

Abstract

Indoleamine 2,3-dioxygenase 1 (IDO1) is a heme-containing dioxygenase enzyme implicated in cancer immune response. This account details the discovery of BMS-986242, a novel IDO1 inhibitor designed for the treatment of a variety of cancers including metastatic melanoma and renal cell carcinoma. Given the substantial interest around this target for cancer immunotherapy, we sought to identify a structurally differentiated clinical candidate that performs comparably to linrodostat (BMS-986205) in terms of both in vitro potency and in vivo pharmacodynamic effect in a mouse xenograft model. On the basis of its preclinical profile, BMS-986242 was selected as a candidate for clinical development.
Keywords: indoleamine 2,3-dioxygenase; IDO1; BMS-986242; immuno-oncology
Human indoleamine 2,3-dioxygenase 1 (IDO1) metabolizes the essential amino acid L-tryptophan (l-trp) into kynurenine via N-formylkynurenine and is expressed in tumor-associated cells such as dendritic cells (DCs) and endothelial cells, as well as tumor cells themselves.1 High levels of IDO1 expression in tumors or draining lymph nodes is a negative prognostic factor for several tumor types including melanoma, colon cancer, brain tumors, ovarian cancer, acute myelogenous leukemia, and others.2 IDO1 expression is upregulated by type I and type II interferons, COX-2 via PGE2 production, and reverse signaling from inhibitory T cell coreceptors including cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) binding to CD80 on DCs.1 Furthermore, IDO1 has been implicated in immune escape in the tumor microenvironment, and inhibition of IDO1 has been shown to counter this effect.3 Many cancer immunotherapies aim to activate T cells within the tumor microenvironment or cause antitumor inflammation which can, itself, induce IDO1 expression. Consequently, these immunotherapies may be increasingly effective when administered in combination with an IDO1 inhibitor.4
IDO1 potentiates a variety of effector pathways. Elevated levels of IDO1 in the tumor microenvironment can cause local depletion of l-trp leading to immune tolerance of tumoral antigens.5 Specifically, local l-trp depletion may cause stress response kinase general control nonderepressible 2 (GCN2) activation.6 Activation of GCN2 can bias differentiation of naive T cells into Treg cells, cause T cell anergy, and inhibit T cell proliferation.7 GCN2 can also alter the phenotype of DCs to produce IL-10, transforming growth factor beta (TGFβ) and other inhibitory cytokines.8l-trp depletion may also cause master amino acid-sensing kinase glucokinase-1 (GLK1) blockade leading to mammalian target of rapamycin complex 1 (mTORC1) and protein kinase C theta (PKC-θ) inhibition.9 Kynurenine produced by IDO1 binds and activates the aryl hydrocarbon receptor (AhR), which encourages Treg differentiation and the formation of DCs with an immunosuppressive phenotype.10
IDO1 is a monomeric heme-containing enzyme that catalyzes the initial, rate-limiting step of L-tryptophan catabolism. Specifically, IDO1 catalyzes pyrrole ring cleavage in D- or l-trp by incorporating both atoms of molecular oxygen (O2) to form N-formyl kynurenine and subsequently kynurenine.11 Other enzymes capable of affecting this transformation include IDO2 and tryptophan 2,3-dioxygenase (TDO). In contrast to IDO1, TDO is tetrameric, enantiospecific (will only catalyze the cleavage of l-trp) and constitutively expressed in the liver and brain.12 TDO is primarily responsible for regulating systemic l-trp levels suggesting that its role may be distinct from IDO1.13 Less is known about IDO2; however, it has 43% sequence homology with IDO1 and reduced affinity for l-trp, implying decreased activity.14
Dozens of IDO1 inhibitors have been reported and assessed preclinically, while several others have been clinically investigated in combination with chemotherapeutics, checkpoint inhibitors, and vaccines.15 The structures of several of these clinical candidates, including linrodostat mesylate (BMS-986205) (1),16a epacadostat (2),16b indoximod (3),16c navoximod (4),16d and PF06840003 (5),16e are shown in Figure 1. While clinical trials with various IDO1 inhibitors have been terminated because of lack of clinical response,17 it has become clear that different IDO1 inhibitors are acting through several discrete mechanisms which could impact pharmacology and clinical outcomes.18 Moreover, linrodostat has a unique mechanism of inhibition targeting apo-IDO1 in contrast to other clinically assessed IDO1 inhibitors (see Figure 1) which likely target holo-IDO1.19
Figure 1.

Clinically investigated IDO1 inhibitors.
Previously, the discovery of linrodostat (1), a heme displacing, cyclohexyl quinoline IDO1 inhibitor, was disclosed.16a This compound demonstrated potent human IDO1 inhibitory activity in HeLa cells and human whole blood that compared favorably with competitor compounds. Currently, linrodostat is in several clinical trials including a phase III study in bladder cancer in combination with nivolumab, as well as phase I and II studies in a variety of tumor types.20 Herein, we will discuss efforts to find a structurally differentiated IDO1 inhibitor resulting in the identification of BMS-986242, which possesses a preclinical PK/PD profile comparable to linrodostat.
An X-ray cocrystal structure of compound 6 (a closely related analogue of BMS-986205) was utilized in the design of new IDO1 inhibitors (Figure 2).19a The key interaction involves hydrogen bonding between the NH of the amide bond and Ser167 as well as the amide carbonyl and His346. There is also a potential hydrogen bonding event between the nitrogen of the quinoline and Arg343. Additionally, the crystal reveals edge to face pi stacking interactions between the cyanophenyl group and Tyr126 as well as between the quinoline ring and Phe270. When the pursuit to identify a structurally differentiated clinical candidate began, we were aware that the aryl amide-containing region of linrodostat was tolerant of a variety of modifications, so we chose to focus our efforts on this portion of the molecule first.
