

Abstract
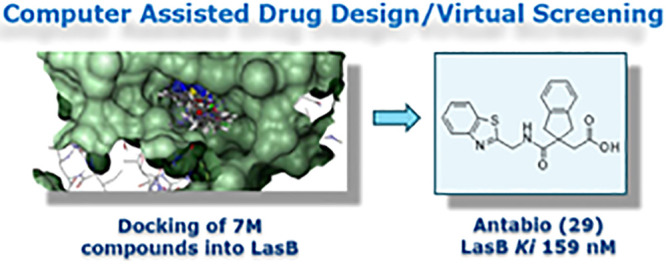
Novel therapies are required to treat chronic bacterial infections in cystic fibrosis (CF) sufferers. The most common pathogen responsible for these infections is Pseudomonas aeruginosa, which persists within the lungs of CF sufferers despite intensive antibiotic treatment. P. aeruginosa elastase (also known as LasB or pseudolysin) is a key virulence determinant that contributes to the pathogenesis and persistence of P. aeruginosa infections in CF patients. The crucial role of LasB in pseudomonal virulence makes it a good target for the development of an adjuvant drug for CF treatment. Herein we discuss the discovery of a new series of LasB inhibitors by virtual screening and computer assisted drug design (CADD) and their optimization leading to compounds 29 and 39 (Ki = 0.16 μM and 0.12 μM, respectively).
Keywords: Cystic fibrosis, Pseudomonas aeruginosa elastase, virtual screening, LasB inhibitors
Cystic fibrosis (CF) is a progressive genetic disease due to inherited mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene, which cause reduced mucociliary clearance in the lungs and commonly results in persistent bacterial lung infections that are recalcitrant to antibiotic treatments. CF is the most common lethal hereditary disease in Caucasian populations. The median incidence of CF among newborns across Europe is 1 in every 3500 births.1 In Europe (35 countries), there were approximately 48000 people living with CF in 2017, whereas 31999 CF patients were registered in the US in 2019.2,3 In recent years, there have been major advances in CF care, including better palliative care and the introduction of CFTR modulator therapies, that have led to improved survival and an increase in median life expectancy to 46 years (www.cff.org);3 however, there is still a critical need for further advances to improve survival and reduce the burden of the disease, particularly with regard to the treatment of bacterial infections.
Most disease-related morbidity and mortality in CF is caused by progressive lung disease as a result of bacterial infection and airway inflammation. The dominant bacterial pathogen colonizing and infecting the lungs of CF patients is the Gram-negative organism Pseudomonas aeruginosa, which is the most common cause of chronic lung infection in CF patients affecting up to 43–46% of the adult CF population.2,3 As the lung becomes colonized, P. aeruginosa is able to adapt to its environment and the pathogen’s capacity for long-term survival improves. A key adaptation is the switch to a biofilm mode of growth on mucosal surfaces and in lung fluids, frequently characterized by production of large quantities of exopolysaccharide material (presenting as a mucoid phenotype), which reduces the effectiveness of phagocytosis and antibiotic therapy.4 The inability to clear the pathogen, even after intensive antibiotic therapy, and the consequent neutrophil-driven innate immune response of the host feed into a decline of lung tissue integrity and function that ultimately necessitates transplantation in later disease stages.
Patients who become colonized with P. aeruginosa show a more rapid decline in lung function and chest radiograph score, reduced weight gain (in children), increased hospitalization rates, and an increased reliance on antibiotic therapy to control the extent of infection.4−6 Median survival is reduced and mortality is increased, resulting in a 2.6-fold increased risk of death.5
P. aeruginosa elastase B (LasB or pseudolysin) is a secreted metalloprotease virulence factor, expressed under the control of a quorum-sensing regulatory mechanism, which contributes to the pathogenesis and persistence of the P. aeruginosa infection. LasB degrades many host proteins involved in both lung structural integrity and the immune response to infection.7,8
The key role of LasB in pseudomonal virulence makes it a potential target for the development of an inhibitor as an adjunct therapy to an antibacterial agent. The concept of inhibiting virulence is an established antimicrobial strategy, but one that has yet to be validated clinically. This approach holds promise in that it seeks to attenuate virulence processes without bactericidal action and, hence, is anticipated to exhibit reduced selective pressure on the emergence of resistant strains. LasB is an attractive target for such a novel antivirulence based therapy as it is expressed as an active protease outside of the bacterial cell and belongs to a target class that is tractable to pharmacological inhibition. For example, protease inhibitors have been the mainstay of antiviral therapy against HIV for over two decades, and protease inhibitors have found clinical application in other antiviral areas, cancers, inflammation, respiratory diseases, and cardiovascular disorders.9,10 Therefore, there is a strong rationale to support that a selective LasB inhibitor drug delivered to the site of lung infection could complement existing antibiotic approaches.11
To date, several structural classes of LasB inhibitors have been reported in the literature, including nonpeptidic compounds.12,13N-Mercaptoacetyl peptides have been described by Cathcart et al. in 2011,14 with the most potent compound, N-mercaptoacetyl-Phe-Tyr-amide, exhibiting Ki = 41 nM against LasB, similar to phosphoramidon (a well characterized metalloprotease inhibitor) (Figure 1).15 However, both N-mercaptoacetyl-Phe-Tyr-amide and phosphoramidon show poor selectivity with respect to mammalian metalloenzymes.16
Figure 1.

