

Abstract
Covalent inhibitors of wild-type HIV-1 reverse transcriptase (CRTIs) are reported. Three compounds derived from catechol diether non-nucleoside inhibitors (NNRTIs) with addition of a fluorosulfate warhead are demonstrated to covalently modify Tyr181 of HIV-RT. X-ray crystal structures for complexes of the CRTIs with the enzyme are provided, which fully demonstrate the covalent attachment, and confirmation is provided by appropriate mass shifts in ESI-TOF mass spectra. The three CRTIs and six noncovalent analogues are found to be potent inhibitors with both IC50 values for in vitro inhibition of WT RT and EC50 values for cytopathic protection of HIV-1-infected human T-cells in the 5–320 nM range.
Keywords: anti-HIV agents, reverse transcriptase inhibitors, covalent inhibitors, fluorosulfates
The HIV/AIDS crisis continues as a major pandemic with worldwide statistics of 37 million people infected, 2 million new infections, and 1 million deaths in 2017, roughly equally split between men and women.1 Considering the rate of new infections and that 40% of the infected people are not receiving drug treatment, the grave problems will continue at high levels for many years. Nevertheless, highly active antiretroviral therapy (HAART) has had significant impact with the annual deaths reduced by half from the peak of 1.95 million in 2006.1 Drugs have been discovered in multiple classes, with nucleoside reverse transcriptase inhibitors (NRTIs), non-nucleoside RTIs (NNRTIs), protease inhibitors (PIs), and integrase inhibitors (INTIs) being the most widely used, typically in three-drug combinations.2 The problems with pill-burden have much improved with the introduction of daily single-tablet regimens (STRs), which have predominantly featured coformulation of two NRTIs and one NNRTI starting with Atripla in 2006, namely, tenofovir disoproxil fumarate (TDF), emtricitabine (FTC), and efavirenz (EVF).3 Recent introductions include Delstrigo, which combines TDF, lamivudine (3TC), and the NNRTI rilpivirine (RPV), the first two-drug maintenance therapy Juluca consisting of the INTI dolutegravir (DTG) and RPV, and Dovato (DTG plus 3TC) for initial therapy in treatment-naïve patients.4,5
In addition to discovery of improved drugs in these classes, there is also much interest in long-acting/extended-release medications for further gains in patient compliance and associated prevention of transmission.6 The most widely investigated approaches are slow-release formulations in injected nanoparticles or in subcutaneous polymer-based implants. A drawback of the former approach is the difficulty in response to possible adverse effects, while implants, though more difficult to administer, can be removed. Our laboratories have pursued development of improved NNRTIs with high-potency against with-type HIV-1 and clinically important variants;7−11 we have reported compounds with excellent pharmacokinetics,12−14 and representative ones have been demonstrated to sustain high plasma levels and efficacy for more than 3 weeks in humanized mice after a single injection of a nanoformulation.13,14
We have also been exploring a new class of anti-HIV agents, covalent inhibitors of HIV-1 reverse transcriptase (CRTIs). The initial efforts successfully targeted clinically problematic Tyr181Cys RT variants.15 Conclusive evidence for the covalent modification of Cys181 included results from mass spectrometry (MS), protein crystallography, and antiviral activity in infected human T-cell assays. The CRTIs were shown to be selective for Cys181 and have lower cytotoxicity than EFV and RPV. Because the NNRTI allosteric site rather than an active site is being targeted, inhibition of active sites in a homologous protein family is not a risk. Indeed, our compounds along with recent G12C KRAS inhibitors are rare examples of targeted covalent allosteric inhibitors, which have enhanced potential for low dosage, low toxicity, and extended duration of action.16,17 Presently, we report design and characterization of the first CRTIs that successfully modify wild-type HIV-1 RT.
