

Abstract
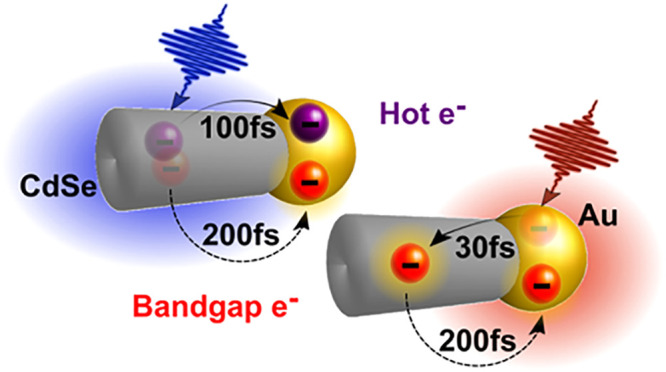
Recently, it was demonstrated that charge separation in hybrid metal–semiconductor nanoparticles (HNPs) can be obtained following photoexcitation of either the semiconductor or of the localized surface plasmon resonance (LSPR) of the metal. This suggests the intriguing possibility of photocatalytic systems benefiting from both plasmon and exciton excitation, the main challenge being to outcompete other ultrafast relaxation processes. Here we study CdSe-Au HNPs using ultrafast spectroscopy with high temporal resolution. We describe the complete pathways of electron transfer for both semiconductor and LSPR excitation. In the former, we distinguish hot and band gap electron transfer processes in the first few hundred fs. Excitation of the LSPR reveals an ultrafast (<30 fs) electron transfer to CdSe, followed by back-transfer from the semiconductor to the metal within 210 fs. This study establishes the requirements for utilization of the combined excitonic–plasmonic contribution in HNPs for diverse photocatalytic applications.
Keywords: hot-electron transfer, localized surface plasmon resonances, photocatalysis, semiconductor nanoparticles, ultrafast optical spectroscopy
Plasmonic photochemistry is an emergent paradigm for various photocatalytic applications including sensing, photodetection, and solar energy conversion.1−3 The ability to generate hot electrons/holes at a metal nanoparticle’s (NP) surface in the form of a localized surface plasmon resonance (LSPR) under a wide range of visible and infrared excitations simply by controlling the NP type, size, and shape is highly appealing in the context of sustainable energy harvesting.4 Conjugating these metal sensitizers to an acceptor such as a semiconductor component or adsorbed molecules permits hot charge carrier transfer across the interface of the heterostructured system.5
Among the mechanisms proposed for this process, two stand out: the conventional indirect plasmon-induced hot-electron transfer (PHET)6−9 and the recently discovered direct plasmon-induced charge transfer (PICT).10−12 In PHET, hot carriers are initially generated in the metal by plasmon decay and then undergo interfacial transfer to the acceptor component. PICT occurs when there is strong interdomain coupling and mixing of the metal and acceptor energy levels. In PICT, plasmon excitation is directly accompanied by a rapid charge separation process that creates an electron in the acceptor region and a hole in the metal.10 In both cases, in comparison to fast thermalization and cooling on unconjugated metallic NPs, a prolonged lifetime of hot electrons following charge separation is essential for initiating chemical reactions at their surfaces and promoting photocatalytic functionality. However, PICT is considered more efficient than PHET since competing processes in the metal domain, such as electron–electron and electron–phonon scattering, are eliminated by the predicted nearly instantaneous charge transfer to the acceptor domain.
Semiconductor–metal HNPs are an interesting potential platform for PICT, having long been considered promising photocatalysts for numerous applications including solar-to-fuel conversion,13,14 photopolymerization,15 and biomedical utilization.16 These applications derive from the unique combined and synergistic photophysical and chemical properties that HNPs offer.17 Among them, first and foremost, is light-induced charge separation, which allows the promotion of redox reactions on their surface following light absorption.13 So far, the majority of the investigations and the applications of such systems in photocatalysis were focused on the photoinduced charge transfer from the semiconductor to the metal domain. Careful studies allowed to decipher the photocatalytic mechanism, while the well-defined morphology and size-control, akin to such HNPs, enabled reaching in-depth understanding and establishment of significant factors for the design of optimal HNPs based photocatalysts.18−23
Combining the effects of plasmonic photocatalysis and excitonic sensitization through the semiconductor bears interesting potential. In particular, the possibility of also exploiting the LSPR excitation of the metal domain to enhance the photocatalytic efficiency, through PICT from the metal domain to the semiconductor, is highly attractive.24 The LSPR of the metal can be used either to increase the local electromagnetic field or to generate charge-separated states. Recently, the latter approach was elegantly demonstrated on CdSe-Au HNPs by Lian and co-workers. A direct PICT mechanism was reported for this HNP model system that manifests strong hybridization of the excitonic and plasmonic states.10 The PICT form of charge separation for this system was argued to happen within the Landau damping time of the plasmon (<20 fs). Experimental evidence of PICT was also found in other systems such as TiO2–Ag25 and TiO2–Au.26 This direct mechanism may improve plasmonic based photocatalytic activity and efficiency of HNPs by extending light harvesting to the red and near-infrared (NIR) region. However, coexisting relaxation pathways and, in particular, back-transfer from the semiconductor to the metal can compete with efficient harvesting of these excited carriers.
