

Abstract

The MEK1 kinase plays a critical role in key cellular processes, and as such, its dysfunction is strongly linked to several human diseases, particularly cancer. MEK1 has consequently received considerable attention as a drug target, and a significant number of small-molecule inhibitors of this kinase have been reported. The majority of these inhibitors target an allosteric pocket proximal to the ATP binding site which has proven to be highly druggable, with four allosteric MEK1 inhibitors approved to date. Despite the significant attention that the MEK1 allosteric site has received, chemotypes which have been shown structurally to bind to this site are limited. With the aim of discovering novel allosteric MEK1 inhibitors using a fragment-based approach, we report here a screening method which resulted in the discovery of multiple allosteric MEK1 binders, one series of which was optimized to sub-μM affinity for MEK1 with promising physicochemical and ADMET properties.
Keywords: MEK1, Kinases, Allosteric, COPD, Fragments, FBDD
The MAP kinase signaling pathway influences many key cellular processes including proliferation and differentiation, and as such, dysregulation of this pathway is known to play a major role in several human diseases, particularly cancer.2,3 Consequently, multiple components of this pathway have been studied intensely as drug targets. In particular, several inhibitors of the MEK1 kinase have now been approved by the FDA and EMA for the treatment of various types of cancer, with additional molecules in clinical and preclinical studies.4−7 Interestingly, the MEK inhibitors approved for clinical use are based on a shared 2,4-dihalogenated anilido motif (Figure 1) and are known or expected to bind to an allosteric pocket adjacent to the ATP binding site of the kinase. The targeting of an allosteric site of a kinase with a small-molecule inhibitor is particularly interesting, as this potentially circumvents the expected selectivity challenges associated with targeting the highly conserved orthosteric ATP binding site.
Figure 1.

Structures of selected approved allosteric MEK1 inhibitors.
While the major focus of MEK1 as a therapeutic target has been for oncology indications, there is additional interest in the inhibition of this enzyme for the treatment of chronic obstructive pulmonary disease (COPD).8−10 While approved MEK1 inhibitors have proven to be highly effective in oncology settings, the differing requirements of COPD small-molecule therapeutics—for example, the desire for rapid systemic clearance of an inhaled drug—suggest that new MEK1 inhibitors with differing physicochemical and ADME properties may be needed, while retaining the benefits of an allosteric binding mode. Interestingly, fragment-based methods11 have been employed as a means to discover allosteric inhibitors of various protein classes,12−17 including kinases,18,19 yet reports of fragment screens targeting MEK1, while successful, have yielded binders of the orthosteric ATP binding site only.20,21 As a consequence, we set out to design and execute a fragment-based screening campaign targeting MEK1 specifically designed to find novel, allosteric inhibitors. These efforts led to the discovery of several novel allosteric MEK1 binders, one series of which was optimized to sub-μM affinity with promising physicochemical and ADMET properties aligned with an inhaled small-molecule therapeutic.
In an effort to find novel allosteric MEK1 binders by fragment-based methods, we first selected a fragment library tailored to the MEK1 allosteric site using virtual screening methods. From a library of around 15k fragments,22 1k were selected based on their complementarity (assessed by ligand–receptor docking studies) to the allosteric site of MEK1.23 A multistep 1D NMR protocol was then used to screen these compounds for binding to MEK1. Fragments were screened in cocktails of 6 compounds against MEK1, with 1 mM AMP-PNP (a nonhydrolyzable analogue of ATP) included as a means to occupy the orthosteric ATP binding site and thereby bias the detection toward allosteric binders. Fragments identified as positive binders under these conditions were then subject to a competition experiment using the known MEK1 allosteric inhibitor binimetinib.6 Using this method, 142 fragments (14%) were identified as potential allosteric binders.
