17, abiraterone acetate, confirms that castration-resistant prostate cancer commonly remains hormone driven. J. Clin. Oncol 26, 4563–4571 [DOI] [PubMed] [Google Scholar]
- 37.de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, Chi KN, Jones RJ, Goodman OB, Saad F, Staffurth JN, Mainwaring P, Harland S, Flaig TW, Hutson TE, Cheng T, Patterson H, Hainsworth JD, Ryan CJ, Sternberg CN, Ellard SL, Fléchon A, Saleh M, Scholz M, Efstathiou E, Zivi A, Bianchini D, Loriot Y, Chieffo N, Kheoh T, Haqq CM, and Scher HI (2011) Abiraterone and Increased Survival in Metastatic Prostate Cancer. N. Engl. J. Med 364, 1995–2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scher HI, Fizazi K, Saad F, Taplin M-E, Sternberg CN, Miller K, de Wit R, Mulders P, Chi KN, Shore ND, Armstrong AJ, Flaig TW, Flechon A, Mainwaring P, Fleming M, Hainsworth JD, Hirmand M, Selby B, Seely L, and de Bono JS (2012) Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med 367, 1187–1197 [DOI] [PubMed] [Google Scholar]
- 39.Tombal B, and van Soest RJ (2017) Prostate cancer: STAMPEDE, LATITUDE and Fernand Labrie’s legacy. Nat. Rev. Urol 14, 588–590 [DOI] [PubMed] [Google Scholar]
- 40.Labrie F, Belanger A, Luu-The V, Labrie C, Simard J, Cusan L, Gomez J, and Candas B (2005) Gonadotropin-releasing hormone agonists in the treatment of prostate cancer. Endocr. Rev 26, 361–379 [DOI] [PubMed] [Google Scholar]
- 41.Thompson IM (2001) Flare Associated with LHRH-Agonist Therapy. Rev. Urol 3 Suppl 3, S10–S14 [PMC free article] [PubMed] [Google Scholar]
- 42.Geller J, Liu J, Albert J, Fay W, Berry CC, and Weis P (1987) Relationship between human prostatic epithelial cell protein synthesis and tissue dihydrotestosterone level. Clin. Endocrinol. (Oxf) 26, 155–161 [DOI] [PubMed] [Google Scholar]
- 43.Liu J, Albert J, and Geller J (1986) Effects of androgen blockade with ketoconazole and megestrol acetate on human prostatic protein patterns. Prostate. 9, 199–205 [DOI] [PubMed] [Google Scholar]
- 44.Liu J, Geller J, Albert J, and Kirshner M (1985) Acute effects of testicular and adrenal cortical blockade on protein synthesis and dihydrotestosterone content of human prostate tissue. J. Clin. Endocrinol. Metab 61, 129–133 [DOI] [PubMed] [Google Scholar]
- 45.Mostaghel EA, Nelson P, Lange PH, Lin DW, Taplin M-E, Balk SP, Ellis WJ, Penning T, Marck B, True LD, Vessella R, and Montgomery RB (2012) Neoadjuvant androgen pathway suppression prior to prostatectomy. J. Clin. Oncol 30, 4520 [Google Scholar]
- 46.Taplin M-E, Montgomery RB, Logothetis C, Bubley GJ, Richie JP, Dalkin BL, Sanda MG, Loda MF, True LD, Troncoso P, Genega EM, Balk SP, Nelson P, Xie W, Haqq CM, Tran N, Liu CS, Kheoh TS, Molina A, and Kantoff P (2012) Effect of neoadjuvant abiraterone acetate (AA) plus leuprolide acetate (LHRHa) on PSA, pathological complete response (pCR), and near pCR in localized high-risk prostate cancer (LHRPC): Results of a randomized phase II study. J. Clin. Oncol 30, 4521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.James ND, de Bono JS, Spears MR, Clarke NW, Mason MD, Dearnaley DP, Ritchie AWS, Amos CL, Gilson C, Jones RJ, Matheson D, Millman R, Attard G, Chowdhury S, Cross WR, Gillessen S, Parker CC, Russell JM, Berthold DR, Brawley C, Adab F, Aung S, Birtle AJ, Bowen J, Brock S, Chakraborti P, Ferguson C, Gale J, Gray E, Hingorani M, Hoskin PJ, Lester JF, Malik ZI, McKinna F, McPhail N, Money-Kyrle J, O’Sullivan J, Parikh O, Protheroe A, Robinson A, Srihari NN, Thomas C, Wagstaff J, Wylie J, Zarkar A, Parmar MKB, and Sydes MR (2017) Abiraterone for Prostate Cancer Not Previously Treated with Hormone Therapy. N. Engl. J. Med 377, 338–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fizazi K, Tran N, Fein L, Matsubara N, Rodriguez-Antolin A, Alekseev BY, Ozguroglu M, Ye D, Feyerabend S, Protheroe A, De Porre P, Kheoh T, Park YC, Todd MB, and Chi KN (2017) Abiraterone plus Prednisone in Metastatic, Castration-Sensitive Prostate Cancer. N. Engl. J. Med 377, 352–360 [DOI] [PubMed] [Google Scholar]
- 49.Fizazi K, Tran N, Fein L, Matsubara N, Rodriguez-Antolin A, Alekseev BY, Özgüroğlu M, Ye D, Feyerabend S, Protheroe A, Sulur G, Luna Y, Li S, Mundle S, and Chi KN (2019) Abiraterone acetate plus prednisone in patients with newly diagnosed high-risk metastatic castration-sensitive prostate cancer (LATITUDE): final overall survival analysis of a randomised, double-blind, phase 3 trial. Lancet Oncol. 10.1016/S1470-2045(19)30082-8 [DOI] [PubMed] [Google Scholar]
- 50.Mostaghel EA, Zhang A, Hernandez S, Marck B, Zhang X, Tamae D, Biehl H, Tretiakova MS, Bartlett J, Burns JF, Dumpit RF, Ang LS, Matsumoto AM, Penning TM, Balk SP, Morrisey C, Corey E, True L, and Nelson PS (2019) Contribution of Adrenal Glands to Intra-tumor Androgens and Growth of Castration Resistant Prostate Cancer. Clin. Cancer Res 25, 426 LP – 439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stuchbery R, McCoy PJ, Hovens CM, and Corcoran NM (2017) Androgen synthesis in prostate cancer: do all roads lead to Rome? Nat. Rev. Urol 14, 49–58 [DOI] [PubMed] [Google Scholar]
- 52.Huhtaniemi R, Oksala R, Knuuttila M, Mehmood A, Aho E, Laajala TD, Nicorici D, Aittokallio T, Laiho A, Elo L, Ohlsson C, Kallio P, Makela S, Mustonen MVJ, Sipila P, and Poutanen M (2018) Adrenals Contribute to Growth of Castration-Resistant VCaP Prostate Cancer Xenografts. Am. J. Pathol 10.1016/j.ajpath.2018.07.029 [DOI] [PubMed] [Google Scholar]
- 53.Lindzey J, Wetsel WC, Couse JF, Stoker T, Cooper R, and Korach KS (1998) Effects of castration and chronic steroid treatments on hypothalamic gonadotropin-releasing hormone content and pituitary gonadotropins in male wild-type and estrogen receptor-alpha knockout mice. Endocrinology. 139, 4092–4101 [DOI] [PubMed] [Google Scholar]
- 54.Bernichtein S, Alevizaki M, and Huhtaniemi I (2008) Is the adrenal cortex a target for gonadotropins? Trends Endocrinol. Metab 19, 231–238 [DOI] [PubMed] [Google Scholar]
- 55.Payne AH, and Hales DB (2004) Overview of Steroidogenic Enzymes in the Pathway from Cholesterol to Active Steroid Hormones. Endocr. Rev 25, 947–970 [DOI] [PubMed] [Google Scholar]
- 56.Touchstone HC, Glazer L, Cooper DY, and Roberts JM (1955) The isolation of delta 4-androstene-11 beta-ol-3,17-dione from human adrenal incubates. J. Clin. Endocrinol. Metab 15, 382–384 [DOI] [PubMed] [Google Scholar]
- 57.Meyer AS, Jeanloz RW, and Pincus G (1953) Chemical transformations of steroids by adrenal perfusion. IV. Dehydroepiandrosterone. J. Biol. Chem 203, 463–468 [PubMed] [Google Scholar]
