

Abstract
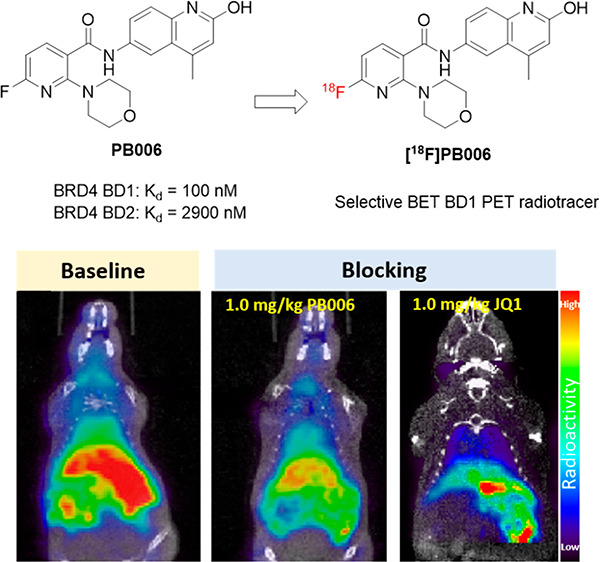
In this paper, we report the design, synthesis, and biological evaluation of the first selective bromodomain and extra-terminal domain (BET) BD1 bromodomains of the PET radiotracer [18F]PB006. The standard compound PB006 showed high affinity and good selectivity toward BRD4 BD1 (Kd = 100 nM and 29-fold selectively for BD1 over BD2) in an in vitro binding assay. PET imaging experiments in rodents were performed to evaluate the bioactivity of [18F]PB006 in vivo. A biodistribution study of [18F]PB006 in mice revealed high radiotracer uptake in peripheral tissues, such as liver and kidney, and moderate radiotracer uptake in the brain. Further blocking studies demonstrated the significant radioactivity decreasing (20–30% reduction compared with baseline) by pretreating unlabeled PB006 and JQ1, suggesting the high binding selectivity and specificity of [18F]PB006. Our study indicated that [18F]PB006 is a potent PET probe selectively targeting BET BD1, and further structural optimization of the radiotracer is still required to improve brain uptake to support neuroepigenetic imaging.
Keywords: epigenetic, BD1, bromodomain, PET, radiotracer, imaging
Epigenetic mechanisms have emerged as promising targets for the development of new therapeutics for various diseases, including inflammation, cancer, and cardiovascular disease, among others.1,2 Multiple enzymes and proteins play pivotal roles in the epigenetic regulation and mediate gene transcription and expression.3,4 Among these proteins, the bromodomain and extra-terminal domain (BET) family of proteins plays an indispensable role in the epigenetic mechanism.5 BET family proteins are a class of bromodomain-containing proteins, which includes four subgroups (BRD2, BRD3, BRD4, and BRDT). Each of the four BRDs is featured as containing two tandem bromodomains (BD1 and BD2) and one extra-terminal bromodomain (ET), which make them different from other bromodomain proteins. By specifically identifying and binding acetylated lysine residues (KAc) on the histone tails, BET family proteins function as gene transcriptional regulators and are thereby known as epigenetic readers.6,7
The BET protein family has attracted increasing interest due to its relationship with many human disease developments, especially cancer. Several BET inhibitors that can cause transcriptional changes by blocking the interactions between BRDs and KAc were discovered for various disease treatments.8 Since the first BET inhibitor, JQ1 (Figure 1), was discovered by Filippakopoulos et al., numerous novel small-molecule BET inhibitors such as isoxazoles, tetrahydroquinolines, and quinoline derivatives have been developed.9−12 Most of the BET inhibitors are designed for various types of cancer treatments, and some of them are now in clinical trials for further investigation. For example, it was found that the JQ1 analogue I-BET762 can inhibit tumor growth by regulating the expression of antiapoptotic gene BCL2 in multiple tumor cells, and it is now in phase I clinical trials for NUT midline carcinoma (NMC) and hematologic disease therapy.13,14 In addition, some evidence has also indicated the BET proteins are involved in multiple central nervous system-related disorders such as Alzheimer’s disease,15,16 Parkinson’s disease,17 and substance use disorder.18
Figure 1.