Phosphoramidon and N-mercaptoacetyl-Phe-Tyr-amide
A novel series of LasB inhibitors was identified through the employment of two complementary virtual screening approaches based on protein structure or ligand conformation. Starting from the publicly available crystal structure of P. aeruginosa LasB complexed with phosphoramidon (PDB ID 3DBK(17)), structure- and ligand-based virtual screens were performed utilizing a 7 million compound external vendor catalogue as the source of commercially available chemical diversity.
Structure-Based Approach
The 3DBK protein structure was prepared using the Protein Preparation Wizard18 in Maestro (version 2015-419). Missing side chains were added using Prime, protonation states were assigned using LigPrep and Epik using a pH value of 7.0, crystallographic waters were removed, and the protein was minimized to RMSD of 0.30. A pharmacophore model was built using Phase20 and included (i) a Zn2+ metal binding motif (deemed essential), (ii) hydrogen bonds to Arg198, Asn112, or Glu111 (at least one required), and (iii) a hydrophobic element to mimic the isobutyl group of phosphoramidon. The pharmacophore was searched against the vendor catalogue, and the resultant hits were docked into 3DBK using Glide.21 Strain energy calculations were performed to remove hits with unfavorable poses (strain energy >4 kcal/mol) before visual inspection of each pose leading to the selection of 120 compounds for purchase.
Ligand-Based Approach
A ligand-based pharmacophore was developed around phosphoramidon using ROCS.22,23 As in the structure-based virtual screen, a Zn2+ motif was deemed essential, and other structural features around phosphoramidon were mapped onto the query. Following shape screening using ROCS, molecules with a Tanimoto_combo score of >1.2 were docked into the 3DBK structure using Glide, assessed for strain energy, and then visually inspected. Using this approach, a further 113 compounds were selected.
Of the 233 compounds identified as of interest from both approaches, 225 compounds were available for purchase, and these were screened at a single concentration (30 μM) for inhibition of LasB in a fluorometric assay using purified LasB enzyme. Six compounds were found to exhibit at least 40% LasB inhibition and were subsequently retested across a range of concentrations (0.4–200 μM) in a dose–response assay, confirming LasB inhibitory activity (IC50 < 50 μM) for four of them. Selectivity studies with rabbit-derived angiotensin converting enzyme (ACE) confirmed that all four LasB inhibitors were inactive against this enzyme (IC50 > 200 μM). One of these four compounds was discarded because of the presence of a hydroxamate functional group, a known zinc-binding group likely to cause promiscuity among other metalloenzymes such as MMPs and therefore liable to cause toxicity issues later in development.24 The structures of the three remaining initial hits, 1, 2, and 3, were confirmed by LCMS and 1H NMR analyses and are shown in Figure 2. Hit 3 was purchased as a racemic mixture. Notably, all three hits were identified following the structure-based virtual screening approach. Corresponding docking modes for these compounds modeled into the LasB binding site and the binding mode of phosphoramidon cocrystallized with LasB protein are presented in Figure 3.
Figure 2.

Structures of three confirmed hits identified from virtual screening.
Figure 3.

Binding modes of phosphoramidon from PDB 3DBK and potential binding modes of compounds 1–3. Docking was performed with Glide (Glide, Schrödinger, LLC, New York, NY, 2015) and the proprietary LasB X-ray structure. All images were created using Maestro (Maestro, Schrödinger, LLC, New York, NY, 2015). (A) Binding mode of phosphoramidon from PDB 3DBK. This image is derived from a proprietary Pseudomonas aeruginosa elastase complex with phosphoramidon solved to a resolution of 1.5 Å. (B) Docking mode of hit compound 1. (C) Docking mode of hit compound 2. (D) Docking mode of hit compound 3.
Compound 1 exhibited modest affinity for LasB (Ki = 18.1 μM, Table 1), probably due to lack of interaction with the Zn2+ binding site of LasB. Indeed, the 1,5-dihydroimidazol-4-one group is an unusual zinc binding group and is likely to bind through a weak interaction via the carbonyl oxygen and adjacent nitrogen of the imidazolone ring (Figure 3B). Additionally, the amide carbonyl potentially creates an important contact with the Arg198 residue.
Table 1. Hit Compounds Identified after First Round Screen.
| compound no. | 1 | 2 | 3 |
| LasB Ki (μM) | 18.1 | 6.87 | 14.8 |
| ACE Ki (μM) | >57 | >57 | >57 |
| follow-ups identified | 15 | 91 | 47 |
| follow-ups selected and tested | 10 | 36 | 18 |
| follow-ups withLasB inhibition >40% @ 30 μM | 3 | 9 | 2 |
Compounds 2 and 3 also showed modest LasB inhibition (Ki = 6.87 μM and 14.8 μM, respectively, Table 1). These are both carboxylic acids, with the carboxylate binding to the Zn2+ through bidentate interactions. The carbonyl functional group of 2 is likely to interact with the Arg198 residue, whereas 3 shows the same interaction through the oxygen atom of the benzofuran ring.
Interestingly, docking studies revealed similar interactions with the lipophilic pocket S1′ of the binding site for all three compounds, involving the 4-bromophenyl, bis-fluorobenzyl, or benzofuran rings of 1, 2 and 3, respectively.
