

Abstract
Introduction:
Melanoma is an aggressive form of skin cancer for which there are no effective drugs for prolonged treatment. The existing kinase inhibitor antiglycolytic drugs (B-Raf serine/threonine kinase or BRAF inhibitors) are effective for a short time followed by a rapid onset of drug resistance.
Presentation of case:
Here, we show that a mitochondria-targeted analog of magnolol, Mito-magnolol (Mito-MGN), inhibits oxidative phosphorylation (OXPHOS) and proliferation of melanoma cells more potently than untargeted magnolol. Mito-MGN also inhibited tumor growth in murine melanoma xenografts. Mito-MGN decreased mitochondrial membrane potential and modulated energetic and mitophagy signaling proteins.
Discussion:
Results indicate that Mito-MGN is significantly more potent than the FDA-approved OXPHOS inhibitor in inhibiting proliferation of melanoma cells.
Conclusion:
These findings have implications in the treatment of melanomas with enhanced OXPHOS status due to metabolic reprogramming or drug resistance.
Keywords: Bioenergetic metabolism, Mitophagy, Melanoma, Mitochondria-targeted agents, Oxidative phosphorylation
1. Introduction
Melanoma is an aggressive disease that accounts for the majority of reported skin cancer deaths [1,2]. Despite increased research and clinical intervention, incidence and mortality rates of melanoma continue to increase [1,2]. Treatment of malignant melanoma has been revolutionized with the approval of kinase inhibitors and neutralizing immune checkpoint antibodies, yet resistance to front-line therapies remains a significant clinical concern [2,3]. Existing standard-of-care drugs provide only short-term benefits, followed by a rapid onset of drug resistance [4]. Presently, there are no effective drugs for prolonged treatment of melanoma. Nearly 50% of melanoma patients express dominant-active mutations in the BRAF serine/threonine kinase proto-oncogene, BRAFV600E kinase, an oncogenic driver mutation responsible for tumor initiation and therapeutic resistance [4–6]. Recently, melanomas have been classified into Warburg-like glycolytic and high oxidative phosphorylation (OXPHOS) types of cancer [7]. Glycolytic melanoma cells, like most glycolytic phenotypes, are sensitive to kinase inhibitors, such as vemurafenib, that decrease glycolytic flux, whereas the high-OXPHOS melanomas are sensitive to OXPHOS inhibition [7–10]. Interestingly, BRAF inhibitors induce metabolic reprogramming of melanoma cancer cells that functionally elevates OXPHOS levels that are correlated with poor prognosis [8].
Metabolic reprogramming from a glycolytic phenotype to an OXPHOS phenotype has been linked with tumorigenesis as well as therapeutic inhibition of oncogenic kinase signaling in cancer cells [7]. Increased mitochondrial biogenesis and upregulated OXPHOS genes accompanied enhanced OXPHOS and mitochondrial respiration in drug-resistant melanomas [11]. The critical dependence of cancer cells on OXPHOS or mitochondrial respiration for energy, survival, and chemosensitivity suggests that cancer-cell-selective inhibitors of OXPHOS may be therapeutically exploited to inhibit tumor growth and prevent or delay resistance [12,13]. Clearly, there is a pressing need to develop a new class of tumor-selective and relatively nontoxic OXPHOS-targeting drugs to be used alone, or in combination with standard-of-care chemotherapeutics, to treat drug-resistant cancers associated with a high OXPHOS status [14–16]. FDA-approved OXPHOS inhibitor (IACS-010759) only modestly inhibits mitochondrial complex I and proliferation [12].
Using mitochondria-targeted agents coupled to the triphenylphosphonium cation (TPP+) (e.g., Mito-Q and Mito-CP), we showed that at submicromolar levels these agents selectively inhibit mitochondrial complex activities in tumor cells and their proliferation [13,17–19]. Dual inhibition of glycolysis with 2-deoxyglucose and mitochondrial function with Mito-CP or Mito-Q caused a synergistic inhibition of tumor cell proliferation [17–20]. In an in vivo mouse xenograft tumor model, upon administration, mitochondria-targeted agents (Mito-Vit-E or Mito-Metformin) accumulated in tumors and resulted in tumor regression [19,21,22].
In this study, we developed a mitochondria-targeted derivative (Mito-magnolol [Mito-MGN]) of a naturally occurring bioactive polyphenolic molecule, magnolol (MGN) (Fig. 1). MGN is the most abundant bioactive component of magnolia extract, a traditional herbal medicine used effectively for centuries in East Asia to treat multiple diseases [23]. Mito-MGN (refers to Mito-MGN10) consists of a TPP+ moiety conjugated to MGN via a 10-carbon alkyl side chain (Fig. 1). We report here that Mito-MGN is significantly more potent than MGN at inhibiting mitochondrial respiration, proliferation, and invasion of melanoma cells, possibly through downregulation of mTOR/AKT signaling accompanied by mitophagy [24]. Mito-MGN represents a new class of mitochondria-targeted polyphenolic drugs that could be used to treat OXPHOS-dependent melanoma.
Fig. 1. Synthetic scheme for conversion of MGN to Mito-MGN and Mito-MGN4.
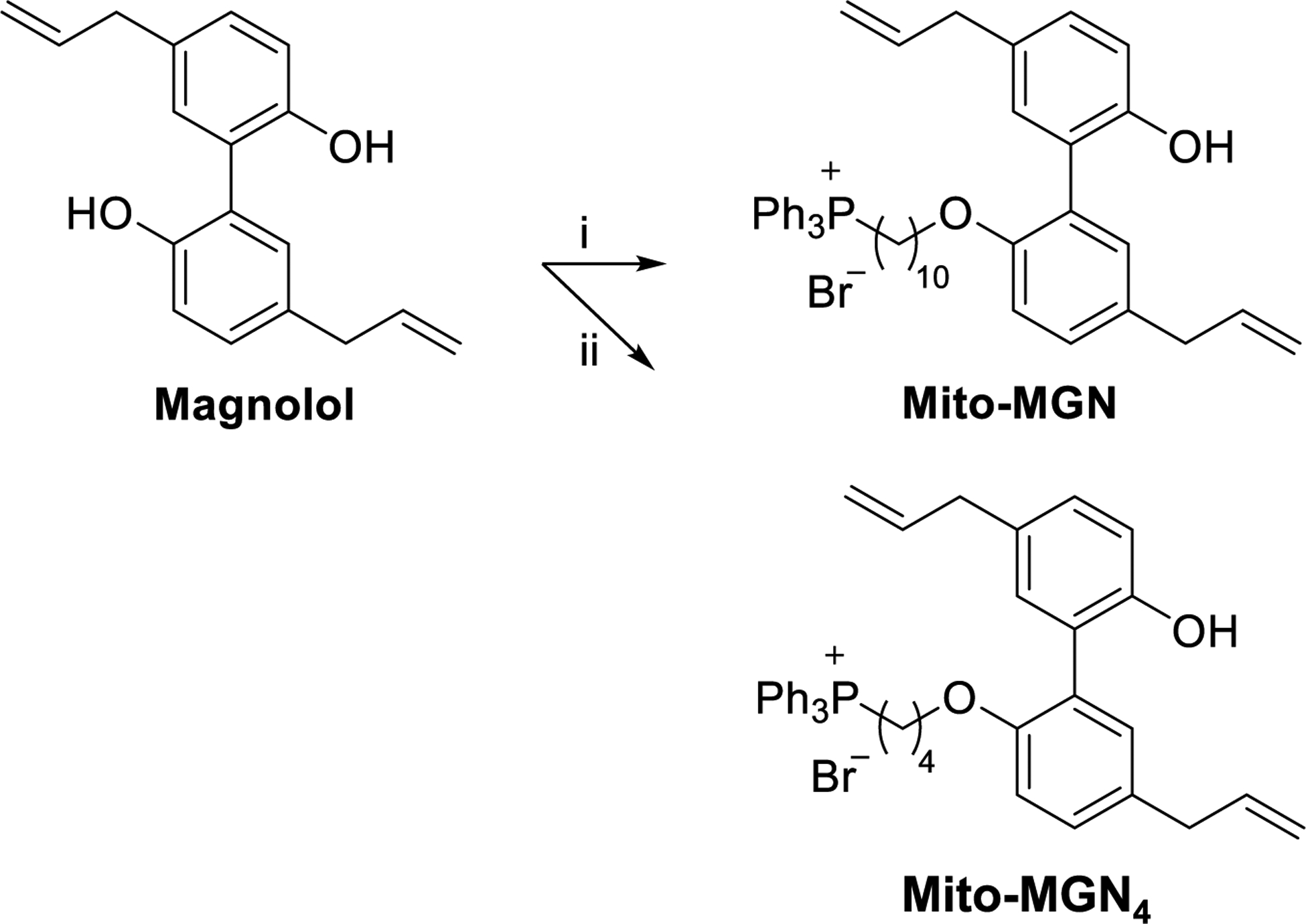
Mito-MGN refers to Mito-MGN10, containing a 10-carbon alkyl linker. The following reagents and conditions were used: i, 10-bromodecyltriphenylphosphonium bromide, DMF, K2CO3, 35 °C, 24 h, 35%. ii, 4-bromobutyltriphenylphosphonium bromide, K2CO3, DMF, 35 °C, 24 h, 41%.
2. Materials and methods
2.1. Mito-MGN synthesis
Mito-MGN (also Mito-MGN10) was prepared as follows: 10-bromodecyltriphenylphosphonium bromide was prepared according to the procedure described by Pan et al. [25]. Briefly, a mixture of triphenylphosphine (1 g, 3.8 mmol; Sigma-Aldrich, St. Louis, MO) and 1,10-dibromodecane (5.7 g, 19 mmol; Alfa Aesar, Haverhill, MA) was heated at 90 °C for 6 h. After cooling, the crude product was purified by flash chromatography (CH2Cl2/EtOH 9:1; obtained from Fisher Scientific, Waltham, MA, and Sigma-Aldrich, respectively) to afford the corresponding phosphonium salt as a white solid (1 g, 47% yield). To a mixture of magnolol (0.2 g, 0.75 mmol; AstaTech Inc., Bristol, PA) and anhydrous potassium carbonate (0.22 g, 1.5 mmol; AstaTech Inc.) in DMF (10 mL; Fisher Scientific) was added 10-bromodecyltriphenylphosphonium bromide (0.42 g, 0.75 mmol) at 0 °C. The mixture was stirred at 35 °C for 24 h. The residue was taken up into water and extracted with CH2Cl2. The organic layer was dried over Na2SO4, and the solvent was removed under reduced pressure. Ether was added to precipitate the crude product. Purification by flash chromatography using the following gradient from CH2Cl2 (100%) to CH2Cl2/EtOH (80/20) as eluent delivered the corresponding Mito-MGN (200 mg, 35% yield).
2.2. Mito-MGN4 synthesis
To a mixture of magnolol (0.5 g, 1.88 mmol), anhydrous potassium carbonate (0.51 g, 3.7 mmol) in DMF (20 mL) was added 4-bromobutyltriphenylphosphonium bromide (0.8, 1.67 mmol; Alfa Aesar) at 0 °C. The mixture was stirred at 35 °C for 24 h. The residue was taken up into water and extracted with CH2Cl2. The organic layer was dried over Na2SO4, and the solvent was removed under reduced pressure. Ether was added to precipitate the crude product. Purification by flash chromatography using the following gradient from CH2Cl2 (100%) to CH2Cl2/EtOH (80/20) as eluent delivered the corresponding Mito-MGN4 (0.45 g, 41% yield) as a white solid.
2.3. Cell culture and treatment conditions
The BRAFV600E expressing UACC-62 melanoma cell line was purchased from AddexBio (San Diego, CA; #C0020003) where it was regularly authenticated. UACC-62 cells were maintained in RPMI-1640 (Gibco, Grand Island, NY; #11875093) containing 10% fetal bovine serum (Omega Scientific, Inc., Tarzana, CA), penicillin (100 U/ml), and streptomycin (100 μg/ml). A375 (ATCC, Manassas, VA; #CRL-1619), and G361 (ATCC; #CRL-1424) BRAF mutant human melanoma cell lines were purchased from ATCC and cultured in RPMI-1640 and McCoy’s 5A (Gibco, Grand Island, NY; #16600082), respectively. Both culture media contained 10% fetal bovine serum and were supplemented with penicillin (100 U/ml) and streptomycin (100 μg/ml).
PLX4720 (Sigma-Aldrich, #553015), an analog of vemurafenib, is a highly selective inhibitor of oncogenic BRAFV600E signaling [7]. To establish drug resistance, UACC-62-wild-type (UACC-62-WT) cells were continuously exposed to increasing concentrations of PLX4720 (0.1–20 μM) for 12 months until logarithmic growth resumed. PLX4720-containing media were replenished twice weekly. The resistant cell line (UACC-62-R) was maintained in routine culture in the presence of 20 μM PLX4720.
2.4. Melanoma cell proliferation measurements
The IncuCyte Live-Cell Imaging system (Essen Bioscience Inc., Ann Arbor, MI) was used to monitor cell proliferation. This system allows for probe-free, noninvasive, and continuous monitoring of cellular confluence changes over several days [25,26]. The phase-contrast images were collected for each time point to determine morphological changes in cells, and changes in cell confluence were used as a surrogate marker of cell proliferation. Cell proliferation was expressed as an increase in percentage of confluence.
UACC-62 cells were pretreated with AMP-activated protein kinase (AMPK) inhibitor Compound C (Sigma-Aldrich, #171260) or control media for 48 h. In a 96-well plate, cells were plated at 4000 cells per well in technical triplicates and left to adhere overnight. Cells were then treated with 0.2 μM Mito-MGN and cell confluency was recorded over a five-day period using the IncuCyte S3 system.