Figure 2.
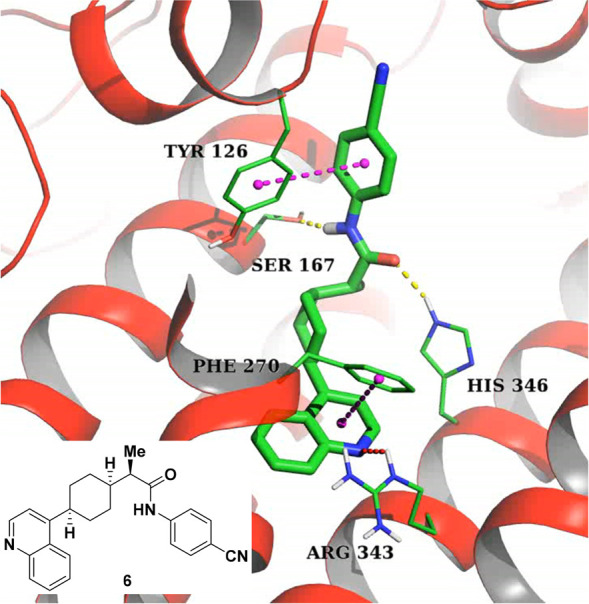
Crystal structure of compound 6 bound to hIDO1.
Expedient exploration of a diverse range of aryl amide isosteres and replacements necessitated a synthetic route wherein the key intermediate would already contain the cyclohexyl-quinoline core with all stereochemistry set. To achieve this, new compounds of generic structure 14 were prepared according to the sequence outlined in Scheme 1 via intermediates 12 or 13. To begin, a previously reported vinyl boronic acid21 was coupled to commercially available 4-chloro-6-fluoroquinoline under standard Suzuki coupling conditions to provide 7. Hydrogenation of 7 with Degussa-type palladium was necessary to avoid over reduction of the quinoline. Subsequent hydrolysis of the ketal gave ketone 8. Diastereoselective reduction of the resulting ketone with sodium borohydride preferentially gave the trans-alcohol (in a 5:1 trans-:cis- ratio) which was then mesylated to yield 9. Next, a one-pot process involving mesylate displacement with di-tert-butyl malonate followed by acetic acid mediated tert-butyl group deprotection with concomitant decarboxylation provided cis-acid 10 containing the necessary stereochemistry around the cyclohexyl ring. The α-methyl group of the acid was installed stereoselectively using an Evan’s oxazolidinone chiral auxiliary. Alkylation of oxazolidinone-containing intermediate 11 with methyl iodide allowed for installation of the methyl group in >20:1 diastereoselectivity in favor of the desired diastereomer. Cleavage of the chiral auxiliary led to intermediate 12. This intermediate could lead to a variety of amide replacements or isosteres using procedures outlined in the Supporting Information provided, as well as other variations not disclosed in this manuscript but exemplified elsewhere.22 Alternatively, this intermediate (12) could be converted to amine 13 via Curtius rearrangement to give another intermediate useful for introducing diversity.22 From intermediates 12 and 13, a variety of amide isosteres and replacements were synthesized (compounds 15–25) (see Supporting Information for details) and assessed in both human HeLa and murine M109 cellular IDO1 inhibition assays where IDO activity can be induced upon treatment with IFNγ as well as a human whole blood (HWB) IDO1 inhibition assay (Table 1).
Scheme 1. Synthetic Route to Cyclohexyl Quinolines.
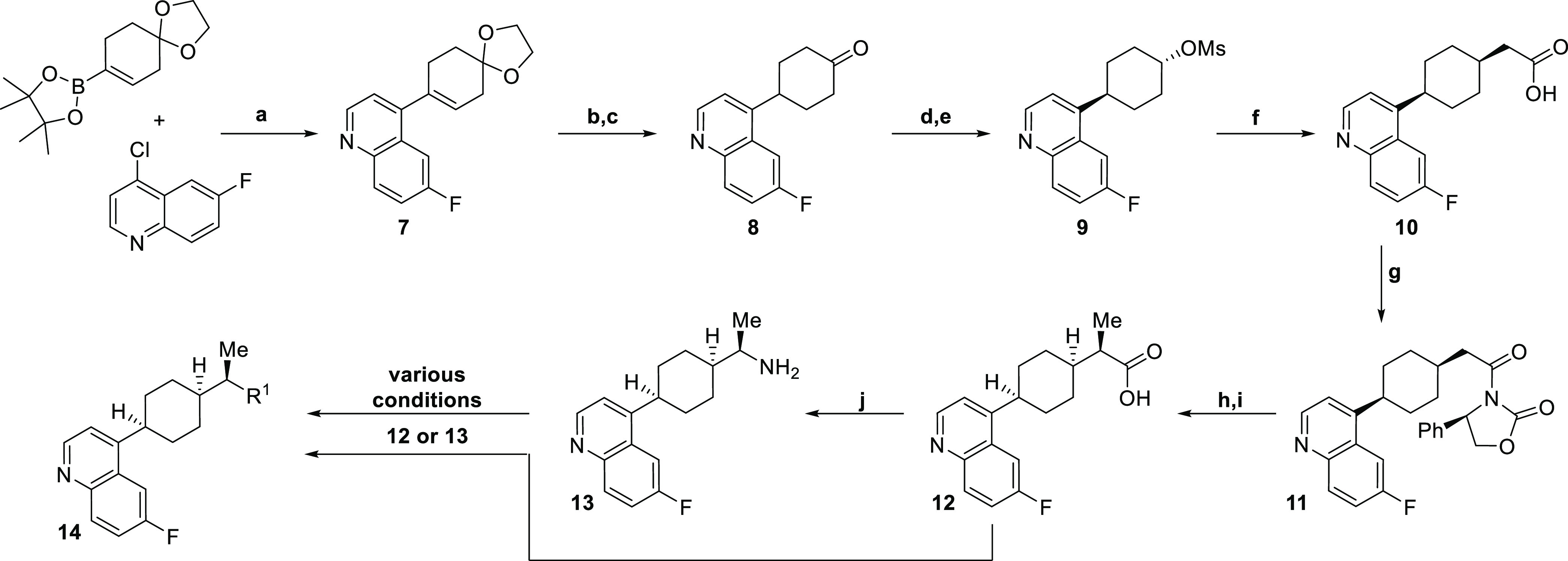
Reagents and conditions: a. Pd(PPh3)4, K2CO3, dioxane, water, 100 °C, 16 h (77%); b. Pd/C (Degussa), NH4HCO2; c. 4 M HCl, Acetone (75% over 2 steps); d. NaBH4 (5:1 trans:cis dr); e. MsCl, Pyr. (76% over 2 steps); f. (i) NaH, di-tBu-malonate, (ii) AcOH, 130 °C (76%); g. PivCl, TEA; lithium-(R)-2-oxo-4-phenyloxazolidin-3-ide (85%); h. NaHMDS, −40 °C; MeI (>20:1 dr) (68%); i. LiOH, H2O2 (82%); j. DPPA, TEA; LiOH, H2O (90%)
Table 1. SAR of Amide Isosteres and Replacements.