Using the three hits identified from the first round of screening as starting points (Table 1), 153 commercially available analogues were identified, of which 64 were purchased for a second round of testing. The follow-up compounds for 1 were chosen to investigate alternative groups to the 4-bromo-2-methylphenyl, while maintaining the 1,5-dihydroimidazol-4-one group as a potential zinc binding group. For 2, the follow-up compounds were selected in order to explore substituents identified as interacting with (i) the S1′ (bis-fluorobenzyl) substituent and (ii) the S2′ (2-methylphenoxy) lipophilic pockets of the LasB binding site. In contrast, follow-ups for 3 were selected with an emphasis on those bearing alternatives to the indane ring system.
All 64 follow-up compounds were tested in duplicate at a single concentration (30 μM). Fourteen compounds showed >40% LasB inhibition, of which three were analogues of 1, two were analogues of 3 and the remaining nine were analogues of 2. As for the initial screening, these 14 compounds were retested in a LasB dose–response assay and in the ACE selectivity assay.
The series of follow-up compounds corresponding to 1 generally exhibited lower LasB inhibition compared to the other two series. Subsequent testing of 1 in a modified LasB assay, in which a low concentration of Triton X-100 (0.01%) was included in the assay buffer, revealed a dramatic loss of inhibitory activity in the presence of detergent, illustrating that 1 was most likely a “promiscuous” hit without genuine catalytic inhibitory activity. Consequently, this series was discarded.
The series related to 2 showed a flat structure–activity relationship (SAR), with all compounds inhibiting LasB to a similar degree and hence little opportunity to optimize the series. In contrast, the indane carboxylic acid series related to 3 showed more potential for structural optimization and offered greater opportunities to develop a novel proprietary series.
Further Investigation of the Indane Carboxylic Acid Series
Following the identification of hit 3 and the encouraging LasB inhibitory activity observed with two of the commercially available analogues, the synthesis of additional analogues of 3 was undertaken in order to confirm the promising properties of this new chemical series.
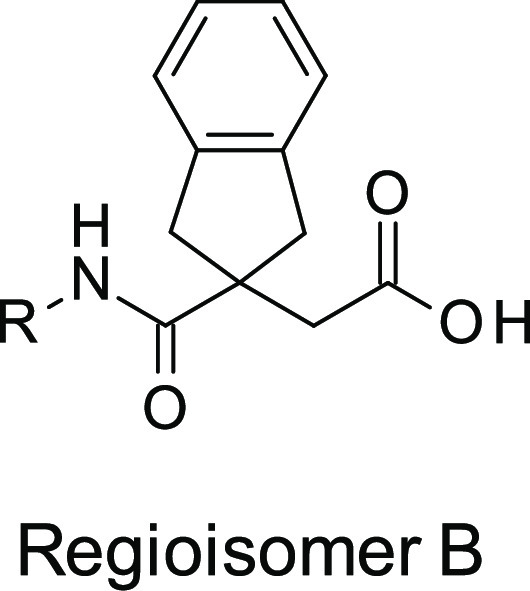
At the beginning of any medicinal chemistry program, it is important to evaluate alternative subseries in order to confirm the best starting point for lead optimization. Hence, it was decided to explore widely around 3, for example, considering the isomeric subseries where the amide linker is next to the indane ring system (named as regioisomer B). Accordingly, we applied “non-regioselective chemistry” in order to produce both regiosiomers from a single reaction (method A, Scheme 1). In this method, the diacid 4 was cyclized with acetyl chloride to produce the corresponding anhydride 5, following literature procedures.25 Reopening the anhydride 5 with various amines yielded mixtures of regioisomer A (6) and regioisomer B (7), which were subsequently separated and purified by standard column chromatography.
Scheme 1. Non-regioselective Ring Opening of Anhydride 4 (Method A).

Both regioisomers were then tested for LasB inhibitory activity using two complementary assays: (i) a LasB inhibition fluorometric assay adapted from Zhu et al.26 and (ii) an elastolytic assay that measures inhibition of the elastolytic activity of secreted LasB in the supernatant of P. aeruginosa cultures, using elastin congo-red (ECR) as substrate. In the ECR elastolytic assay, LasB degrades elastin and releases the congo-red dye into the supernatant, which can then be measured spectrophotometrically; this assay thus uses a more physiological substrate as well as a more physiological preparation of the enzyme, thereby providing greater confidence in the relevance of the observed LasB inhibitory activity (Table 2).
Table 2. LasB Inhibitory Activities of Matched Regiosiomer A and B Pairs from Method Aa.


ND, not determined.