Chemical structures of JQ1 and representative domain-selective BET inhibitors.
Due to the high similarity of amino acid sequences of the BD1 and BD2 bromodomains, most of the known BET inhibitors simultaneously bind to BET BD1 and BD2 without selectivity. However, recent studies indicated that BD1 and BD2 play different roles in the transcriptional control of epigenetics, and the BET inhibitor that targets the specific domain may give a more potent therapeutic effect with fewer side effects than dual BD1/2 inhibition.19−22 As a consequence, there is a lot of interest in developing selective BET BD1 and BD2 inhibitors. For example, Gracias and coworkers found that the selective BET BD1 inhibitor Olinone (Figure 1) rather than the dual BD1/2 inhibitor demonstrated the myelin regenerative efficiency in neurodegeneration disorders.23 Further, Picaud et al. reported the selective BET BD2 inhibitor RVX-208, showing the distinct differences of gene expression alterations compared with pan-BET inhibitors.24 Though considerable efforts have been devoted to the discovery of next-generation BET inhibitors, the development of highly domain-specific BET inhibitors is a huge challenge, and very few inhibitors with high domain selectivity were reported. Furthermore, many fundamental questions such as the mechanisms of action of the two bromodomains and the therapeutic potential of each bromodomain are in need of further investigation. Against this background, there is an urgent need for solutions to these questions.
Positron emission tomography (PET) imaging is a unique tool that can give the answers to these essential questions about BET BD1 and BD2 in living subjects in a noninvasive way.25 With a suitable radiotracer, PET imaging would accelerate BET research to aid investigators in diagnosing disease, monitoring treatment, and in drug development. However, developing the PET radioligand targeting BET BD1 and BD2 with high binding affinity and high selectivity is demanding and challenging. To date, no PET radioligands specifically targeting either BET BD1 or BD2 have been reported for human use or even in the preclinical investigation. Here, we describe the design, radiosynthesis, and biological evaluation of a novel selective BET BD1 PET radiotracer, [18F]PB006. Our studies showed that [18F]PB006 is a potential radioligand that binds BET BD1 selectively and specifically and provides a powerful tool for BET BD1 quantification, visualization, and epigenetic research.
For the development of a PET radiotracer specifically targeting BET BD1, our investigation commenced with the selective BET BD1 inhibitor discovery. Compound 1 (Figure 2) is a morpholine-containing 2-hydroxyquinoline derivative that was first reported in a patent.26 The in vitro binding assay showed that compound 1 exhibited both the high binding affinity and selectivity toward BRD4 BD1 (Kd = 15 nM for BRD4 BD1 and >150-fold selectivity for BRD4 BD1 over BD2), which makes it a suitable candidate to be converted into a PET imaging agent for BD1. For the PET radioligand design, typical methods are radiolabeling the compound with the short-lived isotope such as 11C or 18F. Owing to the presence of pyridine in the structure of 1, we conceived of introducing the 18F at the ortho-position of the nitrogen atom, taking advantage of the electron-absorbing characteristic of the nitrogen atom in the pyridine ring. Accordingly, we designed the standard PB006 by introducing the fluorine atom at the 2-position of pyridine of 1 (Figure 2).
Figure 2.

Chemical structures of selective BET BD1 inhibitor 1, PB006, and [18F]PB006.
The standard compound PB006 and the chlorine-substituted precursor were synthesized through similar synthetic routes (Scheme 1). Key intermediates 2 and 4 were obtained from 2,6-difluoronicotinic acid or 6-chloro-2-fluoronicotinic acid by reacting with morpholine in the presence of LiHMDS in anhydrous tetrahydrofuran at −78 °C for 1 h. Subsequently, the standard compound PB006 and precursor 5 were prepared by the coupling of the intermediates with 6-amino-4-methylquinolin-2-ol in dichloromethane in the presence of EDC at room temperature for 4 h.
Scheme 1. Synthesis Route for the Standard Compound PB006 and the Chlorine-Substituted Precursor.

Reagents and conditions: (i) LiHMDS, THF, −78°C, 1 h; (ii) 6-amino-4-methylquinolin-2-ol, EDC, DCM, room temperature, 4 h.
