

Abstract
Therapeutic modulation of the bile acid-sensing transcription factor farnesoid X receptor (FXR) is an appealing strategy to counteract hepatic and metabolic diseases. Despite the availability of several highly potent FXR agonists structural diversity of FXR modulators is limited, and new ligand scaffolds are needed. Here we report structure–activity relationship elucidation of a new FXR modulator chemotype whose activity can be tuned between agonism and antagonism by two minor structural modifications. Starting from a weak FXR/PPAR agonist, we have developed selective FXR activators and antagonists with nanomolar to low-micromolar potencies and binding affinities. The new FXR ligand chemotype modulates the FXR activity in the native cellular setting, is endowed with favorable metabolic stability, and lacks cytotoxicity. It valuably expands the collection of FXR modulators as a new scaffold for FXR-targeted drug discovery.
Keywords: Farnesoid X receptor, NR1H4, bile acid receptor, nonalcoholic steatohepatitis, NASH
The ligand-activated transcription factor farnesoid X receptor (FXR, NR1H4) is a key endocrine bile acid sensor involved in the regulation of diverse metabolic and inflammatory pathways.1−3 It is particularly found in the liver and intestine and considered to be a very attractive target to treat hepatic and metabolic diseases.4−6 However, despite the enormous attention that FXR has attracted, only a few potent FXR agonist chemotypes (e.g., 1–4,7−11Scheme 1) have been developed and entered clinical trials. The recent drawbacks12 in the development of the leading FXR agonist obeticholic acid (OCA, 2) in the clinical pipeline for the treatment of nonalcoholic steatohepatitis (NASH)13−15 illustrate the strong need for new FXR ligand scaffolds.
Scheme 1. Prominent FXR Agonists and Lead Compound 5.

GW4064 (1), OCA (2), WAY-362450 (3), and fexaramine (4).
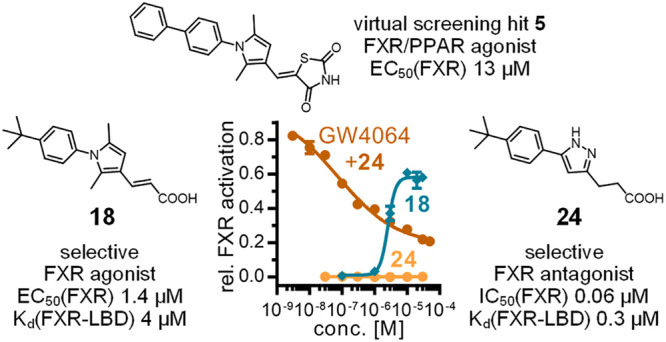
In 2010, Steri et al.16 discovered FXR ligand 5 in a virtual screening campaign. In vitro, 5 displayed FXR agonism with an EC50 value of 13 μM and simultaneously activated the peroxisome proliferator-activated receptor α (PPARα) with EC50 = 1.3 μM. The molecular architecture of 5 differs markedly from those of established FXR agonists such as 1–4, prompting us to study the structure–activity relationship (SAR) of 5 as an FXR ligand and its potential to serve as a template for a new FXR modulator chemotype. Systematic SAR elucidation enabled optimization of the FXR agonist potency and selectivity, and interestingly provided access to potent FXR antagonists, too. Here we report the SAR of FXR modulators derived from 5 and molecular determinants governing agonism or antagonism. With potent and selective orthogonally validated FXR agonists and antagonists, we disclose a structural basis for a new FXR modulator scaffold providing potential access to novel FXR-targeting tools and drugs.
Compounds 6–26 were prepared according to Schemes 2 and 3. Synthesis of pyrrole derivatives 6–20 started from anilines 27–38 (Scheme 2), which were condensed in a Paal–Knorr reaction with hexane-2,5-dione (39) to form pyrrole precursors 40–51. Formylation under Vilsmeier–Haack conditions produced pyrrole-3-carbaldehydes 52–63, and subsequent Knoevenagel condensation with 2,4-thiazolidinedione (64) resulted in the thiazolidinedione series 6–17, which were obtained in the Z-configuration as determined by X-ray powder diffraction (Figure S1). Acrylic acid derivative 18 was generated from pyrrole-3-carbaldehyde 63 by reaction with malonic acid in another Knoevenagel condensation and was obtained in the E-configuration according to NMR coupling constants (Figure S2). Hydrogenation of 18 with PtO2 (Adams’ catalyst) afforded saturated propionic acid analogue 19. To prepare pyrrole-3-carboxylate 20, aldehyde precursor 63 was oxidized with KMnO4.
Scheme 2. Synthesis of FXR Ligands 6–20.