Initially, the indane anhydride 5 was opened nonregioselectively with (5-methylbenzofuran-2-yl) methanamine in order to investigate the impact of the methyl substituent at the α-position of the amide. The two regioisomers 8 and 9 were obtained in a 1:3 approximate ratio, in favor of 9, and were separated by silica gel chromatography.27 Interestingly, 9 (initially the undesired regioisomer, corresponding to regioisomer B) demonstrated greatly superior LasB inhibitory activity (Ki = 0.45 μM and 60% inhibition of elastin hydrolysis at 50 μM) compared to the other regioisomer 8, belonging to the original series of 3 (regioisomer A), which was much less active (Ki > 92 μM). Encouraged by these results, other matched pairs of compounds were synthesized from commercial primary amines to verify that the subseries of regioisomer B was of greater interest for future optimization. This was confirmed for all primary amines investigated. For example, using (1-methylpyrazol-4-yl)methanamine or 2-quinolylmethanamine led, respectively, to 10 (Ki = 4.04 μM) and 11 (Ki = 0.610 μM), which were significantly more potent than their corresponding regiosiomers 12 (Ki > 92 μM) and 13 (Ki = 76.2 μM). At this point in the program, the regioisomer A subseries was abandoned, and all efforts were dedicated to the development of the regioisomer B subseries.
Alternative chemistry was required to develop a synthetic route that would permit the preparation of compounds belonging exclusively to the regioisomer B subseries. The first approach (method B) was to react the anhydride 5 with bulky alcohols, such as benzyl alcohol or 3,4-dimethoxybenzyl alcohol, aiming to obtain a single regioisomer; however, this produced a regioisomeric mixture of isomers (10:1 ratio in favor of the desired ester) that could nevertheless be separated by standard column chromatography or recrystallization, allowing isolation of 14. Amide formation was then performed by standard amide coupling methods to give 15 followed by ester removal by hydrogenation affording regioisomer B (7) (Scheme 2). This approach was used to synthesize the benzofuran analogue 16 as a baseline reference compound to compare to 3, 8, and 9. Despite the fact that the methyl substituent at the 5-position of the benzofuran in 9 led to 3-fold improvement of LasB inhibition compared to the unsubstituted analogue 16 (Table 3), it was decided to work without aromatic substituents in the first instance in order to simplify the synthesis of an initial set of analogues. Although this method B strategy was regioselective to an extent and allowed the preparation of the regioisomer B, it was not truly regiospecific since it still required purification of the two regioisomers obtained after the first step. Consequently, other approaches were investigated.
Scheme 2. Regioselective Synthesis of Regioisomer B (Method B).

(a) Benzyl alcohol, toluene, 100 °C, overnight; (b) R-NH2, HATU, DIPEA, DMF, rt, 2 days; (c) H2, Pd/C.
Table 3. LasB Inhibitory Activities of Regiosiomers B from Methods B and C.
Finally, a fully regiospecific synthesis was developed (method C) starting from methyl ester 17 (Scheme 3).28 Ester 17 was deprotonated with sodium hexamethyldisilazide; then alkylation of the anion with tert-butyl bromoacetate gave the known diester 18.25,27,29 Basic hydrolysis of the methyl ester in the presence of the tert-butyl ester afforded acid 19. Amide formation was performed by standard amide coupling methods to give 20 and the regioisomer B analogues were obtained by ester removal with trifluoroacetic acid.