The binding mode analysis of PB006 was performed to investigate the interaction mode of PB006 for BRD4 BD1 (PDB code: 6C7R) (Figure 3).27 The molecular docking result reveals that the 2-hydroxyquinoline skeleton occupies the lipophilic region, which consists of the Trp81-Pro82-Phe83 triad and is known as the WPF shelf. Additional hydrogen bonding interaction was found between the OH group and Pro82 residue. Of note, the morpholine ring and pyridine ring, functioning as the KAc mimic, entirely occupy the KAc binding pocket and form the hydrophobic interactions with Met149, Ile146, Asp140, Asp144, and Asp145. Crystal structures of BET proteins indicate that the Asp144 is uniquely present in the BET BD1 domains and is substituted by His433 in BD2 domains. It is noteworthy that the morpholine ring is positioned between Asp144 and Asn140, which forms a significant interaction and gives rise to the BD1 bromodomain selectivity. Subsequently, the cocrystal structure of PB006 with the BRD4 BD1 bromodomain indicates that the PB006 has an ideal fit with the BD1 surface, which arranges a structural basis for the high affinity and selectivity binding (Figure 4).
Figure 3.
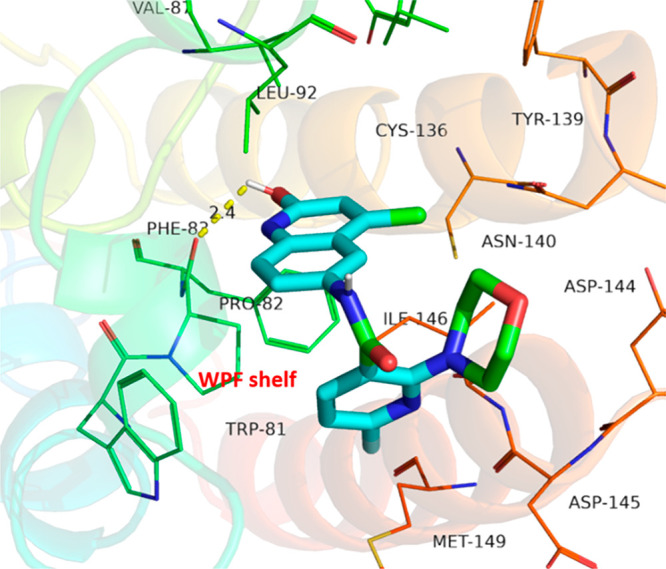
Cocrystal structure of PB006 with the BRD4 BD1 bromodomain (PDB code: 6C7R). The protein is shown as a cartoon, and the ligand and residues are shown in sticks. PB006 occupies the WPF shelf and interacts with BRD4 BD1 bromodomain residues (hydrogen bonds are colored in yellow).
Figure 4.
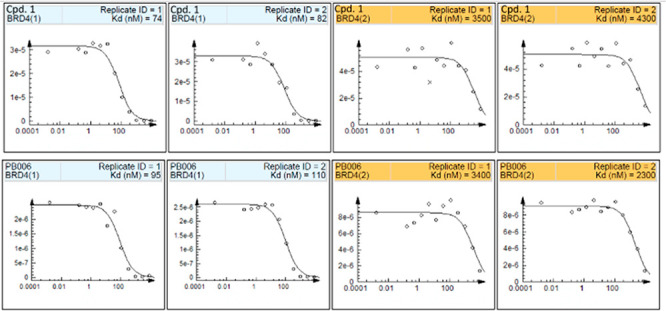
In vitro inhibitory activity against BRD4 (BD1/BD2) of parent compound 1 (up, Kd = 78 nM and 3900 nM for BRD4 BD1 and BRD4 BD2, respectively) and PB006 (below, Kd = 100 nM and 2900 nM for BRD4 BD1 and BRD4 BD2, respectively) (BROMOscan bromodomain competition binding assay).