- 58.Holownia P, Owen EJ, Conway GS, Round J, and Honour JW (1992) Studies to confirm the source of 11 beta-hydroxyandrostenedione. J. Steroid Biochem. Mol. Biol 41, 875–880 [DOI] [PubMed] [Google Scholar]
- 59.Dorfman RI (1954) In vivo metabolism of neutral steroid hormones. J. Clin. Endocrinol. Metab 14, 318–325 [DOI] [PubMed] [Google Scholar]
- 60.Holownia P, Owen EJ, Hampl R, Jacobs HS, and Honour JW (1991) The determination of 11β-hydroxyandrostenedione in human follicular fluid and plasma. J. Steroid Biochem. Mol. Biol 38, 389–398 [DOI] [PubMed] [Google Scholar]
- 61.Rege J, Nakamura Y, Satoh F, Morimoto R, Kennedy MR, Layman LC, Honma S, Sasano H, and Rainey WE (2013) Liquid Chromatography–Tandem Mass Spectrometry Analysis of Human Adrenal Vein 19-Carbon Steroids Before and After ACTH Stimulation. J. Clin. Endocrinol. Metab 98, 1182–1188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xing Y, Edwards MA, Ahlem C, Kennedy M, Cohen A, Gomez-Sanchez CE, and Rainey WE (2011) The effects of ACTH on steroid metabolomic profiles in human adrenal cells. J. Endocrinol 209, 327–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Auchus RJ, and Rainey WE (2004) Adrenarche - physiology, biochemistry and human disease. Clin. Endocrinol. (Oxf) 60, 288–296 [DOI] [PubMed] [Google Scholar]
- 64.Rainey WE, Carr BR, Sasano H, Suzuki T, and Mason JI (2002) Dissecting human adrenal androgen production. Trends Endocrinol. Metab 13, 234–239 [DOI] [PubMed] [Google Scholar]
- 65.Rege J, and Rainey WE (2012) The steroid metabolome of adrenarche. J. Endocrinol 214, 133–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Miller WL, and Auchus RJ (2011) The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr. Rev 32, 81–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Turcu A, Smith JM, Auchus R, and Rainey WE (2014) Adrenal androgens and androgen precursors-definition, synthesis, regulation and physiologic actions. Compr. Physiol 4, 1369–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gilep AA, Sushko TA, and Usanov SA (2011) At the crossroads of steroid hormone biosynthesis: the role, substrate specificity and evolutionary development of CYP17. Biochim. Biophys. Acta 1814, 200–209 [DOI] [PubMed] [Google Scholar]
- 69.Auchus RJ, Lee TC, and Miller WL (1998) Cytochrome b 5 Augments the 17 , 20-Lyase Activity of Human P450c17 without Direct Electron Transfer. J Biol Chem 273, 3158–3165 [DOI] [PubMed] [Google Scholar]
- 70.Saner KJ, Suzuki T, Sasano H, Pizzey J, Ho C, Strauss JF 3rd, Carr BR, and Rainey WE (2005) Steroid sulfotransferase 2A1 gene transcription is regulated by steroidogenic factor 1 and GATA-6 in the human adrenal. Mol. Endocrinol 19, 184–197 [DOI] [PubMed] [Google Scholar]
- 71.Nakamura Y, Xing Y, Hui X-G, Kurotaki Y, Ono K, Cohen T, Sasano H, and Rainey WE (2011) Human adrenal cells that express both 3β-hydroxysteroid dehydrogenase type 2 (HSD3B2) and cytochrome b5 (CYB5A) contribute to adrenal androstenedione production. J. Steroid Biochem. Mol. Biol 123, 122–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nguyen AD, Corbin CJ, Pattison JC, Bird IM, and Conley AJ (2009) The developmental increase in adrenocortical 17,20-lyase activity (biochemical adrenarche) is driven primarily by increasing cytochrome b5 in neonatal rhesus macaques. Endocrinology. 150, 1748–1756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rainey WE, and Nakamura Y (2008) Regulation of the adrenal androgen biosynthesis. J. Steroid Biochem. Mol. Biol 108, 281–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zang T, Taplin M-E, Tamae D, Xie W, Mesaros C, Zhang Z, Bubley G, Montgomery B, Balk SP, Mostaghel EA, Blair IA, and Penning TM (2017) Testicular vs adrenal sources of hydroxy-androgens in prostate cancer. Endocr. Relat. Cancer 24, 393–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Strushkevich N, Gilep AA, Shen L, Arrowsmith CH, Edwards AM, Usanov SA, and Park H-W (2013) Structural insights into aldosterone synthase substrate specificity and targeted inhibition. Mol. Endocrinol 27, 315–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Storbeck K-H, Bloem LM, Africander D, Schloms L, Swart P, and Swart AC (2013) 11beta-Hydroxydihydrotestosterone and 11-ketodihydrotestosterone, novel C19 steroids with androgenic activity: a putative role in castration resistant prostate cancer? Mol. Cell. Endocrinol 377, 135–146 [DOI] [PubMed] [Google Scholar]
- 77.Swart AC, and Storbeck K-H (2015) 11beta-Hydroxyandrostenedione: Downstream metabolism by 11betaHSD, 17betaHSD and SRD5A produces novel substrates in familiar pathways. Mol. Cell. Endocrinol 408, 114–123 [DOI] [PubMed] [Google Scholar]
- 78.Turcu AF, Nanba AT, Chomic R, Upadhyay SK, Giordano TJ, Shields JJ, Merke DP, Rainey WE, and Auchus RJ (2016) Adrenal-derived 11-oxygenated 19-carbon steroids are the dominant androgens in classic 21-hydroxylase deficiency. Eur. J. Endocrinol 174, 601–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schloms L, Storbeck K-H, Swart P, Gelderblom WCA, and Swart AC (2012) The influence of Aspalathus linearis (Rooibos) and dihydrochalcones on adrenal steroidogenesis: quantification of steroid intermediates and end products in H295R cells. J. Steroid Biochem. Mol. Biol 128, 128–138 [DOI] [PubMed] [Google Scholar]
- 80.Barnard M, Quanson JL, Mostaghel E, Pretorius E, Snoep JL, and Storbeck K-H (2018) 11-Oxygenated androgen precursors are the preferred substrates for aldo-keto reductase 1C3 (AKR1C3): Implications for castration resistant prostate cancer. J. Steroid Biochem. Mol. Biol 183, 192–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pretorius E, Arlt W, and Storbeck K-H (2017) A new dawn for androgens: Novel lessons from 11-oxygenated C19 steroids. Mol. Cell. Endocrinol 441, 76–85 [DOI] [PubMed] [Google Scholar]
- 82.O’Reilly MW, Kempegowda P, Walsh M, Taylor AE, Manolopoulos KN, Allwood JW, Semple RK, Hebenstreit D, Dunn WB, Tomlinson JW, and Arlt W (2017) AKR1C3-Mediated Adipose Androgen Generation Drives Lipotoxicity in Women With Polycystic Ovary Syndrome. J. Clin. Endocrinol. Metab 102, 3327–3339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nanba AT, Ren J, Turcu AF, Rainey WE, Auchus RJ, and Rege J (2019) 11-Oxygenated C19 Steroids Do Not Decline with Age in Women. J Clin Endocrinol Metab 7, 2615–2622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rege J, Garber S, Conley AJ, Elsey RM, Turcu AF, Auchus RJ, and Rainey WE (2019) Circulating 11-oxygenated androgens across species. J. Steroid Biochem. Mol. Biol 10.1016/j.jsbmb.2019.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.O’Reilly MW, Kempegowda P, Jenkinson C, Taylor AE, Quanson JL, Storbeck K-H, and Arlt W (2017) 11-Oxygenated C19 Steroids Are the Predominant Androgens in Polycystic Ovary Syndrome. J. Clin. Endocrinol. Metab 102, 840–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Davis SR, Turcu AF, Robinson PJ, and Bell RJ (2019) Exogenous Testosterone Does Not Influence 11-Oxygenated C19 Steroid Concentrations in Healthy Postmenopausal Women. J. Endocr. Soc 3, 670–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Namba A, Rege J, Rainey W, Auchus R, Turcu A (2019) Adrenal Androgen Synthesis in Aging Men. J. Endocr. Soc [Google Scholar]