2.5. Mitochondrial bioenergetic measurements
The mitochondrial bioenergetic function was measured in real time using the Seahorse XF96 Extracellular Flux Analyzer (Agilent, North Billerica, MA). Assays in intact melanoma cells were performed as reported previously [17,18,27]. The oxygen consumption rate (OCR)-based assessment of mitochondrial complex activities was carried out on acutely permeabilized cells in the presence of different mitochondrial substrates (pyruvate/malate [Sigma-Aldrich] for complex I and succinate [Sigma-Aldrich] for complex II) [25,26]. Rotenone, malonate, and antimycin A (Sigma-Aldrich) were used as specific inhibitors of mitochondrial complexes I, II, and III, respectively.
2.6. Mitochondrial membrane potential assessment
Mitochondrial membrane potential was assessed in UACC-62 via staining with the membrane-permeable, positively charged, tetramethylrhodamine ethyl ester (TMRE) fluorescent probe and visualized by flow cytometry [22]. Cells were treated with FCCP (20 μM) or increasing concentrations of Mito-MGN for 24 h. TMRE (200 nM) was added before flow-cytometric analysis using a BD-LSR II flow cytometer (BD Biosciences, San Jose, CA).
2.7. ROS measurements
The intracellular oxidants (reactive oxygen species [ROS]) were measured using the fluorogenic hydroethidine (HE) probe coupled with high-performance liquid chromatography (HPLC)-based profiling of the oxidation products [28–30]. A stock solution of HE (20 mM; Invitrogen, Carlsbad, CA) was prepared in deoxygenated DMSO (dimethyl sulfoxide) and stored in the dark at −80 °C until use. Cells were incubated for 1 h with the probe (10 μM), harvested, processed, and analyzed by HPLC, as described previously [28,31]. To obtain a more complete picture of intracellularly generated oxidants [31,32], we synthesized the appropriate standards as follows. The hydroxylated product, 2-OHE+, a diagnostic marker product of superoxide reaction with HE, was synthesized by reacting HE with Fremy’s salt [33]. The dimeric product (E+-E+), characteristic of HE radical formed by the one-electron oxidation of HE, was prepared by oxidizing HE with excess ferricyanide [34]. The nonspecific oxidation product, E+ formed by two-electron oxidation of HE, was purchased from Sigma-Aldrich. All standards were purified by HPLC.
2.8. Low-temperature EPR measurements
Melanoma cells (60 million cells/dish) were grown in 15-cm dishes until 70% confluency. After treatment with drugs, the medium was removed from the cells, and cells were collected and quickly transferred into electron paramagnetic resonance (EPR) quartz tube as described previously [28,35]. EPR tubes were flash frozen in liquid nitrogen, and samples were kept either at −80 °C or in liquid nitrogen until EPR measurements.
EPR spectroscopy was performed at 9.5 GHz on a Bruker EMX-TDU/L spectrometer equipped with an ER4112-SHQ resonator, a ColdEdge/Bruker S5-L recirculating helium refrigerator (5–100 K), an Oxford MercuryiTC temperature controller and ESR900 cryostat, and an HP 5350B microwave counter [35,36]. Spectra were typically recorded at 12 K, 5.5 mW microwave power, 12 G (1.2 mT) magnetic field, modulation amplitude at 100 kHz, and 1.2 G (0.12 mT) digital field resolution.
2.9. Redox blotting
The redox status of both mitochondrial and cytosolic peroxiredoxins (Prx1 and Prx3, respectively) was assessed by immunoblotting [37,38]. After treatment with drugs, melanoma cells were washed with Hanks’ Balanced Salt Solution and overlaid with thiol-blocking buffer as previously described [25,26]. Whole cell lysates were stored at −80 °C until analysis. Samples were probed using anti-Prx1 and anti-Prx3 antibodies as previously described [25,26].
2.10. Immunoblotting of autophagy-and energy-related signaling
Approximately 15 μg of protein was loaded in precast 4–20% Criterion TGX gels (Bio-Rad Laboratories, Hercules, CA), transferred to a PVDF membrane with the Trans-BlotⓇ Turbo™ system (Bio-Rad Laboratories), blocked, incubated with primary antibodies, and incubated with the secondary antibody. Images were captured via the ChemiDoc Molecular Imager and bands were quantified with ImageLab analysis software (both from Bio-Rad). Expression levels were determined by chemiluminescent immunodetection and normalized to appropriate loading controls. Immunoblotting was performed using commercially available antibodies from Abcam (Cambridge, MA): NDP52 (1:500); BD Biosciences (San Jose, CA): P62 (1:1000); Cell Signaling Technology (Danvers, MA): Beclin-1 (1:750), Caspase 3 (1:500), Caspase 9 (1:1000), GAPDH (1:40,000), LC3 (1:1000), mTOR (1:1000), NBR1 (1:500), phospho-AKTSer473 (1:1000), phospho-mTORSer2448 (1:500), phospho-P70 S6 KinaseThr389 (1:1000), phospho-ULKSer757 (1:1000), RAB7 (1:1000), and TAX1BP1 (1:1000); Novus Biologicals (Littleton, CO): PINK1 (1:250); Proteintech Group, Inc. (Rosemont, IL): Optineurin (1:500); Thermofisher Scientific (Waltham, MA): TFEB (1:500); and Santa Cruz Biotechnology (Dallas, TX): HSP60 (1:5000).
2.11. Melanoma xenograft and CT imaging
A xenograft engraftment model was used to assess tumor progression after treatment with Mito-MGN. Outbred six- to eight-week-old nude mice (Crl:NU-Foxn1nu; #088, Charles River, Frederick, MD) were implanted with 1 × 106 UACC-62 cells injected subcutaneously to the dorsal flank. Tumors were allowed to establish. Starting on day 15, mice were treated three times weekly with 1 mg/tumor Mito-MGN administered via an intratumoral injection in a 50 μL volume, and the control mice were injected with a vehicle of 20% ethanol in phosphate buffered saline. Tumor growth was monitored weekly using computer tomography (micro-CT) imaging (IVIS Spectrum-CT, Perkin Elmer, Hopkinton, MA) and caliper measurements beginning on day 10. On day 58, mice were euthanized, and the wet weight and volume of primary tumors were measured. Toxicity status, including key metabolites of cardiac, hepatic, and renal function, is shown in Table 1. All studies on animals were approved by the Medical College of Wisconsin Institutional Animal Care and Use Committee (approval number: AUA00000076).
Table 1.
Serum levels of key hepatic, cardiac, and kidney metabolites.
| AST [IU/L] | ALT [IU/L] | BUN [mg/dL] | Albumin [g/dL] | Glucose [mg/dL] | Total Protein [g/dL] | |
|---|---|---|---|---|---|---|
| ref (54–269) | ref (26–77) | ref (12–28) | ref (2.8–3.8) | ref (62–175) | ref (4.8–7.0) | |
| Control | 187.7 ± 71.6 | 40.0 ± 22.8 | 24.4 ± 3.6 | 2.9 ± 0.1 | 168 ± 38 | 4.9 ± 0.4 |
| Mito-MGN | 320.0 ± 37.0 | 58.7 ± 25.4 | 24.5 ± 3.9 | 2.7 ± 0.1 | 159.7 ± 18.6 | 4.7 ± 0.3 |
Serum levels assayed in control, non-tumor-bearing C57BL6 mice treated on three times per week for four weeks and serum obtained on day 31. AST, aspartate aminotransferase; ALT, alanine transaminase; BUN, blood urea nitrogen; ref, reference values. Values are mean ± SD, n = 3–5 individual mice.
2.12. Statistical analysis
GraphPad Prism software was used to evaluate statistical differences by treatment. A Student’s t-test was applied for pairwise comparisons. Inhibition of proliferation data was evaluated for statistical significance using an analysis of variance with Tukey’s post hoc test for assessing multiple comparisons. P-values of ≤ 0.05 were considered significant.
3. Results
3.1. Synthesis and characterization of Mito-MGN
Monosubstituted Mito-MGN (also Mito-MGN10) and Mito-MGN4 were prepared by reacting the appropriate bromoalkyl-TPP+ bromide with magnolol in the presence of potassium carbonate in dimethylformamide (Fig. 1). Mito-MGN and Mito-MGN4 were purified by flash chromatography and the structure and purity validated by nuclear magnetic resonance (NMR) (supplemental data and Fig. S1), liquid chromatography-mass spectrometry (LC-MS), and high-resolution mass spectrometry (HRMS) analyses.
3.2. Mito-MGN inhibits cell proliferation and mitochondrial complex I in melanoma cells
Cell proliferation was monitored continuously in real time for eight days under similar treatment conditions using both MGN and Mito-MGN. As shown in Fig. 2A and B, Mito-MGN dose-dependently inhibited proliferation of UACC-62 cells. Fig. 2A also shows representative phase contrast images obtained after six days of incubation in the absence or presence of Mito-MGN. Data presented in Fig. 2B indicate that over a four-log concentration range, Mito-MGN is 100-fold more potent at inhibiting melanoma cell proliferation, as compared with MGN. These results confirm that Mito-MGN potently inhibits (IC50 = =0.3 μM) the proliferation of UACC-62 melanoma cells and is more than 100-fold more potent than its untargeted analog, MGN. In order to fine-tune the structure and potency of Mito-MGN, we compared the relative antiproliferative potencies of a short alkyl side linker Mito-MGN analog (Mito-MGN4, Fig. 1) with Mito-MGN (containing a 10carbon linker) in UACC-62 cells. There was a significant decrease in the potency with regard to the antiproliferative effect as a function of decreasing the alkyl linker length. Mito-MGN4 (IC50 = =2.5 μM, Fig. S4) inhibits the proliferation but is eight-fold less potent than Mito-MGN (or Mito-MGN10). Furthermore, we found that the IC50 value of Mito-MGN4 (24 h treatment) on the inhibition of mitochondrial complex I is 2.4 μM (Fig. S5), which is consistent with its antiproliferative effect.
Fig. 2. Effect of Mito-MGN and MGN on UACC-62 melanoma cell proliferation and bioenergetics.
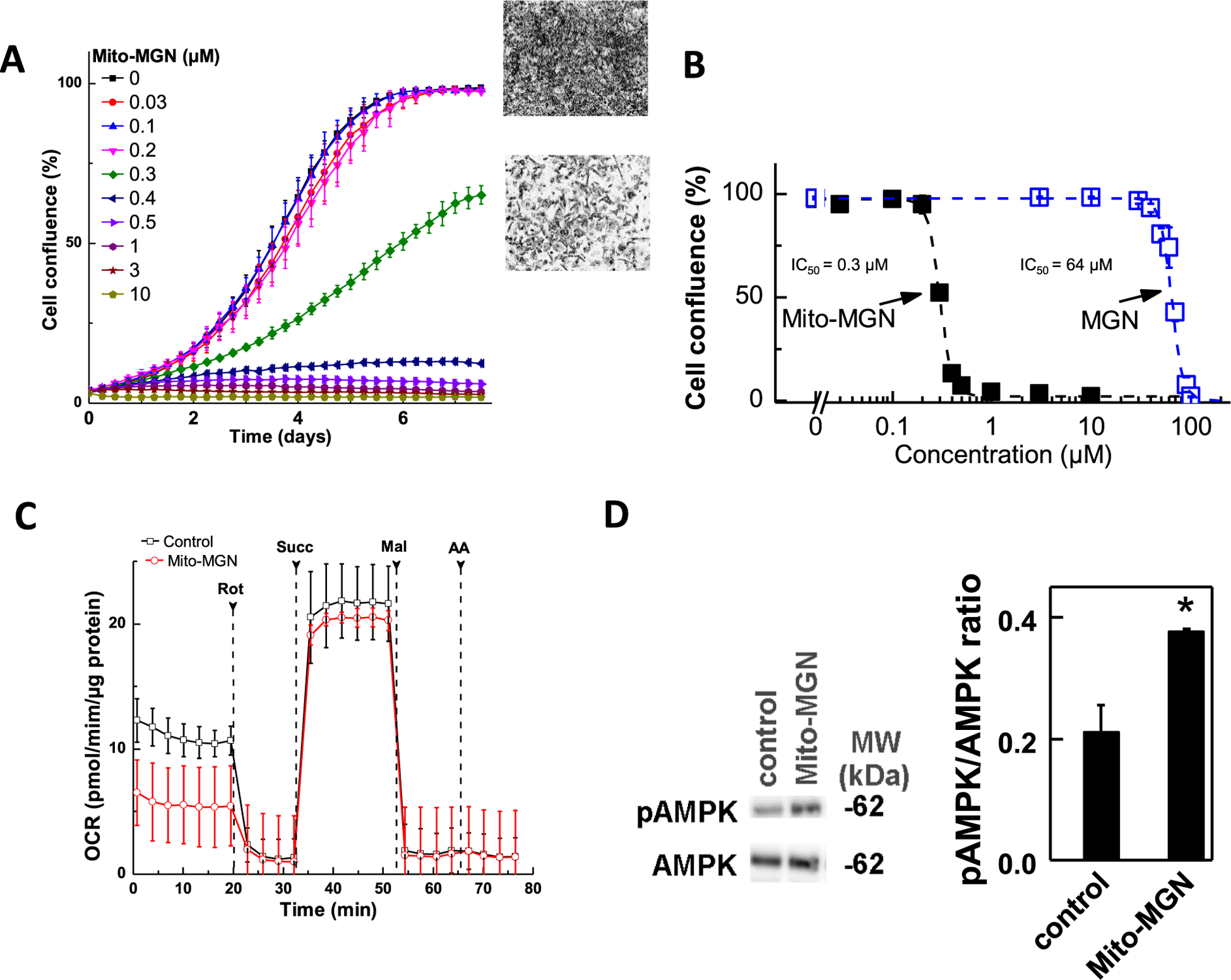
(A) Melanoma cells were cultured in the presence of Mito-MGN and MGN. Cell proliferation was monitored in real time using the IncuCyte imager. (B) Melanoma cell proliferation (measured as cell confluence) quantified from IncuCyte measurements. Data shown indicate the dose-dependent inhibition of confluence with increasing concentration of Mito-MGN and MGN in UACC-62 melanoma cells. The percentage of cell confluence (as control groups reached 98% confluence) is plotted against concentration. Dashed lines represent the fitting curves used to determine the IC50 values. (C) The melanoma cells were treated with Mito-MGN (0.2 μM) for 24 h before mitochondrial complex I and complex II activities were measured. Upon the mitochondrial complex activity measurements, cells were permeabilized and assayed in medium containing 10 mM pyruvate and 1.5 mM malate (substrates for complex I) in mannitol and sucrose buffer using the Seahorse XF96 analyzer. Basal OCR was measured before adding complex I inhibitor, rotenone (Rot, 1 μM). The rotenone inhibitable OCR was used as a measure of mitochondrial complex I activity. Then, succinate (Succ, 10 mM, substrate for complex II) and malonate (Mal, 10 mM, complex II inhibitor) were subsequently injected as indicated. The succinate-driven, malonate-inhibitable OCR was considered as a measure of mitochondrial complex II activity. Finally, antimycin A (AA, 20 μM), a complex III inhibitor, was injected to confirm the complete inhibition of OCR. (D) Effect of the treatment with 0.5 μM Mito-MGN for 24 h on AMPK and p-AMPK in UACC-62 cells.