The in vitro binding assay of compound 1 and PB006 was performed to evaluate its bioactivity for BRD4 BD1 and BD2 (BROMOscan, DiscoverX), and the results are shown in Figure 4. PB006 exhibits favorable binding affinity and selectivity toward BRD4 BD1 (with Kd = 100 nM and 29-fold over BRD4 BD2). Less binding affinity and selectivity of PB006 for BRD4 BD1 compared with compound 1 (Kd = 78 nM and 3900 nM for BRD4 BD1 and BRD4 BD2, respectively) was observed, suggesting that the introduction of fluorine gave a reduction effect on the binding affinity and selectivity for BRD4 BD1. It has been indicated that the domain selectivity inhibition of BRDs can be achieved even from a feeble BET binder such as RVX-208 and Olinone, which exhibit moderate BD1 inhibitory effects and weak domain selectivity and showed significant contrast bioactivity. To develop new PET probes, Bmax is an important property to measure. We measured Bmax of BRD4 in precuneus of healthy controls with the value of 130 nM. We also tested BRD4 expression in disease tissues and found that BRD4 has significantly lower levels in male AUD patients (Figure S3).
[18F]PB006 was synthesized through a straightforward method (Scheme 2). The ortho-position chlorine is easily substituted by the 18F-fluoride ion thanks to the electron-absorbing effect of nitrogen atoms in the pyridine ring. Subsequently, the precursor (5) was reacted with 18F-fluoride in the presence of Kryptofix-222 and K2CO3 in extra-dry DMSO for 10 min at 120 °C for [18F]PB006 preparation. The reaction mixture was then quenched with water containing 0.1% trifluoroacetic acid, and the crude radioproduct was purified by semipreparative HPLC. The reformulation was then conducted by loading collected [18F]PB006 onto a solid-phase exchange (SPE) C-18 cartridge, rinsing with water, eluting with ethanol, and diluting with saline. Including formulation, the whole preparation process of [18F]PB006 took 60–90 min with a good radiochemical yield (RCY) (nondecay corrected radiochemical yield: 24–35%, n = 6) and a high molar activity of 380 GBq/μmol.
Scheme 2. Radiosynthesis of [18F]PB006.

Radiolabeling condition: 5 (precursor, 0.5 mg), K222, K2CO3, DMSO, [18F]F–, 10 min, 120 °C. Radiochemical yield (RCY): 24–35% (non-decay corrected to trapped [18F]F–).
Next, rodent PET imaging was performed to evaluate [18F]PB006 as a potential BET BD1 imaging probe in vivo. Male C57BL/6 mice were used for the PET imaging scan. After administration of [18F]PB006 (3.7–7.4 Mbq of per animal) via intravenous bolus injection, a 60 min dynamic PET imaging scan was performed, followed by a 10 min computed tomography (CT) scan. Dynamic PET data were collected, and the corresponding images were reconstructed. The biodistribution of [18F]PB006 in the organs of interest was initially investigated, and the ratio of radioactivity uptake is defined as the percent injected dose per cubic centimeter in tissue (%ID/cc). As is shown in Figure 5, [18F]PB006 mainly distributed in the blood pool regions outside of the blood–brain barrier (BBB). Among the organs of interest, the highest uptake of [18F]PB006 was found in the liver, and the radioactivity increased gradually as a result of accumulation during the scan. A slow radioactivity accumulation in the kidney was also observed, which indicated the metabolism of [18F]PB006 primarily occurred in the liver and kidney. In the heart, lung, and spleen, [18F]PB006 had a high initial uptake at ∼5 min postinjection (% ID/cc = 12, 8, and 16, respectively) and then washed out steadily during the 60 min scan. Notably, a small amount of radioactivity was found in the bones, which indicated the stability of the fluorine-18 in [18F]PB006. Provirus studies have found that BRDs (BRD2/3/4) are highly distributed in various tissues and organs, especially in the liver, kidney, and lung, which is consistent with our findings.25,28,29 As an important application of PET imaging, it could be used for disease diagnosis such as brain disorders and tumors. Because [18F]PB006 has specific binding in peripheral organs such as the lungs and heart, there is potential to apply this probe for tumor imaging such as NUT midline carcinoma.
Figure 5.

Biodistribution of [18F]PB006 in mice at 5, 15, 30, and 60 min after intravenous administration of radioligand (n = 6 for each time point).