Data reported as average of ≥3 test results. See Supporting Information for description of assay conditions.
Data reported as average of ≥2 test results. See Supporting Information for description of assay conditions.
Percentage of the parent compound (0.5 μM) remaining after a 10 min incubation with 1 mg/mL of HLM or MsLM.
We initiated our efforts by synthesizing compounds such as pyrimidine 15, benzylamine 16, and cyclohexylamine 17. While heterocycles, such as pyrimidine 15, were tolerated in several cases (not shown),22 none were as potent as compound 1, and all showed inferior HWB potency. Benzylamines (like 16) and primary aliphatic amines (not shown) were generally not well tolerated. Of the aliphatic amines explored, cyclohexylamine 17 was the most successful; however, this compound had very poor HWB activity and metabolic stability. Removing the carbonyl group in 1 led to analogue 18. While 18 did maintain a decent level of potency (within ∼3-fold of 1) in the human cellular and whole blood assays, it was >10-fold less potent in the murine M109 cellular assay. Moving the nitrogen over one position as in compound 19 rendered compound 19 significantly less active. Several heterocyclic isosteres were tolerated including benzimidazole 20 which was only 2-fold less potent than 1 in the HeLa and HWB assays. Further exploration of heterocyclic isosteres will be the topic of a future publication. Sulfonamides such as 21 were found to be inactive and although ureas (22) were able to maintain some potency, mediocre HWB potency made them implausible candidates. Finally, several “reverse” amides were synthesized. Two homologated analogues containing four-atom linkers between the cyclohexyl core and the p-chlorophenyl ring (23 and 24) were much less potent than 1 across assay types. In contrast, the “reverse” amide analogue 25 that possessed a three-atom linker between the cyclohexyl core and the p-chlorophenyl ring displayed the same level of cellular potency and was within 2-fold of the HWB potency of linrodostat (1). On the basis of this promising result, SAR around the “reverse” amides was surveyed (Table 2).
Table 2. “Reverse” Amide SAR.

Data reported as average of ≥3 test results. See Supporting Information for description of assay conditions.
Data reported as average of ≥2 test results. See Supporting Information for description of assay conditions.
Percentage of the parent compound (0.5 μM) remaining after a 10 min incubation with 1 mg/mL of HLM or MsLM.
Synthesis of the meta-chloro (26) and ortho-chloro (27) versions of compound 25 revealed an advantage for para-substitution on the phenyl ring both in terms of potency and metabolic stability. Aliphatic reverse amides like 28 and 29 again maintained decent (<25 nM in HeLa) human cellular potency like their “forward” amide versions (i.e., 17), but also suffered from poor HWB activity and metabolic stability. The para-biphenyl analogue 30 had an overall superior in vitro profile based on potency and metabolic stability; however, this compound suffered from extremely poor solubility (<0.001 mg/mL at pH 7.4 and 6.5) and was consequently not progressed. In the course of our efforts to improve the solubility of these compounds, we also found that a wide variety of polar functionalities were tolerated at the meta-position as exemplified by compound 31. Based on the cocrystal structure of 6 shown above, we believe that these polar groups are projecting out of the binding pocket and may be solvent exposed. Unfortunately, molecules like 31 did not improve aqueous solubility (still <0.001 mg/mL at 6.5) and most had very poor metabolic stability. On the basis of this data, “reverse” amide 25 (which was assigned as BMS-986242) was profiled more extensively both in vitro and in vivo.
Further in vitro ADMET profiling (Table 3) revealed that BMS-986242 was more prone to oxidative metabolism and less susceptible to glucuronidation. Biotransformation studies in hepatocytes across species (H, M, R, D, C) identified several oxidative metabolites as well as metabolites where the resulting oxygen was capped as the glucuronide; however, no direct glucuronidation of the parent molecule was observed. Several oxidative metabolites were identified and profiled. They were found to be substantially less potent (>200 nM) than parent in the HWB assay. Intrinsic permeability was satisfactory, and the compound does have a moderate potential for drug–drug interactions based upon CYP inhibition of 2C8, 2C9 and 2C19. While compound BMS-986242 showed 87% hERG inhibition and <20% sodium channel inhibition in a patch clamp assay at a 10 μM concentration at both 1 and 4 Hz, it was also highly protein bound which mitigates this liability. Additionally, no adverse findings were observed in a rat CV telemetry study. In an in vitro safety panel consisting of 40 targets, BMS-986242 showed IC50 > 25 μM for all targets except nAChR a1 (IC50 = 12.3 μM) and nAChR a7 (IC50 > 6 μM with ∼20% max inhibition). The pharmacokinetics of BMS-986242 were investigated in mice, rats, dogs, and monkeys (see Table 4). Despite a seemingly poor in vitro oxidative metabolic stability profile, BMS-986242 had a suitable PK profile across species. Oral bioavailability was ≥39% in all tested species.