In PICT systems, direct experimental observation of the relevant time scales and competing mechanisms is challenging due to their ultrafast nature, as predicted from theoretical analysis.25 Therefore, directly measuring the dynamics of the charge carriers following plasmonic excitation in such hybrid nanosystems is essential for understanding the different mechanisms involved in PICT and designing nanosystems that effectively exploit it for light harvesting. Here we study a model exciton–plasmon photocatalyst system of CdSe-Au hybrid nanorods (NRs) using a combination of ultrafast two-dimensional electronic spectroscopy (2DES) and transient absorption (TA), utilizing pulses with a duration of ≈10 fs in the visible and ≈30 fs in the NIR. The high time resolution enables us to trace and define the rich charge carrier relaxation routes, as summarized in Figure 1b,c, which follows photoexcitation of both the excitonic and the plasmonic components. 2DES allows us to highlight significant differences in carrier relaxation between pristine NRs and the HNPs because of the rapid charge separation processes in the latter. The TA data reveals in detail the dynamics of the charge transfer processes following both CdSe and Au LSPR photoexcitation, which is described employing global analysis. The photophysical processes identified in this study and the corresponding time scales pose the ultimate constraints for utilization of the combined excitonic–plasmonic contribution in such HNPs for diverse photocatalytic applications.
Figure 1.

(a) Linear absorption spectra of bare CdSe NRs and CdSe-Au HNPs in water (black and green lines, respectively) normalized at 2.38 eV. The shaded orange and blue areas represent the spectra of the broadband pulses used in the ultrafast experiments reported here. Right: TEM image of the CdSe-Au NRs. (b) Summary of the ultrafast photophysics of pristine CdSe NRs following broadband photoexcitation. The right-hand side shows the calculated density of electron states in the conduction band, which approximately form three groups. We observe that electrons relax from the high energy group to the intermediate in 110 fs and from the intermediate group to the lowest excited states in 220 fs. (c) Summary of the photophysics of CdSe-Au HNPs following broadband visible and (d) broadband NIR photoexcitation. In the first case, an ultrafast hot-electron transfer to gold on a 115 fs time scale competes with intraband relaxation in the CdSe domain and is followed by a slower (210 fs) transfer of band gap electrons to the gold. For NIR excitation, PICT generates electrons in the CdSe domain with a ≤ 30 fs time constant. This is followed by a back-transfer to the gold in 210 fs.
CdSe NRs were synthesized using a modification of the surfactant controlled growth approach (see Supporting Information for details).27,28 NRs of 8.4 nm diameter and 25.1 nm length were produced by multiple injections of precursors, resulting in a 1.85 eV energy band gap (Figure 1a, black line). In the next step, site-selective Au deposition on the NR apexes was obtained following a recently reported procedure,19 resulting in an average metal domain size of 4.1 ± 0.8 nm, as can be seen from the transmission electron microscopy (TEM) image in Figure 1a. The HNPs and the NRs were then transferred to an aqueous solution by ligand exchange with polyethylenimine, which provided good colloidal stability and was reported as a high-performance surface coating for photocatalytic applications of the HNPs.29 Additional details of the synthesis are provided in the Supporting Information.
Aqueous solutions of CdSe-Au HNPs show a distinct change in their optical properties when compared to CdSe NRs (see Figure 1a). The lowest energy absorption band of the latter is around 1.85 eV, followed by two bands at approximately 2 and 2.3 eV, which roughly corresponds to the split-off valence band.30 These three predominant optical transitions resonant with our visible excitation pulses are consistent with the calculated density of electronic states shown in Figure 1b (Figures S2 and S3 and additional details in the Supporting Information). The hybrid CdSe-Au system still shows distinguishable features of the bare CdSe NRs but is dominated by a broad tail extending into the NIR well below 1 eV. The LSPR of free-standing Au NPs typically peaks around 2.3 eV, so the observed HNP spectrum (green line in Figure 1a) is not a simple combination of its individual constituents. The observed spectral response of the HNPs was previously assigned to a broadening of the gold domain LSPR due to the hybridization with the CdSe energy levels, in contrast with the results observed in other systems, such as CdS-Au HNPs, for which the LSPR and the semiconductor states are spectrally separated and distinct.31
For a complete understanding of the carrier dynamics in the complex HNPs, we compare their response to that of the bare NRs. The ultrafast photophysics of the bare CdSe NRs shows a strong dependence on the excitation photon energy.32−36 Hence, the combination of high temporal resolution and high excitation energy resolution of 2DES is required to fully unravel it.37 2DES is an extension of broadband TA in which a second pump pulse is added, and its time delay with respect to the first pump is controlled with interferometric precision, allowing one to obtain excitation energy resolution through Fourier transform spectroscopy.38,39 The result is a correlation map between the pump and probe photon energies for each pump–probe delay, such that the vertical cut for each point in the pump energy axis corresponds to the narrowband TA spectrum at that delay.