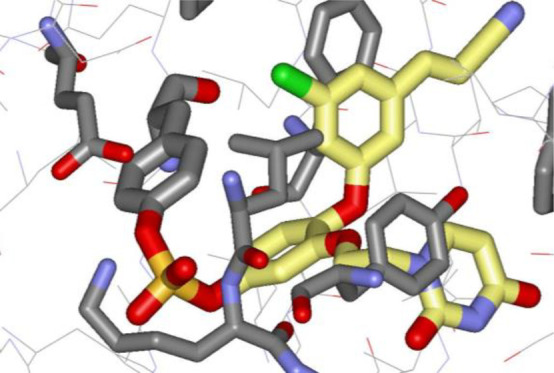
The wild-type (WT) protein presents a greater challenge owing to the lack of cysteine residues in the NNRTI binding site. There are multiple lysines; however, they are solvent-exposed and not expected to be adequately nucleophilic.18 The next choice is the less-explored hydroxyl-containing residues. In this regard, sulfonyl fluoride and fluorosulfate warheads have achieved recent successes with serine and tyrosine targets.19−21 In viewing the crystal structure of one of our potent catechol diether NNRTIs 1 (EC50 = 55 pM) bound to wild-type RT (Figure 1),7,22 replacement of the chlorine in the central ring with a warhead to attach covalently to Tyr181 arose as a possibility. Interest focused on the weakly electrophilic fluorosulfate as the warhead in view of its reported stability in aqueous solution and lesser off-target reactivity than for sulfonyl fluorides.20 We ultimately prepared and tested fluorosulfate-containing analogues of 1 with modifications to the cyanovinyl and uracil containing appendages (2–13) and two sulfonyl fluorides (14, 15), as summarized in Figure 2. The 3-chloro,5-cyanophenoxy substituents, and 1-naphthyloxy and 8-indolizinyloxy alternatives have all been featured in low-nM NNRTIs that we have previously reported.7−9
Figure 1.

Rendering from the crystal structure of the noncovalent inhibitor 1 with HIV-1 reverse transcriptase at 2.85 Å resolution (PDB 4H4M).22 The highlighted distance from the hydroxyl oxygen atom of Tyr181 to the chlorine atom of 1 is 3.6 Å. All carbon atoms of ligands are shown in yellow.
Figure 2.
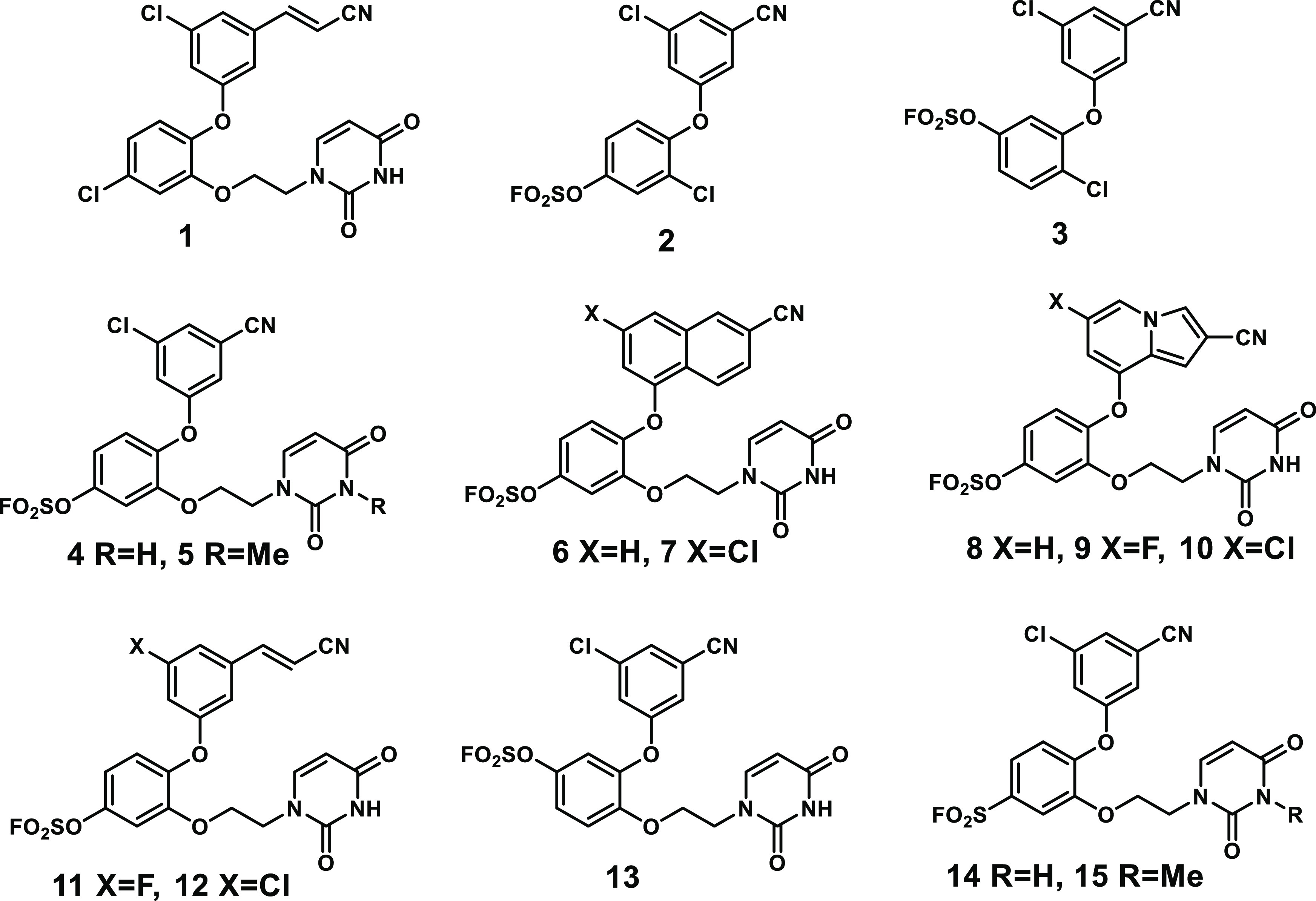
Anti-HIV agents considered here. 1 is a key catechol diether NNRTI; 2–15 are new compounds reported here. 2–13 are potential CRTIs with a fluorosulfate warhead, and 14–15 have a sulfonyl fluoride warhead. 4, 11, and 12 are shown to be CRTIs.
The syntheses of the new compounds are detailed in the Supporting Information. In each case, the fluorosulfate group was introduced in good yield by treating a phenol with sulfuryl fluoride gas and triethylamine in methylene chloride at room temperature for 2 h.23 The sulfonyl fluorides were prepared from aryl iodides using a palladium-catalyzed procedure with 1,4-diazabicyclo[2.2.2]octane-bis(sulfur dioxide) (DABSO) followed by N-fluorobenzenesulfonimide (NFSI) for the fluorination.24 The identity of assayed compounds was confirmed by 1H and 13C NMR, high-resolution mass spectrometry, and ultimately X-ray crystallography for multiple complexes with WT HIV-1 RT; HPLC analyses established purity as >95%.