The binding affinity of these fragments for human full-length MEK1 was then assessed by surface plasmon resonance (SPR) in both the absence and presence of AMP-PNP.24 46 Fragments (5%) displayed similar affinities under both conditions and yielded Kd values in the presence of AMP-PNP of ≤1 mM. These were considered to be likely noncompetitive allosteric MEK1 binders. Only two fragments showed a significant preference for MEK1 in the absence of AMP-PNP (i.e., AMP-PNP competition), indicating that the screening strategy was successful in selecting noncompetitive binders. Given the structural similarity of reported MEK1 inhibitors, the hit rate was surprisingly high yet confirms that this is a highly ligandable binding site. Details of the most interesting hits, based on affinity data, structural novelty, and physicochemical properties, can be found in Table 1.
Table 1. SPR Analysis of Fragment Hits Identified from NMR-Based Screening of MEK1.

Kd values are the geometric mean ± SEM of two or more determinations.
LE (kcal/(mol HA)): 1.37pKd/HA (heavy atom count).
LipE: pKd – log D. SPR pKd in the presence of AMP-PNP used in efficiency metrics. log D measured via shake-flask method in octanol and water at pH 7.4.
We next turned to X-ray crystallography in order to assess the binding mode of the identified AMP-PNP-noncompetitive binders. Crystallization attempts (in the presence of AMP-PNP) of all hits yielded crystal structures of compounds 4 and 6 bound to MEK1, both of which revealed the fragments to bind to the allosteric site as anticipated (Figure 2a,b). Both compounds localize to the allosteric pocket that sits between the ATP binding site and the αC-helix, with both forming strong hydrogen bonds to the backbone NH of residue S212 of the activation loop (A-loop). Compound 6 forms an additional hydrogen bond to the backbone carbonyl of F209, which may explain the improved affinity and ligand efficiency of this hit compared to 4 (Table 1). Comparison of the binding mode of 4 and 6 for MEK1 to that of 2,4-dihalogenated aniline allosteric inhibitors such as cobimetinib revealed clear differences with respect to binding interactions (for example, an absence of contacts from 4 or 6 to K97 or ATP) (Figure 2c), suggesting that compounds 4 and 6 represent novel and differentiated allosteric MEK1 binders.
Figure 2.

Crystal structures of MEK1 bound by compounds 4 (a) and 6 (b). (c) Comparison of binding modes of 6 (blue carbons) and cobimetinib (PDB entry 4AN2(1)) (white carbons) for MEK1. Hydrogen bonds to MEK1 are shown as blue or white dashes for 6 and cobimetinib, respectively. AMP-PNP: adenylyl imidodiphosphate. AMP-PCP: adenylyl methylenediphosphonate.
While we were unable to obtain crystal structures of all fragment hits, the two we were able to obtain indicated clearly that our screening method was indeed able to discover novel allosteric MEK1 binders and encouraged us to also progress alternative scaffolds. Consequently, employing the NNS technology,25 we searched our internal chemical library for close analogues of all of the primary hits, with selected compounds assessed by SPR to measure affinities for MEK1. These efforts yielded two stand-out binders—compound 9, with an affinity of 28 μM, and compound 10, with an affinity of 65 μM (Figure 3).
Figure 3.

Preliminary optimization of MEK1 primary fragment hits. (a) Compound 9 was discovered following analogue searching of our internal chemical library using 5 as a template. A crystal structure was determined for 9 bound to MEK1, confirming an allosteric binding mode (top right panel). (b) Compound 10 was discovered by similar methods as 9 and was used as a template for a library synthesis aimed at further improving affinity for MEK1 (see Scheme 1 and Table 2 for further details).
Crystallization attempts with MEK1 were attempted for both compounds, which yielded a crystal structure of compound 9 (Figure 3a, right panel). This structure shows compound 9 binding to the allosteric pocket, with the interaction seemingly primarily driven by hydrophobic contacts, with polar contacts to the protein limited to indirect interactions mediated via water bridges. Although we were unsuccessful in obtaining structural data for 10, the novelty of this compound (particularly the oxothiomorpholine group), coupled to the synthetic tractability of the scaffold, led us to synthesize a chemical library around the central amide functionality, in an attempt to improve binding affinity for MEK1 (Figure 3b and Scheme 1).