The 2DES differential transmission (ΔT/T) maps for pristine CdSe NRs at representative time delays are shown in Figure 2a–c, with the color scale normalized to the maximum signal among the three maps. All experiments were performed at low fluences to ensure being in a single exciton regime. At 20 fs (Figure 2a), we see that, independently of the pump energy, photobleaching (PB) bands are present at probe energies of 1.85 and 2.27 eV, corresponding to horizontal signatures. As usual, the 2DES signal amplitude is not corrected by the excitation spectrum along the pump energy axis, as that is not readily achievable.40,41 As a result, the exciton peak in the diagonal at 1.85 eV is partially suppressed in comparison with previous reports.32,42,43Figure S5 shows the same data of Figure 2a using a logarithmic color scale, highlighting the weaker positive diagonal signals more clearly. Note that the energy spacing between the lowest energy excitonic transition and the transition at 2.27 eV is 420 meV, which corresponds to the energy difference between the light/heavy hole and spin-orbit split valence band in bulk CdSe.30 Therefore, the transition at 2.27 eV can be approximately assigned to electron–hole transitions of the split-off valence band while sharing the same conduction band energy level as for the 1.85 eV excitonic transition (Figure S4). This coupling is manifested by the observed cross-peaks in Figure 2a.
Figure 2.

2DES maps showing the normalized ΔT/T of CdSe NRs (a–c) at 20, 100, and 2000 fs following visible broadband excitation (pump fluence of 11.6 μJ/cm2) and (d–f) CdSe-Au HNPs at 20, 140, and 460 fs under the same conditions (pump fluence of 20.7 μJ/cm2). While the maps on top show a rising signal at Eprobe = 1.85 eV, the HNPs show a steadily decaying signal. The color map scale is normalized at the maximum signal among the three maps. The cartoons are merely representative; see Figure 1 for TEM data.
For pump energies above 2.0 eV, the band edge PB starts to be accompanied by a negative photoinduced absorption (PIA) signal at probe energies below 1.8 eV, typical of band gap renormalization due to the presence of hot carriers and sometimes attributed to biexciton absorption in CdSe.44−47 We also note that, at pump energies above 2 eV, PB signals extend from the band gap up to the diagonal region, showing the presence of carriers that have not yet relaxed from the initial excitation. At 100 fs (Figure 2b), we already observe a significant decrease of PB signals at high pump and probe energies (partially obscured near 2.25 eV due to the overlapping cross-peak from band gap electrons, see Figure S6), accompanied by an increase in the band edge PB and a decrease of the subgap PIA at the corresponding pump energies, reflecting the transient signatures of carrier cooling. Still, a PB band around the probe energy of 2 eV persists. At 2000 fs (Figure 2c), all signatures of hot carriers have disappeared, and for all pump energies, PB signals are observed at the band gap and at 2.27 eV, corresponding to the split-off band. At later times, no further evolution is observed apart from the well-known signal decay on the ns time scale.
The 2DES data summarized in Figure 2a–c include many more time delays between −100 and 2000 fs, and the kinetics of all coordinates in the 2DES maps were analyzed employing a global fitting algorithm.48 Two subpicosecond exponential components, along with a long-living component, sufficed to fit the full 2DES data set (Figure S6). The two decay components extracted from the global analysis are assigned to a two-step relaxation of hot carriers to the band edge (pathways i and ii in Figure 1b). At early times, the ≈110 fs component shows matching kinetics between the decay of the PB of the higher energy transitions (2.27 eV) and the buildup of the PB of the intermediate energy transitions (2 eV) (Figure S6a). This match strongly suggests a relaxation pathway of hot electrons from the high energy to the intermediate energy transitions. Subsequently, a buildup of the PB signal is seen both at the band edge energy and the higher energy transitions, alongside the decay of the intermediate energy transitions on a ≈220 fs time scale (Figure S6b). This indicates a relaxation pathway from the intermediate energies to the band edge, which is coupled with the higher energy transition from the split-off valence band. The observed two-step relaxation process and the associated time scales are in agreement with previous works on spherical CdSe NPs.32,33
The 2DES maps of CdSe-Au HNPs under similar photoexcitation conditions are shown in Figure 2d–f, with the color scale normalized to the maximum signal on that data set. Significant differences are observed for the HNPs compared to the bare NRs. The signal level at time zero is substantially lower as is expected because hybrid samples of similar optical density include a large absorption component of the broadened LSPR of gold in contact with CdSe, which gives a very weak transient response. This is consistent with previous studies.10 The 2DES map at 20 fs in Figure 2d is very similar to that in Figure 2a, further confirming that the transient response at early times reflects mainly direct CdSe excitation. However, the signal evolution follows a drastically different path, with the entire 2DES line shape quickly decaying to zero, and no noticeable relaxation to the band edge (Figure 2e,f). This is a remarkable result, in strong contrast with the CdSe NRs, for which the kinetics strongly depended on the excitation energy, suggesting that charge transfer processes thoroughly outcompete internal relaxation in the CdSe domain. As all signals decay independently of the excitation photon energy, 2DES is no longer an absolute requirement, allowing us to use broadband TA to further scrutinize the dynamics.
Broadband TA experiments, performed with the same ultrashort pulses used in 2DES, are compared for bare NRs in Figure 3a–c and for HNPs in Figure 3d–f. The TA maps as a function of probe photon energy and pump–probe delay for bare NRs (Figure 3a) and HNPs (Figure 3d) are consistent with the evolution observed in the 2DES data both kinetically (Figure 3b,e) and spectrally (Figure 3c,f). The data was analyzed employing a global fit algorithm using Glotaran.49 For bare CdSe NRs, the TA fit recovered similar components as the 2DES fit, namely, 110 and 220 fs (see Figure 3b, Figures S6 and S10a). For the HNPs, the transient signatures from gold are expected to be very small due to the strong broadening of the LSPR of the tips. Indeed, the spectra in Figures 3c at 14 fs (blue) and Figure 3f at 20 fs are almost coincident (see also Figure S8), demonstrating the dominance of CdSe signatures in TA spectra.