Table 3. In Vitro ADMET Profile of BMS-986242 (25).
| parameter | BMS-986242 (25) |
|---|---|
| met. stability CYP (T1/2 min) | 14 (H), 4 (M), 10 (R), 10 (D), 2 (C) |
| met. stability UGT (T1/2 min) | >120 (H), >120 (M), 93 (R), 33 (D), >120 (C) |
| PAMPA (pH 7.4) | 2830 nm/sec |
| Caco (a-b:b-a) | 88:55 nm/sec |
| human CYP IC50 (μM) | 2C8:1.39; 2C9:0.74; 2C19:3.37; others >15 |
| hERG patch clamp | 87% inh at 10 μM |
| Na+ patch clamp | <20% inh at 10 μM (1 and 4 Hz) |
| Ca2+ patch clamp IC50 | >25 μM |
| protein binding (% free): h, m | <0.3, <0.2 |
| PXR-TA EC50 | >50 μM |
Table 4. Pharmacokinetic Parameters for BMS-986242 (25).
| parameter | mousea | rata | doga | cynoa |
|---|---|---|---|---|
| dose (mg/kg) iv/po | 0.5b/6c | 0.5b/2c | 1b/5c | 1b/5c |
| CL (mL/min/kg) iv | 30 | 3.70 | 5.1 | 12 |
| Vss (L/kg) iv | 17 | 0.97 | 3.87 | 1 |
| AUCtotal (μM·h) iv/po | 0.69/12.6 | 5.6/28.2 | 8.7/17.4 | 3.4/7.1 |
| t1/2 (h) iv | 8 | 4 | 19 | 2 |
| Fpo (%) | 152 | 127 | 39 | 42 |
Data reported as average of ≥3 animals.
70% PEG 400; 30% water.
5% ethanol; 55% PEG 400; 20% propylene glycol; 20% TPGS.
Pharmacodynamic effects were studied in a nu/nu mouse xenograft model: mice were implanted with SKOV3 cells (human ovarian cancer cell line) 2 weeks prior to treatment (see Table 5 for results, see Supporting Information for protocol details). Compound exposure in the tumor was then monitored (PK), and pharmacodynamic (PD) effects were quantified by observing kynurenine (a downstream product of tryptophan metabolism catalyzed by IDO1) levels in the tumor. BMS-986242 (3, 10, and 30 mpk QD PO), linrodostat (60 mpk QD PO), and epacadostat (100 mpk BID PO) were all examined in this mouse xenograft model. BMS-986242 (25) exhibited dose-proportional exposure and a statistically significant reduction in kynurenine concentration in the tumor at all three doses. In this model, the pharmacodynamic effect of a 10 mpk QD dose of BMS-986242 (25) was comparable to a 60 mpk QD dose of linrodostat (1) and a 100 mpk BID dose of epacadostat (2) despite having lower levels of exposure (21.1 vs 34.9 vs 48.0 μM*h, respectively). Also of note, the 30 mpk dose of BMS-986242 provided significantly higher levels of exposure than the 60 mpk dose of linrodostat (60.1 vs 34.9 μM*h respectively)
Table 5. PK–PD Study of BMS-986242 (25) vs Linrodostat vs Epacadostat in Human SKOV3 Xenograft Tumor Mouse Model.
| compound | dose (mg/kg) | PKa | PDb |
|---|---|---|---|
| BMS-986242 (25) | 3c | 9.6 | 45% |
| BMS-986242 (25) | 10c | 21.1 | 58% |
| BMS-986242 (25) | 30c | 61.0 | 59% |
| linrodostat (1) | 60c | 34.9 | 61% |
| epacadostat (2) | 100d | 48.0 | 54% |
PK is AUC (0–24 h) in μM*h measured in the tumor.
PD is percent kynurenine AUEC (0–24 h) reduction measured in the tumor. %Kyn reduction was measured at steady state after the 5th dose, calculated as the area under Kyn concentration–time curve from 0 to 24 h and compared with that of vehicle control.
QD PO dosing.
BID PO dosing.
In summary, we have identified BMS-986242 (25) as a structurally differentiated clinical candidate for IDO1 inhibition with robust human and mouse in vitro activity as well as dose proportional exposure and significant PD effect in vivo in a mouse xenograft model. BMS-986242 elicited a PD effect comparable to linrodostat (1) and epacadostat (2) in a PD model of tumor kynurenine reduction at a lower level of exposure. Given the promising preclinical safety and pharmacodynamic profile of BMS-986242, it was progressed into a phase I/II study in combination with nivolumab.23
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00668.