Scheme 1. General Synthetic Route of Compounds 11–22.

Reagents and conditions: (a) MeI, K2CO3, DMF, rt, 3 h. (b) Cysteamine, MeOH, rt, 2 h. (c) K2CO3, MeOH, 60 °C, 5 h. (d) NaOH, H2O, MeOH, rt, 3 h. (e) Aromatic amine, HATU, DIPEA, DMF, rt, 3 h.
Final library compounds were prepared in high overall yield from commercially available 2-bromosuccinic acid (I). The synthesis commenced by converting building block I into its corresponding methyl diester II by treatment with iodomethane and potassium carbonate in DMF in moderate yield (53%). The 3-oxothiomorpholine core was successfully constructed with a one-pot, two-step procedure in good yield (59%): reaction with cysteamine in MeOH furnished precursor III, and ring closure was subsequently accomplished by adding potassium carbonate and heating the resultant solution at 60 °C for 5 h. Saponification of methyl ester IV with aqueous NaOH in MeOH afforded carboxylic acid V, which was then converted, with appropriate amines, into the corresponding desired final compounds employing HATU as the peptide coupling agent in DMF (Scheme 1). Final compounds were then assessed by SPR to measure binding affinity for MEK1 (Table 2).
Table 2. Affinity and Efficiency Metrics of Analogues of 10 against MEK1.
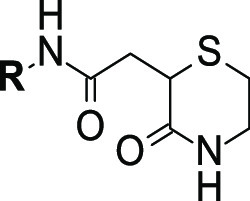

Kd values are the geometric mean ± SEM of two or more determinations.
LE (kcal/(mol HA)): 1.37pKd/HA (heavy atom count).
LipE: pKd – log D. SPR pKd in the presence of AMP-PNP used in efficiency metrics. log D measured via shake-flask method in octanol and water at pH 7.4.
n.d.: not determined.
Interestingly, removal of both chloro atoms (11) resulted in a 3-fold increase in affinity, with further analogues strongly indicating substitution with moderately bulky groups at the ortho and meta positions to be detrimental to affinity for MEK1 (12 and 13). Substitution of the para position, however, appeared to result in favorable affinity gains for MEK1, with fluoro (21), chloro (22), methoxy (14), and ethoxy (16) substitutions all resulting in significant (up to 30-fold) improvements in affinity. Larger substituents at the para position were also tolerated (17 and 20), as were polar groups such as phenols (19), although to a lesser degree.
Focusing on the most ligand and lipophilic efficient compound (14), the enantiomers 23 and 24 were separated, and their absolute stereochemical configuration was assigned by comparison of experimental and measured vibrational circular dichroism (VCD) spectra (full details can be found in the Supporting Information). The affinities for MEK1 were assessed by SPR (Table 3), and this highlighted that the (S) enantiomer (23) has a clear preference to bind MEK1 over its antipode 24, with Kd values of 0.3 and 30 μM for 23 and 24, respectively.
Table 3. Affinity and Efficiency Metrics of 4-Methoxyanilide Derivatives against MEK1.


Kd values are the geometric mean ± SEM of two or more determinations.
LE (kcal/(mol HA)): 1.37pKd/HA (heavy atom count).
LipE: pKd – log D. SPR pKd in the presence of AMP-PNP used in efficiency metrics. log D measured via shake-flask method in octanol and water at pH 7.4.
(μL/(min mg)).
(μL/(min 1E6)).
Papp: (*1 × 10–6 cm/s).