To evaluate the characteristic of specific binding of [18F]PB006, the blocking studies were conducted by pretreating unlabeled PB006 (self-blocking, 1.0 mg/kg) and JQ1 (nonselective BET inhibitor, 1.0 mg/kg) 5 min prior to radiotracer administration. Results of the blocking studies and time–activity curve of regions of interest are presented in Figure 6. We noticed that the uptake of [18F]PB006 was significantly blocked by the pretreatment of PB006 (at an average of 30% reduction in each organ of interest) and JQ1 (at an average of 20% reduction in each organ of interest), which demonstrated the high binding specificity as well as high stability of [18F]PB006.
Figure 6.
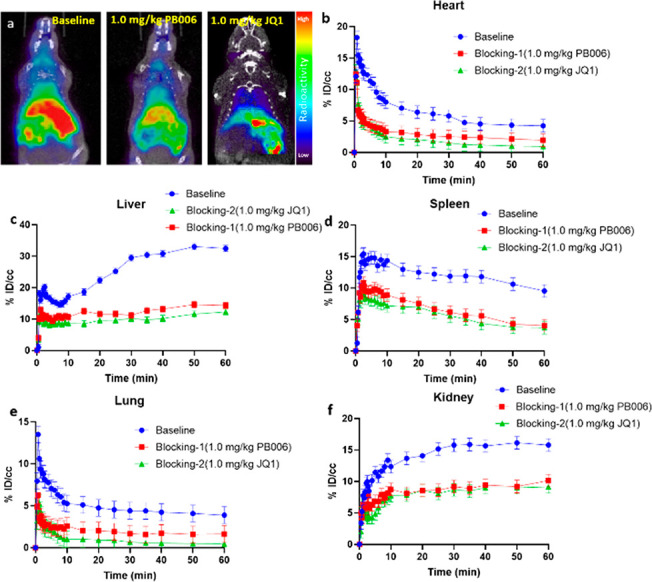
(a) The mice body PET-CT imaging of [18F]PB006 and time–activity curves of (b) heart, (c) liver, (d) spleen, (e) lung, and (f) kidney, demonstrating uptake of radioligand for baseline and blocking studies (n = 6).
Followed by the encouraging results in the peripheral regions, we took a more nuanced and in-depth look at the bioperformance of [18F]PB006 in the brain (Figure 7). The whole-brain analysis showed that [18F]PB006 reached the peak with a maximum of 0.8%ID/cc in the first few minutes after injection and sustained binding over the scanning time (60 min). Meanwhile, the distinct reduction of radioactivity uptake was also observed in the blocking studies, suggesting the binding specificity. One of the reasons for the low brain uptake of [18F]PB006 is the relatively high lipophilicity (LogD7.4 = 3.8), which is out of the preferred BBB penetration range values (1.5–3.5).30 Considering the moderate uptake in the brain, further structural optimization of the radiotracer may be required to improve brain penetration to support neuroepigenetic imaging.
Figure 7.

Mice brain PET-CT imaging of [18F]PB006 focuses on the brain (20–60 min after intravenous administration (i.v.)) and baseline and blocking time–activity curve of [18F]PB006 (n = 6).
In summary, the first selective BET BD1 PET radioligand [18F]PB006 was designed, synthesized, and evaluated by the PET imaging experiment in rodents. The standard compound PB006 showed high affinity and selectivity toward BRD4 BD1 in our in vitro binding assay. Biodistribution study of [18F]PB006 in mice revealed high radiotracer uptake in peripheral tissues, such as liver and kidney, and moderate radiotracer uptake in the brain. The binding specificity of [18F]PB006 was further evaluated by the blocking studies with unlabeled PB006 and JQ1 pretreatment in the rodent PET scan. The results of blocking studies indicated the high selectivity and specificity toward BRD4 BD1 of [18F]PB006. In conclusion, [18F]PB006 is a promising selective BET BD1 PET probe for epigenetic imaging, and further investigation will focus on the structural optimization and nonhuman primate study.
Acknowledgments
This research was supported by NIH grant DA050860 and pilot funding from Martinos Center (C.W.). The author P.B. gratefully acknowledges financial support by the China Scholarship Council (CSC) during the training at the Martinos Center. The authors are grateful to Helen Deng as well as the Martinos Center radiopharmacy and imaging staff for help with nonhuman primate experiments.