Figure 3.

(a) TA map of CdSe nanorods in aqueous solution using the visible pulses from Figure 1a as a pump and probe (pump fluence of 11.6 μJ/cm2). (b) TA kinetics at selected photon energies (dots) and fits to the data (lines). (c) TA spectra at selected time delays. (d–f) Same as panels a–c for CdSe-Au under the same excitation conditions (pump fluence of 20.7 μJ/cm2). (g–i) Same as panels a–c for CdSe-Au using the near-infrared pulse in Figure 1 centered at 1.3 eV as a pump (fluence of 135 μJ/cm2) and the visible one as a probe. The cartoons are merely representative; see Figure 1 for TEM data.
Three exponential components were required to fit the kinetics in the HNPs, at 114, 212, and 1210 fs (Figure S10b). It is important to note that, although both NRs and HNPs include a fast component of approximately 110 fs, the spectral evolution is drastically different. For the NRs, Figure 3c shows a decreasing PB between 1.95 and 2.2 eV accompanied by a rising PB at 1.86 eV, whereas Figure 3f shows that, for the hybrids, the PB decreases at all probe energies. Therefore, although the relaxation of hot carriers within the CdSe manifold is possible (pathways i+ii in Figure 1c), this observation confirms that the dominant decay pathway in HNPs is hot-electron transfer to the gold domain (pathway 2 in Figure 1c), as already inferred from 2DES. The origin of the transferred electrons at this time scale is predominantly from high energy excited states, as can be deduced from Figure 3f. At 20 fs, the sharp PIA at 1.78 eV marks the presence of hot electrons, and at 80 fs, this feature is mostly gone, demonstrating the association of hot electrons transfer with the 114 fs time scale. The 212 fs time scale corresponds then to the transfer of band gap electrons to the gold domain (pathway 3 in Figure 1c).
These data experimentally resolve the ultrafast electron transfer from CdSe to gold, which has been previously inferred from the gold-like picosecond relaxation observed in TA performed with ∼150 fs pulses.9,44,50 In particular, Okuhata et al. very recently proposed the distinction between band gap and hot-electron transfer kinetics,51 which we directly observe here. We also note that the electron transfer process depends on the density of states available in the gold domain and can be controlled by changing the size of the gold tips.19,51,52 Lastly, the 1210 fs component corresponds to a full decay of the remaining signal, with only a weak PIA feature around 1.79 eV remaining afterward. This is the typical time scale of electron–phonon cooling in the gold domain,53 and the weak remaining PIA is assigned to a red-shift of the CdSe band gap following heating of the lattice.
To directly resolve the PICT pathway, we perform TA with NIR excitation (using the orange spectrum in Figure 1a as a pump and the blue one as a probe), thus photoexciting selectively only the broadened gold LSPR region. The pump pulses are centered at ≈1.3 eV and compressed to ≈30 fs at the sample position. The TA maps around time zero include substantial cross-phase modulation caused by the cuvette windows and by the sample. Nonetheless, the TA spectrum at 70 fs (dashed blue line in Figure 3i) shows that the typical cross-phase modulation oscillations are centered around an overall positive baseline, which is quite similar to the TA spectrum at 150 fs (purple line in Figure 3i), after the cross-phase modulation artifact has subsided, which includes a pronounced peak at 1.86 eV (Figures S6 and S7d). We can thus conclude that electron injection in the CdSe conduction band by the PICT mechanism is already completed at 70 fs.
Global analysis of the data including a model to fit the cross-phase modulation convoluted with the instrumental response function yielded a 30 fs formation time from the excitation to the first discernible spectrum (pathway 1 in Figure 1d). This can be considered as an upper limit for the PICT charge transfer time scale, in agreement with the original hypothesis by Wu et al.10The remaining components found by the global analysis of the data in Figure 3g–i were 212 fs and 1.8 ps (Figure S8c). The 212 fs component matches the time constant of the band gap electron transfer from CdSe to gold following visible excitation (pathway 3 in Figure 1c). Moreover, the decay shown in Figure 3h,i consists mainly of a recovery of the CdSe band edge PB signal at 1.86 eV. Hence, we attribute it to a back-transfer of electrons generated by the PICT mechanism in CdSe to gold (pathway 3 in Figure 1d). Considering the high density of states in the metal, there will be an entropic drive toward the back-transfer, which explains this equilibration. The TA spectra then decay in 1.8 ps, a time scale consistent with electron–phonon cooling, and only a weak PIA at 1.79 eV remains, once again reflecting the heating of the CdSe lattice following the rapid dissipation of the 1.3 eV pump energy. Further, we note that, from the TA spectra at 500 and 2500 fs in Figure 3i, we can assign the broad PB band from 1.95 to 2.25 eV to the gold tips, as at these times, no hot electrons should be present in CdSe to act as a confounding signal in this range. We also conclude that the PICT mechanism mostly generates band gap electrons in CdSe, since no hot electron back-transfer to the gold at 110 fs is observed. Furthermore, the spectral shape of Figure 3i at early times differs substantially from those of Figure 3c,f, which in turn match each other (see also Figure S8), demonstrating a different distribution of carriers.