With improved affinity, in vitro physicochemical and ADMET properties of these compounds were then assessed (Table 3). Eutomer 23 was shown to possess good aqueous solubility (355 μM), good permeability (as measured by the Caco2 assay), and, encouragingly, no activity against the hERG ion channel (IC50 > 40 μM). Furthermore, in vitro assessment of metabolic stability in human liver microsomes (HLMs) and rat hepatocytes (rHeps) indicated relatively low stability compared to orally administered small molecules. For inhaled administration, low metabolic stability may be advantageous, by limiting systemic exposure.26,27
Early assessment of potential significant off-target interactions was carried out on top compound 23 against a safety panel (Eurofins SafetyScreen panel) of 22 off-targets, comprising enzymes, receptors, and ion channels (e.g., COX2, PDEs, INSR, GABA, NMDA, CaV-L), as well as a subpanel of 139 different kinases (Thermofisher kinome subpanel). Compound 23 did not show any significant off-target activity, with IC50 values >100 μM (SafetyScreen panel) and percentage inhibitions <20% at 1.0 μM (kinome subpanel) against all targets tested.
Having established promising affinity and ADMET properties, we next sought to understand the binding mode of compound 23 to MEK1. Pleasingly, we were able to determine a crystal structure of 23 in complex with MEK1 to a resolution of 1.7 Å, revealing the compound to bind to the desired allosteric site of the kinase (Figure 4a). The phenyl group occupies a hydrophobic cavity formed by residues L118, F209, V127, and M143. In this orientation, the ortho and meta positions are sterically constrained—the meta particularly so—correlating well with affinity data measured for the library compounds 12 and 13 (Table 2). The methoxy group protrudes into a smaller cavity, occupied by displaceable water molecules, explaining the tolerance for slightly larger substituents in this position. The oxothiomorpholine group is positioned close to the imidodiphosphate group of AMP-PNP, forming a hydrogen bond via its nitrogen to a phosphate oxygen. 23 forms two further hydrogen bonds to the protein—one to the side chain of D208 and another to the side chain of K97 (both catalytic residues). The hydrogen bond to D208, formed from the linker amide NH of 23, appears to be a particularly key contact, with the N-methyl variant of 23 (25) essentially losing all binding affinity for the kinase. We note, however, that steric clashes and/or conformational changes may also contribute to the loss of activity of 25.
Figure 4.

(a) Crystal structure of MEK1 bound by compound 23. Comparison of binding modes of 23 (b) and cobimetinib (c) for MEK1. Parts b and c are shown in the same orientation (RMSD of overlay: 1.092 Å2 for Cαs). The structure shown in part c is for PDB entry 4AN2.1 AMP-PNP: adenylyl imidodiphosphate. AMP-PCP: adenylyl methylenediphosphonate.
Comparison of the binding mode of 23 to MEK1 to that of cobimetinib shows that the methoxy-phenyl group of 23 occupies the same position as the 2,4-dihalogen group of cobimetinib. Polar contacts to K97 and the terminal phosphate of the bound ATP analogue also appear to be a shared feature of both compounds; however, the hydrogen bond formed from 23 to the catalytic residue D208 does not feature in the binding mode of cobimetinib (Figure 4c). A broader inspection of available crystal structures of MEK1 bound by allosteric inhibitors indicates interactions with D208 to be rare. Targeting conserved catalytic residues is of particular interest as such residues would be expected to be less likely to be the site of acquired-resistance mutation in oncology settings due to their crucial enzymatic function. In addition, the presence of electron-rich sulfur atoms within allosteric MEK1 inhibitors also appears rare, and we were able to identify few examples of such compounds bound to MEK1 in the Protein Data Bank (e.g., PDB entries 3EQH(22) and 3SLS(28)). Indeed, while the scarcity of sulfur-containing MEK1 inhibitors may well be intentional due to metabolic liabilities, high systemic metabolism is a desirable property of an inhaled small-molecule therapeutic. Therefore, 23 represents a promising starting point for further optimization toward an inhaled MEK1 therapeutic inhibitor.