Glossary
Abbreviations
- PET
positron emission tomography
- CT
computed tomography
- BBB
blood–brain barrier
- RCY
radiochemical yield
- DMSO
dimethyl sulfoxide
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00650.
Experimental procedure details on the synthesis of compounds, HPLC and NMR spectral data, in vitro binding assays, and animal procedures (PDF)
The authors declare no competing financial interest.
This paper was originally published ASAP on January 8, 2021, with an error in Figure 1. The corrected version was reposted on January 14, 2021.
Supplementary Material
References
- Samanta S.; Rajasingh S.; Cao T.; Dawn B.; Rajasingh J. Epigenetic dysfunctional diseases and therapy for infection and inflammation. Biochim. Biophys. Acta, Mol. Basis Dis. 2017, 1863 (2), 518–528. 10.1016/j.bbadis.2016.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skvortsova K.; Iovino N.; Bogdanovic O. Functions and mechanisms of epigenetic inheritance in animals. Nat. Rev. Mol. Cell Biol. 2018, 19 (12), 774–790. 10.1038/s41580-018-0074-2. [DOI] [PubMed] [Google Scholar]
- Copeland R. A.; Olhava E. J.; Scott M. P. Targeting epigenetic enzymes for drug discovery. Curr. Opin. Chem. Biol. 2010, 14 (4), 505–10. 10.1016/j.cbpa.2010.06.174. [DOI] [PubMed] [Google Scholar]
- Doroshow D. B.; Eder J. P.; LoRusso P. M. BET inhibitors: a novel epigenetic approach. Ann. Oncol 2017, 28 (8), 1776–1787. 10.1093/annonc/mdx157. [DOI] [PubMed] [Google Scholar]
- Gallenkamp D.; Gelato K. A.; Haendler B.; Weinmann H. Bromodomains and their pharmacological inhibitors. ChemMedChem 2014, 9 (3), 438–64. 10.1002/cmdc.201300434. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P.; Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nat. Rev. Drug Discovery 2014, 13 (5), 337–56. 10.1038/nrd4286. [DOI] [PubMed] [Google Scholar]
- Cochran A. G.; Conery A. R.; Sims R. J. 3rd Bromodomains: a new target class for drug development. Nat. Rev. Drug Discovery 2019, 18 (8), 609–628. 10.1038/s41573-019-0030-7. [DOI] [PubMed] [Google Scholar]
- Zhang G.; Smith S. G.; Zhou M. M. Discovery of Chemical Inhibitors of Human Bromodomains. Chem. Rev. 2015, 115 (21), 11625–68. 10.1021/acs.chemrev.5b00205. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P.; Qi J.; Picaud S.; Shen Y.; Smith W. B.; Fedorov O.; Morse E. M.; Keates T.; Hickman T. T.; Felletar I.; Philpott M.; Munro S.; McKeown M. R.; Wang Y.; Christie A. L.; West N.; Cameron M. J.; Schwartz B.; Heightman T. D.; La Thangue N.; French C. A.; Wiest O.; Kung A. L.; Knapp S.; Bradner J. E. Selective inhibition of BET bromodomains. Nature 2010, 468 (7327), 1067–73. 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnier J. M.; Sharp P. P.; Burns C. J. BET bromodomain inhibitors: a patent review. Expert Opin. Ther. Pat. 2014, 24 (2), 185–99. 10.1517/13543776.2014.859244. [DOI] [PubMed] [Google Scholar]
- Chan K. H.; Zengerle M.; Testa A.; Ciulli A. Impact of Target Warhead and Linkage Vector on Inducing Protein Degradation: Comparison of Bromodomain and Extra-Terminal (BET) Degraders Derived from Triazolodiazepine (JQ1) and Tetrahydroquinoline (I-BET726) BET Inhibitor Scaffolds. J. Med. Chem. 2018, 61 (2), 504–513. 10.1021/acs.jmedchem.6b01912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosmini R.; Nguyen V. L.; Toum J.; Simon C.; Brusq J. M.; Krysa G.; Mirguet O.; Riou-Eymard A. M.; Boursier E. V.; Trottet L.; Bamborough P.; Clark H.; Chung C. W.; Cutler L.; Demont E. H.; Kaur R.; Lewis A. J.; Schilling M. B.; Soden P. E.; Taylor S.; Walker A. L.; Walker M. D.; Prinjha R. K.; Nicodeme E. The discovery of I-BET726 (GSK1324726A), a potent tetrahydroquinoline ApoA1 up-regulator and selective BET bromodomain inhibitor. J. Med. Chem. 2014, 57 (19), 8111–31. 10.1021/jm5010539. [DOI] [PubMed] [Google Scholar]