Scheme 3. Regiospecific Synthesis of Regioisomer B (Method C).

(a) NaHMDS, tert-butyl bromoacetate, THF, −60 °C, 1 h; (b) KOH, EtOH/H2O 2:1, rt, 3.5 h; (c) R-NH2, HATU, DIPEA, DMF, rt, 2 days; (d) TFA, DCM, rt, 1 h.
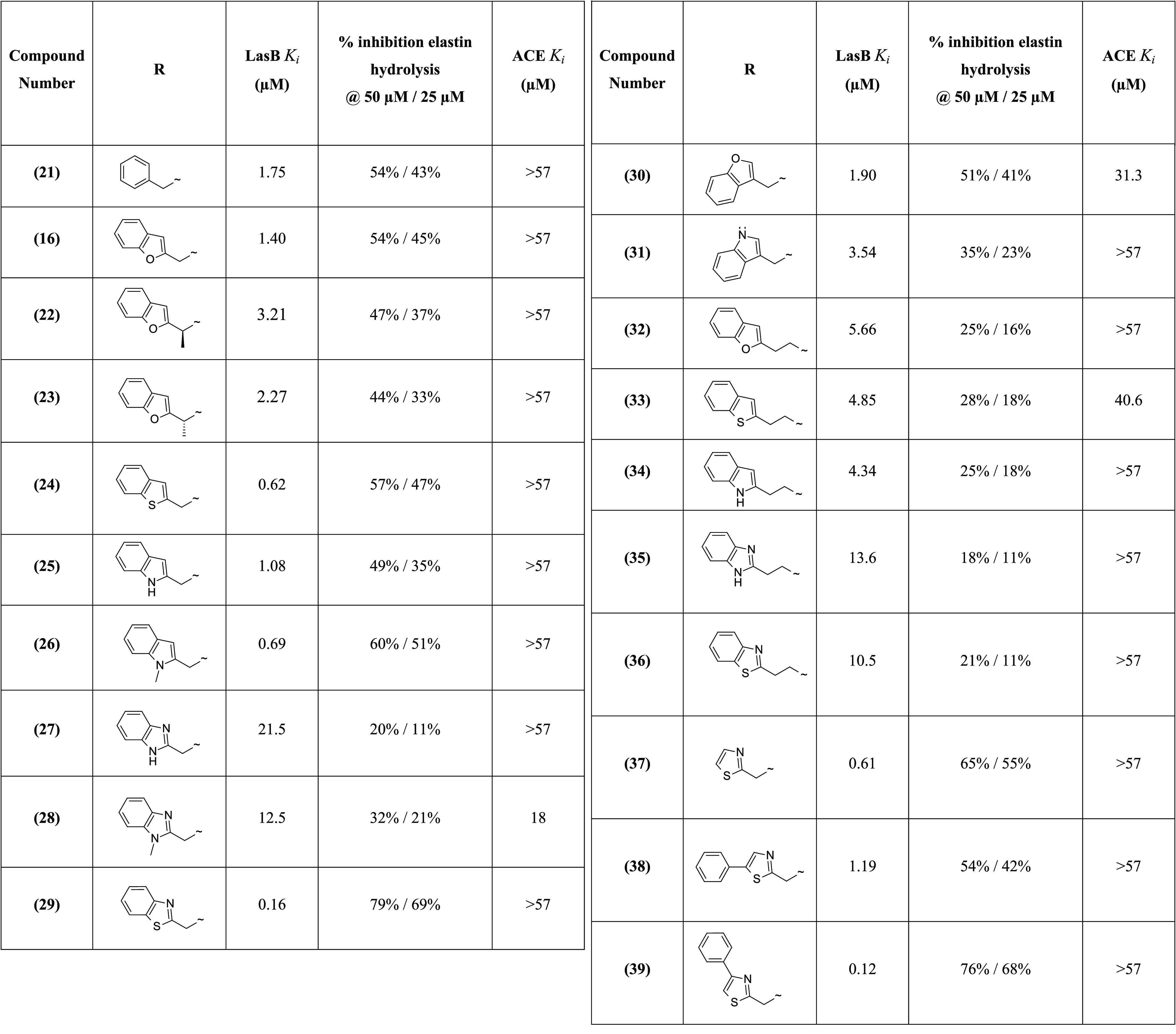
The first compound synthesized using method C was the benzylamide derivative 21, considered as “baseline” compound, allowing a simple comparison to hit 3, with the phenyl ring system providing a starting point for optimization. Although the phenyl ring improved LasB inhibition by approximately 2-fold compared to the simple N-methylpyrazole 10, 21 showed slightly lower activity (Ki = 1.75 μM) compared to 9 and 11 (Tables 2 and 3), with 5-methylbenzofuran and quinoline bis-aromatic groups, respectively, replacing phenyl. These first results led us to investigate bis-aromatic systems.
It was noted that introduction of a methyl substituent at the α position of the benzofuran of 16 reduced the LasB inhibition. The (S)- and (R)-enantiomers 22 and 23 were specifically synthesized (Scheme 4), and the LasB inhibitory activity of these compounds was decreased by 2-fold (Ki = 3.21 μM and 2.27 μM, respectively) compared to 16 (Table 3).
Scheme 4. Synthesis of 22 and 23.

(a) NH2OH·HCl, NaOH, EtOH/H2O, rt, 1 h; (b) Zn, AcOH, rt, 16 h; (c) 19, EDC·HCl, HOBt, TEA, DMF, rt, 16 h; (d) TFA, TEA, DCM, rt, 1 h.
Bis-aromatic systems such as simple benzothiophene 24, indole 25, and N-methylindole 26 analogues were then synthesized using method C. Interestingly, benzothiophene 24 (Ki = 0.62 μM) and N-methylindole 26 (Ki = 0.69 μM) showed improved LasB inhibition, compared to benzofuran and indole analogues 16 and 25, by 2-fold in both the fluorometric and ECR elastolytic assays (Table 3).
Additionally, other bis-aromatic groups with two heteroatoms instead of one in the ring system were investigated. The first attempts to synthesize the N-methylbenzimidazole analogue 28 using method C failed due to the poor quality of the commercial reagent (1-methyl-1H-benzo[d]imidazol-2-yl)methanamine); this reagent was then synthesized as previously reported.27 The addition of an extra nitrogen atom into 25 and 26, with benzimidazole and N-methylbenzimidazole analogues 27 and 28, was detrimental to the LasB inhibitory activity, causing ∼20-fold reduction of inhibition (Table 3). In contrast, the benzothiazole analogue 29 showed greater LasB inhibitory activity compared to 24, with a 4-fold improvement of inhibition after introduction of the nitrogen atom into the benzothiophene ring. Compound 29 exhibited excellent potency (Ki = 0.16 μM) confirmed by a level of elastin hydrolysis inhibition not achieved thus far (79% inhibition at 50 μM) (Table 3).
The linker between the amide functionality and the bis-aromatic ring system was also modified, initially with a series of simple derivatives for which the linker was attached at the C-2 of the ring system instead of the C-1. However, this led to a decrease in LasB inhibition for the benzofuran 30 and the indole 31 analogues. Results were similar for a series of analogues with different lengths of the linker (Table 3). For example, the two-carbon linker derivatives showed lower LasB inhibition than the corresponding one-carbon analogues for benzofuran 32, benzothiophene 33, indole 34, benzimidazole 35, and benzothiazole 36 analogues, compared to analogues 16, 24, 25, 27, and 29, respectively (Table 3).