We have reported here a fragment-based screening campaign targeting MEK1 kinase which was designed to specifically discover novel allosteric binders by screening with the ATP binding site blocked. These efforts led to the discovery of several novel allosteric MEK1 binders. These were confirmed by X-ray crystal structures, and one series was optimized to lead compound 23 with sub-μM binding affinity and promising physicochemical and in vitro ADMET properties for the further optimization toward an inhaled small-molecule therapeutic. The same approach may likely be used to find Type III inhibitors in few other kinases. The structural origin of the existence of the allosteric pocket for binding of Type III kinase inhibitors was studied in a recent paper by Zhao et al. (2017).29 The authors predicted that, apart from the MEK proteins, this type of allosteric pocket might also exist in only another 15 human kinases.29,30
Acknowledgments
We thank Pharmaron China for supporting the synthetic work.
Glossary
Abbreviations
- SPR
surface plasmon resonance
- COPD
chronic obstructive pulmonary disease
- MEK1
dual specificity mitogen-activated protein kinase kinase 1
- AMP-PNP
adenylyl-imidodiphosphate
- FBDD
fragment-based drug discovery
- DMF
N,N-dimethylformamide
- HATU
hexafluorophosphate azabenzotriazole tetramethyl uronium
- DIPEA
N,N-diisopropylethylamine.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00563.
Full details of all materials and methods used in this work including protein production, SPR, NMR, X-ray crystallography, VCD, and chemical synthesis (PDF)
Author Contributions
All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Notes
Accession Codes: Atomic coordinates and structure factors have been deposited in the Protein Data Bank with accession codes 7B7R (4), 7B3M (6), 7B94 (9), and 7B9L (23).
Supplementary Material
References
- Rice K. D.; Aay N.; Anand N. K.; Blazey C. M.; Bowles O. J.; Bussenius J.; Costanzo S.; Curtis J. K.; Defina S. C.; Dubenko L.; Engst S.; Joshi A. A.; Kennedy A. R.; Kim A. I.; Koltun E. S.; Lougheed J. C.; Manalo J. C.; Martini J. F.; Nuss J. M.; Peto C. J.; Tsang T. H.; Yu P.; Johnston S. Novel Carboxamide-Based Allosteric MEK Inhibitors: Discovery and Optimization Efforts toward XL518 (GDC-0973). ACS Med. Chem. Lett. 2012, 3, 416–421. 10.1021/ml300049d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E. K.; Choi E. J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta, Mol. Basis Dis. 2010, 1802, 396–405. 10.1016/j.bbadis.2009.12.009. [DOI] [Google Scholar]
- Roberts P. J.; Der C. J. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007, 26, 3291–3310. 10.1038/sj.onc.1210422. [DOI] [PubMed] [Google Scholar]
- Akinleye A.; Furqan M.; Mukhi N.; Ravella P.; Liu D. MEK and the inhibitors: from bench to bedside. J. Hematol. Oncol. 2013, 6, 27. 10.1186/1756-8722-6-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caunt C. J.; Sale M. J.; Smith P. D.; Cook S. J. MEK1 and MEK2 inhibitors and cancer therapy: the long and winding road. Nat. Rev. Cancer 2015, 15, 577–592. 10.1038/nrc4000. [DOI] [PubMed] [Google Scholar]
- Cheng Y.; Tian H. Current Development Status of MEK Inhibitors. Molecules 2017, 22, 1551. 10.3390/molecules22101551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; Adjei A. A. The clinical development of MEK inhibitors. Nat. Rev. Clin. Oncol. 2014, 11, 385–400. 10.1038/nrclinonc.2014.83. [DOI] [PubMed] [Google Scholar]