- Mirguet O.; Gosmini R.; Toum J.; Clement C. A.; Barnathan M.; Brusq J. M.; Mordaunt J. E.; Grimes R. M.; Crowe M.; Pineau O.; Ajakane M.; Daugan A.; Jeffrey P.; Cutler L.; Haynes A. C.; Smithers N. N.; Chung C. W.; Bamborough P.; Uings I. J.; Lewis A.; Witherington J.; Parr N.; Prinjha R. K.; Nicodeme E. Discovery of epigenetic regulator I-BET762: lead optimization to afford a clinical candidate inhibitor of the BET bromodomains. J. Med. Chem. 2013, 56 (19), 7501–15. 10.1021/jm401088k. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Yang C. Y.; Wang S. The making of I-BET762, a BET bromodomain inhibitor now in clinical development. J. Med. Chem. 2013, 56 (19), 7498–500. 10.1021/jm4014407. [DOI] [PubMed] [Google Scholar]
- Benito E.; Ramachandran B.; Schroeder H.; Schmidt G.; Urbanke H.; Burkhardt S.; Capece V.; Dean C.; Fischer A. The BET/BRD inhibitor JQ1 improves brain plasticity in WT and APP mice. Transl. Psychiatry 2017, 7 (9), e1239. 10.1038/tp.2017.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magistri M.; Velmeshev D.; Makhmutova M.; Patel P.; Sartor G. C.; Volmar C. H.; Wahlestedt C.; Faghihi M. A. The BET-Bromodomain Inhibitor JQ1 Reduces Inflammation and Tau Phosphorylation at Ser396 in the Brain of the 3xTg Model of Alzheimer’s Disease. Curr. Alzheimer Res. 2016, 13 (9), 985–995. 10.2174/1567205013666160427101832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figge D. A.; Standaert D. G. Dysregulation of BET proteins in levodopa-induced dyskinesia. Neurobiol. Dis. 2017, 102, 125–132. 10.1016/j.nbd.2017.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartor G. C.; Powell S. K.; Brothers S. P.; Wahlestedt C. Epigenetic Readers of Lysine Acetylation Regulate Cocaine-Induced Plasticity. J. Neurosci. 2015, 35 (45), 15062–72. 10.1523/JNEUROSCI.0826-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baud M. G. J.; Lin-Shiao E.; Cardote T.; Tallant C.; Pschibul A.; Chan K. H.; Zengerle M.; Garcia J. R.; Kwan T. T.; Ferguson F. M.; Ciulli A. Chemical biology. A bump-and-hole approach to engineer controlled selectivity of BET bromodomain chemical probes. Science 2014, 346 (6209), 638–641. 10.1126/science.1249830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilan O.; Rioja I.; Knezevic K.; Bell M. J.; Yeung M. M.; Harker N. R.; Lam E. Y. N.; Chung C. W.; Bamborough P.; Petretich M.; Urh M.; Atkinson S. J.; Bassil A. K.; Roberts E. J.; Vassiliadis D.; Burr M. L.; Preston A. G. S.; Wellaway C.; Werner T.; Gray J. R.; Michon A. M.; Gobbetti T.; Kumar V.; Soden P. E.; Haynes A.; Vappiani J.; Tough D. F.; Taylor S.; Dawson S. J.; Bantscheff M.; Lindon M.; Drewes G.; Demont E. H.; Daniels D. L.; Grandi P.; Prinjha R. K.; Dawson M. A. Selective targeting of BD1 and BD2 of the BET proteins in cancer and immuno-inflammation. Science 2020, 368, 387. 10.1126/science.aaz8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugle M.; Lucas X.; Weitzel G.; Ostrovskyi D.; Breit B.; Gerhardt S.; Einsle O.; Gunther S.; Wohlwend D. 4-Acyl Pyrrole Derivatives Yield Novel Vectors for Designing Inhibitors of the Acetyl-Lysine Recognition Site of BRD4(1). J. Med. Chem. 2016, 59 (4), 1518–30. 10.1021/acs.jmedchem.5b01267. [DOI] [PubMed] [Google Scholar]