On the one hand, our results provide the time scale for the PICT process and reveal subsequent electron back-transfer processes; on the other hand, they set some conditions and constraints for possible exploitation of PICT generated charge carriers for photocatalytic applications in such energy-hybridized systems. Extraction of the electrons generated in the semiconductor via PICT needs to be extremely rapid to compete with the 210 fs back-transfer to the metal. This could perhaps be achievable by a suitable electron-accepting ligand on the surface or via a secondary metal domain with suitable band alignment to receive such electrons. In photodetector applications, similarly, the PICT electrons generated through NIR excitation need to be harvested rapidly prior to the back-transfer. Therefore, the design and realization of an efficient hybrid system that harvests the NIR plasmonic excitation must consider the complete photoinduced mechanism in order to avoid losses to undesired pathways.
In summary, we have experimentally unraveled the ultrafast photophysics of CdSe NRs and CdSe-Au HNPs. Using 2DES, we were able to describe in detail the two-step relaxation of hot carriers in CdSe NRs and demonstrate that CdSe-Au HNPs show entirely different dynamics, which we have unraveled using high time resolution TA. We show that, upon NIR excitation of the gold LSPR, electrons are transferred to the CdSe domain within 30 fs, confirming the PICT pathway. Owing to the high time resolution, we were able to observe a fast back-transfer of electrons to the metal domain on a ≈200 fs time scale, unresolved by previous experiments. Further, we also show that following visible excitation of CdSe-Au HNPs, hot electrons and band gap electrons are transferred to the gold on different time scales, with the band gap electrons matching the back-transfer time seen after PICT. Our data provides a complete picture of the ultrafast photophysics of CdSe-Au hybrid nanorods and sets the kinetic ground rules for designing combined exciton–plasmon-induced photocatalysts and photoactive systems.
Acknowledgments
The authors thank Prof. Elisabetta Collini and Dr. Andrea Volpato for sharing the 2DES global analysis software. The research was partially supported by the Israel Science Foundation (1363/18, U.B.). U.B. Thanks the Alfred & Erica Larisch Memorial Chair. G.C. acknowledges the support from the PRIN 2017 Project 201795SBA3–HARVEST. U.B. and G.C. acknowledge support from Horizon 2020 (654148, Laserlab-Europe).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.nanolett.0c04614.
Sample preparation and characterization, calculations, description of the ultrafast experiments, and supplementary plots (PDF)
Author Contributions
Y.B.S., U.B., and G.C. conceived the research. F.V.A.C., T.N., and G.C. designed the ultrafast experiments, which were performed by F.V.A.C. and T.N. F.V.A.C., Y.B.S., T.N., and M.R. performed the data analysis. Y.B.S. prepared and characterized the samples. Y.E.P. performed the calculations. The paper was written by F.V.A.C. and Y.B.S. with input from all authors. U.B. and G.C. supervised and coordinated the project. F.V.A.C. and Y.B.S. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Zhang Y.; He S.; Guo W.; Hu Y.; Huang J.; Mulcahy J. R.; Wei W. D. Surface-Plasmon-Driven Hot Electron Photochemistry. Chem. Rev. 2018, 118 (6), 2927–2954. 10.1021/acs.chemrev.7b00430. [DOI] [PubMed] [Google Scholar]
- Tang H.; Chen C. J.; Huang Z.; Bright J.; Meng G.; Liu R. S.; Wu N. Plasmonic Hot Electrons for Sensing, Photodetection, and Solar Energy Applications: A Perspective. J. Chem. Phys. 2020, 152 (22), 220901. 10.1063/5.0005334. [DOI] [PubMed] [Google Scholar]
- Wolff C. M.; Frischmann P. D.; Schulze M.; Bohn B. J.; Wein R.; Livadas P.; Carlson M. T.; Jäckel F.; Feldmann J.; Würthner F.; Stolarczyk J. K. All-in-One Visible-Light-Driven Water Splitting by Combining Nanoparticulate and Molecular Co-Catalysts on CdS Nanorods. Nat. Energy 2018, 3 (10), 862–869. 10.1038/s41560-018-0229-6. [DOI] [Google Scholar]