Assays were performed to assess both the in vitro inhibition of recombinant WT HIV-1 RT and for protection of human MT-2 T-cells that were infected with the IIIB strain of HIV-1. The details are unchanged from prior reports.15,22 Briefly, a PicoGreen-based EnzChek Reverse Transcriptase Assay Kit (Thermo Fisher Scientific, Inc., E22064) was used to determine RT activity in the presence of inhibitors in a 96-well plate format. The enzyme was preincubated with the test compounds or DMSO control, followed by addition of the substrate solution including an r(A)350 template and d(T)16 primer. After 30 min of incubation at room temperature, EDTA was used to quench the reaction. PicoGreen reagent was then added, and product formation was detected with a plate reader using excitation and emission at 485 and 520 nm. The measurements were made in triplicate, and the results were normalized to DMSO controls to determine IC50 values. The MT-2 T-cell assay is a standard that has been extensively applied.7−15 The cells are infected with the IIIB strain of HIV-1, and the measurements are made in triplicate to yield EC50 values as the dose required to achieve 50% protection of the infected MT-2 cells and CC50 values for inhibition of MT-2 cell growth by 50%.
The assay results for the 15 compounds in Figure 2 along with two FDA-approved NNRTI drugs, nevirapine (NEV) and rilpivirine (RPV), are recorded in Table 1. Some overall observations are that the trends in the IC50 and EC50 values are generally similar with a wider range of EC50 results; the IC50 values for the new compounds 3–12 are mostly between those for NEV and RPV; 6–10 have EC50 values below 100 nM, and the new compounds, in spite of bearing an electrophilic warhead, are all less cytotoxic than RPV. Taking a more detailed look, it may be first noted that 1 is an extraordinarily potent NNRTI with an IC50 of 3 nM and EC50 of 55 pM.7,22 For the new compounds, we began with 2 and 3 by considering truncation of the uracilylpropoxy substituent and placement of the fluorosulfate group either para or meta to the phenoxy group. However, neither compound showed good activity, so the subsequent analogues retained the uracil-containing appendage.