Given the strong effects of Mito-MGN on UACC-62 cells, we examined its impact on additional human melanoma cells. Both MGN and Mito-MGN were broadly effective at halting melanoma proliferation as both A375 and G361 human melanoma cells were inhibited (Figs. 3 and 4). As shown in Fig. 3, Mito-MGN was more than 150 times more potent than MGN in inhibiting A375 melanoma cell proliferation. The antiproliferative effects of MGN and Mito-MGN in G361 melanoma cells are shown in Fig. 4 (panels A–D). The IC50 values to inhibit G361 cell proliferation for MGN and Mito-MGN correspond to 38 μM and 0.22 μM, respectively (Fig. 4). Clearly, in all human melanoma cells tested, both MGN and Mito-MGN efficiently inhibited cell proliferation, but Mito-MGN is more than 100-fold more potent than MGN.
Fig. 3. Effects of MGN and Mito-MGN on proliferation and mitochondrial complex I activity of A375 melanoma cells.
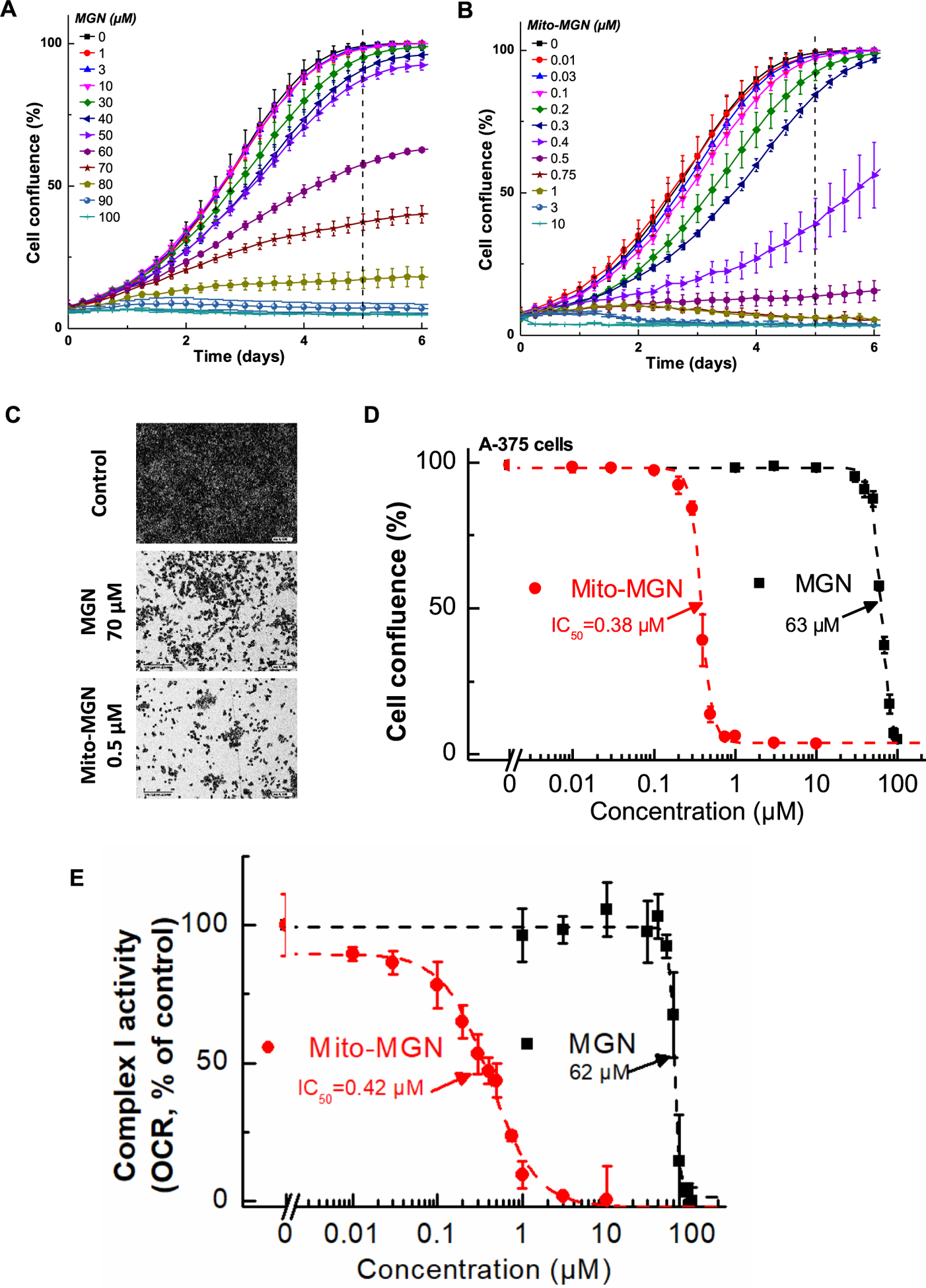
(A, B) A375 melanoma cells were treated with MGN (A) or Mito-MGN (B). Cell proliferation was monitored in real time using the IncuCyte imager. (C) Representative phase contrast images (10×) of three replicate experiments. (D) The dependence of the cell confluence (as control groups reach 98% confluence) as a function of MGN and Mito-MGN concentrations. (E) Effect of MGN and Mito-MGN on the activity of mitochondrial complex I. A375 cells were pretreated with MGN and Mito-MGN for 24 h. The mitochondrial complex I OCR values are plotted against concentrations of MGN and Mito-MGN. Dashed lines represent the fitting curves used for determination of the IC50 values. Data shown are the means ± SD.
Fig. 4. Effect of MGN and Mito-MGN on the proliferation of G361 melanoma cells.
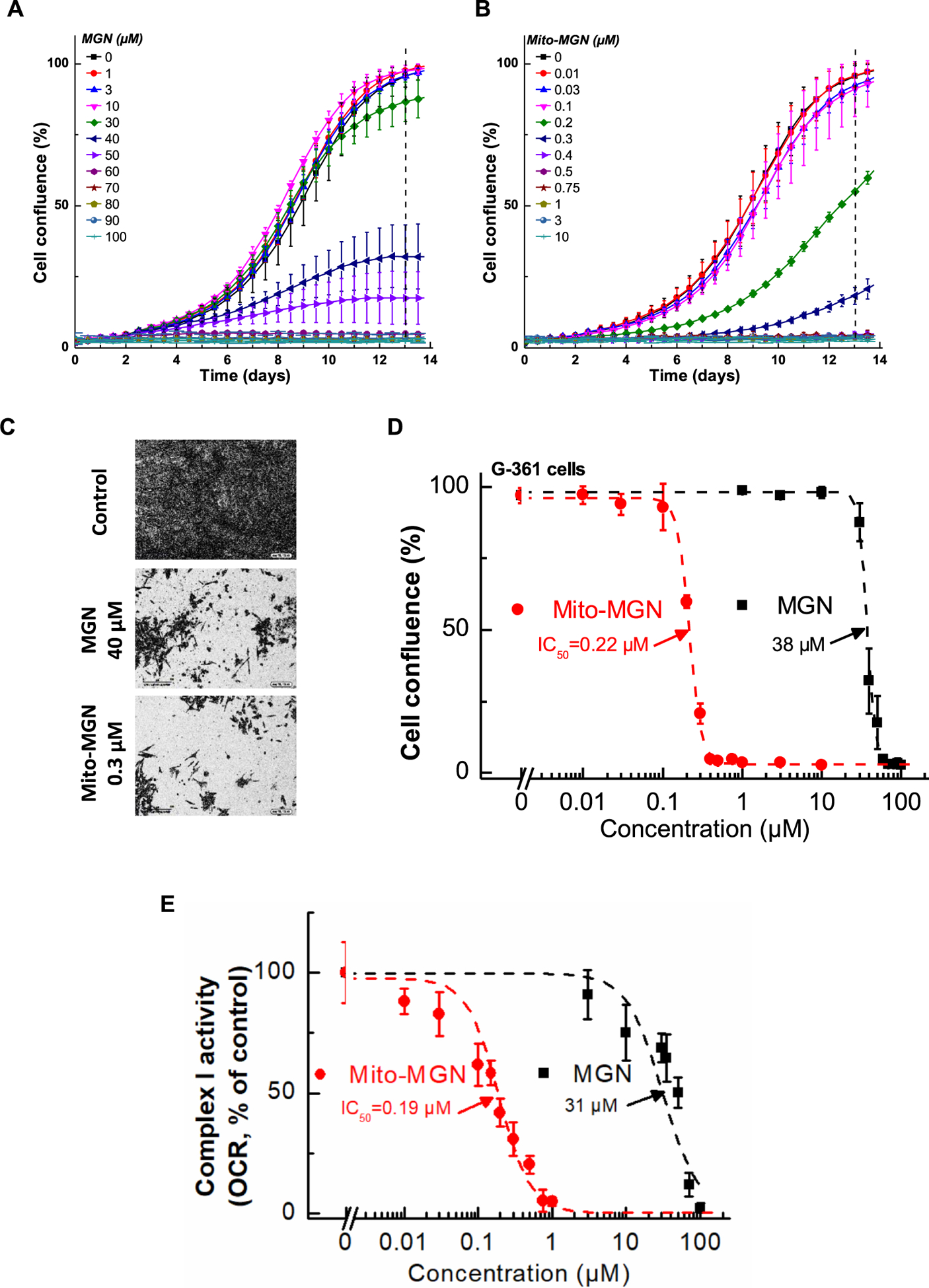
(A,B) G361 melanoma cells were treated with MGN (A) or Mito-MGN (B). Cell proliferation was monitored in real time using the IncuCyte imager. (C) Representative phase contrast images (10×). (D) Cells were treated with MGN or Mito-MGN, and cell growth was monitored continuously using the IncuCyte instrument. The cell confluence (as control groups reach 98% confluence) is plotted against concentration. Dashed lines represent the fitting curves used for determination of the IC50 values. (E) Effect of MGN and Mito-MGN on the activity of mitochondrial complex I. G361 cells were pretreated with MGN and Mito-MGN for 24 h. The mitochondrial complex I OCR values are plotted against the concentrations of MGN and Mito-MGN. Dashed lines represent the fitting curves used to determine the IC50 values.
We have previously reported that targeting metformin to mitochondria leads to inhibition of mitochondrial respiration by blocking complex I activity. Therefore, we asked if the inhibition of mitochondrial respiration by Mito-MGN may be responsible for its effects on melanoma cell proliferation. Using the Seahorse XF96 extracellular flux analyzer, we measured the mitochondrial complex activity in melanoma cells. The effect of Mito-MGN on the activities of mitochondrial complexes I and II of the UACC-62 melanoma cells is shown in Fig. 2C. Mito-MGN inhibited pyruvate/malate-driven, rotenone-inhibitable OCR (complex I activity) but did not affect the succinate-driven, malonate-inhibitable OCR (complex II activity). Treatment of A375 or G361 melanoma cells with MGN and Mito-MGN inhibited complex Idependent mitochondrial respiration. The IC50 values to inhibit complex I-dependent OCR for MGN and Mito-MGN in A375 cells were 62 μM and 0.42 μM, respectively (Fig. 3E), and in G361 cells were 31 μM and 0.19 μM, respectively (Fig. 4E). Those IC50 values are very close to the determined IC50 values of MGN and Mito-MGN in cell proliferation assays. These data suggest a direct correlation between the OXPHOS inhibition and antiproliferative activity of MGN and Mito-MGN [25].
At concentrations inhibiting proliferation of UACC-62 melanoma cells, we observed activation of the AMPK signaling pathway. AMPK, a master intracellular energy sensor, typically is activated under mitochondrial stress (complex I inhibition) due to induction of bioenergetic and/or redox stress. Mito-MGN treatment induced AMPK Thr172 phosphorylation (Fig. 2D), indicating activation of the AMPK pathway.
3.3. Mito-MGN decreases mitochondrial membrane potential
Given the potent inhibitory effects of Mito-MGN on mitochondrial complex I activity, we sought to test the effect of Mito-MGN on mitochondrial membrane potential. The TMRE probe was taken up into metabolically active mitochondria and quantified using flow cytometry (Fig. 5). Upon depolarization or stress-induced damage to mitochondrial membranes, TMRE was released into the cell cytosol and extracellular medium and this is reflected by a decrease in intracellular TMRE fluorescence intensity. Consistent with its inhibitory effects on complex I and proliferation, Mito-MGN dose-dependently decreased TMRE fluorescence, a proxy measurement of mitochondrial membrane potential in UACC-62 cells (Fig. 5A and B). FCCP, a potent mitochondrial OXPHOS uncoupler, was used as a positive control. These results are consistent with previous findings for other mitochondria-targeted compounds in cancer cells [22,39].