Reagents and conditions: (a) p-TSA, toluene, reflux, 16 h, 32–97%; (b) POCl3, DMF, rt, 1 h, 30–90%; (c) 2,4-thiazolidinedione (64), piperidine, 110 °C, 1 h, 18–75%; (d) malonic acid, β-alanine, pyridine, 90 °C, 2 h, 34%; (e) PtO2, H2, MeOH, rt, 18 h, 3%; (f) KMnO4, acetone/H2O, 0 °C to rt, 18 h, 20%. R1 groups are shown in Table 1.
Scheme 3. Synthesis of FXR Ligands 21–26.

Reagents and conditions: (a) I2, Oxone, triflic acid, 100 °C, 5 h, 14%; (b) LiOH, EtOH/H2O, rt, 18 h, 12%; (c) DIPEA, DMA, rt, 18 h, quant.; (d) H2SO4, 80 °C, 0.5 h, 5%; (e) LiHMDS, THF, −20 °C to rt, 18 h, 29–32%; (f) hydrazine or methylhydrazine, NaOAc, AcOH, 80 °C, 2 h, 8–85%; (g) NaN3, DMF, 80 °C, 18 h, 50%; (h) CuSO4, sodium ascorbate, H2O/t-BuOH, rt, 18 h, 41%; (i) LiOH, EtOH/H2O, rt, 15 min, quant.
Analogues 21–26 comprising alternative heterocycles were prepared according to Scheme 3. Oxazole derivative 21 was synthesized by cyclization17 of propiophenone 65 with methyl 3-cyanopropionate (66) in the presence of triflic acid to afford 67 and subsequent saponification to give 21. Oxadiazole 22 was synthesized by the reaction of phenylhydrazide 68 with succinic anhydride (69) to give 70, which was then cyclized under acidic conditions to afford 22. For the preparation of pyrazoles 23, 24, and 26, acetophenone 71 was reacted in the presence of LiHMDS with succinic anhydride (69) to give 72 or with maleic anhydride (73) to give 74, which were subsequently cyclized with methylhydrazine or hydrazine under acidic conditions to afford 23, 24, or 26. The yields of pyrazoles 23, 24, and 26 varied due to purification issues. 26 was obtained in the E-configuration (Figure S2). For the preparation of triazole 25, ethyl 3-bromopropionate (75) was treated with sodium azide to obtain ethyl 3-azidopropionate (76). Copper(I)-catalyzed Huisgen 1,3-dipolar cycloaddition of 76 and alkyne 77 yielded triazole 78, and alkaline ester hydrolysis produced 25.
FXR modulation by 5–26 was evaluated in a cellular (HEK293T) reporter gene assay based on a hybrid receptor composed of the ligand binding domain (LBD) of human FXR and the DNA binding domain of the yeast protein Gal4. A Gal4-responsive firefly luciferase construct served as the reporter gene, and constitutively expressed renilla luciferase was cotransfected to monitor cell proliferation, transfection efficiency, and test compound toxicity. To evaluate competitive antagonism, compounds were also studied for FXR modulation in the presence of 1 μM GW4064 (1) in this setting. FXR modulation by selected compounds was further assessed in a full-length FXR reporter gene assay wherein the human FXR response element from the promoter region of the FXR-regulated gene bile salt export protein (BSEP) governs reporter gene expression. This assay captures the natural cellular conditions more realistically since it involves the full human FXR:RXR heterodimer. Additionally, selected compounds were profiled for selectivity over related nuclear receptors in equivalent hybrid assays.
As a first step in systematically studying the SAR of 5 as an FXR agonist, we evaluated the biphenyl motif. For structural simplification, we downsized the biphenyl motif to a phenyl substituent and systematically introduced a methyl group at each position (6–8) to probe the potential for optimization by extension (Table 1). Among these three tolyl analogues 6–8, only the para isomer 8 retained weak FXR agonist activity, suggesting a preference for 4-substituted phenyl residues on the pyrrole nitrogen. Hence, we subsequently studied structurally diverse substituents at the 4-position of the phenyl motif. 4-Methoxy analogue 9 failed to activate FXR, whereas replacement of the 4-methyl substituent in 8 by a 4-trifluoromethyl moiety in 10 markedly enhanced the potency to a low EC50 value of 2.6 μM. 4-Trifluoromethoxy (11) and 4-chloro (12) substitution were favored as well, despite low activation efficacy. Dimethylaniline 13 was inactive, whereas the bulkier morpholino derivative 14 achieved an EC50 value of 1 μM and considerable relative activation efficacy of 28%. A 4-phenoxy substituent (15) and the 2-naphthyl analogue 16 retained low-micromolar potency, but the activation efficacy dropped markedly. The 4-tert-butyl group (17), which is a well-established FXR ligand motif,18−20 revealed the most favorable profile with an EC50 value of 2.6 μM and a relative activation efficacy of 58%.
Table 1. In Vitro Activity of 5–17 on FXR in a Hybrid Reporter Gene Assaya.