- Brongersma M. L.; Halas N. J.; Nordlander P. Plasmon-Induced Hot Carrier Science and Technology. Nat. Nanotechnol. 2015, 10 (1), 25–34. 10.1038/nnano.2014.311. [DOI] [PubMed] [Google Scholar]
- Cortés E.; Xie W.; Cambiasso J.; Jermyn A. S.; Sundararaman R.; Narang P.; Schlücker S.; Maier S. A. Plasmonic Hot Electron Transport Drives Nano-Localized Chemistry. Nat. Commun. 2017, 8, 1–10. 10.1038/ncomms14880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govorov A. O.; Zhang H.; Gun’Ko Y. K. Theory of Photoinjection of Hot Plasmonic Carriers from Metal Nanostructures into Semiconductors and Surface Molecules. J. Phys. Chem. C 2013, 117 (32), 16616–16631. 10.1021/jp405430m. [DOI] [Google Scholar]
- Tian Y.; Tatsuma T. Mechanisms and Applications of Plasmon-Induced Charge Separation at TiO2 Films Loaded with Gold Nanoparticles. J. Am. Chem. Soc. 2005, 127 (20), 7632–7637. 10.1021/ja042192u. [DOI] [PubMed] [Google Scholar]
- Zhou L.; Zhang C.; McClain M. J.; Manjavacas A.; Krauter C. M.; Tian S.; Berg F.; Everitt H. O.; Carter E. A.; Nordlander P.; Halas N. J. Aluminum Nanocrystals as a Plasmonic Photocatalyst for Hydrogen Dissociation. Nano Lett. 2016, 16 (2), 1478–1484. 10.1021/acs.nanolett.5b05149. [DOI] [PubMed] [Google Scholar]
- Wu K.; Rodríguez-Córdoba W. E.; Yang Y.; Lian T. Plasmon-Induced Hot Electron Transfer from the Au Tip to CdS Rod in CdS-Au Nanoheterostructures. Nano Lett. 2013, 13 (11), 5255–5263. 10.1021/nl402730m. [DOI] [PubMed] [Google Scholar]
- Wu K.; Chen J.; Mcbride J. R.; Lian T. Efficient Hot-Electron Transfer by a Plasmon-Induced Interfacial Charge-Transfer Transition. Science 2015, 349 (6248), 632–635. 10.1126/science.aac5443. [DOI] [PubMed] [Google Scholar]
- Boerigter C.; Aslam U.; Linic S. Mechanism of Charge Transfer from Plasmonic Nanostructures to Chemically Attached Materials. ACS Nano 2016, 10 (6), 6108–6115. 10.1021/acsnano.6b01846. [DOI] [PubMed] [Google Scholar]
- Boerigter C.; Campana R.; Morabito M.; Linic S. Evidence and Implications of Direct Charge Excitation as the Dominant Mechanism in Plasmon-Mediated Photocatalysis. Nat. Commun. 2016, 7, 10545. 10.1038/ncomms10545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Shahar Y.; Banin U. Hybrid Semiconductor–Metal Nanorods as Photocatalysts. Top. Curr. Chem. 2016, 374 (4), 1–26. 10.1007/s41061-016-0052-0. [DOI] [PubMed] [Google Scholar]
- Wu K.; Lian T. Quantum Confined Colloidal Nanorod Heterostructures for Solar-to-Fuel Conversion. Chem. Soc. Rev. 2016, 45 (14), 3781–3810. 10.1039/C5CS00472A. [DOI] [PubMed] [Google Scholar]
- Pawar A. A.; Halivni S.; Waiskopf N.; Ben-Shahar Y.; Soreni-Harari M.; Bergbreiter S.; Banin U.; Magdassi S. Rapid Three-Dimensional Printing in Water Using Semiconductor-Metal Hybrid Nanoparticles as Photoinitiators. Nano Lett. 2017, 17 (7), 4497–4501. 10.1021/acs.nanolett.7b01870. [DOI] [PubMed] [Google Scholar]
- Waiskopf N.; Ben-Shahar Y.; Galchenko M.; Carmel I.; Moshitzky G.; Soreq H.; Banin U. Photocatalytic Reactive Oxygen Species Formation by Semiconductor-Metal Hybrid Nanoparticles. Toward Light-Induced Modulation of Biological Processes. Nano Lett. 2016, 16 (7), 4266–4273. 10.1021/acs.nanolett.6b01298. [DOI] [PubMed] [Google Scholar]
- Banin U.; Ben-Shahar Y.; Vinokurov K. Hybrid Semiconductor-Metal Nanoparticles: From Architecture to Function. Chem. Mater. 2014, 26 (1), 97–110. 10.1021/cm402131n. [DOI] [Google Scholar]
- Amirav L.; Alivisatos A. P. Photocatalytic Hydrogen Production with Tunable Nanorod Heterostructures. J. Phys. Chem. Lett. 2010, 1 (7), 1051–1054. 10.1021/jz100075c. [DOI] [Google Scholar]
- Ben-shahar Y.; Scotognella F.; Kriegel I.; Moretti L.; Cerullo G.; Rabani E.; Banin U. With Hybrid Semiconductor-Metal Nanorods. Nat. Commun. 2016, 7, 1–7. 10.1038/ncomms10413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu K.; Chen Z.; Lv H.; Zhu H.; Hill C. L.; Lian T. Hole Removal Rate Limits Photodriven H2 Generation Efficiency in CdS-Pt and CdSe/CdS-Pt Semiconductor Nanorod-Metal Tip Heterostructures. J. Am. Chem. Soc. 2014, 136 (21), 7708–7716. 10.1021/ja5023893. [DOI] [PubMed] [Google Scholar]