Table 1. Measured Activities in the Enzyme Inhibition (IC50) and Infected T-Cell Assays (EC50), and Cytotoxicity (CC50).
| compd | IC50 (nM)a | EC50 (nM)b | CC50 (μM)c |
|---|---|---|---|
| 1 | 3.0 | 0.055 | 10 |
| 2 | 4900 | >50000 | 50 |
| 3 | 833 | 9200 | 78 |
| 4 | 103 | 280 | 66 |
| 5 | 246 | 320 | 40 |
| 6 | 82.3 | 58 | 62 |
| 7 | 14.7 | 37 | 15 |
| 8 | 164 | 5 | 75 |
| 9 | 64 | 7 | 55 |
| 10 | 42.4 | 26 | 26 |
| 11 | 150 | 270 | 42 |
| 12 | 119 | 160 | 45 |
| 13 | ND | 14000 | 100 |
| 14 | 5000 | 17000 | 90 |
| 15 | 375 | 2700 | 60 |
| nevirapine | 1060 | 110 | >100 |
| rilpivirine | 38 | 0.67 | 8 |
For 50% inhibition of reverse transcription (nM).
For 50% protection in MT-2 cells (nM).
For 50% inhibition of MT-2 cell growth (μM).
With compound 4, the basic design from Figure 1 was followed with subtraction of the vinyl group. As the compounds were being synthesized and assayed, crystal structures for the complexes with WT RT were being sought and were eventually obtained for 4–11, as detailed below. The crystallographic and MS results clearly showed that 4 was the first CRTI to covalently modify Tyr181 to form the biaryl sulfate. Under the standard assay procedures, 4 also showed good potency, with IC50 and EC50 values of 103 and 280 nM.
Additional compounds 6–12 were prepared to evaluate the effects of modifications to the phenoxy substituent, which makes aryl–aryl contacts with Tyr188 and Trp229 (Figure 1). Small changes to the core structure are expected to cause repositioning that affects the disposition and contact of Tyr181 and the fluorosulfate group. 6–12 turned out to all be potent inhibitors, with 7, 9, and 10 having sub-100 nM IC50 values, and the indolizines 8 and 9 having striking EC50 values of 5 and 7 nM. The activity results were surprisingly favorable, as it was unclear that it would be possible to replace the central chlorine atom in 1 with the much bulkier and polar fluorosulfate group without severe loss of potency. In addition to 4, the crystallographic and MS data show that 11 and 12 are covalent inhibitors, while the others are noncovalent NNRTIs.
A few more compounds were prepared. Model building of structures indicated that both meta and para isomers as for 2 and 3 might be viable. Thus, the meta analogue 13 of the CRTI 4 was synthesized, but it was found to be a comparatively weak NNRTI with an EC50 of 14 μM. The sulfonyl fluoride 14 corresponding to the fluorosulfate 4 was also prepared and found to be a weak inhibitor; modeling suggests that the increased bulk closer to the central ring of the inhibitor leads to steric conflict with Lys101. 15, the N-methyl analogue of 14, was also prepared and tested. Although it showed notable improvement over 14, the IC50 and EC50 results were much higher than for 4 and 6–10. For 4 and 5, the opposite pattern was found with the N-methylated 5 being less active, possibly reflecting a boost from the CRTI character of 4.
It may be noted that the three CRTIs (4, 11, 12) all had similar IC50 values (103–150 nM) and EC50 values (160–280 nM). It is generally expected that covalent inhibitors show increasing potency with increasing time as the concentration of free protein declines, while noncovalent inhibition should not be affected as soon as the binding equilibrium is achieved.15,16,25 To check this, the dependence of the enzymatic assay data on incubation time was examined for 1–48 h. For the present compounds, a different pattern was generally observed with the covalent inhibitors not showing incubation-time dependence and with the noncovalent ones showing loss of potency over time. For example, the measured IC50 for 4 was 103 nM with the usual 1 h incubation and it was 163 nM after a 48 h incubation, while the corresponding results for 8 were 164 and 1115 nM, and for 9 they were 64 and 429 nM. Though further kinetic analyses are warranted, a possible explanation is that there is some degradation of the inhibitors over the 2 days as reflected in the increasing IC50 values for the noncovalent inhibitors, while this effect is being offset by the covalent inhibitors removing free protein. A few compounds were also tested for inhibition of recombinant RT protein bearing the clinically common Tyr181Cys mutation. For the indolizines 8, 9, and 10, very similar IC50 values were obtained for the WT (164, 64, 42 nM) and mutant (202, 63, 41 nM) proteins, respectively, while the CRTI 4 showed greater inhibitory potency for the variant (45 nM) than the WT enzyme (82 nM). These results are encouraging, and further study of resistance profiles is of interest, but beyond the present focus.