- 88.Koh E, Noda T, Kanaya J, and Namiki M (2002) Differential expression of 17beta-hydroxysteroid dehydrogenase isozyme genes in prostate cancer and noncancer tissues. Prostate. 53, 154–159 [DOI] [PubMed] [Google Scholar]
- 89.Mitsiades N, Sung CC, Schultz N, Danila DC, He B, Eedunuri VK, Fleisher M, Sander C, Sawyers CL, and Scher HI (2012) Distinct patterns of dysregulated expression of enzymes involved in androgen synthesis and metabolism in metastatic prostate cancer tumors. Cancer Res. 72, 6142–6152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pfeiffer MJ, Smit FP, Sedelaar JPM, and Schalken JA (2011) Steroidogenic enzymes and stem cell markers are upregulated during androgen deprivation in prostate cancer. Mol. Med 17, 657–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Stanbrough M, Bubley GJ, Ross K, Golub TR, Rubin MA, Penning TM, Febbo PG, and Balk SP (2006) Increased Expression of Genes Converting Adrenal Androgens to Testosterone in Androgen-Independent Prostate Cancer. Cancer Res. 66, 2815–2826 [DOI] [PubMed] [Google Scholar]
- 92.Stanbrough M, Bubley GJ, Ross K, Golub TR, Rubin M. a., Penning TM, Febbo PG, and Balk SP (2006) Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res. 66, 2815–2825 [DOI] [PubMed] [Google Scholar]
- 93.Fung K-M, Samara ENS, Wong C, Metwalli A, Krlin R, Bane B, Liu CZ, Yang JT, Pitha JV, Culkin DJ, Kropp BP, Penning TM, and Lin H-K (2006) Increased expression of type 2 3alpha-hydroxysteroid dehydrogenase/type 5 17beta-hydroxysteroid dehydrogenase (AKR1C3) and its relationship with androgen receptor in prostate carcinoma. Endocr. Relat. Cancer 13, 169–180 [DOI] [PubMed] [Google Scholar]
- 94.Ko H-K, Berk M, Chung Y-M, Willard B, Bareja R, Rubin M, Sboner A, and Sharifi N (2018) Loss of an Androgen-Inactivating and Isoform-Specific HSD17B4 Splice Form Enables Emergence of Castration-Resistant Prostate Cancer. Cell Rep. 22, 809–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hofland J, van Weerden WM, Dits NFJ, Steenbergen J, van Leenders GJLH, Jenster G, Schroder FH, and de Jong FH (2010) Evidence of limited contributions for intratumoral steroidogenesis in prostate cancer. Cancer Res. 70, 1256–1264 [DOI] [PubMed] [Google Scholar]
- 96.Liu C, Armstrong CM, Lou W, Lombard A, Evans CP, and Gao AC (2017) Inhibition of AKR1C3 Activation Overcomes Resistance to Abiraterone in Advanced Prostate Cancer. Mol. Cancer Ther 16, 35–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Liu C, Lou W, Zhu Y, Yang JC, Nadiminty N, Gaikwad NW, Evans CP, and Gao AC (2015) Intracrine Androgens and AKR1C3 Activation Confer Resistance to Enzalutamide in Prostate Cancer. Cancer Res. 75, 1413 LP – 1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Fankhauser M, Tan Y, Macintyre G, Haviv I, Hong MKH, Nguyen A, Pedersen JS, Costello AJ, Hovens CM, and Corcoran NM (2014) Canonical androstenedione reduction is the predominant source of signaling androgens in hormone-refractory prostate cancer. Clin. Cancer Res 20, 5547–5557 [DOI] [PubMed] [Google Scholar]
- 99.Hamid ARAH, Pfeiffer MJ, Verhaegh GW, Schaafsma E, Brandt A, Sweep FCGJ, Sedelaar JPM, and Schalken JA (2013) Aldo-keto reductase family 1 member C3 (AKR1C3) is a biomarker and therapeutic target for castration-resistant prostate cancer. Mol. Med 18, 1449–1455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jernberg E, Thysell E, Bovinder Ylitalo E, Rudolfsson S, Crnalic S, Widmark A, Bergh A, and Wikstrom P (2013) Characterization of prostate cancer bone metastases according to expression levels of steroidogenic enzymes and androgen receptor splice variants. PLoS One. 8, e77407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Powell K, Semaan L, Conley-LaComb MK, Asangani I, Wu Y-M, Ginsburg KB, Williams J, Squire JA, Maddipati KR, Cher ML, and Chinni SR (2015) ERG/AKR1C3/AR Constitutes a Feed-Forward Loop for AR Signaling in Prostate Cancer Cells. Clin. Cancer Res 21, 2569–2579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tian Y, Zhao L, Zhang H, Liu X, Zhao L, Zhao X, Li Y, and Li J (2014) AKR1C3 overexpression may serve as a promising biomarker for prostate cancer progression. Diagn. Pathol 9, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Song C, and Chen H (2018) Predictive significance of TMRPSS2-ERG fusion in prostate cancer: a meta-analysis. Cancer Cell Int. 18, 177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yepuru M, Wu Z, Kulkarni A, Yin F, Barrett CM, Kim J, Steiner MS, Miller DD, Dalton JT, and Narayanan R (2013) Steroidogenic enzyme AKR1C3 is a novel androgen receptor-selective coactivator that promotes prostate cancer growth. Clin. Cancer Res 19, 5613–5625 [DOI] [PubMed] [Google Scholar]
- 105.Bonkhoff H, Stein U, Aumuller G, and Remberger K (1996) Differential expression of 5 alpha-reductase isoenzymes in the human prostate and prostatic carcinomas. Prostate. 29, 261–267 [DOI] [PubMed] [Google Scholar]
- 106.Iehle C, Radvanyi F, Gil Diez de Medina S, Ouafik LH, Gerard H, Chopin D, Raynaud JP, and Martin PM (1999) Differences in steroid 5alpha-reductase iso-enzymes expression between normal and pathological human prostate tissue. J. Steroid Biochem. Mol. Biol 68, 189–195 [DOI] [PubMed] [Google Scholar]
- 107.Thomas LN, Lazier CB, Gupta R, Norman RW, Troyer DA, O’Brien SP, and Rittmaster RS (2005) Differential alterations in 5alpha-reductase type 1 and type 2 levels during development and progression of prostate cancer. Prostate. 63, 231–239 [DOI] [PubMed] [Google Scholar]
- 108.Thomas LN, Douglas RC, Vessey JP, Gupta R, Fontaine D, Norman RW, Thompson IM, Troyer DA, Rittmaster RS, and Lazier CB (2003) 5alpha-reductase type 1 immunostaining is enhanced in some prostate cancers compared with benign prostatic hyperplasia epithelium. J. Urol 170, 2019–2025 [DOI] [PubMed] [Google Scholar]