- Kalisman P.; Houben L.; Aronovitch E.; Kauffmann Y.; Bar-Sadan M.; Amirav L. The Golden Gate to Photocatalytic Hydrogen Production. J. Mater. Chem. A 2015, 3 (39), 19679–19682. 10.1039/C5TA05784A. [DOI] [Google Scholar]
- Simon T.; Bouchonville N.; Berr M. J.; Vaneski A.; Adrovic A.; Volbers D.; Wyrwich R.; Döblinger M.; Susha A. S.; Rogach A. L.; Jäckel F.; Stolarczyk J. K.; Feldmann J. Redox Shuttle Mechanism Enhances Photocatalytic H2 Generation on Ni-Decorated CdS Nanorods. Nat. Mater. 2014, 13 (11), 1013–1018. 10.1038/nmat4049. [DOI] [PubMed] [Google Scholar]
- Simon T.; Carlson M. T.; Stolarczyk J. K.; Feldmann J. Electron Transfer Rate vs Recombination Losses in Photocatalytic H2 Generation on Pt-Decorated CdS Nanorods. ACS Energy Lett. 2016, 1 (6), 1137–1142. 10.1021/acsenergylett.6b00468. [DOI] [Google Scholar]
- Ratchford D. C. Plasmon-Induced Charge Transfer: Challenges and Outlook. ACS Nano 2019, 13 (12), 13610–13614. 10.1021/acsnano.9b08829. [DOI] [PubMed] [Google Scholar]
- Ma J.; Gao S. Plasmon-Induced Electron-Hole Separation at the Ag/TiO2(110) Interface. ACS Nano 2019, 13 (12), 13658–13667. 10.1021/acsnano.9b03555. [DOI] [PubMed] [Google Scholar]
- Moon S. Y.; Song H. C.; Gwag E. H.; Nedrygailov I. I.; Lee C.; Kim J. J.; Doh W. H.; Park J. Y. Plasmonic Hot Carrier-Driven Oxygen Evolution Reaction on Au Nanoparticles/TiO2 Nanotube Arrays. Nanoscale 2018, 10 (47), 22180–22188. 10.1039/C8NR05144E. [DOI] [PubMed] [Google Scholar]
- Peng Z. A.; Peng X. Nearly Monodisperse and Shape-Controlled CdSe Nanocrystals via Alternative Routes: Nucleation and Growth. J. Am. Chem. Soc. 2002, 124 (13), 3343–3353. 10.1021/ja0173167. [DOI] [PubMed] [Google Scholar]
- Hung A. M.; Konopliv N. A.; Cha J. N. Solvent-Based Assembly of CdSe Nanorods in Solution. Langmuir 2011, 27 (20), 12322–12328. 10.1021/la2019956. [DOI] [PubMed] [Google Scholar]
- Ben-Shahar Y.; Scotognella F.; Waiskopf N.; Kriegel I.; Dal Conte S.; Cerullo G.; Banin U. Effect of Surface Coating on the Photocatalytic Function of Hybrid CdS-Au Nanorods. Small 2015, 11 (4), 462–471. 10.1002/smll.201402262. [DOI] [PubMed] [Google Scholar]
- Shan W.; Song J. J.; Luo H.; Furdyna J. K. Determination of the Fundamental and Split-off Band Gaps in Zinc-Blende CdSe by Photomodulation Spectroscopy. Phys. Rev. B: Condens. Matter Mater. Phys. 1994, 50 (11), 8012–8015. 10.1103/PhysRevB.50.8012. [DOI] [PubMed] [Google Scholar]
- Shaviv E.; Schubert O.; Alves-Santos M.; Goldoni G.; Di Felice R.; Vallée F.; Del Fatti N.; Banin U.; Sönnichsen C. Absorption Properties of Metal-Semiconductor Hybrid Nanoparticles. ACS Nano 2011, 5 (6), 4712–4719. 10.1021/nn200645h. [DOI] [PubMed] [Google Scholar]
- Caram J. R.; Zheng H.; Dahlberg P. D.; Rolczynski B. S.; Griffin G. B.; Dolzhnikov D. S.; Talapin D. V.; Engel G. S. Exploring Size and State Dynamics in CdSe Quantum Dots Using Two-Dimensional Electronic Spectroscopy. J. Chem. Phys. 2014, 140 (2014), 084701. 10.1063/1.4865832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Righetto M.; Bolzonello L.; Volpato A.; Amoruso G.; Panniello A.; Fanizza E.; Striccoli M.; Collini E. Deciphering Hot- and Multi-Exciton Dynamics in Core-Shell QDs by 2D Electronic Spectroscopies. Phys. Chem. Chem. Phys. 2018, 20 (27), 18176–18183. 10.1039/C8CP02574F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrett J. W.; Yi C.; Stoll T.; Rehault J.; Oriana A.; Branchi F.; Cerullo G.; Knappenberger K. L. Dissecting Charge Relaxation Pathways in CdSe/CdS Nanocrystals Using Femtosecond Two-Dimensional Electronic Spectroscopy. Nanoscale 2017, 9, 4572–4577. 10.1039/C7NR00654C. [DOI] [PubMed] [Google Scholar]
- Stoll T.; Branchi F.; Réhault J.; Scotognella F.; Tassone F.; Kriegel I.; Cerullo G. Two-Dimensional Electronic Spectroscopy Unravels Sub-100 Fs Electron and Hole Relaxation Dynamics in Cd-Chalcogenide Nanostructures. J. Phys. Chem. Lett. 2017, 8 (10), 2285–2290. 10.1021/acs.jpclett.7b00682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenngren N.; Abdellah M.; Zheng K.; Al-Marri M. J.; Zigmantas D.; Zidek K.; Pullerits T. Hot Electron and Hole Dynamics in Thiol-Capped CdSe Quantum Dots Revealed by 2D Electronic Spectroscopy. Phys. Chem. Chem. Phys. 2016, 18, 26199–26204. 10.1039/C6CP04190F. [DOI] [PubMed] [Google Scholar]