For the crystallography, recombinant WT RT52A enzyme was used with cocrystallization methods, as previously described.8−10,15,22 Structures were obtained for the complexes with 4–12 at resolutions of 2.60–2.73 Å. The first covalent structure was obtained for 4, as illustrated in Figure 3. The covalent bond between the Tyr181 oxygen atom and the sulfur of the warhead is 1.5 Å, and there are hydrogen bonds for both the side chain and backbone nitrogen atoms of Lys101 with sulfate oxygen atoms. There is also a hydrogen bond between an uracil oxygen atom and the backbone nitrogen atom of Lys103. The distortion of the binding site is slight in comparison with related noncovalent structures. For example, the distances between the Cβ atoms for the triangle made by the key aromatic residues Tyr188–Trp229–Phe227 are 8.4, 4.7, and 8.5 Å for the structure with 4 (Figure 3) and 8.5, 5.7, and 9.3 for 1 (Figure 1).
Figure 3.

Renderings from the 2.70 Å crystal structure for 4 covalently bound to wild-type HIV-1 reverse transcriptase. (A) Closeup highlighting the covalent bond between the oxygen atom of Tyr181 and the sulfate sulfur atom of 4. (B) View highlighting three hydrogen bonds between the backbone and side chain nitrogen atoms of Lys101 and two sulfate oxygen atoms (PDB 7KRD).
As an example of a structure for one of the noncovalent inhibitors with an intact fluorosulfate group, the complex for 6 bound to WT RT is illustrated in Figure 4. In this case, the distance between the tyrosine oxygen and sulfur atoms is 3.8 Å and the shortest contact between the backbone nitrogen atom of Lys101 and an oxygen atom of the warhead is 4.0 Å versus 3.4 Å for 4. The Cβ–Cβ distances for the Tyr188–Trp229–Phe227 triangle are also expanded slightly to 8.7, 4.9, and 8.7 Å.
Figure 4.

Rendering from the 2.73 Å crystal structure for 6 noncovalently bound to wild-type HIV-1 reverse transcriptase. The highlighted distance between the Tyr181 oxygen atom and fluorosulfate sulfur atom is 3.8 Å (PDB 7KRE).
As a further example of a covalent complex, the structure for 12 is provided in Figure 5A. The side chains for Lys101, Lys102, and Lys103 are particularly well resolved in this structure and reveal a striking hydrogen-bonded network for the ammonium groups of Lys101 and Lys103 and the backbone nitrogen of Lys 101 with all four oxygens of the sulfate group. In both this structure and the one for 11 (not illustrated), the covalent bond length is 1.7 Å between the tyrosine oxygen atom and the sulfur atom of the sulfate group. Again, there is no significant distortion of the binding site as reflected in the Cβ–Cβ distances for the Tyr188–Trp229–Phe227 triangle of 8.4, 4.7, and 8.5 Å. Furthermore, concerning the conformation of the diphenyl sulfate substructure, three conformational energy minima are normally expected for dialkyl or diaryl sulfates corresponding to G(gauche)G, GT(trans), and TT for the two C–O–S–O dihedral angles with relative energies of ca. 0.0, 1.0, and 3.0 kcal/mol, respectively (Figure 5B).26 With use of the BOSS program and OPLS/CM1A force field, these results are well reproduced.27,28 For the covalent complexes of 4, 11, and 12, the inhibitor C–O–S–O dihedral angle is G; however, the Tyr181 C–O–S–O dihedral angle is closer to syn at ca. 10°. Thus, there is some strain relative to a GG conformation; it is roughly estimated to be 5 kcal/mol with the OPLS/CM1A force field from constrained energy-minimizations for diphenyl sulfate. Such strain is readily offset by formation of the new covalent O–S covalent bond and elimination of the fluoride ion. Far more strained aryl sulfates and sulfonates have been well studied such as catechol sulfate.29
Figure 5.