Fig. 5. Mitochondrial depolarization in Mito-MGN-treated melanoma cells.
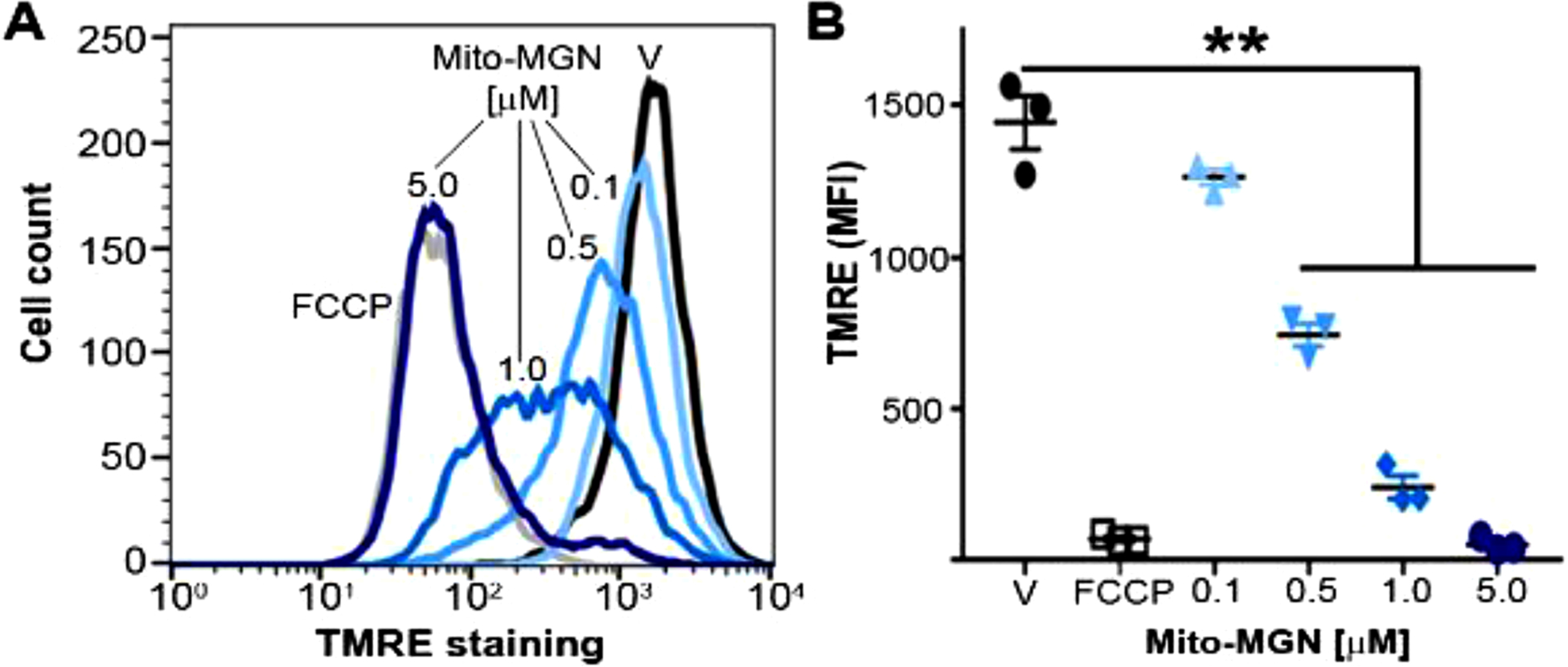
(A) Representative quantification of the cellular TMRE uptake in UACC-62 melanoma cells. (B) Mean fluorescence intensity (MFI) of TMRE in Mito-MGN-treated (24 h) and vehicle-treated UACC-62 cells. Values are mean ± SEM, n = 3, biological replicates, **P ≤ 0.01.
3.4. Mito-MGN induces mitophagy and modulates energetic signaling in a caspase-independent manner
Previous reports suggest that disruption of the mitochondrial membrane potential by mitochondria-targeted compounds activates AMPK signaling and autophagy in cancer cells [22,39]. To investigate the role of mitophagy (mitochondrial autophagy), lysates were isolated from vehicle and Mito-MGN (0.5 μM) treated UACC-62 cells after 24 h and 48 h incubation and probed for mitophagy-linked proteins. Immunoblot analysis of specific mitophagy adaptor proteins is presented in Fig. 6A. LC3, a microtubule-associated protein light chain, is crucial for cargo recruitment and the lipidated form, LC3-II, is a canonical marker of autophagy. Results show that in melanoma cell lines treated with Mito-MGN, there was an increase in the percentage of LC3-II and a decrease in P62 receptor levels by 48 h, indicative of autophagy induction and completion of the autophagic degradative process, respectively. The ratios of LC3-II:LC3-I in cells treated with vehicle and with Mito-MGN at 24 h and 48 h are 1.5, 3.2, and 6.1.
Fig. 6. Modulation of autophagy- and energy-regulated proteins in Mito-MGN-treated melanoma cells.
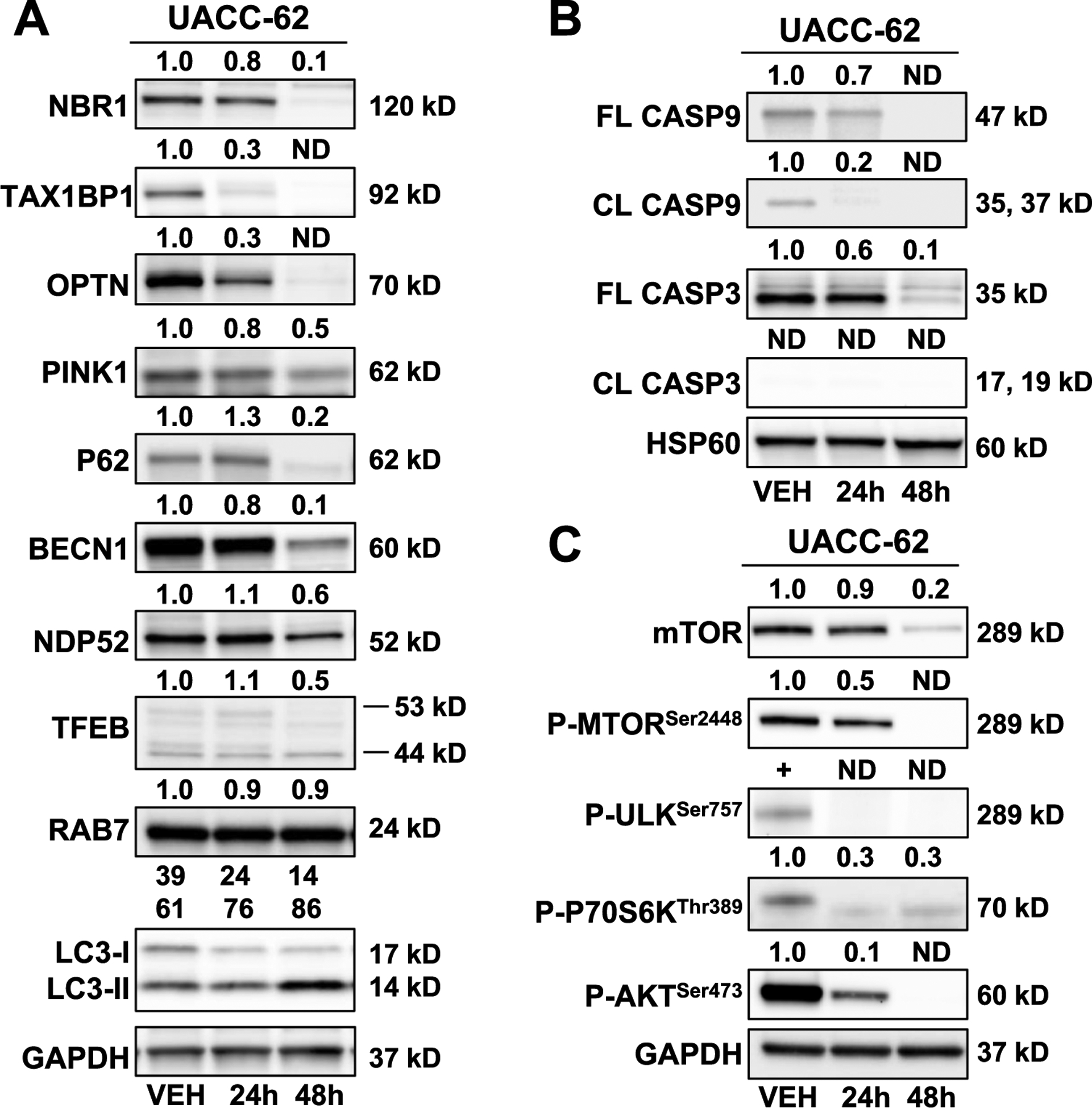
(A) Mito-MGN (0.5 μM) modulated autophagy and specific mitophagy-linked proteins in UACC-62 melanoma cell lines. (B) Mito-MGN (0.5 μM) did not induce caspase activation or cleavage in UACC-62 melanoma cell lines. (C) Mito-MGN (0.5 μM) modulated AKT/mTOR signaling cascades in UACC-62 melanoma cell lines over time.
Next, specific ubiquitin-binding mitophagy receptors were evaluated in melanoma cell lines. UACC-62 cells treated with Mito-MGN showed decreased levels of a number of mitophagy adaptor proteins including NBR1, TAX1BP1, OPTN, and NDP52 at both 24 h and 48 h. PINK1 was assessed as a marker of PINK1-Parkin-dependent mitophagy. PINK1 levels were reduced by Mito-MGN treatment at 48 h. PINK1 has a role in mitophagy, chemoresistance, and bioenergetics and more recently has been recognized as an AKT signaling regulator [40,41]. Mito-MGN treatment did not induce the autophagy regulator BECN1 or RAB7, an indicator of late-autophagy, over the treatment time course. These results indicate that Mito-MGN-induced mitophagy is regulated by nonclassical autophagic pathways, independent of Beclin-1.
TFEB is an upstream regulator of AKT/mTOR signaling, interacts with P62 and PINK/Parkin signaling, and is upregulated during oxidative stress, linking this transcriptional node to altered energetics, metabolic reprogramming, and selective autophagy. In addition to autophagy and specific mitophagy linked protein changes, we determined that Mito-MGN does not induce caspase 3 or 9 activation in UACC-62 (Fig. 6B), as reported in other agents targeting resistant cancer cell lines via mitophagy activation [40]. Together, these results suggest that Mito-MGN induces autophagy via a nonclassical pathway but not apoptosis in melanoma cells.
Next, because the autophagy cascade is known to be sensitive to the energy status of the cell [42], we investigated how Mito-MGN modulates energy signaling, and we assessed several members of the mTOR/AKT pathway (Fig. 6C). In UACC-62 cells treated with Mito-MGN, there was nearly a complete loss of phospho-mTORSer2448, phospho-ULKSer757, and phospho-AKTSer473 by 48 h post treatment. PhosphoP70S6KThr389 levels decreased following Mito-MGN treatment. These data suggest that the mTOR/AKT signaling pathway is downregulated by Mito-MGN in melanoma cells.
Further support for the involvement of mitophagy in the Mito-MGN-mediated antiproliferative effects in melanoma cells was obtained using hydroxychloroquine (HCQ) (Fig. 7). HCQ, a lysotropic chloroquine derivative, has been shown to inhibit autophagy [43]; Mito-MGN alone inhibited cell proliferation (Fig. 7A). In the presence of 10 μM HCQ, the antiproliferative effects of Mito-MGN were significantly reversed (Fig. 7A and B). Cumulatively, these data suggest that Mito-MGN-induced mitophagy is responsible for the antiproliferative effects of Mito-MGN.
Fig. 7. HCQ autophagy inhibitor blocks the antiproliferative effects of Mito-MGN.