- 109.Soderstrom TG, Bjelfman C, Brekkan E, Ask B, Egevad L, Norlen BJ, and Rane A (2001) Messenger ribonucleic acid levels of steroid 5 alpha-reductase 2 in human prostate predict the enzyme activity. J. Clin. Endocrinol. Metab 86, 855–858 [DOI] [PubMed] [Google Scholar]
- 110.Luo J, Dunn TA, Ewing CM, Walsh PC, and Isaacs WB (2003) Decreased gene expression of steroid 5 alpha-reductase 2 in human prostate cancer: implications for finasteride therapy of prostate carcinoma. Prostate. 57, 134–139 [DOI] [PubMed] [Google Scholar]
- 111.Bjelfman C, Soderstrom TG, Brekkan E, Norlen BJ, Egevad L, Unge T, Andersson S, and Rane A (1997) Differential gene expression of steroid 5 alpha-reductase 2 in core needle biopsies from malignant and benign prostatic tissue. J. Clin. Endocrinol. Metab 82, 2210–2214 [DOI] [PubMed] [Google Scholar]
- 112.Andriole G, Bostwick D, Brawley O, Gomella L, Marberger M, Tindall D, Breed S, Somerville M, and Rittmaster R (2004) Chemoprevention of prostate cancer in men at high risk: rationale and design of the reduction by dutasteride of prostate cancer events (REDUCE) trial. J. Urol 172, 1314–1317 [DOI] [PubMed] [Google Scholar]
- 113.Mostaghel EA (2013) Steroid hormone synthetic pathways in prostate cancer. Transl. Androl. Urol 2, 212–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sharifi N (2012) The 5alpha-androstanedione pathway to dihydrotestosterone in castration-resistant prostate cancer. J. Investig. Med 60, 504–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chang K-H, Ercole CE, and Sharifi N (2014) Androgen metabolism in prostate cancer: from molecular mechanisms to clinical consequences. Br. J. Cancer 111, 1249–1254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chang K-H, Li R, Papari-Zareei M, Watumull L, Zhao YD, Auchus RJ, and Sharifi N (2011) Dihydrotestosterone synthesis bypasses testosterone to drive castration-resistant prostate cancer. Proc. Natl. Acad. Sci. U. S. A 108, 13728–13733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Knuuttila M, Yatkin E, Kallio J, Savolainen S, Laajala TD, Aittokallio T, Oksala R, Hakkinen M, Keski-Rahkonen P, Auriola S, Poutanen M, and Makela S (2014) Castration induces up-regulation of intratumoral androgen biosynthesis and androgen receptor expression in an orthotopic VCaP human prostate cancer xenograft model. Am. J. Pathol 184, 2163–2173 [DOI] [PubMed] [Google Scholar]
- 118.Dai C, Chung Y-M, Kovac E, Zhu Z, Li J, Magi-Galluzzi C, Stephenson AJ, Klein EA, and Sharifi N (2017) Direct Metabolic Interrogation of Dihydrotestosterone Biosynthesis from Adrenal Precursors in Primary Prostatectomy Tissues. Clin. Cancer Res 23, 6351 LP – 6362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Taplin M-E, Montgomery B, Logothetis CJ, Bubley GJ, Richie JP, Dalkin BL, Sanda MG, Davis JW, Loda M, True LD, Troncoso P, Ye H, Lis RT, Marck BT, Matsumoto AM, Balk SP, Mostaghel EA, Penning TM, Nelson PS, Xie W, Jiang Z, Haqq CM, Tamae D, Tran N, Peng W, Kheoh T, Molina A, and Kantoff PW (2014) Intense androgen-deprivation therapy with abiraterone acetate plus leuprolide acetate in patients with localized high-risk prostate cancer: results of a randomized phase II neoadjuvant study. J. Clin. Oncol 32, 3705–3715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Tamae D, Mostaghel E, Montgomery B, Nelson PS, Balk SP, Kantoff PW, Taplin M-E, and Penning TM (2015) The DHEA-sulfate depot following P450c17 inhibition supports the case for AKR1C3 inhibition in high risk localized and advanced castration resistant prostate cancer. Chem. Biol. Interact 234, 332–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Penning TM (2018) Vitamins and Hormones in Vitamins and Hormones, 108th Ed., pp. 310–325, Elsevier [Google Scholar]
- 122.Cho E, Montgomery RB, and Mostaghel EA (2014) Minireview: SLCO and ABC transporters: a role for steroid transport in prostate cancer progression. Endocrinology. 155, 4124–4132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Arakawa H, Nakanishi T, Yanagihara C, Nishimoto T, Wakayama T, Mizokami A, Namiki M, Kawai K, and Tamai I (2012) Enhanced expression of organic anion transporting polypeptides (OATPs) in androgen receptor-positive prostate cancer cells: possible role of OATP1A2 in adaptive cell growth under androgen-depleted conditions. Biochem. Pharmacol 84, 1070–1077 [DOI] [PubMed] [Google Scholar]
- 124.Green SM, Kaipainen A, Bullock K, Zhang A, Lucas JM, Matson C, Banks WA, and Mostaghel EA (2017) Role of OATP transporters in steroid uptake by prostate cancer cells in vivo. Prostate Cancer Prostatic Dis. 20, 20–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yang M, Xie W, Mostaghel E, Nakabayashi M, Werner L, Sun T, Pomerantz M, Freedman M, Ross R, Regan M, Sharifi N, Figg WD, Balk S, Brown M, Taplin M-E, Oh WK, Lee G-SM, and Kantoff PW (2011) SLCO2B1 and SLCO1B3 may determine time to progression for patients receiving androgen deprivation therapy for prostate cancer. J. Clin. Oncol 29, 2565–2573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sharifi N, Hamada A, Sissung T, Danesi R, Venzon D, Baum C, Gulley JL, Price DK, Dahut WL, and Figg WD (2008) A polymorphism in a transporter of testosterone is a determinant of androgen independence in prostate cancer. BJU Int. 102, 617–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hamada A, Sissung T, Price DK, Danesi R, Chau CH, Sharifi N, Venzon D, Maeda K, Nagao K, Sparreboom A, Mitsuya H, Dahut WL, and Figg WD (2008) Effect of SLCO1B3 haplotype on testosterone transport and clinical outcome in caucasian patients with androgen-independent prostatic cancer. Clin. Cancer Res 14, 3312–3318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wright JL, Kwon EM, Ostrander EA, Montgomery RB, Lin DW, Vessella R, Stanford JL, and Mostaghel EA (2011) Expression of SLCO transport genes in castration-resistant prostate cancer and impact of genetic variation in SLCO1B3 and SLCO2B1 on prostate cancer outcomes. Cancer Epidemiol. Biomarkers Prev 20, 619–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Selcer KW, Kabler H, Sarap J, Xiao Z, and Li P-K (2002) Inhibition of steryl sulfatase activity in LNCaP human prostate cancer cells. Steroids. 67, 821–826 [DOI] [PubMed] [Google Scholar]
- 130.Dalla Valle L, Toffolo V, Nardi A, Fiore C, Armanini D, Belvedere P, and Colombo L (2007) The expression of the human steroid sulfatase-encoding gene is driven by alternative first exons. J. Steroid Biochem. Mol. Biol 107, 22–29 [DOI] [PubMed] [Google Scholar]