Following the excellent LasB inhibitory activity of benzothiazole analogue 29, additional derivatives bearing the thiazole scaffold were synthesized using method C.28 For the synthesis of 39, the corresponding amino reagent 40 was generated by reaction between thioamide 41 and 2-bromo-1-phenyl-ethanone followed by Boc cleavage (Scheme 5). Significantly, LasB inhibition was maintained for the simple thiazole analogue 37, with Ki = 0.61 μM, showing only a 4-fold reduction compared to 29 (Table 3). Interestingly, the two phenylthiazole derivatives showed different activities against the targeted protein depending on the position of the phenyl substituent on the thiazole ring. Indeed, the potency of 5-phenyl-thiazole analogue 38 (Ki = 1.19 μM) was decreased 10-fold compared to the 4-phenyl-thiazole analogue 39 (Ki = 0.12 μM) (Table 3).
Scheme 5. Synthesis of 39.

(a) 2-Bromo-1-phenyl-ethanone, EtOH, rt, 16 h; (b) TFA, DCM, reflux, 1 h; (c) 16, HATU, DIPEA, DMF, rt, 2 h; (d) TFA, DCM, rt, 2.5 h.
The majority of the compounds in this chemical series showed selectivity for LasB over ACE (Table 3). This was most apparent for the two most potent LasB inhibitors 29 and 39, which exhibited greater than 356-fold selectivity. The selectivity of these two compounds was also evaluated with respect to three human matrix metalloproteinases, MMP-1, -2, and -13, since these proteases perform a variety of roles in pulmonary immunity and hence their inhibition could have undesirable (or at least uncertain) consequences. MMP IC50 values were all >10 μM indicating a high degree of selectivity also against these metalloenzymes.
A crystal structure of the LasB–29 complex was obtained at a maximum resolution of 1.75 Å (see Supporting Information for methods and data collection and refinement statistics). As anticipated, compound 29 was found to bind to a Zn2+ atom in the binding site of LasB through its carboxylic acid functionality. In addition, the indane moiety was found to sit in the S1′ lipophilic pocket as hypothesized from additional docking studies undertaken on the regioisomer B sub-series (Figure 4). Stabilizing interactions were found between compound 29 and certain LasB residues, namely, (i) between the oxygen and nitrogen atoms of the amide, forming a strong bidentate interaction to Arg198 and Asn112, respectively, (ii) the benzothiazole sitting in a groove adjacent to the S2′ pocket, stabilized by π-stacking interaction to Phe129 (Figure 5), and (iii) the nitrogen atom of the benzothiazole ring, which forms part of a water-mediated network extending to the base of the shallow S2′ pocket. As shown in Figure 6, when 29 is overlaid with phosphoramidon (PDB ID 3DBK(17)), the indole ring and the isobutyl group of phosphoramidon are, respectively, superposed with the benzothiazole and the indane rings of 29, this latter group having a more extensive interaction with the S1′ lipophilic pocket of the LasB binding site compared to the isobutyl.
Figure 4.

View of the LasB active site with (A) and without (B) compound 29.
Figure 5.

Key interactions between 29 and LasB binding site.
Figure 6.
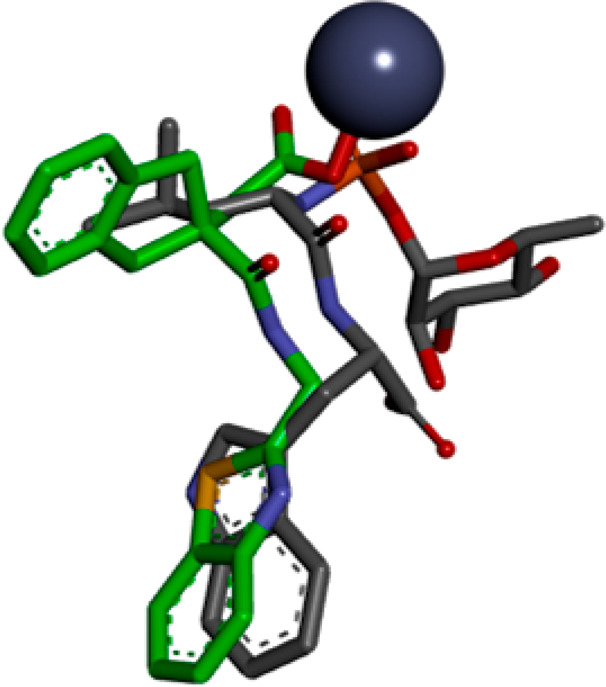
Compound 29 (green) overlaid with phosphoramidon (gray) within the LasB binding site.
In summary, a rational approach involving virtual screening of 7 million commercially available compound structures and supported by computer assisted drug design (CADD) led to the identification of hit 3 and ultimately to the discovery of a novel indane carboxylate LasB inhibitor series. The LasB inhibitory activity of this series was then improved through medicinal chemistry approaches. After the synthesis of several compound pairs by nonregioselective chemistry, the initial subseries of hit 3 analogues was abandoned in favor of its corresponding regioisomer B subseries, which consistently demonstrated superior LasB inhibition. This regioisomer B subseries was then further improved by investigating alternative aromatic ring systems and linkers, leading to the identification of compounds 29 and 39, which exhibited excellent LasB inhibition against both purified and secreted LasB preparations. Indane carboxylic acid 29 was successfully cocrystallized with LasB protein, and X-ray crystallography data confirmed that the compound forms key interactions in the LasB active site.