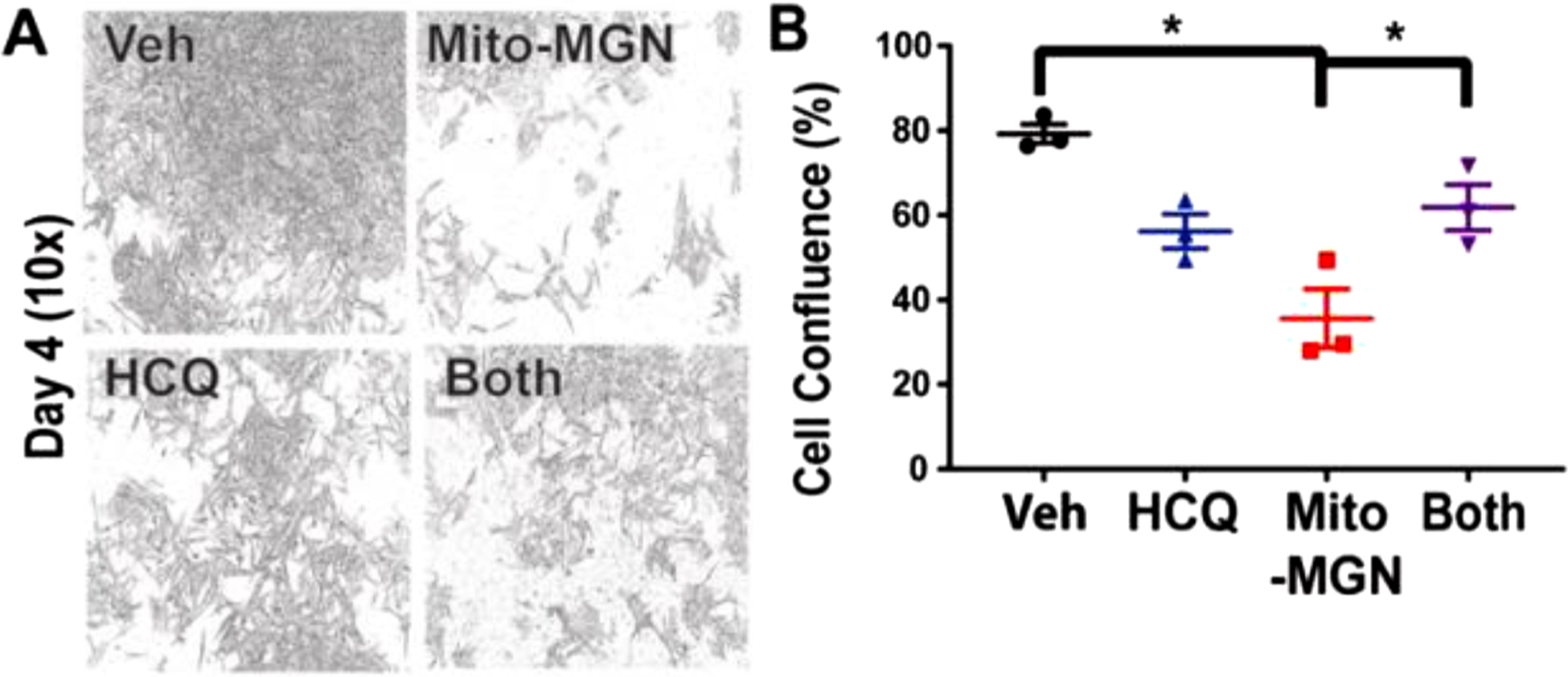
(A) Representative photomicrographs at 10× magnification of cells treated with vehicle alone, Mito-MGN, HCQ, and HCQ plus Mito-MGN on day 4. (B) Quantification of cell confluence in HCQ (10 μM), Mito-MGN (0.5 μM), or HCQ plus Mito-MGN combination-treated melanoma cells. Values are mean ± SD, n = 3–6, biological replicates, P ≤ 0.05.
3.5. Mito-MGN induces redox changes, ROS formation, and oxidation of mitochondrial peroxiredoxin in melanoma cells
The release of TMRE from the mitochondrion with Mito-MGN suggests a mechanical compromise of the mitochondrial membrane, or transmembrane components, and consequent partial membrane depolarization (Fig. 5). To further understand Mito-MGN-induced redox changes and formation of mitochondrial ROS, a cryogenic-temperature EPR approach was used. This probe-free method monitors the redox status of mitochondrial electron-transferring metalloprotein redox centers and changes in expression, or ROS-mediated chemical modification, of other associated metabolic metalloenzymes [28,35,36,44–46]. EPR spectra (recorded at 12 K) of UACC-62 melanoma cells with and without Mito-MGN incubation exhibited signals in the g = 2 region due to the oxidatively inactivated [3Fe4S]+ cluster of aconitase (Acn) and the [3Fe4S]+ S3 cluster of complex II (S3), and a composite signal due to multiple overlapping g2 and g3 resonances from reduced low-potential [2Fe2S]+ and [4Fe4S]+ clusters of complexes I and II (labeled FeS in Fig. 8A) [35]. The most striking difference between the two spectra is a six-fold increase in the Acn signal in the Mito-MGN-treated cells. This signal arises from the oxidative removal of the unique noncovalently bound Fea ion from the native, active [4Fe4S]2+ cluster to give the EPR-active [3Fe4S]+ cluster that is ROS-mediated and is likely due directly to increased superoxide production [47,48]. Additionally, the intensity of the FeS signal diminished by a factor of about three upon Mito-MGN treatment, indicating an increase in the (negative-valued) mitochondrial redox potential (i.e., a decrease in the stored electrochemical potential energy). Low amplitude signals from the g3 resonances of the lowest potential redox centers, the N3 and N4 clusters of complex I (in the region labeled N4b in Fig. 8A), are largely obscured by the MI ==|−5/2〉 highest-field hyperfine resonance of the M = | ± 1/2〉 Kramers doublet of S = 5/2 Mn(II). The identification of a three-fold diminution of the composite EPR signal of iron-sulfur clusters upon Mito-MGN incubation reveals a significant loss of electrochemical potential. The clusters contributing to this signal have a wide range of redox potentials, but a cautious estimate is that the mitochondrial redox potential collapses from −320 mV to −340 mV in untreated cells, determined by the NAD+/NADH couple, to about −100 mV to −150 mV upon incubation with Mito-MGN [49,50]. This observation is entirely consistent with the complete loss of the complex I N4 signal.
Fig. 8. Measurement of Mito-MGN-induced redox changes and intracellular oxidants in melanoma cells.
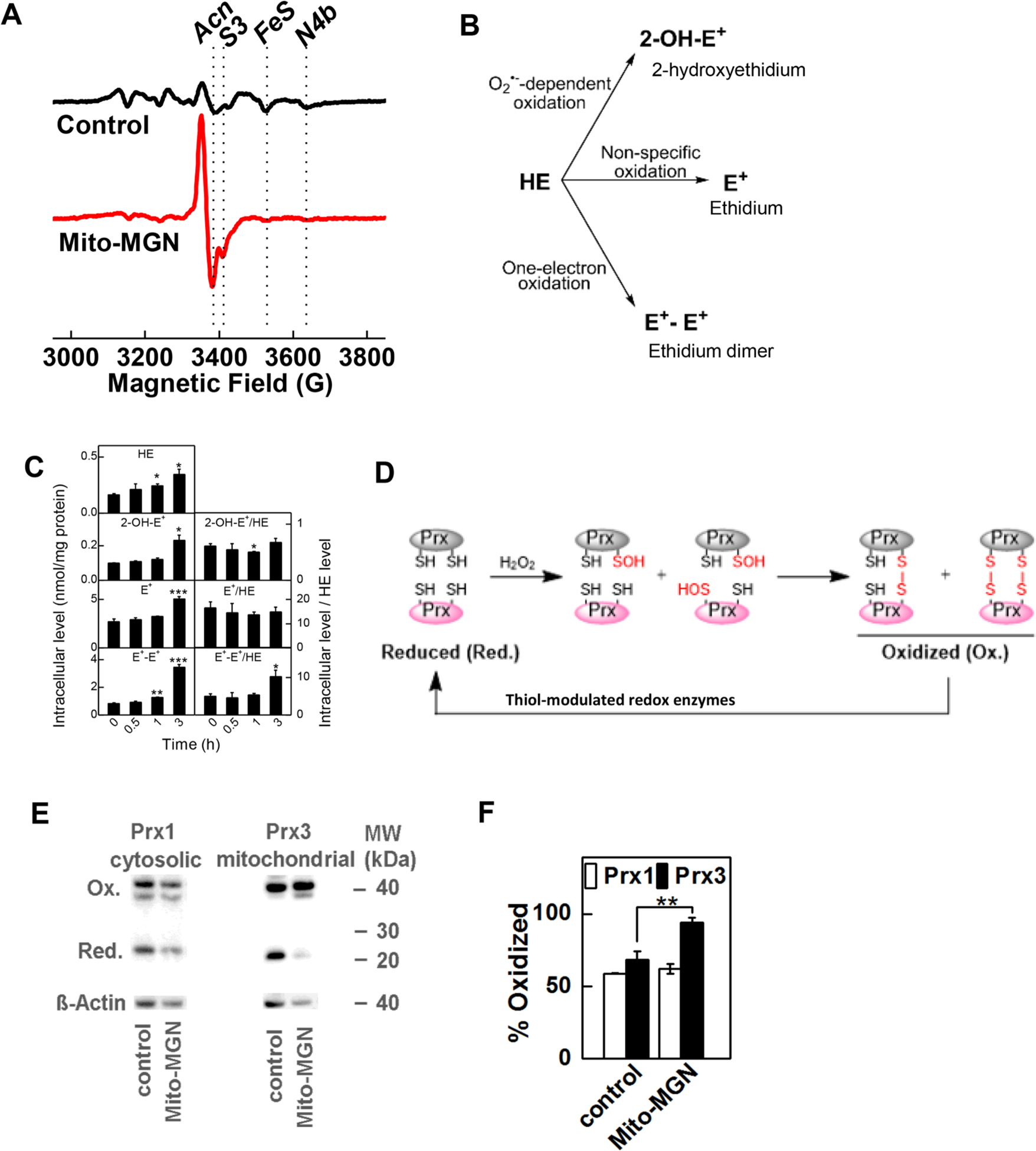
(A) UACC-62 melanoma cells were analyzed by low-temperature EPR to monitor redox signals. UACC-62 cells were treated with Mito-MGN (0.5 μM) for 24 h and cell lysates were analyzed by EPR. Spectrum (indicated in red) shows increased level of oxidized Acn. The other minor absorptions shown for control include S3 attributable to complexes I and II and to reduced iron sulfur centers (Fe-S, 4Fe-4S). (B) Schematic of ROS-dependent formation of oxidation products of HE. (C) Mito-MGN time-dependently enhanced the intracellular oxidation products of HE. The melanoma cells were treated for 0.5, 1, and 3 h with Mito-MGN (0.5 μM) followed by 1 h incubation with HE probe. (D) Diagram showing the redox cycle of Prx enzymes. (E) Melanoma cells were treated with Mito-MGN (0.5 μM) for 24 h, and the extent of Prx1 and Prx3 oxidation was determined by redox western blotting. (F) Densitometric analysis of the Prx immunoblots. Values are mean ± SD, n = 3, *P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001.
Inhibition of mitochondrial complex I may also stimulate the excess production of ROS, which might in turn hinder cell proliferation [21]. The redox probe, HE, was used to monitor oxidants generated in melanoma cells treated with Mito-MGN. Fig. 8B shows the various oxidation products of HE that were monitored [51,52]. Mito-MGN also induced the iron or peroxidase-dependent oxidation of HE as evidenced by enhanced intracellular formation of the dimeric product (E+-E+) and ethidium cation (E+) (Fig. 8B and C). Mito-MGN enhanced oxidation of HE, suggesting that peroxidatic oxidation of HE occurred even after a 3 h treatment (Fig. 8C). Mito-MGN induced a significant increase in 2-OH-E+ and nonspecific oxidation products in cells after a 24 h treatment (Fig. S2).
Peroxiredoxins (Prxs) are abundant endogenous thiol peroxidases that act as compartment-specific sensors of hydrogen peroxide in mitochondria (Prx3) and cytosol (Prx1) [53]. Prxs rapidly react with hydrogen peroxide (k > 106 M−1s−1), forming oxidized Prx that is recycled back to the reduced form in the presence of NADPH and thioredoxin/thioredoxin reductase enzymatic systems (Fig. 8D) [47,54]. Prx3 accounts for nearly 90% of mitochondrial peroxidase activity [53]. We determined that Mito-MGN stimulated the oxidation of mitochondrial Prx3, while cytosolic Prx1 remained almost unaffected, suggesting that oxidants were induced selectively in the mitochondria (Fig. 8E and F).
3.6. Mito-MGN inhibits tumor growth
Having shown that Mito-MGN inhibits proliferation in cultured melanoma cells in vitro, we sought to examine its in vivo efficacy in a murine model of melanoma. Mice were orthotopically engrafted with UACC-62 cells and treated twice per week with vehicle or 1 mg/tumor of Mito-MGN injected intratumorally. Tumors were routinely monitored in vivo using micro-CT imaging, and the representative image obtained on day 58 from control and treated mice xenografts reflects a marked reduction in tumor size in Mito-MGN-treated mice (Fig. 9A). Tumor weight was measured in excised tumors (Fig. 9B). These experiments demonstrate that Mito-MGN greatly inhibits melanoma progression in a murine xenograft model.
Fig. 9. Mito-MGN inhibits melanoma tumor progression in vivo.