- 131.Nakamura Y, Suzuki T, Fukuda T, Ito A, Endo M, Moriya T, Arai Y, and Sasano H (2006) Steroid sulfatase and estrogen sulfotransferase in human prostate cancer. Prostate. 66, 1005–1012 [DOI] [PubMed] [Google Scholar]
- 132.Chang K-H, Li R, Kuri B, Lotan Y, Roehrborn CG, Liu J, Vessella R, Nelson PS, Kapur P, Guo X, Mirzaei H, Auchus RJ, and Sharifi N (2013) A gain-of-function mutation in DHT synthesis in castration-resistant prostate cancer. Cell. 154, 1074–1084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Agarwal N, Hahn AW, Gill DM, Farnham JM, Poole AI, and Cannon-Albright L (2017) Independent Validation of Effect of HSD3B1 Genotype on Response to Androgen-Deprivation Therapy in Prostate Cancer. JAMA Oncol. 3, 856–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Shiota M, Fujimoto N, Imada K, Kashiwagi E, Takeuchi A, Inokuchi J, Tatsugami K, Kajioka S, Uchiumi T, and Eto M (2018) Independent validation of missense polymorphism in HSD3B1 in Japanese men treated with primary androgen-deprivation therapy for metastatic prostate cancer. J. Clin. Oncol 36, 179 [Google Scholar]
- 135.Wu G, Huang S, Nastiuk KL, Li J, Gu J, Wu M, Zhang Q, Lin H, and Wu D (2015) Variant allele of HSD3B1 increases progression to castration-resistant prostate cancer. Prostate. 75, 777–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hearn JWD, AbuAli G, Reichard CA, Reddy CA, Magi-Galluzzi C, Chang K-H, Carlson R, Rangel L, Reagan K, Davis BJ, Karnes RJ, Kohli M, Tindall D, Klein EA, and Sharifi N (2016) HSD3B1 and resistance to androgen-deprivation therapy in prostate cancer: a retrospective, multicohort study. Lancet. Oncol 17, 1435–1444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hearn JWD, Xie W, Nakabayashi M, Almassi N, Reichard CA, Pomerantz M, Kantoff PW, and Sharifi N (2018) Association of HSD3B1 Genotype With Response to Androgen-Deprivation Therapy for Biochemical Recurrence After Radiotherapy for Localized Prostate Cancer. JAMA Oncol. 4, 558–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Almassi N, Reichard C, Li J, Russell C, Perry J, Ryan CJ, Friedlander T, and Sharifi N (2018) HSD3B1 and Response to a Nonsteroidal CYP17A1 Inhibitor in Castration-Resistant Prostate Cancer. JAMA Oncol. 4, 554–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Hettel D, and Sharifi N (2018) HSD3B1 status as a biomarker of androgen deprivation resistance and implications for prostate cancer. Nat. Rev. Urol 15, 191–196 [DOI] [PubMed] [Google Scholar]
- 140.Alyamani M, Emamekhoo H, Park S, Taylor J, Almassi N, Upadhyay S, Tyler A, Berk MP, Hu B, Hwang TH, Figg WD, Peer CJ, Chien C, Koshkin VS, Mendiratta P, Grivas P, Rini B, Garcia J, Auchus RJ, and Sharifi N (2018) HSD3B1(1245A>C) variant regulates dueling abiraterone metabolite effects in prostate cancer. J. Clin. Invest 128, 3333–3340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Hahn AW, Gill DM, Nussenzveig RH, Poole A, Farnham J, Cannon-Albright L, and Agarwal N (2018) Germline Variant in HSD3B1 (1245 A > C) and Response to Abiraterone Acetate Plus Prednisone in Men With New-Onset Metastatic Castration-Resistant Prostate Cancer. Clin. Genitourin. Cancer 16, 288–292 [DOI] [PubMed] [Google Scholar]
- 142.Gao X, Dai C, Huang S, Tang J, Chen G, Li J, Zhu Z, Zhu X, Zhou S, Gao Y, Hou Z, Fang Z, Xu C, Wang J, Wu D, Sharifi N, and Li Z (2018) Functional silencing of HSD17B2 in prostate cancer promotes disease progression. Clin. Cancer Res 10.1158/1078-0432.CCR-18-2392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, Chen Y, Mohammad TA, Chen Y, Fedor HL, Lotan TL, Zheng Q, De Marzo AM, Isaacs JT, Isaacs WB, Nadal R, Paller CJ, Denmeade SR, Carducci MA, Eisenberger MA, and Luo J (2014) AR-V7 and Resistance to Enzalutamide and Abiraterone in Prostate Cancer. N. Engl. J. Med 371, 1028–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Berge EO, Staalesen V, Straume AH, Lillehaug JR, and Lonning PE (2010) Chk2 splice variants express a dominant-negative effect on the wild-type Chk2 kinase activity. Biochim. Biophys. Acta 1803, 386–395 [DOI] [PubMed] [Google Scholar]
- 145.Dehm SM, Schmidt LJ, Heemers HV, Vessella RL, and Tindall DJ (2008) Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 68, 5469–5477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Peterfy M, Phan J, and Reue K (2005) Alternatively spliced lipin isoforms exhibit distinct expression pattern, subcellular localization, and role in adipogenesis. J. Biol. Chem 280, 32883–32889 [DOI] [PubMed] [Google Scholar]
- 147.Tazi J, Bakkour N, and Stamm S (2009) Alternative splicing and disease. Biochim. Biophys. Acta - Mol. Basis Dis 1792, 14–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Sharp A, Coleman I, Yuan W, Sprenger C, Dolling D, Rodrigues DN, Russo JW, Figueiredo I, Bertan C, Seed G, Riisnaes R, Uo T, Neeb A, Welti J, Morrissey C, Carreira S, Luo J, Nelson PS, Balk SP, True LD, de Bono JS, and Plymate SR (2019) Androgen receptor splice variant-7 expression emerges with castration resistance in prostate cancer. J. Clin. Invest 129, 192–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Todenhofer T, Azad A, Stewart C, Gao J, Eigl BJ, Gleave ME, Joshua AM, Black PC, and Chi KN (2017) AR-V7 Transcripts in Whole Blood RNA of Patients with Metastatic Castration Resistant Prostate Cancer Correlate with Response to Abiraterone Acetate. J. Urol 197, 135–142 [DOI] [PubMed] [Google Scholar]
- 150.Welti J, Rodrigues DN, Sharp A, Sun S, Lorente D, Riisnaes R, Figueiredo I, Zafeiriou Z, Rescigno P, de Bono JS, and Plymate SR (2016) Analytical Validation and Clinical Qualification of a New Immunohistochemical Assay for Androgen Receptor Splice Variant-7 Protein Expression in Metastatic Castration-resistant Prostate Cancer. Eur. Urol 70, 599–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Zhu Y, Sharp A, Anderson CM, Silberstein JL, Taylor M, Lu C, Zhao P, De Marzo AM, Antonarakis ES, Wang M, Wu X, Luo Y, Su N, Nava Rodrigues D, Figueiredo I, Welti J, Park E, Ma X-J, Coleman I, Morrissey C, Plymate SR, Nelson PS, de Bono JS, and Luo J (2018) Novel Junction-specific and Quantifiable In Situ Detection of AR-V7 and its Clinical Correlates in Metastatic Castration-resistant Prostate Cancer. Eur. Urol 73, 727–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Qu Y, Dai B, Ye D, Kong Y, Chang K, Jia Z, Yang X, Zhang H, Zhu Y, and Shi G (2015) Constitutively active AR-V7 plays an essential role in the development and progression of castration-resistant prostate cancer. Sci. Rep 5, 7654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Hörnberg E, Ylitalo