- Faivre E. J.; McDaniel K. F.; Albert D. H.; Mantena S. R.; Plotnik J. P.; Wilcox D.; Zhang L.; Bui M. H.; Sheppard G. S.; Wang L.; Sehgal V.; Lin X.; Huang X.; Lu X.; Uziel T.; Hessler P.; Lam L. T.; Bellin R. J.; Mehta G.; Fidanze S.; Pratt J. K.; Liu D.; Hasvold L. A.; Sun C.; Panchal S. C.; Nicolette J. J.; Fossey S. L.; Park C. H.; Longenecker K.; Bigelow L.; Torrent M.; Rosenberg S. H.; Kati W. M.; Shen Y. Selective inhibition of the BD2 bromodomain of BET proteins in prostate cancer. Nature 2020, 578 (7794), 306–310. 10.1038/s41586-020-1930-8. [DOI] [PubMed] [Google Scholar]
- Gacias M.; Gerona-Navarro G.; Plotnikov A. N.; Zhang G.; Zeng L.; Kaur J.; Moy G.; Rusinova E.; Rodriguez Y.; Matikainen B.; Vincek A.; Joshua J.; Casaccia P.; Zhou M. M. Selective chemical modulation of gene transcription favors oligodendrocyte lineage progression. Chem. Biol. 2014, 21 (7), 841–854. 10.1016/j.chembiol.2014.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picaud S.; Wells C.; Felletar I.; Brotherton D.; Martin S.; Savitsky P.; Diez-Dacal B.; Philpott M.; Bountra C.; Lingard H.; Fedorov O.; Muller S.; Brennan P. E.; Knapp S.; Filippakopoulos P. RVX-208, an inhibitor of BET transcriptional regulators with selectivity for the second bromodomain. Proc. Natl. Acad. Sci. U. S. A. 2013, 110 (49), 19754–9. 10.1073/pnas.1310658110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai P.; Bai S.; Placzek M. S.; Lu X.; Fiedler S. A.; Ntaganda B.; Wey H.-Y.; Wang C. Positron Emission Tomography Probe for Orexin Receptors Neuroimaging. Molecules 2020, 25 (5), 1018. 10.3390/molecules25051018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seitzberg J. G.; Kronborg T.; Poljak V.; Friberg G.; Teuber L.. Compounds active towards bromodomains. US20170349607A1, 2017.
- Zhao Y.; Zhou B.; Bai L.; Liu L.; Yang C. Y.; Meagher J. L.; Stuckey J. A.; McEachern D.; Przybranowski S.; Wang M.; Ran X.; Aguilar A.; Hu Y.; Kampf J. W.; Li X.; Zhao T.; Li S.; Wen B.; Sun D.; Wang S. Structure-Based Discovery of CF53 as a Potent and Orally Bioavailable Bromodomain and Extra-Terminal (BET) Bromodomain Inhibitor. J. Med. Chem. 2018, 61 (14), 6110–6120. 10.1021/acs.jmedchem.8b00483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meloche J.; Potus F.; Vaillancourt M.; Bourgeois A.; Johnson I.; Deschamps L.; Chabot S.; Ruffenach G.; Henry S.; Breuils-Bonnet S.; Tremblay E.; Nadeau V.; Lambert C.; Paradis R.; Provencher S.; Bonnet S. Bromodomain-Containing Protein 4: The Epigenetic Origin of Pulmonary Arterial Hypertension. Circ. Res. 2015, 117 (6), 525–35. 10.1161/CIRCRESAHA.115.307004. [DOI] [PubMed] [Google Scholar]
- Donati B.; Lorenzini E.; Ciarrocchi A. BRD4 and Cancer: going beyond transcriptional regulation. Mol. Cancer 2018, 17 (1), 164. 10.1186/s12943-018-0915-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike V. W. PET radiotracers: crossing the blood-brain barrier and surviving metabolism. Trends Pharmacol. Sci. 2009, 30 (8), 431–40. 10.1016/j.tips.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.