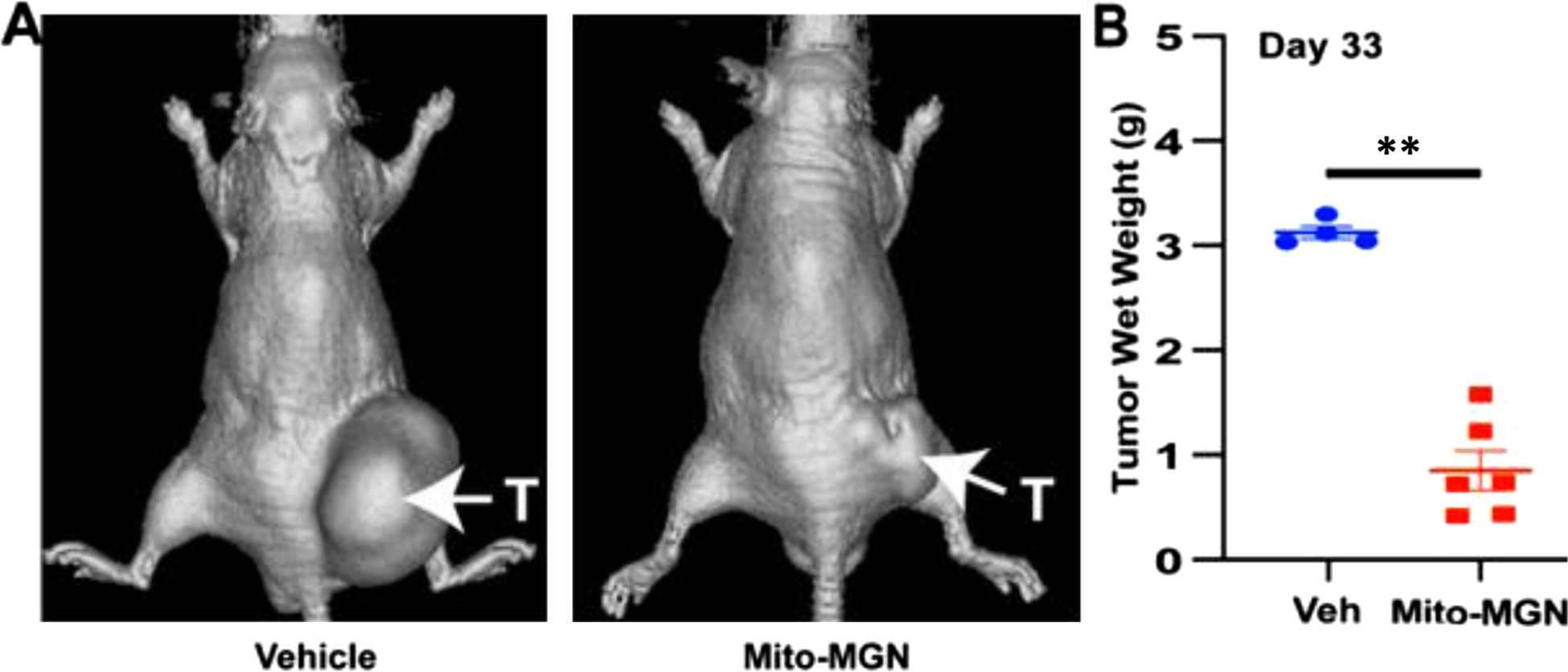
(A) Representative micro-CT images from UACC-62 xenografted (T - tumor) mice treated three times per week with vehicle (left) or 1 mg/tumor Mito-MGN (right). Data representative of four to six mice per group. (B) Quantification of tumor wet weight measured ex vivo on day 58. Values are mean ± SEM, n = 4–6, ** P ≤ 0.01 determined using an ANOVA with Tukey’s post hoc test.
4. Discussion
Despite advances in treating melanoma patients with emerging immune-based therapies, a pronounced metabolic reprogramming renders melanoma resistant to adjunct cytotoxic therapies used in combination. With nearly half of all melanomas exhibiting BRAF mutations, BRAF inhibitors provide a strong initial tumor regression in melanoma patients. However, most patients develop resistance to the treatment, resulting in disease progression and there is still a pressing need to develop new therapies, for use either as single agents or in combination with checkpoint blockade immunotherapy. In this study, using a panel of three melanoma cancer cell lines, we report that Mito-MGN, a mitochondria-targeted derivative of a naturally occurring polyphenolic compound, MGN, inhibits complex I-dependent OXPHOS and proliferation of melanoma cells. Further, mechanistic studies revealed that Mito-MGN decreases mitochondrial membrane potential, resulting in stimulation of mitochondrial autophagy or mitophagy, and induces redox and bioenergetic changes (promotes oxidative environment). In vivo studies indicate that Mito-MGN inhibits tumor growth in a melanoma mouse xenograft model.
Conventional inhibitors of mitochondrial electron transport complexes, such as cyanide, oligomycin, rotenone, and 2,4-dinitrophenol, are not specific for cancer cell mitochondria and exhibit low therapeutic indices due to their toxicity to normal cells. Mitochondria-targeted novel drugs have much lower toxicity and therefore could be developed as potent OXPHOS-modulating drugs. A fundamental property of cancer mitochondria is their higher (more negative inside) matrix transmembrane potential [55,56]. This property was exploited in strategic targeting of drugs to mitochondria [17,19,26]. We have previously developed mitochondria-targeted cationic drugs that selectively sequester in the mitochondrial matrix and membranes of tumor cells due to increased negative mitochondrial membrane potential [17,21,26]. TPP+-containing mitochondria-targeted agents are potent and selective inhibitors of OXPHOS in tumor cells [12,21]. Lipophilic and cationic TPP+-containing drugs diffuse across cell membranes, accumulate selectively in tumor tissue, and lack the toxicity associated with traditional OXPHOS inhibitors [25,26].
We quantitatively established that compared with the untargeted parent molecule, TPP+-containing compounds are typically 100–1000 times more potent in inhibiting tumor cell proliferation [17,21,26]. Mitochondria-targeted TPP+-containing agents also inhibited mitochondrial respiration at concentrations similar to those needed to inhibit proliferation. Inhibition of mitochondrial electron transport, especially complex I, has been proposed as a predominant mechanism responsible for the antiproliferative effects [12,25,26]. Aerobic glycolysis (the Warburg effect) is the preferred pathway to generate energy to sustain the rapid proliferation of tumor cells [15]. However, we showed that dual targeting of glycolytic and OXPHOS metabolism synergistically inhibits tumor cell proliferation [17,19]. Dual targeting of glycolysis and mitochondrial respiration is equally effective in BRAFV600E mutant melanoma cells [57].
The role of autophagy in tumor initiation, progression, and metastasis is rather paradoxical. Evidence suggests that autophagy has an inhibitory role in tumor initiation but may provide a growth advantage to established cancer and confer therapeutic resistance [58,59]. However, the precise role of selective forms of autophagy, in established cancers in the context of BRAF mutant melanoma, is far from resolved. Mitophagy is a specialized form of autophagy that involves selective degradation and removal of mitochondrial proteins via the macro-autophagic pathway. Depending on the cancer type and micro-environment, autophagy can be either tumor suppressive or oncogenic [60,61]. In response to a modest increase of mitochondrial stress induced by mitochondria-targeted agents (Mito-Q, Mito-Metformin, or Mito-Lonidamine), activation of autophagy and an increase in cell death was reported in breast, colon, and lung cancer cells and in mice xenografts [22,26,62]. In these studies, selective inhibition of mitochondrial complexes, particularly complex I, led to enhanced ROS formation and redox modulation in mitochondria, resulting in altered redox signaling, enhanced mitophagy, and inhibition of cancer cell proliferation and viability [22,26]. In contrast, a recent report suggests that mitochondrial complex I activity is essential for maximal autophagic activation and that mitochondrial inhibitors such as phenformin suppress mitophagy in human embryonic kidney 293 cells [63]. Also, a recent report showed that treatment with chloroquine that inhibited lysosome formation suppressed pancreatic ductal adenocarcinoma cell proliferation [64].
Inhibition of mitochondrial complex I within the electron transport chain causes stimulation of ROS including superoxide and hydrogen peroxide [65]. Consistent with previous reports [25,26], Mito-MGN induced superoxide and other oxidants generated from hydrogen peroxide and peroxidase. Mito-MGN stimulated oxidation of mitochondrial peroxiredoxin, Prx3, but did not affect the redox state of the cytosolic Prx1. The redox state of peroxiredoxins serves as endogenous indicator of mitochondrial and cytosolic redox stress. Therefore, increased oxidation of Prx3 suggests the enhanced formation of mitochondrial hydrogen peroxide in Mito-MGN-treated cells.
To determine the temporal relationship between ROS and autophagy induced by Mito-MGN, we monitored the formation of the specific product of the superoxide reaction with HE (2-OH-E+), the nonspecific two-electron oxidation product (E+), and the dimeric product (E+-E+) formed by the recombination of two HE-derived radicals in cells treated with Mito-MGN over a period of 24 h (Figs. 8C and S2). Under these conditions, we observed a slight increase in LC3-II and PINK1 protein levels (not shown). Although the temporal study showed an increase in both oxidant formation and autophagy as early as 4 h after incubation with Mito-MGN, the causal relationship still remains to be determined.
Targeting OXPHOS is emerging as a promising approach in cancer therapy [66]. Most OXPHOS inhibitors tested so far target mitochondrial respiration and tumor cell proliferation. Recent reports indicate cancer-cell-specific and highly potent complex I/complex II inhibitor (e.g., Mito-Lonidamine) and complex V inhibitor (e.g., Gboxin) [26,67]. Atovaquone and arsenic trioxide are FDA-approved drugs that inhibit mitochondrial complex III and complex IV in tumor cells [68,69]. Mitochondria-targeted agents were shown to activate lysosomal-dependent mitophagy in tumor cells [70]. Thus, it is conceivable that these different types of OXPHOS inhibitors, either alone or in combination, could be used as potent drugs in cancer treatment. A head-to-head comparison between the FDA-approved OXPHOS inhibitor (IACS-010759) and Mito-MGN indicates that Mito-MGN is more than 50-fold more potent than IACS-010759 at inhibiting melanoma cell proliferation (Fig. S3).
Previous reports suggest that the potency of mitochondria-targeted agents is critically dependent on the alkyl side chain length [21]. As shown in Fig. 1, Mito-MGN (also Mito-MGN10 with a 10-carbon side chain) potently inhibits (IC50 = 0.3 μM) the proliferation of UACC-62 melanoma cells. We compared the relative antiproliferative potencies of Mito-MGN10 with another Mito-MGN analog containing a shorter alkyl side chain with a four-carbon side chain (Mito-MGN4) in UACC-62 cells. As shown in Fig. S4, there was a significant decrease (eight-fold) in the antiproliferative potency of Mito-MGN4 (IC50 = 2.5 μM), as compared with the long chain Mito-MGN. The IC50 value of Mito-MGN4 (24 h treatment) to inhibit the mitochondrial complex I-dependent oxygen consumption was calculated to be 2.4 μM (Fig. S5); that is consistent with its antiproliferative potency. These findings further reinforce the view that fine-tuning of the alkyl linker length is crucial to maximizing the optimal antiproliferative effects of TPP+-conjugated mitochondria-targeted therapeutics.
Chemotherapy-induced metabolic reprogramming—from glycolysis to OXPHOS—is proposed as a viable mechanism of decreasing chemotherapeutic efficacy in tumor cells [7–10,71]. An increase in OXPHOS was attributed to observed drug resistance in many cancers, including melanoma, colon, prostate, and leukemia. The emerging findings that resistant tumor cells rely more on mitochondrial OXPHOS and respiration than glycolysis have important therapeutic implications [7,12,21]. Drug-resistant melanoma cells with compromised glycolytic mechanism switch to enhanced OXPHOS and, therefore, remain sensitive to OXPHOS inhibition. We generated a UACC-62 human melanoma cell line that is resistant to the BRAF kinase inhibitor, vemurafenib analog PLX4720 (UACC-62-R), by sustained propagation in the presence of a low dose of drug over time. Proliferation assays in the presence of PLX4720 demonstrated that our newly generated UACC-62-R cell line was approximately six times more resistant to the drug compared with UACC-62-WT cells (Fig. S6A). The IC50 values of 4.3 μM and 26.2 μM for wild type and resistant cells, respectively, confirmed the differential sensitivity of UACC-62-WT cells compared with UACC-62-R cells (Fig. S6A). Next, we determined whether Mito-MGN inhibits the UACC-62-R cells. As shown in Fig. S6B, Mito-MGN dose-dependently inhibited proliferation of both UACC-62-WT (IC50 ==0.3 μM) and UACC-62-R cells (IC50 ==0.29 μM). Melanoma cells harboring BRAF oncogenic mutations preferentially use aerobic glycolysis (i.e., the Warburg effect) to meet their energy demands. However, cells with acquired resistance to BRAF inhibitors exhibit metabolic reprogramming, i.e., decreased glycolysis and compensatory sensitivity to OXPHOS inhibition. Using a Seahorse imaging technique, we measured the glycolytic and OXPHOS pathways in UACC-62-WT and UACC-62-R cells. As shown in Fig. S6E, the two-dimensional map for the OCR (representing OXPHOS status) and proton production rate (PPR, representing glycolysis status) demonstrated a 50% increase in the OCR and an 11% decrease in PPR in UACC-62-R cells. Treatment with Mito-MGN inhibited mitochondrial respiration in both cell lines, regardless of the sensitivity to PLX4720 kinase inhibitor (Fig. S6E).
We recognize the study limitations. For instance, several unanswered questions remain with regard to the exact mechanism of action: Is complex I inhibition required for Mito-MGN-induced inhibition of cell proliferation tumor/growth? Is induction of autophagy/mitophagy required for the antiproliferative effects of Mito-MGN? Clearly, specific inhibition of mitochondrial respiratory chain protein (complex I) and/or autophagy-inducing genes in melanoma cells will further clarify the molecular mechanism of action of Mito-MGN. In agreement with our prior report [21], inhibition of AMPK with Compound C restored melanoma cell proliferation in cells treated with antiproliferative doses of Mito-MGN (Fig. S7). Although those data support a key role for AMPK in the anti-tumor effects of Mito-MGN, additional in vivo studies, including post-treatment tissue analysis of the mTOR pathway, AMPK, and Prx 1 and Prx 3 levels, would provide key correlative support for the mechanism established using reductionist cell culture models.
5. Conclusion
In summary, the mitochondria-targeted agent, Mito-MGN, potently inhibited proliferation of melanoma cells and tumor growth in mice xenografts. Potential anti-melanoma therapeutic mechanisms of Mito-MGN include inhibition of mitochondrial complex I-mediated respiration, decreased mitochondrial membrane potential, enhanced ROS formation and mitochondrial peroxiredoxin oxidation, modulation of energy signaling pathways, and/or stimulation of autophagy-related signaling in melanoma cells (Fig. 10).
Fig. 10.