EB, Crnalic S, Antti H, Stattin P, Widmark A, Bergh A, and Wikström P (2011) Expression of androgen receptor splice variants in prostate cancer bone metastases is associated with castration-resistance and short survival. PLoS One. 6, e19059–e19059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Auvin S, Öztürk H, Abaci YT, Mautino G, Meyer-Losic F, Jollivet F, Bashir T, de Thé H, and Sahin U (2019) A molecule inducing androgen receptor degradation and selectively targeting prostate cancer cells. Life Sci. alliance 2, e201800213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Yu Z, Chen S, Sowalsky AG, Voznesensky OS, Mostaghel EA, Nelson PS, Cai C, and Balk SP (2014) Rapid induction of androgen receptor splice variants by androgen deprivation in prostate cancer. Clin. Cancer Res 20, 1590–1600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Armstrong AJ, Halabi S, Luo J, Nanus DM, Giannakakou P, Szmulewitz RZ, Danila DC, Healy P, Anand M, Rothwell CJ, Rasmussen J, Thornburg B, Berry WR, Wilder RS, Lu C, Chen Y, Silberstein JL, Kemeny G, Galletti G, Somarelli JA, Gupta S, Gregory SG, Scher HI, Dittamore R, Tagawa ST, Antonarakis ES, and George DJ (2019) Prospective Multicenter Validation of Androgen Receptor Splice Variant 7 and Hormone Therapy Resistance in High-Risk Castration-Resistant Prostate Cancer: The PROPHECY Study. J. Clin. Oncol 37, 1120–1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Taplin M-E, Antonarakis ES, Ferrante KJ, Horgan K, Blumenstein BA, Saad F, Luo J, and De Bono JS (2017) Clinical factors associated with AR-V7 detection in ARMOR3-SV, a randomized trial of galeterone (Gal) vs enzalutamide (Enz) in men with AR-V7+ metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol 35, 5005 [Google Scholar]
- 158.Bubley GJ, and Balk SP (2017) Association Between Androgen Receptor Splice Variants and Prostate Cancer Resistance to Abiraterone and Enzalutamide. J. Clin. Oncol 35, 2103–2105 [DOI] [PubMed] [Google Scholar]
- 159.Clarke NW, Ali A, Ingleby FC, Hoyle A, Amos CL, Brawley CD, Calvert J, Chowdhury S, Cook A, Cross W, Dearnaley DP, Douis H, Gilbert D, Gillessen S, Jones RJ, Langley RE, MacNair A, Malik Z, Mason MD, Matheson D, Millman R, Parker CC, Ritchie AWS, Rush H, Russell JM, Brown J, Beesley S, Birtle A, Capaldi L, Gale J, Lydon A, Nikapota A, Omlin A, O’Sullivan JM, Parikh O, Protheroe A, Rudman S, Srihari NN, Simms M, Tanguay JS, Investigators S, Tolan S, Wagstaff J, Wallace J, Wylie J, Zarkar A, Sydes MR, Parmar MKB, and James ND (2019) Addition of docetaxel to hormonal therapy in low- and high-burden metastatic hormone sensitive prostate cancer: long-term survival results from the STAMPEDE trial. Ann. Oncol 10.1093/annonc/mdz396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Turcu AF, and Auchus RJ (2017) Clinical significance of 11-oxygenated androgens. Curr. Opin. Endocrinol. Diabetes. Obes 24, 252–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Turcu AF, Nanba AT, and Auchus RJ (2018) The Rise, Fall, and Resurrection of 11-Oxygenated Androgens in Human Physiology and Disease. Horm. Res. Paediatr 89, 284–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Dovio A, Sartori ML, De Francia S, Mussino S, Perotti P, Saba L, Abbadessa G, Racca S, and Angeli A (2009) Differential expression of determinants of glucocorticoid sensitivity in androgen-dependent and androgen-independent human prostate cancer cell lines. J. Steroid Biochem. Mol. Biol 116, 29–36 [DOI] [PubMed] [Google Scholar]
- 163.Page N, Warriar N, and Govindan MV (1994) 11 beta-Hydroxysteroid dehydrogenase and tissue specificity of androgen action in human prostate cancer cell LNCaP. J. Steroid Biochem. Mol. Biol 49, 173–181 [DOI] [PubMed] [Google Scholar]
- 164.Albiston AL, Obeyesekere VR, Smith RE, and Krozowski ZS (1994) Cloning and tissue distribution of the human 11 beta-hydroxysteroid dehydrogenase type 2 enzyme. Mol. Cell. Endocrinol 105, R11–7 [DOI] [PubMed] [Google Scholar]
- 165.Pelletier G, Luu-The V, Li S, Ouellet J, and Labrie F (2004) Cellular localization of mRNA expression of enzymes involved in the formation and inactivation of hormonal steroids in the mouse prostate. J. Histochem. Cytochem 52, 1351–1356 [DOI] [PubMed] [Google Scholar]
- 166.du Toit T, Bloem LM, Quanson JL, Ehlers R, Serafin AM, and Swart AC (2017) Profiling adrenal 11beta-hydroxyandrostenedione metabolites in prostate cancer cells, tissue and plasma: UPC(2)-MS/MS quantification of 11beta-hydroxytestosterone, 11keto-testosterone and 11keto-dihydrotestosterone. J. Steroid Biochem. Mol. Biol 166, 54–67 [DOI] [PubMed] [Google Scholar]
- 167.Gent R, du Toit T, Bloem LM, and Swart AC (2019) The 11beta-hydroxysteroid dehydrogenase isoforms: pivotal catalytic activities yield potent C11-oxy C19 steroids with 11betaHSD2 favouring 11-ketotestosterone, 11-ketoandrostenedione and 11-ketoprogesterone biosynthesis. J. Steroid Biochem. Mol. Biol 189, 116–126 [DOI] [PubMed] [Google Scholar]
- 168.Pretorius E, Africander DJ, Vlok M, Perkins MS, Quanson J, and Storbeck K-H (2016) 11-Ketotestosterone and 11-Ketodihydrotestosterone in Castration Resistant Prostate Cancer: Potent Androgens Which Can No Longer Be Ignored. PLoS One. 11, e0159867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Yazawa T, Uesaka M, Inaoka Y, Mizutani T, Sekiguchi T, Kajitani T, Kitano T, Umezawa A, and Miyamoto K (2008) Cyp11b1 is induced in the murine gonad by luteinizing hormone/human chorionic gonadotropin and involved in the production of 11-ketotestosterone, a major fish androgen: conservation and evolution of the androgen metabolic pathway. Endocrinology. 149, 1786–1792 [DOI] [PubMed] [Google Scholar]
- 170.Campana C, Rege J, Turcu AF, Pezzi V, Gomez-Sanchez CE, Robins DM, and Rainey WE (2016) Development of a novel cell based androgen screening model. J. Steroid Biochem. Mol. Biol 156, 17–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Attard G, Reid AHM, A’Hern R, Parker C, Oommen NB, Folkerd E, Messiou C, Molife LR, Maier G, Thompson E, Olmos D, Sinha R, Lee G, Dowsett M, Kaye SB, Dearnaley D, Kheoh T, Molina A, and de Bono JS (2009) Selective inhibition of CYP17 with abiraterone acetate is highly active in the treatment of castration-resistant prostate cancer. J. Clin. Oncol 27, 3742–3748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Auchus RJ, Yu MK, Nguyen S, and Mundle SD (2014) Use of prednisone with abiraterone acetate in metastatic castration-resistant prostate cancer. Oncologist. 19, 1231–1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, and Gwyther SG (2000) New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J. Natl. Cancer Inst 92, 205–216 [DOI] [PubMed] [Google Scholar]