- Jonas D. M. Two-Dimensional Femtosecond Spectroscopy. Annu. Rev. Phys. Chem. 2003, 54 (1), 425–463. 10.1146/annurev.physchem.54.011002.103907. [DOI] [PubMed] [Google Scholar]
- Rehault J.; Maiuri M.; Oriana A.; Cerullo G. Two-Dimensional Electronic Spectroscopy with Birefringent Wedges. Rev. Sci. Instrum. 2014, 85, 123107. 10.1063/1.4902938. [DOI] [PubMed] [Google Scholar]
- Nuernberger P.; Ruetzel S.; Brixner T. Multidimensional Electronic Spectroscopy of Photochemical Reactions. Angew. Chem., Int. Ed. 2015, 54 (39), 11368–11386. 10.1002/anie.201502974. [DOI] [PubMed] [Google Scholar]
- Camargo F. V. A.; Grimmelsmann L.; Anderson H. L.; Meech S. R.; Heisler I. A. Resolving Vibrational from Electronic Coherences in Two-Dimensional Electronic Spectroscopy: The Role of the Laser Spectrum. Phys. Rev. Lett. 2017, 118, 1–6. 10.1103/PhysRevLett.118.033001. [DOI] [PubMed] [Google Scholar]
- Turner D. B. Standardized Specifications of 2D Optical Spectrometers. Results Chem. 2019, 1, 100001. 10.1016/j.rechem.2019.100001. [DOI] [Google Scholar]
- Griffin G. B.; Ithurria S.; Dolzhnikov D. S.; Linkin A.; Talapin D. V.; Engel G. S. Two-Dimensional Electronic Spectroscopy of CdSe Nanoparticles at Very Low Pulse Power. J. Chem. Phys. 2013, 138, 014705. 10.1063/1.4772465. [DOI] [PubMed] [Google Scholar]
- Seiler H.; Palato S.; Kambhampati P. Investigating Exciton Structure and Dynamics in Colloidal CdSe Quantum Dots with Two-Dimensional Electronic Spectroscopy. J. Chem. Phys. 2018, 149, 074702. 10.1063/1.5037223. [DOI] [PubMed] [Google Scholar]
- Ghosh T.; Aharon S.; Shpatz A.; Etgar L.; Ruhman S. Reflectivity Effects on Pump-Probe Spectra of Lead Halide Perovskites: Comparing Thin Films versus Nanocrystals. ACS Nano 2018, 12 (6), 5719–5725. 10.1021/acsnano.8b01570. [DOI] [PubMed] [Google Scholar]
- Wong C. Y.; Scholes G. D. Biexcitonic Fine Structure of CdSe Nanocrystals Probed by Polarization-Dependent Two-Dimensional Photon Echo Spectroscopy. J. Phys. Chem. A 2011, 115 (16), 3797–3806. 10.1021/jp1079197. [DOI] [PubMed] [Google Scholar]
- Seiler H.; Palato S.; Sonnichsen C.; Baker H.; Kambhampati P. Seeing Multiexcitons through Sample Inhomogeneity : Bandedge Biexciton Stucture in CdSe Nanocrystals Revealed by 2D Electronic Spectroscopy. Nano Lett. 2018, 18 (5), 2999–3006. 10.1021/acs.nanolett.8b00470. [DOI] [PubMed] [Google Scholar]
- Price M. B.; Butkus J.; Jellicoe T. C.; Sadhanala A.; Briane A.; Halpert J. E.; Broch K.; Hodgkiss J. M.; Friend R. H.; Deschler F. Hot-Carrier Cooling and Photoinduced Refractive Index Changes in Organic–Inorganic Lead Halide Perovskites. Nat. Commun. 2015, 6, 8420. 10.1038/ncomms9420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpato A.; Bolzonello L.; Meneghin E.; Collini E. Global Analysis of Coherence and Population Dynamics in 2D Electronic Spectroscopy. Opt. Express 2016, 24 (21), 24773–24785. 10.1364/OE.24.024773. [DOI] [PubMed] [Google Scholar]
- Snellenburg J. J.; Laptenok S.; Seger R.; Mullen K. M.; van Stokkum I. H. M. Glotaran: A Java-Based Graphical User Interface for the R Package TIMP. J. Stat. Softw. 2012, 49 (3), 1–22. 10.18637/jss.v049.i03. [DOI] [Google Scholar]
- Yu P.; Wen X.; Lee Y. C.; Lee W. C.; Kang C. C.; Tang J. Photoinduced Ultrafast Charge Separation in Plexcitonic CdSe/Au and CdSe/Pt Nanorods. J. Phys. Chem. Lett. 2013, 4 (21), 3596–3601. 10.1021/jz401985k. [DOI] [Google Scholar]
- Okuhata T.; Katayama T.; Tamai N. Ultrafast and Hot Electron Transfer in CdSe QD-Au Hybrid Nanostructures. J. Phys. Chem. C 2020, 124, 1099–1107. 10.1021/acs.jpcc.9b09042. [DOI] [Google Scholar]
- Liu Y.; Chen Q.; Cullen D. A.; Xie Z.; Lian T. Efficient Hot Electron Transfer from Small Au Nanoparticles. Nano Lett. 2020, 20 (6), 4322–4329. 10.1021/acs.nanolett.0c01050. [DOI] [PubMed] [Google Scholar]
- Link S.; El-Sayed M. A. Spectral Properties and Relaxation Dynamics of Surface Plasmon Electronic Oscillations in Gold and Silver Nanodots and Nanorods. J. Phys. Chem. B 1999, 103 (40), 8410–8426. 10.1021/jp9917648. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.