Maximum relative activation corresponds to the activity of 1 μM GW4064 (1). Inactive indicates that there was no statistically significant reporter activation at the indicated concentration. Data are reported as mean ± SEM, n ≥ 3.
After elucidating the SAR of the phenyl substituent, we probed further optimization in the acidic side chain. The thiazolidinedione residue in 5–17 is a common bioisostere for carboxylic acids and an important motif in PPARγ agonists.21−23 Thus, we replaced the thiazolidinedione in 17 by a carboxylic acid, resulting in 3-(pyrrol-3-yl)acrylic acid 18, which slightly gained in FXR agonist potency (EC50 = 1.4 μM) compared with 17 (Table 2 and Figure 1). Reduction of the double bond in 18 gave 3-(pyrrol-3-yl)propionic acid 19, which retained FXR agonist activity but with a 10-fold loss in potency and lower activation efficacy. Interestingly, the shortened pyrrole-3-carboxylic acid analogue 20 continued this trend and failed to activate FXR but exhibited competitive FXR antagonism (IC50 = 8 μM).
Table 2. In Vitro Activity of 17–20 on FXR in a Hybrid Reporter Gene Assaya.

Maximum relative activation (max. rel. act.) and remaining activity (remain. act.) correspond to the activity of 1 μM GW4064 (1). Data are reported as mean ± SEM, n ≥ 3. Kd values were obtained from ITC with recombinant FXR LBD.
Figure 1.