- 174.Gupta S, Nordquist LT, Fleming MT, Berry WR, Zhang J, Ervin SL, Eisner JR, Baskin-Bey ES, and Shore ND (2018) Phase I Study of Seviteronel, a Selective CYP17 Lyase and Androgen Receptor Inhibitor, in Men with Castration-Resistant Prostate Cancer. Clin. Cancer Res 24, 5225–5232 [DOI] [PubMed] [Google Scholar]
- 175.Chandrasekar T, Yang JC, Gao AC, and Evans CP (2015) Mechanisms of resistance in castration-resistant prostate cancer (CRPC). Transl. Androl. Urol 4, 365–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Efstathiou E, Titus M, Wen S, Hoang A, Karlou M, Ashe R, Tu SM, Aparicio A, Troncoso P, Mohler J, and Logothetis CJ (2015) Molecular characterization of enzalutamide-treated bone metastatic castration-resistant prostate cancer. Eur. Urol 67, 53–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Attard G, Reid AHM, Auchus RJ, Hughes BA, Cassidy AM, Thompson E, Oommen NB, Folkerd E, Dowsett M, Arlt W, and de Bono JS (2012) Clinical and biochemical consequences of CYP17A1 inhibition with abiraterone given with and without exogenous glucocorticoids in castrate men with advanced prostate cancer. J. Clin. Endocrinol. Metab 97, 507–516 [DOI] [PubMed] [Google Scholar]
- 178.Moll JM, Kumagai J, van Royen ME, Teubel WJ, van Soest RJ, French PJ, Homma Y, Jenster G, de Wit R, and van Weerden WM (2019) A bypass mechanism of abiraterone-resistant prostate cancer: Accumulating CYP17A1 substrates activate androgen receptor signaling. Prostate. 79, 937–948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Boudadi K, and Antonarakis ES (2016) Resistance to Novel Antiandrogen Therapies in Metastatic Castration-Resistant Prostate Cancer. Clin. Med. Insights. Oncol 10, 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Cai C, and Balk SP (2011) Intratumoral androgen biosynthesis in prostate cancer pathogenesis and response to therapy. Endocr. Relat. Cancer 18, R175–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Li Z, Bishop AC, Alyamani M, Garcia JA, Dreicer R, Bunch D, Liu J, Upadhyay SK, Auchus RJ, and Sharifi N (2015) Conversion of abiraterone to D4A drives anti-tumour activity in prostate cancer. Nature. 523, 347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Li Z, Alyamani M, Li J, Rogacki K, Abazeed M, Upadhyay SK, Balk SP, Taplin M-E, Auchus RJ, and Sharifi N (2016) Redirecting abiraterone metabolism to fine-tune prostate cancer anti-androgen therapy. Nature. 533, 547–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Mostaghel EA, Marck BT, Plymate SR, Vessella RL, Balk S, Matsumoto AM, Nelson PS, and Montgomery RB (2011) Resistance to CYP17A1 inhibition with abiraterone in castration-resistant prostate cancer: induction of steroidogenesis and androgen receptor splice variants. Clin. Cancer Res 17, 5913–5925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Bruno RD, Vasaitis TS, Gediya LK, Purushottamachar P, Godbole AM, Ates-Alagoz Z, Brodie AMH, and Njar VCO (2011) Synthesis and biological evaluations of putative metabolically stable analogs of VN/124–1 (TOK-001): head to head anti-tumor efficacy evaluation of VN/124–1 (TOK-001) and abiraterone in LAPC-4 human prostate cancer xenograft model. Steroids. 76, 1268–1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Njar VCO, and Brodie AMH (2015) Discovery and development of Galeterone (TOK-001 or VN/124–1) for the treatment of all stages of prostate cancer. J. Med. Chem 58, 2077–2087 [DOI] [PubMed] [Google Scholar]
- 186.Soifer HS, Souleimanian N, Wu S, Voskresenskiy AM, Collak FK, Cinar B, and Stein CA (2012) Direct regulation of androgen receptor activity by potent CYP17 inhibitors in prostate cancer cells. J. Biol. Chem 287, 3777–3787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Kwegyir-Afful AK, Bruno RD, Purushottamachar P, Murigi FN, and Njar VCO (2016) Galeterone and VNPT55 disrupt Mnk-eIF4E to inhibit prostate cancer cell migration and invasion. FEBS J. 283, 3898–3918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Alyamani M, Li Z, Berk M, Li J, Tang J, Upadhyay S, Auchus RJ, and Sharifi N (2017) Steroidogenic Metabolism of Galeterone Reveals a Diversity of Biochemical Activities. Cell Chem. Biol 24, 825–832.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Taplin M-E, and Montgomery RB (2014) ARMOR2: Galeterone in progressive CRPC patients who have failed oral therapy. J. Clin. Oncol 32, 71 [Google Scholar]
- 190.Montgomery B, Eisenberger MA, Rettig MB, Chu F, Pili R, Stephenson JJ, Vogelzang NJ, Koletsky AJ, Nordquist LT, Edenfield WJ, Mamlouk K, Ferrante KJ, and Taplin M-E (2016) Androgen Receptor Modulation Optimized for Response (ARMOR) Phase I and II Studies: Galeterone for the Treatment of Castration-Resistant Prostate Cancer. Clin. Cancer Res 22, 1356–1363 [DOI] [PubMed] [Google Scholar]
- 191.McKay RR, Werner L, Fiorillo M, Roberts J, Heath EI, Bubley GJ, Montgomery RB, and Taplin M-E (2017) Efficacy of Therapies After Galeterone in Patients With Castration-resistant Prostate Cancer. Clin. Genitourin. Cancer 15, 463–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Taplin M-E, Antonarakis ES, Ferrante KJ, Horgan K, Blumenstein B, Saad F, Luo J, and de Bono JS (2019) Androgen Receptor Modulation Optimized for Response-Splice Variant: A Phase 3, Randomized Trial of Galeterone Versus Enzalutamide in Androgen Receptor Splice Variant-7-expressing Metastatic Castration-resistant Prostate Cancer. Eur. Urol 10.1016/j.eururo.2019.08.034 [DOI] [PubMed] [Google Scholar]
- 193.Rafferty SW, Eisner JR, Moore WR, Schotzinger RJ, and Hoekstra WJ (2014) Highly-selective 4-(1,2,3-triazole)-based P450c17a 17,20-lyase inhibitors. Bioorg. Med. Chem. Lett 24, 2444–2447 [DOI] [PubMed] [Google Scholar]
- 194.Norris JD, Ellison SJ, Baker JG, Stagg DB, Wardell SE, Park S, Alley HM, Baldi RM, Yllanes A, Andreano KJ, Stice JP, Lawrence SA, Eisner JR, Price DK, Moore WR, Figg WD, and McDonnell DP (2017) Androgen receptor antagonism drives cytochrome P450 17A1 inhibitor efficacy in prostate cancer. J. Clin. Invest 127, 2326–2338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Maity SN, Titus MA, Gyftaki R, Wu G, Lu J-F, Ramachandran S, Li-Ning-Tapia EM, Logothetis CJ, Araujo JC, and Efstathiou E (2016) Targeting of CYP17A1 Lyase by VT-464 Inhibits Adrenal and Intratumoral Androgen Biosynthesis and Tumor Growth of Castration Resistant Prostate Cancer. Sci. Rep 6, 35354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Toren PJ, Kim S, Pham S, Mangalji A, Adomat H, Guns EST, Zoubeidi A, Moore W, and Gleave ME (2015) Anticancer activity of a novel selective CYP17A1 inhibitor in preclinical models of castrate-resistant prostate cancer. Mol. Cancer Ther 14, 59–69 [DOI] [PubMed] [Google Scholar]