Biological assays, pharmacokinetic studies, in vivo pharmacokinetic-pharmacodynamic study, experimental procedures, and analytical data for key compounds (PDF)
Author Present Address
† X.Z.: Kymera Therapeutics, 200 Arsenal Yards Blvd., Suite 230, Watertown, MA 02472
Author Present Address
‡ J.G.-B.: Pfizer Inc., 10555 Science Center Dr, San Diego, CA 92121
Author Present Address
⊥ T.-A.L.: Black Diamond Therapeutics, One Main Street, 10th Floor, Cambridge, MA 02142
Author Present Address
¶ D.D.: Enyo Pharma, BIOSERRA 1 Bâtiment B, 60 avenue Rockefeller, 69008 Lyon, France
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Katz J. B.; Muller A. J.; Prendergast G. C. Indoleamine 2,3-dioxygenase in T-cell tolerance and tumoral immune escape. Immunol. Rev. 2008, 222, 206–221. 10.1111/j.1600-065X.2008.00610.x. [DOI] [PubMed] [Google Scholar]
- a Munn D. H.; Sharma M. D.; Hou D.; Baban B.; Lee J. R.; Antonia S. J.; Messina J. L.; Chandler P.; Koni P. A.; Mellor A. J. Expression of indoleamine 2,3-dioxygenase by plasmacytoid dendritic cells in tumor-draining lymph nodes. J. Clin. Invest. 2004, 114, 280–290. 10.1172/JCI21583. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Gerlini G.; Di Gennaro P.; Mariotti G.; Urso C.; Chiarugi A.; Pimpinelli N.; Borgognoni L. Indoleamine 2,3-dioxygenase+ cells correspond to the BDCA2+ plasmacytoid dendritic cells in human melanoma sentinel nodes. J. Invest. Dermatol. 2010, 130, 898–901. 10.1038/jid.2009.307. [DOI] [PubMed] [Google Scholar]; c Speeckaert R.; Vermaelen K.; van Geel N.; Autier P.; Lambert J.; Haspeslagh M.; Van Gele M.; Thielemans K.; Neyns B.; Roche N.; Verbeke N.; Deron P.; Speeckaert M.; Brochez L. Indoleamine 2,3-dioxygenase, a new prognostic marker in sentinel lymph nodes of melanoma patients. Eur. J. Cancer 2012, 48, 2004–2011. 10.1016/j.ejca.2011.09.007. [DOI] [PubMed] [Google Scholar]; d Ferdinande L.; Decaestecker C.; Verset L.; Mathieu A.; Moles Lopez X.; Negulescu A. M.; Van Maerken T.; Salmon I.; Cuvelier C. A.; Demetter P. Clinicopathological significance of indoleamine 2,3-dioxygenase 1 expression in colorectal cancer. Br. J. Cancer 2012, 106, 141–147. 10.1038/bjc.2011.513. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Brandacher G.; Perathoner A.; Ladurner R.; Schneeberger S.; Obrist P.; Winkler C.; Werner E. R.; Werner-Felmayer G.; Weiss H. G.; Göbel G.; Margreiter R.; Königsrainer A.; Fuchs D.; Amberger A. Prognostic value of indoleamine 2,3-dioxygenase expression in colorectal cancer: effect on tumor-infiltrating T cells. Clin. Cancer Res. 2006, 12, 1144–1151. 10.1158/1078-0432.CCR-05-1966. [DOI] [PubMed] [Google Scholar]; f Zhai L.; Lauing K. L.; Chang A. L.; Dey M.; Qian J.; Cheng Y.; Lesniak M. S.; Wainwright D. A. The role of IDO in brain tumor immunotherapy. J. Neuro-Oncol. 2015, 123, 395–403. 10.1007/s11060-014-1687-8. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Okamoto A.; Nikaido T.; Ochiai K.; Takakura S.; Saito M.; Aoki Y.; Ishii N.; Yanaihara N.; Yamada K.; Takikawa O.; Kawaguchi R.; Isonishi S.; Tanaka T.; Urashima M. Indoleamine 2,3-dioxygenase serves as a marker of poor prognosis in gene expression profiles of serous ovarian cancer cells. Clin. Cancer Res. 2005, 11, 6030–6039. 10.1158/1078-0432.CCR-04-2671. [DOI] [PubMed] [Google Scholar]; h Chamuleau M. E.; Van de Loosdrecht A. A.; Hess C. J.; Janssen J. J.; Zevenbergen A.; Delwel R.; Valk P. J.; Löwenberg B.; Ossenkoppele G. J. High INDO (indoleamine 2,3-dioxygenase) mRNA level in blasts of acute myeloid leukemic patients predicts poor clinical outcome. Haematologica 2008, 93, 1894–1898. 10.3324/haematol.13112. [DOI] [PubMed] [Google Scholar]; i Folgiero V.; Goffredo B. M.; Filippini P.; Masetti R.; Bonanno G.; Caruso R.; Bertaina V.; Mastronuzzi A.; Gaspari S.; Zecca M.; Torelli G. F.; Testi A. M.; Pession A.; Locatelli F.; Rutella S. Indoleamine 2,3-dioxygenase 1 (IDO1) activity in leukemia blasts correlates with poor outcome in childhood acute myeloid leukemia. Oncotarget 2014, 5, 2052–2064. 10.18632/oncotarget.1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller A. J.; Malachowski W. P.; Prendergast G. C. Indoleamine 2,3-dioxygenase in cancer: targeting pathological immune tolerance with small-molecule inhibitors. Expert Opin. Ther. Targets 2005, 9, 831–849. 10.1517/14728222.9.4.831. [DOI] [PubMed] [Google Scholar]