In vitro profiling of 17 and 18. (a) Chemical structures of 17 and 18. (b) ITC demonstrated binding of 17 and 18 to the recombinant FXR LBD. (c) Dose–response curves of 17 and 18 on FXR in a Gal4 hybrid reporter gene assay. Curves for GW4064 (1) and CDCA are shown for comparison. Data are the mean ± SEM relative FXR activation compared to 1 μM 1, n ≥ 3. (d) 18 induced expression of the FXR-regulated genes small heterodimer partner (SHP) and bile salt export protein (BSEP) in hepatocytes (HepG2) with similar efficacy as CDCA. Data are the mean ± SEM relative mRNA expression, n = 3: *, p < 0.05; **, p < 0.01 (t test vs DMSO). (e) Selectivity profiles of 17 and 18 on related nuclear receptors. The heat map shows mean relative activation compared with the reference agonist (FXR, 1; PPARα, GW7647; PPARγ, pioglitazone; PPARδ, L165,041; RXRα, bexarotene; RARα, tretinoin), n = 3. (f) Dose–response curves of 18 on FXR, PPARα, and RXRα. Data are the mean ± SEM relative activation compared with 1 μM of the respective reference agonist, n ≥ 3.
Overall, 17 and 18 evolved as the most favored descendants of 5 and were therefore further characterized (Figure 1). Binding of 17 and 18 to the recombinant FXR LBD was confirmed by isothermal titration calorimetry (ITC) with Kd values of 9 and 4 μM, respectively (Figure 1b). Comparison with common FXR agonists (Figure 1c) revealed potency and efficacy profiles of 17 and 18 that were between those of the endogenous FXR ligand CDCA and the reference agonist 1. In human hepatocytes (HepG2), 18 (10 μM) induced mRNA expression of the FXR-regulated genes small heterodimer partner (SHP) and BSEP with similar efficacies as the natural agonist CDCA (30 μM), demonstrating FXR agonism in a native cellular setting (Figure 1d). Profiling on related lipid-sensing nuclear receptors (LXRs, PPARs, RXRα, RARα; Figure 1e) demonstrated favorable selectivity for both compounds. Dose–response profiling of 18 on PPARα and RXRα up to 100 μM (Figure 1f) demonstrated that PPARα agonism was fully eliminated and revealed weak agonism on RXRα (EC50 = 16.4 ± 0.1 μM, 22 ± 1% eff.). Because of their low molecular weights, 17 and 18 even possessed superior ligand efficiency metrics compared with the reference FXR agonist 1 (LE: 1, 0.27; 17, 0.31; 18, 0.36; LLE: 1, <0; 17, 0.77; 18, 1.0),24 highlighting their value as leads for medicinal chemistry.
Molecular docking of 18 in the ligand binding site of CDCA-bound (PDB ID 4qe6(18)) and 1-bound (PDB ID 3dct(25)) FXR cocrystal structures (Figure 2) suggested a typical binding mode with a contact to Arg335 as an exclusive polar interaction. The compound favorably spans the sustained FXR ligand binding site with the dimethyl decoration of the pyrrole placed in a lipophilic environment that also accommodates the chlorine substituent of 1 and the D ring of CDCA. The tert-butyl motif protrudes toward helix 12 but not far enough to cause conformational changes, as it has been observed for weaker partial agonists.18,19 However, the terminus of the FXR ligand binding site, which is occupied by the hammerhead structure of 1 and important for strong agonism of 1 and analogues, is not addressed by 18, rationalizing the intermediate FXR activation efficacy of this novel chemotype. Further optimization potential might therefore be present in the tert-butylphenyl region of 18. Supporting this assumption, molecular docking of the extended morpholino derivative 14 (Figure S3) indicated further binding site occupation and suggested a potential polar contact of the morpholine oxygen to Tyr361 at the end of the pocket, which also forms a hydrogen bond with the natural ligand CDCA.
Figure 2.
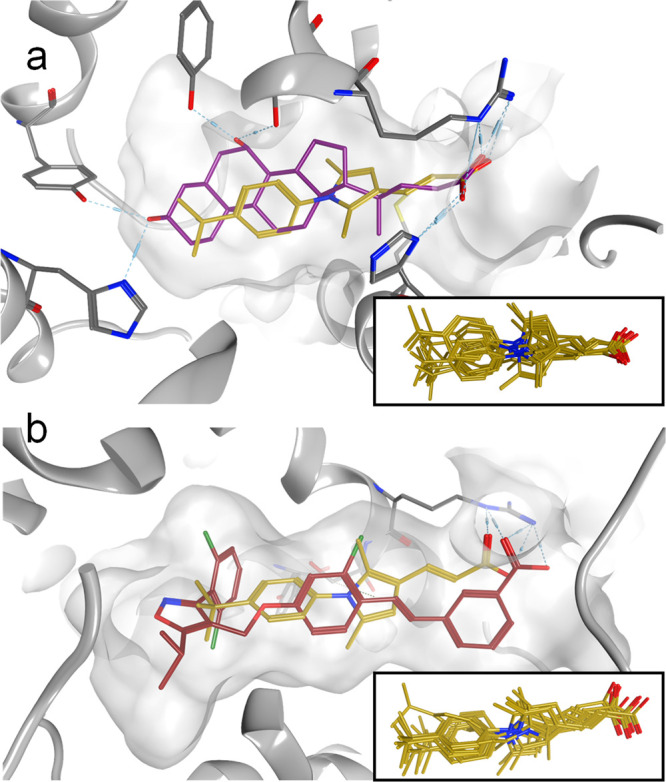
Molecular docking of 18 (yellow) to the ligand binding sites of the FXR cocrystal structures in complex with (a) CDCA (purple, PDB ID 4qe6) and (b) 1 (red, PDB ID 3dct). The inserts show overlays of the top 10 binding poses for each structure.
As the remaining structural feature of this FXR ligand chemotype, we then addressed the SAR of the central heterocyclic component (21–25; Table 3). Replacing the 2,5-dimethylpyrrole of the partial agonist 19 by a 4-methyloxazole in 21 led to a loss of FXR agonism and resulted in a weak antagonist (IC50 = 13.9 μM). 1,3,4-Oxadiazole analogue 22 lacking the methyl decorations revealed a submicromolar IC50 value for competitive FXR antagonism. 1-Methyl-1H-pyrazole analogue 23 was less potent but demonstrated remarkably enhanced antagonist efficacy, and unsubstituted pyrazole derivative 24 was both potent and effective in competitive FXR antagonism (Figure 3). Incorporation of a third nitrogen atom in 1H-1,2,3-triazole derivative 25 retained strong FXR antagonism but with lower potency than for pyrazole 24. To further confirm this interesting SAR, we prepared and characterized 26 comprising the antagonism-driving pyrazole heterocycle of 24 and the agonism-favored acrylate motif of 18 (Table 4). 26 turned out to be a very weak FXR agonist (<5% activation at 30 μM) and did not counteract FXR activation by 1. Thus, only two minor structural variations—replacement of the dimethylpyrrole by a pyrazole and reduction of the acrylate motif—govern the activity type of the FXR ligand chemotype between the potent FXR agonism of 18 and the pronounced antagonist activity of 24.
Table 3. In Vitro Activity of 21–25 on FXR in a Hybrid Reporter Gene Assaya.