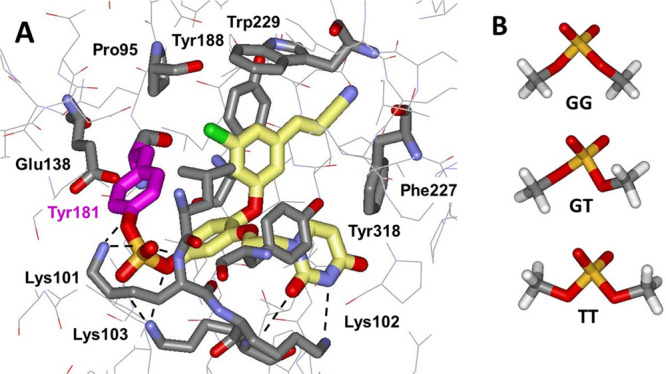
(A) Rendering from the 2.63 Å crystal structure for 12 covalently bound to wild-type HIV-1 reverse transcriptase. Note the network of hydrogen bonds for the sulfate group with the ammonium groups of Lys101 and Lys103 and the backbone NH of Lys101 (PDB 7KRC). (B) The three conformations of dimethylsulfate.
The crystallographic conclusions on the covalent modification of Tyr181 by 4, 11, and 12 were fully supported by electrospray ionization–time-of-flight mass spectrometry (ESI-TOF MS), which was used to determine the molecular weight of the intact protein and to examine potential covalent modification reflected by increases in mass. The same procedures were followed as in the previous work.15 WT RT 20 μM was incubated alone or with 0.5 mM inhibitor in 1.5% DMSO for 6 days at 4 °C. Samples were prepared by reverse-phase HPLC and injected into a Bruker IMPACT II ESI-TOF mass spectrometer (Bruker Inc., Billerica, MA) operated in positive ion mode. As shown in Figure 6A, the top two panels show a reference sample of WT RT with peaks at 63 907.5 and 50 027.8 Da, corresponding to the p51 and p66 subunits of RT. The bottom panel shows representative spectra after the incubation of RT with 11. As p66 contains the catalytic and NNRTI binding sites, a mass shift for only this subunit would be expected upon introduction of the covalent inhibitors. As illustrated in this bottom panel, no shift is observed for the p51 subunit. For spectra showing the p66 subunit, a peak corresponding to the unmodified p66 is observed at 63 097.5 as well an additional peak at 64 378.8 with a mass shift of 471 corresponding to the covalent addition of 11. Similarly, peaks are observed at 64 370 (mass shift of 461) for 4 and 64 395.5 (mass shift 487) for 12. The calculated and observed masses for each p66 and p51 species are summarized in Figure 6B.
Figure 6.

Representative spectra for ESI TOF MS analysis of RT covalent catalysis. (A) (top) spectra for RT p66 and p51 subunits; (bottom) spectra of RT p66 and p51 subunits after incubation with 11. (B) Summary of calculated and observed masses for RT with 4, 11, and 12.
In summary, a new class of anti-HIV agents, which covalently modify HIV-1 reverse transcriptase is under investigation. In this work, the first successful covalent inhibition of the wild-type protein is reported by taking advantage of the NNRTI binding site and modification of known noncovalent inhibitors with introduction of an electrophilic warhead. As there are no cysteine residues in the vicinity of the binding site, the less precedented covalent modification of a tyrosine, specifically Tyr181, was pursued. Success was demonstrated through protein crystallography and mass spectrometry for three compounds, 4, 11, and 12, which incorporate a fluorosulfate warhead. These compounds and six analogues were found to be nanomolar inhibitors of HIV-RT through both an in vitro enzyme-inhibition assay and an infected T-cell assay. Further work is investigating potential ramifications of the covalent inhibition for induction of resistance and considering additional residues as targets in the NNRTI binding site.
Acknowledgments
Gratitude is expressed to the National Institutes of Health (AI44616, GM32136, GM49551, AI155072) for research support. Crystal screening was conducted with support in the Yale Macromolecular X-ray Core Facility (1S10OD018007-01). This work is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P41 GM103403). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02–06CH11357. This research used the FMX beamline of the National Synchrotron Light Source II, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DESC0012704. The Life Science Biomedical Technology Research resource is primarily supported by the NIH, NIGMS through a Biomedical Technology Research Resource P41 grant (P41 GM111244), and by the DOE Office of Biological and Environmental Research (KP1605010).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00612.
Accession Codes
The crystal structures have been deposited in the RCSB Protein Data Bank with the PDB codes 7KRD, 7KRE, 7KRF, and 7KRC, respectively.