- Baturcam E.; Vollmer S.; Schlüter H.; Maciewicz R. A.; Kurian N.; Vaarala O.; Ludwig S.; Cunoosamy D. M. MEK inhibition drives anti-viral defence in RV but not RSV challenged human airway epithelial cells through AKT/p70S6K/4E-BP1 signalling. Cell Commun. Signaling 2019, 17, 1–19. 10.1186/s12964-019-0378-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurian N.; Cohen T. S.; Öberg L.; De Zan E.; Skogberg G.; Vollmer S.; Baturcam E.; Svanberg P.; Bonn B.; Smith P. D.; Vaarala O.; Cunoosamy D. M. Dual Role For A MEK Inhibitor As A Modulator Of Inflammation And Host Defense Mechanisms With Potential Therapeutic Application In COPD. Int. J. Chronic Obstruct. Pulm. Dis. 2019, 14, 2611–2624. 10.2147/COPD.S211619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P.; Wu Y.; Li M.; Qiu X.; Bai X.; Zhao X. AS-703026 Inhibits LPS-Induced TNFα Production through MEK/ERK Dependent and Independent Mechanisms. PLoS One 2015, 10, e0137107. 10.1371/journal.pone.0137107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamoree B.; Hubbard R. E. Current perspectives in fragment-based lead discovery (FBLD). Essays Biochem. 2017, 61, 453–464. 10.1042/EBC20170028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopher J. A.; Aves S. J.; Bennett K. A.; Doré A. S.; Errey J. C.; Jazayeri A.; Marshall F. H.; Okrasa K.; Serrano-Vega M. J.; Tehan B. G.; Wiggin G. R.; Congreve M. Fragment and Structure-Based Drug Discovery for a Class C GPCR: Discovery of the mGlu5 Negative Allosteric Modulator HTL14242 (3-Chloro-5-[6-(5-fluoropyridin-2-yl)pyrimidin-4-yl]benzonitrile. J. Med. Chem. 2015, 58, 6653–6664. 10.1021/acs.jmedchem.5b00892. [DOI] [PubMed] [Google Scholar]
- Coutard B.; Decroly E.; Li C.; Sharff A.; Lescar J.; Bricogne G.; Barral K. Assessment of Dengue virus helicase and methyltransferase as targets for fragment-based drug discovery. Antiviral Res. 2014, 106, 61–70. 10.1016/j.antiviral.2014.03.013. [DOI] [PubMed] [Google Scholar]
- Orgován Z.; Ferenczy G. G.; Keserű G. M. Fragment-Based Approaches for Allosteric Metabotropic Glutamate Receptor (mGluR) Modulators. Curr. Top. Med. Chem. 2019, 19, 1768–1781. 10.2174/1568026619666190808150039. [DOI] [PubMed] [Google Scholar]
- Scott D. E.; Coyne A. G.; Hudson S. A.; Abell C. Fragment-based approaches in drug discovery and chemical biology. Biochemistry 2012, 51, 4990–5003. 10.1021/bi3005126. [DOI] [PubMed] [Google Scholar]
- Tiefenbrunn T.; Forli S.; Happer M.; Gonzalez A.; Tsai Y.; Soltis M.; Elder J. H.; Olson A. J.; Stout C. D. Crystallographic fragment-based drug discovery: use of a brominated fragment library targeting HIV protease. Chem. Biol. Drug Des. 2014, 83, 141–148. 10.1111/cbdd.12227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance N. R.; Gakhar L.; Spies M. A. Allosteric Tuning of Caspase-7: A Fragment-Based Drug Discovery Approach. Angew. Chem., Int. Ed. 2017, 56, 14443–14447. 10.1002/anie.201706959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C.; Zhang X.; Zhang N.; Zhou Y.; Sun G.; Zhao L.; Zhong R. Identification and Biological Evaluation of CK2 Allosteric Fragments through Structure-Based Virtual Screening. Molecules 2020, 25, 237. 10.3390/molecules25010237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoepfer J.; Jahnke W.; Berellini G.; Buonamici S.; Cotesta S.; Cowan-Jacob S. W.; Dodd S.; Drueckes P.; Fabbro D.; Gabriel T.; Groell J. M.; Grotzfeld R. M.; Hassan A. Q.; Henry C.; Iyer V.; Jones D.; Lombardo F.; Loo A.; Manley P. W.; Pellé X.; Rummel G.; Salem B.; Warmuth M.; Wylie A. A.; Zoller T.; Marzinzik A. L.; Furet P. Discovery of Asciminib (ABL001), an Allosteric Inhibitor of the Tyrosine Kinase Activity of BCR-ABL1. J. Med. Chem. 2018, 61, 8120–8135. 10.1021/acs.jmedchem.8b01040. [DOI] [PubMed] [Google Scholar]