This novel and tractable series of LasB inhibitors, represented by compounds 29 and 39, shows strong potential for further optimization of both LasB inhibition (aided by X-ray crystallographic data) and physicochemical properties (solubility, stability, ADME, and pharmacokinetic profile) with the aim of delivering a novel antivirulence drug candidate for the treatment of P. aeruginosa infections in cystic fibrosis patients.
Acknowledgments
We are grateful to Dr. John Dixon and Dr. Simon Hodgson for constructive discussion relating to the development of the program. We are grateful to Charles River Laboratories for the molecular modelling support, including virtual screening, docking experiments, and X-ray crystallography, and also for purification of the LasB protein for biological studies.
Glossary
Abbreviations
- Boc
tert-butyloxycarbonyl
- HATU
1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate
- NaHMDS
sodium bis(trimethylsilyl)amide
- DCM
dichloromethane
- EDC
N-(3-(dimethylamino)propyl)-N′-ethylcarbodiimide hydrochloride
- HOBt
hydroxybenzotriazole
- TEA
triethylamine
- DIPEA
N,N-diisopropylethylamine
- DMF
N,N-dimethylformamide
- THF
tetrahydrofuran
- TFA
trifluoroacetic acid
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00554.
General methods for the synthesis and characterization of all compounds, methods for enzymatic assays, and X-ray crystallography (PDF)
Accession Codes
The coordinates and structure factors of LasB in complex with compound 29 were deposited to the Protein Data Bank under accession code 7AJR.
Author Contributions
S.L., D.D., J.S., and D.P. designed the molecules and performed the medicinal chemistry; M.B. performed the docking studies and the computer assisted virtual screening; J.S., T.M., M.J., A.F., P.K., S.K.M., B.P., R.R., V.K., N.P., and S.P. synthesized the molecules; N.S., J.C., L.B., M.L., and M.E. devised the biological screening strategies and tested the molecules; M.B. generated and analyzed the X-ray structure data; S.L., D.D., and M.E. wrote the paper.
We thank the Wellcome Trust for funding this program (application no. 106310/Z/14/).
The authors declare no competing financial interest.
Supplementary Material
References
- Southern K. W.; Munck A.; Pollitt R.; Travert G.; Zanolla L.; Dankert-Roelse J.; Castellani C. A survey of newborn screening for cystic fibrosis in Europe. J. Cystic Fibrosis 2007, 6, 57–65. 10.1016/j.jcf.2006.05.008. [DOI] [PubMed] [Google Scholar]
- European cystic fibrosis society report 2017. https://www.ecfs.eu/sites/default/files/general-content-files/working-groups/ecfs-patient-registry/At-a-Glance_2017_ECFSPR.pdf. (accessed on January 11, 2021).
- Cystic fibrosis foundation patient registry highlights 2019. https://www.cff.org/Research/Researcher-Resources/Patient-Registry/Cystic-Fibrosis-Foundation-Patient-Registry-Highlights.pdf. (accessed on January 11, 2021).
- Gaspar M. C.; Couet W.; Olivier J. C.; Pais A. A.; Sousa J. J. Pseudomonas aeruginosa infection in cystic fibrosis lung disease and new perspectives of treatment: a review. Eur. J. Clin. Microbiol. Infect. Dis. 2013, 32, 1231–1252. 10.1007/s10096-013-1876-y. [DOI] [PubMed] [Google Scholar]
- Hauser A. R.; Jain M.; Bar-Meir M.; McColley S. A. Clinical significance of microbial infection and adaptation in cystic fibrosis. Clin. Microbiol. Rev. 2011, 24, 29–70. 10.1128/CMR.00036-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoiby N. Recent advances in the treatment of Pseudomonas aeruginosa infections in cystic fibrosis. BMC Med. 2011, 9, 32. 10.1186/1741-7015-9-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Zhao H.-L.; Ran L.-Y.; et al. Mechanistic insights into elastin degradation by pseudolysin, the major virulence factor of the opportunistic pathogen Pseudomonas aeruginosa. Sci. Rep. 2015, 5, 9936. 10.1038/srep09936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez-Cuartin G.; Smith A.; Abo-Leyah H.; Rodrigo-Troyano A.; Perea L.; Vidal S.; Plaza V.; Fardon T. C.; Sibila O.; Chalmers J. D. Anti-Pseudomonas aeruginosa IgG antibodies and chronic airway infection in bronchiectasis. Respir. Med. 2017, 128, 1–6. 10.1016/j.rmed.2017.05.001. [DOI] [PubMed] [Google Scholar]
- Sable P. N. Protease Inhibitors: A review. Indian Drugs 2013, 50, 5–19. [Google Scholar]
- Leung D.; Abbenante G.; Fairlie D. P. Protease inhibitors: Current status and future prospects. J. Med. Chem. 2000, 43, 305–341. 10.1021/jm990412m. [DOI] [PubMed] [Google Scholar]