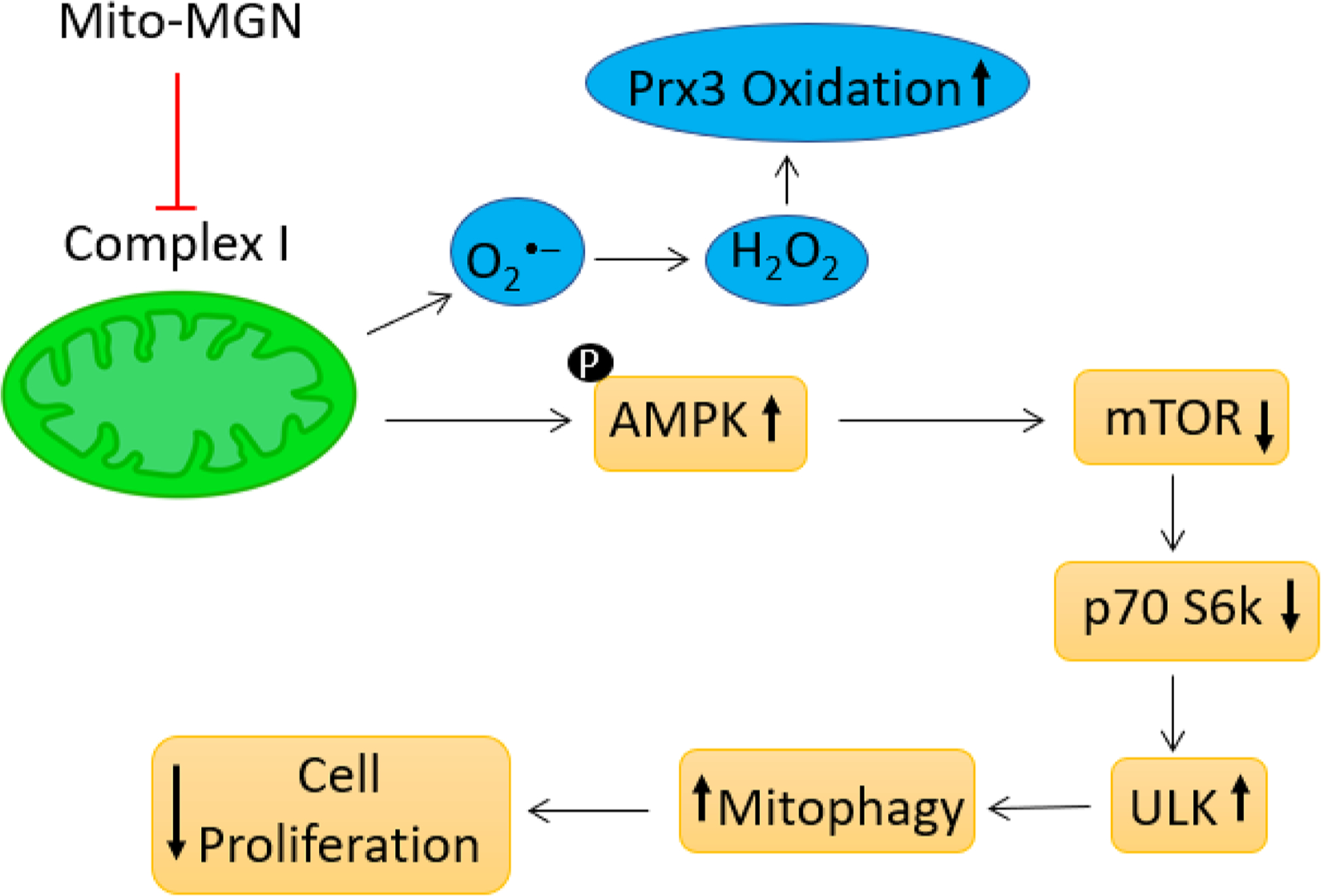
Proposed mechanism of action of Mito-MGN relating mitophagy induction to inhibition of melanoma cell proliferation.
Supplementary Material
Acknowledgments
This work was supported in part by the National Institutes of Health under grant U01 CA178960 to M.B.D. and B.K. and grant R01 CA226279 to M.B.D. Measurements of cell bioenergetic function, mitochondrial complex I and II activities and peroxiredoxins oxidation status were performed in the Medical College of Wisconsin Cancer Center Redox & Bioenergetics Shared Resource. Low-temperature EPR was supported in part by NSF Major Research Instrumentation award CHE-1532168 to B.B. Autophagy work was supported by the University of Michigan Rogel Cancer Center funds awarded to L.A.K.
Footnotes
Declaration of Competing Interest
G.C., M.H., J.Z., and B.K. are co-inventors in the patent application “Mito-magnolol compounds and methods of synthesis and use thereof” (WO2020124019A1).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.ctarc.2020.100210.
References
- [1].Batus M, Waheed S, Ruby C, Petersen L, Bines SD, Kaufman HL, Optimal management of metastatic melanoma: current strategies and future directions, Am. J. Clin. Dermatol 14 (2013) 179–194, 10.1007/s40257-013-0025-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Miller KD, Siegel RL, Lin CC, Mariotto AB, Kramer JL, Rowland JH,Stein KD, Alteri R, A Jemal, Cancer treatment and survivorship statistics, 2016, CA Cancer J. Clin 66 (2016) 271–289, 10.3322/caac.21349. [DOI] [PubMed] [Google Scholar]
- [3].Gide TN, Wilmott JS, Scolyer RA, Long GV, Primary and acquired resistance to immune checkpoint inhibitors in metastatic melanoma, Clin. Cancer Res 24 (2018) 1260–1270, 10.1158/1078-0432.Ccr-17-2267. [DOI] [PubMed] [Google Scholar]
- [4].Abildgaard C, Dahl C, Basse AL, Ma T, Guldberg P, Bioenergetic modulation with dichloroacetate reduces the growth of melanoma cells and potentiates their response to BRAFV600E inhibition, J. Transl. Med 12 (2014) 247, 10.1186/s12967-014-0247-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Domingues B, Lopes JM, Soares P, Populo H, Melanoma treatment in review, Immunotargets Ther 7 (2018) 35–49, 10.2147/itt.S134842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Tsao H, Goel V, Wu H, Yang G, Haluska FG, Genetic interaction between NRAS and BRAF mutations and PTEN/MMAC1 inactivation in melanoma, J. Invest. Dermatol 122 (2004) 337–341, 10.1046/j.0022-202X.2004.22243.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Vazquez F, Lim JH, Chim H, Bhalla K, Girnun G, Pierce K, Clish CB,Granter SR, Widlund HR, Spiegelman BM, Puigserver P, PGC1alpha expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress, Cancer Cell 23 (2013) 287–301, 10.1016/j.ccr.2012.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Roesch A, Vultur A, Bogeski I, Wang H, Zimmermann KM, Speicher D, Korbel C, Laschke MW, Gimotty PA, Philipp SE, Krause E, Patzold S, Villanueva J, Krepler C, Fukunaga-Kalabis M, Hoth M, Bastian BC, Vogt T, Herlyn M, Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1B(high) cells, Cancer Cell 23 (2013) 811–825, 10.1016/j.ccr.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hardeman KN, Peng C, Paudel BB, Meyer CT, Luong T, Tyson DR, Young JD, Quaranta V, P Fessel J, Dependence on glycolysis sensitizes BRAF-mutated melanomas for increased response to targeted BRAF inhibition, Sci. Rep 7 (2017) 42604, 10.1038/srep42604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Haq R, Shoag J, Andreu-Perez P, Yokoyama S, Edelman H, Rowe GC,Frederick DT, Hurley AD, Nellore A, Kung AL, Wargo JA, Song JS,Fisher DE, Arany Z, Widlund HR, Oncogenic BRAF regulates oxidative metabolism via PGC1alpha and MITF, Cancer Cell 23 (2013) 302–315, 10.1016/j.ccr.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zhang G, Frederick DT, Wu L, Wei Z, Krepler C, Srinivasan S, Chae YC, Xu X, Choi H, Dimwamwa E, Ope O, Shannan B, Basu D, Zhang D, Guha M, Xiao M, Randell S, Sproesser K, Xu W, Liu J, Karakousis GC, Schuchter LM,Gangadhar TC, Amaravadi RK, Gu M, Xu C, Ghosh A, Xu W, Tian T, Zhang J, Zha S, Liu Q, Brafford P, Weeraratna A, Davies MA, Wargo JA, Avadhani NG, Lu Y, Mills GB, Altieri DC, Flaherty KT, Herlyn M, Targeting mitochondrial biogenesis to overcome drug resistance to MAPK inhibitors, J. Clin. Invest 126 (2016) 1834–1856, 10.1172/jci82661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Molina JR, Sun Y, Protopopova M, Gera S, Bandi M, Bristow C, McAfoos T, Morlacchi P, Ackroyd J, Agip AA, Al-Atrash G, Asara J, Bardenhagen J, Carrillo CC, Carroll C, Chang E, Ciurea S, Cross JB, Czako B, Deem A, Daver N, de Groot JF, Dong JW, Feng N, Gao G, Gay J, Do MG, Greer J, Giuliani V, Han J, Han L, Henry VK, Hirst J, Huang S, Jiang Y, Kang Z, Khor T, Konoplev S, Lin YH, Liu G, Lodi A, Lofton T, Ma H, Mahendra M, Matre P, Mullinax R, Peoples M, Petrocchi A, Rodriguez-Canale J, Serreli R, Shi T, Smith M, Tabe Y, Theroff J, Tiziani S, Xu Q, Zhang Q, Muller F, DePinho RA, Toniatti C, Draetta GF, Heffernan TP, Konopleva M, Jones P, Di Francesco ME, Marszalek JR, An inhibitor of oxidative phosphorylation exploits cancer vulnerability, Nat. Med 24 (2018) 1036–1046, 10.1038/s41591-018-0052-4. [DOI] [PubMed] [Google Scholar]
- [13].Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, Kalyanaraman B, Mutlu GM, Budinger GR, Chandel NS, Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity, Proc. Natl. Acad. Sci. USA 107 (2010) 8788–8793, 10.1073/pnas.1003428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Luengo A, Gui DY, Vander Heiden MG, Targeting metabolism for cancer therapy, Cell Chem. Biol 24 (2017) 1161–1180, 10.1016/j.chembiol.2017.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Vander Heiden MG, Cantley LC, Thompson CB, Understanding the Warburg effect: the metabolic requirements of cell proliferation, Science 324 (2009) 1029–1033, 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Weinberg SE, Chandel NS, Targeting mitochondria metabolism for cancer therapy, Nat. Chem. Biol 11 (2015) 9–15, 10.1038/nchembio.1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Cheng G, Zielonka J, Dranka BP, McAllister D, Mackinnon AC, Jr J Joseph, B. Kalyanaraman, Mitochondria-targeted drugs synergize with 2-deoxyglucose to trigger breast cancer cell death, Cancer Res. 72 (2012) 2634–2644, 10.1158/0008-5472.can-11-3928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Cheng G, Zielonka J, McAllister D, Tsai S, Dwinell MB, Kalyanaraman B, Profiling and targeting of cellular bioenergetics: inhibition of pancreatic cancer cell proliferation, Br. J. Cancer 111 (2014) 85–93, 10.1038/bjc.2014.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Cheng G, Zielonka J, McAllister DM, Mackinnon AC Jr, Joseph J, Dwinell MB, Kalyanaraman B, Mitochondria-targeted vitamin E analogs inhibit breast cancer cell energy metabolism and promote cell death, BMC Cancer 13 (2013) 285, 10.1186/1471-2407-13-285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Hong SK, Starenki D, Wu PK, Park JI, Suppression of B-Raf(V600E) melanoma cell survival by targeting mitochondria using triphenyl-phosphonium-conjugated nitroxide or ubiquinone, Cancer Biol. Ther 18 (2017) 106–114, 10.1080/15384047.2016.1250987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Cheng G, Zielonka J, Ouari O, Lopez M, McAllister D, Boyle K, Barrios CS, Weber JJ, Johnson BD, Hardy M, Dwinell MB, Kalyanaraman B, Mitochondria-targeted analogues of metformin exhibit enhanced antiproliferative and radio-sensitizing effects in pancreatic cancer cells, Cancer Res. 76 (2016) 3904–3915, 10.1158/0008-5472.Can-15-2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Boyle KA, Van Wickle J, Hill RB, Marchese A, Kalyanaraman B, Dwinell MB, Mitochondria-targeted drugs stimulate mitophagy and abrogate colon cancer cell proliferation, J. Biol. Chem 293 (2018) 14891–14904, 10.1074/jbc.RA117.001469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Sarrica A, Kirika N, Romeo M, Salmona M, Diomede L, Safety and toxicology of magnolol and honokiol, Planta Med. 84 (2018) 1151–1164, 10.1055/a-0642-1966. [DOI] [PubMed] [Google Scholar]
- [24].Vara-Perez M, Felipe-Abrio B, Agostinis P, Mitophagy in cancer: a tale of adaptation, Cells 8 (2019), 10.3390/cells8050493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Pan J, Lee Y, Cheng G, Zielonka J, Zhang Q, Bajzikova M, Xiong D, Tsaih S−W, Hardy M, Flister M, Olsen CM, Wang Y, Vang O, Neuzil J, Myers CR, Kalyanaraman B, You M, Mitochondria-targeted honokiol confers a striking inhibitory effect on lung cancer via inhibiting complex I activity, iScience 3 (2018) 192–207, 10.1016/j.isci.2018.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Cheng G, Zhang Q, Pan J, Lee Y, Ouari O, Hardy M, Zielonka M, Myers CR, Zielonka J, Weh K, Chang AC, Chen G, Kresty L, Kalyanaraman B, You M, Targeting lonidamine to mitochondria mitigates lung tumorigenesis and brain metastasis, Nat. Commun 10 (2019) 2205, 10.1038/s41467-019-10042-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Nicholls DG, Darley-Usmar VM, Wu M, Jensen PB, Rogers GW, Ferrick DA, Bioenergetic profile experiment using C2C12 myoblast cells, J. Vis. Exp (2010) 2511, 10.3791/2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Cheng G, Zielonka M, Dranka B, Kumar SN, Myers CR, Bennett B, Garces AM, Dias Duarte Machado LG, Thiebaut D, Ouari O, Hardy M, Zielonka J, Kalyanaraman B, Detection of mitochondria-generated reactive oxygen species in cells using multiple probes and methods: potentials, pitfalls, and the future, J. Biol. Chem 293 (2018) 10363–10380, 10.1074/jbc.RA118.003044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Zielonka J, Vasquez-Vivar J, Kalyanaraman B, Detection of 2-hydroxyethidium in cellular systems: a unique marker product of superoxide and hydroethidine, Nat. Protoc 3 (2008) 8–21, 10.1038/nprot.2007.473. [DOI] [PubMed] [Google Scholar]
- [30].Zhao H, Kalivendi S, Zhang H, Joseph J, Nithipatikom K, Vasquez-Vivar J, Kalyanaraman B, Superoxide reacts with hydroethidine but forms a fluorescent product that is distinctly different from ethidium: potential implications in intracellular fluorescence detection of superoxide, Free Radic. Biol. Med 34 (2003) 1359–1368. [DOI] [PubMed] [Google Scholar]
- [31].Kalyanaraman B, Dranka BP, Hardy M, Michalski R, Zielonka J, HPLC-based monitoring of products formed from hydroethidine-based fluorogenic probes - The ultimate approach for intra- and extracellular superoxide detection, Biochimica et Biophysica Acta 1840 (2014) 739–744, 10.1016/j.bbagen.2013.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Zielonka J, Zielonka M, Sikora A, Adamus J, Joseph J, Hardy M, Ouari O, Dranka BP, Kalyanaraman B, Global profiling of reactive oxygen and nitrogen species in biological systems: high-throughput real-time analyses, J. Biol. Chem 287 (2012) 2984–2995, 10.1074/jbc.M111.309062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zielonka J, Zhao H, Xu Y, Kalyanaraman B, Mechanistic similarities between oxidation of hydroethidine by Fremy’s salt and superoxide: stopped-flow optical and EPR studies, Free Radic. Biol. Med 39 (2005) 853–863, 10.1016/j.freeradbiomed.2005.05.001. [DOI] [PubMed] [Google Scholar]