- Amaning K.; Lowinski M.; Vallee F.; Steier V.; Marcireau C.; Ugolini A.; Delorme C.; Foucalt F.; McCort G.; Derimay N.; Andouche C.; Vougier S.; Llopart S.; Halland N.; Rak A. The use of virtual screening and differential scanning fluorimetry for the rapid identification of fragments active against MEK1. Bioorg. Med. Chem. Lett. 2013, 23, 3620–3626. 10.1016/j.bmcl.2013.04.003. [DOI] [PubMed] [Google Scholar]
- Linke P.; Amaning K.; Maschberger M.; Vallee F.; Steier V.; Baaske P.; Duhr S.; Breitsprecher D.; Rak A. An Automated Microscale Thermophoresis Screening Approach for Fragment-Based Lead Discovery. J. Biomol. Screening 2016, 21, 414–421. 10.1177/1087057115618347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller N.; Spadola L.; Cowen S.; Patel J.; Schönherr H.; Cao Q.; McKenzie A.; Edfeldt F.; Rabow A.; Goodnow R. An improved model for fragment-based lead generation at AstraZeneca. Drug Discovery Today 2016, 21, 1272–1283. 10.1016/j.drudis.2016.04.023. [DOI] [PubMed] [Google Scholar]
- The high-performance ligand–receptor docking program Glide SP (Schrodinger) and the X-ray crystallographic structure of human MEK1 with bound inhibitor RDEA119, Mg2+, and ATP (PDB entry 3E8N) were used to screen the 15 000 fragment library and identify the top 1000 virtual hits that best fit into the allosteric site.
- For SPR studies, Escherichia coli-expressed human full-length MEK1 with an N-terminal avi tag was attached to a streptavidin-coated sensor chip following biotinylation. Compounds were diluted from either 10 or 100 mM DMSO stocks to obtain a top concentration of 100 or 1000 μM. 3-fold serial dilutions were then made to obtain 7 or 10 dose–response series. AMP-PNP (1 mM) was added to each well when determining affinities in the presence of AMP-PNP.
- Stanton D. T.; Morris T. W.; Roychoudhury S.; Parker C. N. Application of nearest-neighbor and cluster analyses in pharmaceutical lead discovery. J. Chem. Inf. Comput. Sci. 1999, 39, 21–27. 10.1021/ci9801015. [DOI] [PubMed] [Google Scholar]
- Borghardt J. M.; Kloft C.; Sharma A. Inhaled Therapy in Respiratory Disease: The Complex Interplay of Pulmonary Kinetic Processes. Can. Respir. J. 2018, 2018, 1–11. 10.1155/2018/2732017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper A. E.; Ferguson D.; Grime K. Optimisation of DMPK by the inhaled route: challenges and approaches. Curr. Drug Metab. 2012, 13, 457–473. 10.2174/138920012800166571. [DOI] [PubMed] [Google Scholar]
- Meier C.; Brookings D. C.; Ceska T. A.; Doyle C.; Gong H.; McMillan D.; Saville G. P.; Mushtaq A.; Knight D.; Reich S.; Pearl L. H.; Powell K. A.; Savva R.; Allen R. A. Engineering human MEK-1 for structural studies: A case study of combinatorial domain hunting. J. Struct. Biol. 2012, 177, 329–334. 10.1016/j.jsb.2012.01.002. [DOI] [PubMed] [Google Scholar]
- Zhao Z.; Xie L.; Bourne P. E. Insights into the binding mode of MEK type-III inhibitors. A step towards discovering and designing allosteric kinase inhibitors across the human kinome. PLoS One 2017, 12, e0179936 10.1371/journal.pone.0179936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M. I.; Hunt J. P.; Herrgard S.; Ciceri P.; Wodicka L. M.; Pallares G.; Hocker M.; Treiber D. K.; Zarrinkar P. P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.