Author Contributions
∥ J.A.I., H.N., and N.B. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Frank T. D.; et al. Estimates of global, regional, and national incidence, prevalence, and mortality of HIV, 1980–2017, and Forecasts to 2030. Lancet HIV 2019, 6, e831–e859. 10.1016/S2352-3018(19)30196-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Clercq E.; Li G. Approved Antiviral Drugs over the Past 50 Years. Clin. Microbiol. Rev. 2016, 29, 695–747. 10.1128/CMR.00102-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebaaly J. C.; Kelley D. Single-Tablet Regimens for the Treatment of HIV-1 Infection. Ann. Pharmacother. 2017, 51, 332–344. 10.1177/1060028016682531. [DOI] [PubMed] [Google Scholar]
- Wang Y.; De Clercq E.; Li G. The Current and Emerging Nonnucleoside Reverse Transcriptase Inhibitors (NNRTIs) for HIV-1 Treatment. Expert Opin. Drug Metab. Toxicol. 2019, 15, 813–829. 10.1080/17425255.2019.1673367. [DOI] [PubMed] [Google Scholar]
- Sancar F. Two-Drug Regimen for HIV. JAMA 2019, 321, 1862. 10.1001/jama.2019.6188. [DOI] [PubMed] [Google Scholar]
- Gulick R. M.; Flexner C. Long-Acting HIV Drugs for Treatment and Preventation. Annu. Rev. Med. 2019, 70, 137–150. 10.1146/annurev-med-041217-013717. [DOI] [PubMed] [Google Scholar]
- Bollini M.; Domaoal R. A.; Thakur V. V.; Gallardo-Macias R.; Spasov K. A.; Anderson K. S.; Jorgensen W. L. Computationally-Guided Optimization of a Docking Hit to Yield Catechol Diethers as Potent Ani-HIV Agents. J. Med. Chem. 2011, 54, 8582–8591. 10.1021/jm201134m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W.-G.; Gallardo-Macias R.; Frey K. M.; Spasov K. A.; Bollini M.; Anderson K. S.; Jorgensen W. L. Picomolar Inhibitors of HIV Reverse Transcriptase Featuring Bicyclic Replacement of a Cyanovinylphenyl Group. J. Am. Chem. Soc. 2013, 135, 16705–16713. 10.1021/ja408917n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W.-G.; Gallardo-Macias R.; Frey K. M.; Spasov K. A.; Bollini M.; Anderson K. S.; Jorgensen W. L. Picomolar Inhibitors of HIV Reverse Transcriptase: Design and Crystallography of Naphthyl Phenyl Ethers. ACS Med. Chem. Lett. 2014, 5, 1259–1262. 10.1021/ml5003713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W. G.; Chan A. H.; Spasov K. A.; Anderson K. A.; Jorgensen W. L. Design, Conformation, and Crystallography of 2-Naphthyl Phenyl Ethers as Potent Anti-HIV Agents. ACS Med. Chem. Lett. 2016, 7, 1156–1160. 10.1021/acsmedchemlett.6b00390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W. L. Computer-aided Discovery of Anti-HIV Agents. Bioorg. Med. Chem. 2016, 24, 4768–4778. 10.1016/j.bmc.2016.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudalkar S. N.; Beloor J.; Chan A. H.; Lee W. G.; Jorgensen W. L.; Kumar P.; Anderson K. S. Structural and Preclinical Studies of Computationally Designed Non-Nucleoside Reverse Transcriptase Inhibitors for Treating HIV infection. Mol. Pharmacol. 2017, 91, 383–391. 10.1124/mol.116.107755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudalkar S. N.; Beloor J.; Quijano E.; Spasov K. A.; Lee W.-G.; Cisneros J. A.; Saltzman W. M.; Kumar P.; Jorgensen W. L.; Anderson K. S. From In Silico Hit to Long-Acting Late-Stage Preclinical Candidate to Combat HIV Infection. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E802–E811. 10.1073/pnas.1717932115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudalkar S. N.; Ullah I.; Bertoletti N.; Mandl H. K.; Cisneros J. A.; Beloor J.; Chan A. H.; Quijano E.; Saltzman W. M.; Jorgensen W. L.; Kumar P.; Anderson K. S. Structural and Pharmacological Evaluation of a Novel Non-nucleoside Reverse Transcriptase Inhibitor as a Promising Long Acting Nanoformulation for Treating HIV. Antiviral Res. 2019, 167, 110–116. 10.1016/j.antiviral.2019.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan A. H.; Lee W.-G.; Spasov K. A.; Cisneros J. A.; Kudalkar S. N.; Petrova Z. O.; Buckingham A. B.; Anderson K. S.; Jorgensen W. L. Covalent inhibitors for eradication of drug-resistant HIV-1 reverse transcriptase: from design to protein crystallography. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 9725–9730. 10.1073/pnas.1711463114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussinov R.; Tsai C.-J. The Design of Covalent Allosteric Drugs. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 249–267. 10.1146/annurev-pharmtox-010814-124401. [DOI] [PubMed] [Google Scholar]