- 197.McConnell JD, Roehrborn CG, Bautista OM, Andriole GLJ, Dixon CM, Kusek JW, Lepor H, McVary KT, Nyberg LMJ, Clarke HS, Crawford ED, Diokno A, Foley JP, Foster HE, Jacobs SC, Kaplan SA, Kreder KJ, Lieber MM, Lucia MS, Miller GJ, Menon M, Milam DF, Ramsdell JW, Schenkman NS, Slawin KM, and Smith JA (2003) The long-term effect of doxazosin, finasteride, and combination therapy on the clinical progression of benign prostatic hyperplasia. N. Engl. J. Med 349, 2387–2398 [DOI] [PubMed] [Google Scholar]
- 198.Roehrborn CG, Barkin J, Siami P, Tubaro A, Wilson TH, Morrill BB, and Gagnier RP (2011) Clinical outcomes after combined therapy with dutasteride plus tamsulosin or either monotherapy in men with benign prostatic hyperplasia (BPH) by baseline characteristics: 4-year results from the randomized, double-blind Combination of Avodart and Tamsulo. BJU Int. 107, 946–954 [DOI] [PubMed] [Google Scholar]
- 199.Dorsam J, and Altwein J (2009) 5alpha-Reductase inhibitor treatment of prostatic diseases: background and practical implications. Prostate Cancer Prostatic Dis. 12, 130–136 [DOI] [PubMed] [Google Scholar]
- 200.Becher E, Roehrborn CG, Siami P, Gagnier RP, Wilson TH, and Montorsi F (2009) The effects of dutasteride, tamsulosin, and the combination on storage and voiding in men with benign prostatic hyperplasia and prostatic enlargement: 2-year results from the Combination of Avodart and Tamsulosin study. Prostate Cancer Prostatic Dis. 12, 369–374 [DOI] [PubMed] [Google Scholar]
- 201.Thompson IM, Goodman PJ, Tangen CM, Lucia MS, Miller GJ, Ford LG, Lieber MM, Cespedes RD, Atkins JN, Lippman SM, Carlin SM, Ryan A, Szczepanek CM, Crowley JJ, and Coltman CAJ (2003) The influence of finasteride on the development of prostate cancer. N. Engl. J. Med 349, 215–224 [DOI] [PubMed] [Google Scholar]
- 202.Thompson IMJ, Goodman PJ, Tangen CM, Parnes HL, Minasian LM, Godley PA, Lucia MS, and Ford LG (2013) Long-term survival of participants in the prostate cancer prevention trial. N. Engl. J. Med 369, 603–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Andriole GL, Bostwick DG, Brawley OW, Gomella LG, Marberger M, Montorsi F, Pettaway CA, Tammela TL, Teloken C, Tindall DJ, Somerville MC, Wilson TH, Fowler IL, and Rittmaster RS (2010) Effect of dutasteride on the risk of prostate cancer. N. Engl. J. Med 362, 1192–1202 [DOI] [PubMed] [Google Scholar]
- 204.Hamilton RJ, and Freedland SJ (2011) 5-α reductase inhibitors and prostate cancer prevention: where do we turn now? BMC Med. 9, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Wallerstedt A, Strom P, Gronberg H, Nordstrom T, and Eklund M (2018) Risk of Prostate Cancer in Men Treated With 5alpha-Reductase Inhibitors-A Large Population-Based Prospective Study. J. Natl. Cancer Inst 110, 1216–1221 [DOI] [PubMed] [Google Scholar]
- 206.Montgomery B, Tretiakova MS, Joshua AM, Gleave ME, Fleshner N, Bubley GJ, Mostaghel EA, Chi KN, Lin DW, and Sanda M (2017) Neoadjuvant enzalutamide prior to prostatectomy. Clin. Cancer Res 23, 2169–2176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Pinto-Bazurco Mendieta MAE, Hu Q, Engel M, and Hartmann RW (2013) Highly Potent and Selective Nonsteroidal Dual Inhibitors of CYP17/CYP11B2 for the Treatment of Prostate Cancer To Reduce Risks of Cardiovascular Diseases. J. Med. Chem 56, 6101–6107 [DOI] [PubMed] [Google Scholar]
- 208.Hu Q, Jagusch C, Hille UE, Haupenthal J, and Hartmann RW (2010) Replacement of Imidazolyl by Pyridyl in Biphenylmethylenes Results in Selective CYP17 and Dual CYP17/CYP11B1 Inhibitors for the Treatment of Prostate Cancer. J. Med. Chem 53, 5749–5758 [DOI] [PubMed] [Google Scholar]
- 209.Penning TM, Burczynski ME, Jez JM, Hung CF, Lin HK, Ma H, Moore M, Palackal N, and Ratnam K (2000) Human 3alpha-hydroxysteroid dehydrogenase isoforms (AKR1C1-AKR1C4) of the aldo-keto reductase superfamily: functional plasticity and tissue distribution reveals roles in the inactivation and formation of male and female sex hormones. Biochem. J 351, 67–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Liedtke AJ, Adeniji AO, Chen M, Byrns MC, Jin Y, Christianson DW, Marnett LJ, and Penning TM (2013) Development of potent and selective indomethacin analogues for the inhibition of AKR1C3 (Type 5 17beta-hydroxysteroid dehydrogenase/prostaglandin F synthase) in castrate-resistant prostate cancer. J. Med. Chem 56, 2429–2446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Lovering AL, Ride JP, Bunce CM, Desmond JC, Cummings SM, and White SA (2004) Crystal structures of prostaglandin D(2) 11-ketoreductase (AKR1C3) in complex with the nonsteroidal anti-inflammatory drugs flufenamic acid and indomethacin. Cancer Res. 64, 1802–1810 [DOI] [PubMed] [Google Scholar]
- 212.Pippione AC, Giraudo A, Bonanni D, Carnovale IM, Marini E, Cena C, Costale A, Zonari D, Pors K, Sadiq M, Boschi D, Oliaro-Bosso S, and Lolli ML (2017) Hydroxytriazole derivatives as potent and selective aldo-keto reductase 1C3 (AKR1C3) inhibitors discovered by bioisosteric scaffold hopping approach. Eur. J. Med. Chem 139, 936–946 [DOI] [PubMed] [Google Scholar]
- 213.Pippione AC, Carnovale IM, Bonanni D, Sini M, Goyal P, Marini E, Pors K, Adinolfi S, Zonari D, Festuccia C, Wahlgren WY, Friemann R, Bagnati R, Boschi D, Oliaro-Bosso S, and Lolli ML (2018) Potent and selective aldo-keto reductase 1C3 (AKR1C3) inhibitors based on the benzoisoxazole moiety: application of a bioisosteric scaffold hopping approach to flufenamic acid. Eur. J. Med. Chem 150, 930–945 [DOI] [PubMed] [Google Scholar]
- 214.Verma K, Gupta N, Zang T, Wangtrakluldee P, Srivastava SK, Penning TM, and Trippier PC (2018) AKR1C3 Inhibitor KV-37 Exhibits Antineoplastic Effects and Potentiates Enzalutamide in Combination Therapy in Prostate Adenocarcinoma Cells. Mol. Cancer Ther 17, 1833–1845 [DOI] [PubMed] [Google Scholar]
- 215.Wangtrakuldee P, Adeniji AO, Zang T, Duan L, Khatri B, Twenter BM, Estrada MA, Higgins TF, Winkler JD, and Penning TM (2019) A 3-(4-nitronaphthen-1-yl) amino-benzoate analog as a bifunctional AKR1C3 inhibitor and AR antagonist: Head to head comparison with other advanced AKR1C3 targeted therapeutics. J. Steroid Biochem. Mol. Biol 10.1016/j.jsbmb.2019.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]