- a Gu T.; Rowswell-Turner R. B.; Kilinc M. O.; Egilmez N. K. Cancer Res. 2010, 70, 129–138. 10.1158/0008-5472.CAN-09-3170. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Grohmann U.; Orabona C.; Fallarino F.; Vacca C.; Calcinaro F.; Falorni A.; Candeloro P.; Belladonna M. L.; Bianchi R.; Fioretti M. C.; Puccetti P. CTLA-4-Ig regulates tryptophan catabolism in vivo. Nat. Immunol. 2002, 3, 1097–1101. 10.1038/ni846. [DOI] [PubMed] [Google Scholar]
- Muller A. J.; Prendergast G. C. Indoleamine 2,3-Dioxygenase in Immune Suppression and Cancer. Curr. Curr. Cancer Drug Targets 2007, 7, 31–40. 10.2174/156800907780006896. [DOI] [PubMed] [Google Scholar]
- Munn D. H.; Sharma M. D.; Baban B.; Harding H. P.; Zhang Y.; Ron D.; Mellor A. L. GCN2 Kinase in T Cells Mediates Proliferative Arrest and Anergy Induction in Response to Indoleamine 2,3-Dioxygenase. Immunity 2005, 22, 633–642. 10.1016/j.immuni.2005.03.013. [DOI] [PubMed] [Google Scholar]
- Fallarino F.; Grohmann U.; You S.; McGrath B. C.; Cavener D. R.; Vacca C.; Orabona C.; Bianchi R.; Belladonna M. L.; Volpi C.; Santamaria P.; Fioretti M. C.; Puccetti P. The Combined Effects of Tryptophan Starvation and Tryptophan Catabolites Down-Regulate T Cell Receptor ζ-Chain and Induce a Regulatory Phenotype in Naive T Cells. J. Immunol. 2006, 176, 6752–6761. 10.4049/jimmunol.176.11.6752. [DOI] [PubMed] [Google Scholar]
- Liu H.; Huang L.; Bradley J.; Liu K.; Bardhan K.; Ron D.; Mellor A. L.; Munn D. H.; McGaha T. L. GCN2-Dependent Metabolic Stress Is Essential for Endotoxemic Cytokine Induction and Pathology. Mol. Cell. Biol. 2014, 34, 428–438. 10.1128/MCB.00946-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metz R.; Rust S.; DuHadaway J. B.; Mautino M. R.; Munn D. H.; Vahanian N. N.; Link C. J.; Prendergast G. C. IDO inhibits a tryptophan sufficiency signal that stimulates mTOR: A novel IDO effector pathway targeted by D-1-methyl-tryptophan. Oncoimmunology 2012, 1, 1460–1468. 10.4161/onci.21716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Quintana F. J.; Murugaiyan G.; Farez M. F.; Mitsdoerffer M.; Tukpah A.-M.; Burns E. J.; Weiner H. L. An endogenous aryl hydrocarbon receptor ligand acts on dendritic cells and T cells to suppress experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 20768–20773. 10.1073/pnas.1009201107. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Jaronen M.; Quintana F. J. Immunological relevance of the coevolution of IDO1 and AHR. Front. Immunol. 2014, 5, 521–528. 10.3389/fimmu.2014.00521. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Manlapat A. K.; Kahler D. K.; Chandler P. R.; Munn D. H.; Mellor A. L. Cell-autonomous control of interferon type I expression by indoleamine 2,3-dioxygenase in regulatory CD19+ dendritic cells. Eur. J. Immunol. 2007, 37, 1064–1071. 10.1002/eji.200636690. [DOI] [PubMed] [Google Scholar]
- Sugimoto H.; Oda S.-I.; Otsuki T.; Hino T.; Yoshida T.; Shiro Y. Crystal structure of human indoleamine 2,3-dioxygenase: Catalytic mechanism of O2 incorporation by a heme-containing dioxygenase. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 2611–2616. 10.1073/pnas.0508996103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Shimizu T.; Nomiyama S.; Hirata F.; Hayaishi O. Indoleamine 2,3-Dioxygenase. J. Biol. Chem. 1978, 253, 4700–4706. 10.1016/S0021-9258(17)30447-7. [DOI] [PubMed] [Google Scholar]; b Haber R.; Bessette D.; Hulihan-Giblin B.; Durcan M. J.; Goldman D. Identification of Tryptophan 2,3-Dioxygenase RNA in Rodent Brain. J. Neurochem. 1993, 60, 1159–1162. 10.1111/j.1471-4159.1993.tb03269.x. [DOI] [PubMed] [Google Scholar]
- a Greene L. I.; Bruno T. C.; Christenson J. L.; D’Alessandro A.; Culp-Hill R.; Torkko K.; Borges V. F.; Slansky J. E.; Richer J. K. A Role for tryptophan-2,3-dioxygenase in CD8 Tcell suppression and evidence of tryptophan catabolism in breast cancer patient plasma. Mol. Cancer Res. 2019, 17, 131–139. 10.1158/1541-7786.MCR-18-0362. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Pilotte L.; Larrieu P.; Stroobant V.; Colau D.; Dolusič E.; Fredé rick R.; De Plaen E.; Uyttenhove C.; Wouters J.; Masereel B.; Van den Eynde B. J. Reversal of tumoral immune resistance by inhibition of tryptophan 2,3-dioxygenase. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 2497–2502. 10.1073/pnas.1113873109. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Zhai L.; Spranger S.; Binder D. C.; Gritsina G.; Lauing K. L.; Giles F. J.; Wainwright D. A. Molecular Pathways: Targeting IDO1 and Other Tryptophan Dioxygenases for Cancer Immunotherapy. Clin. Cancer Res. 2015, 21, 5427–5433. 