- McCormick F. Sticking It to KRAS: Covalent Inhibitors Enter the Clinic. Cancer Cell 2020, 37, 3–4. 10.1016/j.ccell.2019.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettinger J.; Jones K.; Cheeseman M. D. Lysine-Targeting Covalent Inhibitors. Angew. Chem., Int. Ed. 2017, 56, 15200–15209. 10.1002/anie.201707630. [DOI] [PubMed] [Google Scholar]
- Chen W.; Dong J.; Plate L.; Mortenson D. E.; Brighty G. J.; Li S.; Liu Y.; Galmozzi A.; Lee P. S.; Hulce J. J.; Cravatt B. F.; Saez E.; Powers E. T.; Wilson I. A.; Sharpless K. B.; Kelly J. W. Arylfluorosulfates Inactivate Intracellular Lipid Binding Protein(s) through Chemoselective SuFFex Reaction with a Binding Site Tyr Residue. J. Am. Chem. Soc. 2016, 138, 7353–7364. 10.1021/jacs.6b02960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadeyi O. O.; Hoth L. R.; Choi C.; Feng X.; Gopalsamy A.; Hett E. C.; Kyne R.E. Jr.; Robinson R. P.; Jones L. H. Covalent Enzyme Inhibition through Fluorosulfate Modification of a Noncatalytic Serine Residue. ACS Chem. Biol. 2017, 12, 2015–2020. 10.1021/acschembio.7b00403. [DOI] [PubMed] [Google Scholar]
- Jones L. H. Emerging Utility of Fluorosulfate Chemical Probes. ACS Med. Chem. Lett. 2018, 9, 584–586. 10.1021/acsmedchemlett.8b00276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey K. M.; Bollini M.; Mislak A. C.; Cisneros J. A.; Gallardo-Macias R.; Jorgensen W. L.; Anderson K. S. Crystal Structures of HIV-1 Reverse Transcriptase with Picomolar Inhibitors Reveal Key Interactions for Drug Design. J. Am. Chem. Soc. 2012, 134, 19501–19503. 10.1021/ja3092642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J.; Krasnova L.; Finn M. G.; Sharpless K. B. Sulfur(VI) Fluoride Exchange (SuFEx): Another Good Reaction for Click Chemsitry. Angew. Chem., Int. Ed. 2014, 53, 9430–9448. 10.1002/anie.201309399. [DOI] [PubMed] [Google Scholar]
- Davies A. T.; Curto J. M.; Bagley S. W.; Willis M. C. One-pot Palladium-catalyzed Synthesis of Sulfonyl Fluorides for Aryl Bromides. Chem. Sci. 2017, 8, 1233–1237. 10.1039/C6SC03924C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Cesco S.; Kurian J.; Dufresne C.; Mittermaier A. K.; Moitessier N. Covalent Inhibitors Design and Discovery. Eur. J. Med. Chem. 2017, 138, 96–114. 10.1016/j.ejmech.2017.06.019. [DOI] [PubMed] [Google Scholar]
- Borba A.; Gómez-Zavaglia A.; Simões P. N. N. L.; Fausto R. Matrix-isolation FT-IR Spectra and Theoretical study of Dimethyl Sulfate. Spectrochim. Acta, Part A 2005, 61, 1461–1470. 10.1016/j.saa.2004.10.050. [DOI] [PubMed] [Google Scholar]
- Jorgensen W. L.; Tirado-Rives J. Potential Energy Functions for Atomic-Level Simulations of Water and Organic and Biomolecular Systems. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 6665–6670. 10.1073/pnas.0408037102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodda L. S.; Cabeza de Vaca I.; Tirado-Rives J.; Jorgensen W. L. LigParGen Web Server: An Automatic OPLS-AA Parameter Generator for Organic Ligands. Nucleic Acids Res. 2017, 45, W331–W336. 10.1093/nar/gkx312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser E. T. Enzymatic and Nonenzymatic Reactions of Cyclic Sulfonate and Sulfate Esters. Acc. Chem. Res. 1970, 3, 145–151. 10.1021/ar50029a001. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.