- Galdino A. C. M.; Viganor L.; De Castro A. A.; Da Cunha E. F. F.; Mello T. P.; Mattos L. M.; Pereira M. D.; Hunt M. C.; O’Shaughnessy M.; Howe O.; Devereux M.; McCann M.; Ramalho T. C.; Branquinha M. H.; Santos A. L. S. Disarming Pseudomonas aeruginosa virulence by the inhibitory action of 1,10-phenanthroline-5,6-dione-based compounds: elastase B (LasB) as a chemotherapeutic target. Front. Microbiol. 2019, 10, 1701. 10.3389/fmicb.2019.01701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garner A. L.; Struss A. K.; Fullagar J. L.; Agrawal A.; Moreno A. Y.; Cohen S. M.; Janda K. D. 3-Hydroxy-1-alkyl-2-methylpyridine-4(1H)-thiones: Inhibition of the Pseudomonas aeruginosa Virulence Factor LasB. ACS Med. Chem. Lett. 2012, 3, 668–672. 10.1021/ml300128f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullagar J. L.; Garner A. L.; Struss A. K.; Day J. A.; Martin D. P.; Yu J.; Cai X.; Janda K. D.; Cohen S. M. Antagonism of a zinc metalloprotease using a unique metal-chelating scaffold: tropolones as inhibitors of P. aeruginosa elastase. Chem. Commun. 2013, 49, 3197–3199. 10.1039/c3cc41191e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cathcart G. R.; Quinn D.; Greer B.; Harriott P.; Lynas J. F.; Gilmore B. F.; Walker B. Novel inhibitors of the Pseudomonas aeruginosa virulence factor lasB: A potential therapeutic approach for the attenuation of virulence mechanisms in pseudomonal infection. Antimicrob. Agents Chemother. 2011, 55, 2670–2678. 10.1128/AAC.00776-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morihara K.; Tsuzuki H. Phosphoramidon as an inhibitor of elastase from Pseudomonas Aeruginosa. Jpn. J. Exp. Med. 1978, 48, 81–84. [PubMed] [Google Scholar]
- In house inhibitory activity against ACE obtained for phosphoramidon: Ki (ACE) = 3.3 μM.
- Overgaard M. T.; McKay D. B.. Structure of the elastase of Pseudomonas aeruginosa complexed with phosphoramidon. RCSB Protein Data Bank. 10.2210/pdb3dbk/pdb, deposited June 1, 2008. [DOI]
- Madhavi Sastry G. M.; Adzhigirey M.; Day T.; Annabhimoju R.; Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided Mol. Des. 2013, 27, 221–234. 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- Schrödinger, Release 2015-4, Schrödinger, LLC, New York, NY, 2015.
- Dixon S. L.; Smondyrev A. M.; Rao S. N. Phase: A novel approach to pharmacophore modeling and 3D database searching. Chem. Biol. Drug Des. 2006, 67, 370–372. 10.1111/j.1747-0285.2006.00384.x. [DOI] [PubMed] [Google Scholar]
- Friesner R. A.; Banks J. L.; Murphy R. B.; Halgren T. A.; Klicic J. J.; Mainz D. T.; Repasky M. P.; Knoll E. H.; Shaw D. E.; Shelley M.; Perry J. K.; Francis P.; Shenkin P. S. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- ROCS 3.2.0: OpenEye Scientific Software, Santa Fe, NM, http://www.eyesopen.com (accessed on January 11, 2021).
- Hawkins P. C. D.; Skillman A. G.; Nicholls A. Comparison of shape-matching and docking as virtual screening tools. J. Med. Chem. 2007, 50, 74–82. 10.1021/jm0603365. [DOI] [PubMed] [Google Scholar]
- Shen S.; Kozikowski P. A. Why hydroxamates may not be the best histone deacetylase inhibitors – what some may have forgotten or would rather forget?. ChemMedChem 2016, 11, 15–21. 10.1002/cmdc.201500486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell I. M.; Stump C. A.. Monocyclic anilide spirolactam CGRP receptor. WO200629153, 2006.
- Zhu J.; Cai X.; Harris T. L.; Gooyit M.; Wood M.; Lardy M.; Janda K. D. Disarming Pseudomonas aeruginosa virulence factor lasB by leveraging a Caenorhabditis elegans infection model. Chem. Biol. 2015, 22, 483–491. 10.1016/j.chembiol.2015.03.012. [DOI] [PubMed] [Google Scholar]
- Leiris S.; Davies D. T.; Everett M.; Sprynski N.; Sutton J. M.; Bodnarchuk M. S.; Pallin T. D.; Cridland A. P.; Blench T. J.; Clark D. E.; Elliott L. R.. Chemical compounds as antibiotics. WO2018172423, 2018.
- Tomiyama T.; Wakabayashi S.; Yokota M. Synthesis and biological activity of novel carbacyclins having bicyclic substituents on the omega-chain. J. Med. Chem. 1989, 32, 1988–1996. 10.1021/jm00128a049. [DOI] [PubMed] [Google Scholar]
- Robinson R. P.; Ragan J. A.; Cronin B. J.; Donahue K. M.; Lopresti-Morrow L. L.; Mitchell P. G.; Reeves L. M.; Yocum S. A. Inhibitors of MMP-1: an examination of P1′ Cα gem-disubstitution in the succinamide hydroxamate series. Bioorg. Med. Chem. Lett. 1996, 6, 1719–1724. 10.1016/0960-894X(96)00303-4. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Overgaard M. T.; McKay D. B.. Structure of the elastase of Pseudomonas aeruginosa complexed with phosphoramidon. RCSB Protein Data Bank. 10.2210/pdb3dbk/pdb, deposited June 1, 2008. [DOI]