10.1158/1078-0432.CCR-15-0420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Pantouris G.; Serys M.; Yuasa H. J.; Ball H. J.; Mowat C. G. Human indoleamine 2,3-dioxygenase-2 has substrate specificity and inhibition characteristics distinct from those of indoleamine 2,3-dioxygenase-1. Amino Acids 2014, 46, 2155–2163. 10.1007/s00726-014-1766-3. [DOI] [PubMed] [Google Scholar]; b D’Amato N. C.; Rogers T. J.; Gordon M. A.; Greene L. I.; Cochrane D. R.; Spoelstra S.; Nemkov T. G.; D’Alessandro A.; Hansen K. C.; Richer J. K. A TDO2-AhR signaling axis facilitates anoikis resistance and metastasis in triple-negative breast cancer. Cancer Res. 2015, 75, 4651–4664. 10.1158/0008-5472.CAN-15-2011. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Merlo L. M.; DuHadaway J. B.; Grabler S.; Prendergast G. C.; Muller A. J.; Mandik-Nayak L. IDO2 modulates T cell-dependent autoimmune responses through a B cell-intrinsic mechanism. J. Immunol. 2016, 196, 4487–4497. 10.4049/jimmunol.1600141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Weng T.; Qiu X.; Wang J.; Li Z.; Bian J. Recent discovery of indoleamine-2,3-dioxygenase 1 inhibitors targeting cancer immunotherapy. Eur. J. Med. Chem. 2018, 143, 656–669. 10.1016/j.ejmech.2017.11.088. [DOI] [PubMed] [Google Scholar]; b Coletti A.; Greco F. A.; Dolciami D.; Camaioni E.; Sardella R.; Pallotta M. T.; Volpi C.; Orabona C.; Grohmann U.; Macchiarulo A. Advances in indoleamine 2,3-dioxygenase 1 medicinal chemistry. MedChemComm 2017, 8, 1378–1392. 10.1039/C7MD00109F. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Prendergast G. C.; Malachowski W. P.; DuHadaway J. B.; Muller A. J. Discovery of IDO1 inhibitors: from bench to bedside. Cancer Res. 2017, 77, 6795–6811. 10.1158/0008-5472.CAN-17-2285. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Röhrig U. F.; Majjigapu S. R.; Vogel P.; Zoete V.; Michielin O. Challenges in the discovery of indoleamine 2,3-dioxygenase 1 (IDO1) inhibitors. J. Med. Chem. 2015, 58, 9421–9437. 10.1021/acs.jmedchem.5b00326. [DOI] [PubMed] [Google Scholar]; e Dounay A. B.; Tuttle J. B.; Verhoest P. R. Challenges and opportunities in the discovery of new therapeutics targeting the kynurenine pathway. J. Med. Chem. 2015, 58, 8762–8782. 10.1021/acs.jmedchem.5b00461. [DOI] [PubMed] [Google Scholar]
- a Balog A.; Lin T.-A.; Maley E.; Gullo-Brown J.; Hamza Kandoussi E.; Zeng J.; Hunt J. T. Preclinical Characterization of Linrodostat Mesylate, a Novel, Potent, and Selective Oral Indoleamine 2,3-Dioxygenase 1 Inhibitor. Mol. Cancer Ther. 2020, molcanther.0251.2020. 10.1158/1535-7163.MCT-20-0251. [DOI] [PubMed] [Google Scholar]; b Yue E. W.; Sparks R.; Polam P.; Modi D.; Douty B.; Wayland B.; Glass B.; Takvorian A.; Glenn J.; Zhu W.; Bower M.; Liu S.; Leffet L.; Wang Q.; Bowman K. J.; Hansbury M. J.; Wei M.; Li Y.; Wynn R.; Burn T. C.; Koblish H. K.; Fridman J. S.; Emm R.; Scherle P. A.; Metcalf B.; Combs A. P. INCB24360 (Epacadostat), a Highly Potent and Selective Indoleamine-2,3-dioxygenase 1 (IDO1) Inhibitor for Immuno-oncology. ACS Med. Chem. Lett. 2017, 8, 486–491. 10.1021/acsmedchemlett.6b00391. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Soliman H. H.; Antonia S.; Sullivan D.; Vanahanian N.; Link C. Overcoming coming tumor antigen anergy in human malignancies using the novel indeolamine 2,3-dioxygenase (IDO) enzyme inhibitor, 1-methyl-D-tryptophan (1MT). J. Clin. Oncol. 2009, 27, 3004. 10.1200/jco.2009.27.15_suppl.3004. [DOI] [Google Scholar]; d Kumar S.; Waldo J. P.; Jaipuri F. A.; Marcinowicz A.; Van Allen C.; Adams J.; Kesharwani T.; Zhang S.; Metz R.; Oh A. J.; Harris S. F.; Mautino M. R. Discovery of Clinical Candidate (1R,4r)-4-((R)-2-((S)-6- Fluoro-5H-imidazo[5,1-a]isoindol-5-yl)-1-hydroxyethyl)cyclohexan1-ol (Navoximod), a Potent and Selective Inhibitor of Indoleamine 2,3-Dioxygenase 1. J. Med. Chem. 2019, 62, 6705–6733. 10.1021/acs.jmedchem.9b00662. [DOI] [PubMed] [Google Scholar]; e Crosignani S.; Bingham P.; Bottemanne P.; Cannelle H.; Cauwenberghs S.; Cordonnier M.; Dalvie D.; Deroose F.; Feng J. L.; Gomes B.; Greasley S.; Kaiser S. E.; Kraus M.; Negrerie M.; Maegley K.; Miller N.; Murray B. W.; Schneider M.; Soloweij J.; Stewart A. E.; Tumang J.; Torti V. R.; Van Den Eynde B.; Wythes M. Discovery of a novel and selective indoleamine 2,3-dioxygenase (IDO-1) inhibitor 3-(5-Fluoro-1H-indol-3-yl)pyrrolidine-2,5-dione (EOS200271/PF-06840003) and its characterization as a potential clinical candidate. J. Med. Chem. 2017, 60, 9617–9629. 10.1021/acs.jmedchem.7b00974. [DOI] [PubMed] [Google Scholar]