- [34].Zielonka J, Srinivasan S, Hardy M, Ouari O, Lopez M, Vasquez-Vivar J,Avadhani NG, Kalyanaraman B, Cytochrome c-mediated oxidation of hydroethidine and mito-hydroethidine in mitochondria: identification of homo- and heterodimers, Free Radic. Biol. Med 44 (2008) 835–846, 10.1016/j.freeradbiomed.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kalyanaraman B, Cheng G, Zielonka J, Bennett B, Low-temperature EPR spectroscopy as a probe-free technique for monitoring oxidants formed in tumor cells and tissues: implications in drug resistance and OXPHOS-targeted therapies, Cell Biochem. Biophys 77 (2019) 89–98, 10.1007/s12013-018-0858-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Bennett B, Helbling D, Meng H, Jarzembowski J, Geurts AM, Friederich MW, Van Hove JL, Lawlor MW, Dimmock DP, Potentially diagnostic electron paramagnetic resonance spectra elucidate the underlying mechanism of mitochondrial dysfunction in the deoxyguanosine kinase deficient rat model of a genetic mitochondrial DNA depletion syndrome, Free Radic. Biol. Med 92 (2016) 141–151, 10.1016/j.freeradbiomed.2016.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Cox AG, Pullar JM, Hughes G, Ledgerwood EC, Hampton MB, Oxidation of mitochondrial peroxiredoxin 3 during the initiation of receptor-mediated apoptosis, Free Radic. Biol. Med 44 (2008) 1001–1009, 10.1016/j.freeradbiomed.2007.11.017. [DOI] [PubMed] [Google Scholar]
- [38].Myers JM, Myers CR, The effects of hexavalent chromium on thioredoxin reductase and peroxiredoxins in human bronchial epithelial cells, Free Radic. Biol. Med 47 (2009) 1477–1485, 10.1016/j.freeradbiomed.2009.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Sun C, Liu X, Di C, Wang Z, Mi X, Liu Y, Zhao Q, Mao A, Chen W, Gan L, Zhang H, MitoQ regulates autophagy by inducing a pseudo-mitochondrial membrane potential, Autophagy 13 (2017) 730–738, 10.1080/15548627.2017.1280219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Yao N, Wang C, Hu N, Li Y, Liu M, Lei Y, Chen M, Chen L, Chen C, Lan P, Chen W, Chen Z, Fu D, Ye W, Zhang D, Inhibition of PINK1/Parkin-dependent mitophagy sensitizes multidrug-resistant cancer cells to B5G1, a new betulinic acid analog, Cell Death Dis. 10 (2019) 232, 10.1038/s41419-019-1470-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Furlong RM, Lindsay A, Anderson KE, Hawkins PT, Sullivan AM, O’Neill C, The Parkinson’s disease gene PINK1 activates Akt via PINK1 kinase-dependent regulation of the phospholipid PI(3,4,5)P3, J. Cell Sci (2019) 132, 10.1242/jcs.233221. [DOI] [PubMed] [Google Scholar]
- [42].Alers S, Loffler AS, Wesselborg S, Stork B, Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks, Mol. Cell Biol 32 (2012) 2–11, 10.1128/mcb.06159-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Mauthe M, Orhon I, Rocchi C, Zhou X, Luhr M, Hijlkema KJ, Coppes RP, Engedal N, Mari M, Reggiori F, Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion, Autophagy 14 (2018) 1435–1455, 10.1080/15548627.2018.1474314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Orme-Johnson NR, Hansen RE, Beinert H, Electron paramagnetic resonance-detectable electron acceptors in beef heart mitochondria, J. Biol. Chem 249 (1974) 1928–1939. [PubMed] [Google Scholar]
- [45].Beinert H, [11]EPR spectroscopy of components of the mitochondrial electron-transfer system, Methods Enzymol 54 Academic Press, 1978, pp. 133–150. [DOI] [PubMed] [Google Scholar]
- [46].Kennedy MC, Antholine WE, Beinert H, An EPR investigation of the products of the reaction of cytosolic and mitochondrial aconitases with nitric oxide, J. Biol. Chem 272 (1997) 20340–20347. [DOI] [PubMed] [Google Scholar]
- [47].Ogusucu R, Rettori D, Munhoz DC, Netto LE, Augusto O, Reactions of yeast thioredoxin peroxidases I and II with hydrogen peroxide and peroxynitrite: rate constants by competitive kinetics, Free Radic. Biol. Med 42 (2007) 326–334, 10.1016/j.freeradbiomed.2006.10.042. [DOI] [PubMed] [Google Scholar]
- [48].Beinert H, Kennedy MC, Stout CD, Aconitase as ironminus signSulfur protein, enzyme, and iron-regulatory protein, Chem. Rev 96 (1996) 2335–2374, 10.1021/cr950040z. [DOI] [PubMed] [Google Scholar]
- [49].Medvedev ES, Couch VA, Stuchebrukhov AA, Determination of the intrinsic redox potentials of FeS centers of respiratory complex I from experimental titration curves, Biochim. Biophys. Acta 1797 (2010) 1665–1671, 10.1016/j.bbabio.2010.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Ohnishi T, Nakamaru-Ogiso E, Were there any “misassignments” among iron-sulfur clusters N4, N5 and N6b in NADH-quinone oxidoreductase (complex I)? Biochim. Biophys. Acta 1777 (2008) 703–710, 10.1016/j.bbabio.2008.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Robinson KM, Janes MS, Pehar M, Monette JS, Ross MF, Hagen TM,Murphy MP, Beckman JS, Selective fluorescent imaging of superoxide in vivo using ethidium-based probes, Proc. Natl. Acad. Sci. USA 103 (2006) 15038–15043, 10.1073/pnas.0601945103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Hardy M, Zielonka J, Karoui H, Sikora A, Michalski R, Podsiadly R, Lopez M, Vasquez-Vivar J, Kalyanaraman B, Ouari O, Detection and characterization of reactive oxygen and nitrogen species in biological systems by monitoring species-specific products, Antioxid. Redox Signal 28 (2017), 10.1089/ars.2017.7398 14116–11432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Andreyev AY, Kushnareva YE, Murphy AN, Starkov AA, Mitochondrial ROS metabolism: 10 years later, Biochemistry (Mosc.) 80 (2015) 517–531, 10.1134/s0006297915050028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Winterbourn CC, Biological production, detection, and fate of hydrogen peroxide, Antioxid. Redox Signal 29 (2018) 541–551, 10.1089/ars.2017.7425. [DOI] [PubMed] [Google Scholar]
- [55].Liberman EA, Topaly VP, Tsofina LM, Jasaitis AA, Skulachev VP, Mechanism of coupling of oxidative phosphorylation and the membrane potential of mitochondria, Nature 222 (1969) 1076–1078. [DOI] [PubMed] [Google Scholar]
- [56].Lichtshtein D, Kaback HR, Blume AJ, Use of a lipophilic cation for determination of membrane potential in neuroblastoma-glioma hybrid cell suspensions, Proc. Natl. Acad. Sci. USA 76 (1979) 650–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Wei L, Yu Y, Shen Y, Wang MC, Min W, Vibrational imaging of newly synthesized proteins in live cells by stimulated Raman scattering microscopy, Proc. Natl. Acad. Sci. USA 110 (2013) 11226–11231, 10.1073/pnas.1303768110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Rosenfeldt MT, Ryan KM, The multiple roles of autophagy in cancer, Carcinogenesis 32 (2011) 955–963, 10.1093/carcin/bgr031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].White E, DiPaola RS, The double-edged sword of autophagy modulation in cancer, Clin. Cancer Res 15 (2009) 5308–5316, 10.1158/1078-0432.Ccr-07-5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Cai J, Li R, Xu X, Zhang L, Lian R, Fang L, Huang Y, Feng X, Liu X, Li X, Zhu X, Zhang H, Wu J, Zeng M, Song E, He Y, Yin Y, Li J, Li M, CK1alpha suppresses lung tumour growth by stabilizing PTEN and inducing autophagy, Nat. Cell Biol 20 (2018) 465–478, 10.1038/s41556-018-0065-8. [DOI] [PubMed] [Google Scholar]
- [61].Humpton TJ, Alagesan B, DeNicola GM, Lu D, Yordanov GN, Leonhardt CS, Yao MA, Alagesan P, Zaatari MN, Park Y, Skepper JN, Macleod KF,Perez-Mancera PA, Murphy MP, Evan GI, Vousden KH, A Tuveson D, Oncogenic KRAS induces NIX-mediated mitophagy to promote pancreatic cancer, Cancer Discov. 9 (2019) 1268–1287, 10.1158/2159-8290.Cd-18-1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Rao VA, Klein SR, Bonar SJ, Zielonka J, Mizuno N, Dickey JS, Keller PW, Joseph J, Kalyanaraman B, Shacter E, The antioxidant transcription factor Nrf2 negatively regulates autophagy and growth arrest induced by the anticancer redox agent mitoquinone, J. Biol. Chem 285 (2010) 34447–34459, 10.1074/jbc.M110.133579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Thomas HE, Zhang Y, Stefely JA, Veiga SR, Thomas G, Kozma SC,Mercer CA, Mitochondrial complex I activity is required for maximal autophagy, Cell Rep. 24 (2018) 2404–2417, 10.1016/j.celrep.2018.07.101 e2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Elliott IA, Dann AM, Xu S, Kim SS, Abt ER, Kim W, Poddar S, Moore A, Zhou L, Williams JL, Capri JR, Ghukasyan R, Matsumura C, Tucker DA, Armstrong WR, Cabebe AE, Wu N, Li L, Le TM, Radu CG, Donahue TR, Lysosome inhibition sensitizes pancreatic cancer to replication stress by aspartate depletion, Proc. Natl. Acad. Sci. USA 116 (2019) 6842–6847, 10.1073/pnas.1812410116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Murphy MP, How mitochondria produce reactive oxygen species, Biochem. J 417 (2009) 1–13, 10.1042/bj20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Ashton TM, McKenna WG, Kunz-Schughart LA, Higgins GS, Oxidative phosphorylation as an emerging target in cancer therapy, Clin. Cancer Res 24 (2018) 2482–2490, 10.1158/1078-0432.Ccr-17-3070. [DOI] [PubMed] [Google Scholar]
- [67].Shi Y, Lim SK, Liang Q, Iyer SV, Wang HY, Wang Z, Xie X, Sun D, Chen YJ, Tabar V, Gutin P, Williams N, De Brabander JK, Parada LF, Gboxin is an oxidative phosphorylation inhibitor that targets glioblastoma, Nature 567 (2019) 341–346, 10.1038/s41586-019-0993-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Diepart C, Karroum O, Magat J, Feron O, Verrax J, Calderon PB, Gregoire V, Leveque P, Stockis J, Dauguet N, Jordan BF, Gallez B, Arsenic trioxide treatment decreases the oxygen consumption rate of tumor cells and radiosensitizes solid tumors, Cancer Res. 72 (2012) 482–490, 10.1158/0008-5472.Can-11-1755. [DOI] [PubMed] [Google Scholar]
- [69].Fiorillo M, Lamb R, Tanowitz HB, Mutti L, Krstic-Demonacos M, Cappello AR, Martinez-Outschoorn UE, Sotgia F, Lisanti MP, Repurposing atovaquone: targeting mitochondrial complex III and OXPHOS to eradicate cancer stem cells, Oncotarget 7 (2016) 34084–34099, 10.18632/oncotarget.9122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Biel TG, Rao VA, Mitochondrial dysfunction activates lysosomal-dependent mitophagy selectively in cancer cells, Oncotarget 9 (2018) 995–1011, 10.18632/oncotarget.23171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Saito Rde F, Tortelli TC Jr., Jacomassi MD, Otake AH, Chammas R, Emerging targets for combination therapy in melanomas, FEBS Lett. 589 (2015) 3438–3448, 10.1016/j.febslet.2015.09.022. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.