- a Gangadhar T. C.; Hamid O.; Smith D. C.; Bauer T. M.; Waser J. S.; Luke J. J.; Balmanoukian A. S.; Kaufman D. R.; Zhao Y.; Maleski J.; Leopold L.; Gajewski T. F. Preliminary results from a phase I/II study of epacadostat (INCB024360) in combination with pembrolizumab in patients with selected advanced cancers. J. Immunother. Cancer 2015, 3 (Suppl 2), 07. 10.1186/2051-1426-3-S2-O7. [DOI] [Google Scholar]; b A Phase 3 Study of Pembrolizumab + Epacadostat or Placebo in Subjects With Unresectable or Metastatic Melanoma (Keynote-252/ECHO-301). ClinicalTrials.gov Identifier: NCT02752074. Last update: Sept. 4, 2019 (accessed on Oct. 23, 2019).; c Long G. V.; Dummer R.; Hamid O.; Gajewski T.; Caglevic C.; Dalle S.; Arance A.; Carlino M. S.; Grob J.-J.; Kim T. M.; Demidov L. V.; Robert C.; Larkin J. M. G.; Anderson J.; Maleski J. E.; Jones M. M.; Diede S. J.; Mitchell T. C. Epacadostat (E) plus pembrolizumab (P) versus pembrolizumab alone in patients (pts) with unresectable or metastatic melanoma: Results of the phase 3 ECHO-301/KEYNOTE-252 study. 2018 ASCO Annual Meeting. J. Clin. Oncol. 2018, 36, 108. 10.1200/JCO.2018.36.15_suppl.108. [DOI] [Google Scholar]; (suppl 15; abstr 108).; d Sondak V. K.; Khushalani N. Y. Echoes of a failure: what lessons can we learn?. Lancet Oncol. 2019, 20, 1037–1039. 10.1016/S1470-2045(19)30312-2. [DOI] [PubMed] [Google Scholar]; e Garber K. A new cancer immunotherapy suffers a setback. Science 2018, 360, 588. 10.1126/science.360.6389.588. [DOI] [PubMed] [Google Scholar]; f Komiya T.; Huang C. H. Updates in the clinical development of epacadostat and other indoleamine 2,3-dioxygenase 1 inhibitors (IDO1) for human cancer. Front. Oncol. 2018, 8, 423–429. 10.3389/fonc.2018.00423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Röhrig U. F.; Reynaud A.; Majjigapu S. R.; Vogel P.; Pojer F.; Zoete V. Inhibition Mechanisms of Indoleamine 2,3-Dioxygenase 1 (IDO1). J. Med. Chem. 2019, 62, 8784–8795. 10.1021/acs.jmedchem.9b00942. [DOI] [PubMed] [Google Scholar]
- a Nelp M. T.; Kates P. A.; Hunt J. T.; Newitt J. A.; Balog A.; Maley D.; Zhu X.; Abell L.; Allentoff A.; Borzilleri R.; Lewis H. A.; Lin Z.; Seitz S. P.; Yan C.; Groves J. T. Immune-modulating enzyme indoleamine 2,3-dioxygenase is effectively inhibited by targeting its apo-form. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 3249–3254. 10.1073/pnas.1719190115. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Pham K. N.; Yeh S.-R. Mapping the Binding Trajectory of a Suicide Inhibitor in Human Indoleamine 2,3-Dioxygenase 1. J. Am. Chem. Soc. 2018, 140, 14538–14541. 10.1021/jacs.8b07994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a A Study of Chemo Only Versus Chemo Plus Nivo With or Without BMS-986205, Followed by Post- Surgery Therapy With Nivo or Nivo and BMS-986205 in Patients With MIBC. ClinicalTrials.gov Identifier: NCT03661320.; b Sonpavde G.; Necchi A.; Gupta S.; Steinberg G. D.; Gschwend J. E.; Van Der Heijden M. S.; Garzon N.; Ibrahim M.; Raybold B.; Liaw D.; Rutstein M.; Galsky M. D. ENERGIZE: a Phase III study of neoadjuvant chemotherapy alone or with nivolumab with/without linrodostat mesylate for muscle-invasive bladder cancer. Future Oncol. 2020, 16 (2), 4359–4368. 10.2217/fon-2019-0611. [DOI] [PubMed] [Google Scholar]; c Tabernero J.; Luke J. J.; Joshua A. M.; Varga A. I.; Moreno V.; Desai J.; Markman B.; Gomez-Roca C. A.; De Braud F. G.; Patel S. P. BMS-986205, an indoleamine 2,3-dioxygenase 1 inhibitor (IDO1i), in combination with nivolumab (NIVO): Updated safety across all tumor cohorts and efficacy in pts with advanced bladder cancer (advBC). J. Clin. Oncol. 2018, 36, 4512. 10.1200/JCO.2018.36.15_suppl.4512. [DOI] [Google Scholar]
- Benarous R.; Chevreuil F.; Ledoussal B.; Chasset S.; Le Strat F.. Preparation of substituted thienylacetic acids as inhibitors of viral replication. WO 2014053666 A1. April 10, 2014.
- a Zhang L.; Cherney E. C.; Balog A. J.; Zhu X.. Inhibitors of indoleamine 2,3-dioxygenase and methods of their use. WO 2018039518 A1; March 1, 2018.; b Cherney E. C.; Shan W.; Zhang L.; Nara S. J.; Huang A.; Balog A. J.. Inhibitors of indoleamine 2,3-dioxygenase and methods of their use. WO 2018039512 A. March 1, 2018.
- An Investigational Immuno-Therapy Study of Experimental Medication BMS-986242 Given in Combination With Nivolumab in Patients With Advanced Cancer. ClinicalTrials.gov Identifier: NCT03351231.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.