Maximum relative activation (max. rel. act.) and remaining activity (remain. act.) correspond to the activity of 1 μM GW4064 (1). Data are reported as mean ± SEM, n ≥ 3.
Figure 3.

In vitro profiling of 24. (a) Chemical structure and activity data for 24 on FXR. (b) Dose–response curve of 24 on FXR in a hybrid reporter gene assay. 24 caused no FXR activation up to 30 μM concentration (gray curve) but antagonized GW4064-induced FXR activity (black curve). Data are the mean ± SEM relative FXR activation compared with 1 μM GW4064 (1), n ≥ 3. (c) Control experiment. 24 strongly antagonized GW4064-induced FXR activity when the two compounds were coincubated (16 h), but no effect was observed when 24 was added 1 h before lysis and luminescence measurement. The box plot shows mean and min.–max., n = 4. **, p < 0.01. (d) 24 robustly antagonized CDCA-induced activation of the human FXR:RXR heterodimer in HepG2 cells. Data are reported as mean ± SEM, n = 3. (e) ITC demonstrated binding of 24 to the recombinant FXR LBD with a Kd of 0.3 μM. (f) Selectivity profiling of 24 revealed no agonism or antagonism on related nuclear receptors. The heat map shows mean relative activation compared with the respective reference agonist, n = 3.
Table 4. In Vitro Activity of 18, 19, 24, and 26 on FXR in a Hybrid Reporter Gene Assaya.

Maximum relative activation (max. rel. act.) and remaining activity (remain. act.) correspond to the activity of 1 μM GW4064 (1). Data are reported as mean ± SEM, n ≥ 3.
As a control experiment, we repeated the hybrid reporter gene assay but added 24 only 1 h before lysis and luminescence measurement to cells treated with 1 for full FXR activation (Figure 3c). In this setting, no effect of 24 on the reporter activity was observed, in contrast to coincubation of 24 and 1, which demonstrated a strong decrease in reporter activity. Thus, the observed activity of 24 was FXR-mediated and not an artifact of luciferase inhibition. Moreover, 24 robustly antagonized CDCA-induced activation of the human FXR:RXR heterodimer in HepG2 cells, as observed with a full-length FXR reporter construct bearing the human FXR response element from the promoter region of the FXR target gene BSEP to govern reporter activity (Figure 3d). ITC further confirmed direct interaction of 24 with the recombinant FXR LBD with submicromolar binding affinity (Kd = 0.3 μM; Figure 3e), and profiling on related nuclear receptors characterized 24 as a selective FXR antagonist (Figure 3f).
From the systematic SAR elucidation of the new FXR ligand chemotype, 18 and 24 evolved as the most active FXR modulators, prompting us to further evaluate their potential as tool compounds for pharmacology and as leads for medicinal chemistry (Figure 4). The pyrrole- and pyrazole-based FXR ligands exhibited remarkably higher stability against microsomal degradation than the reference FXR agonist 1 (Figure 4a) and were also superior in terms of lipophilicity (18, alogP 4.84; 24, alogP 3.99; 1, alogP 7.41).26 Furthermore, 18 and 24 were nontoxic in human hepatocytes (HepG2) up to 100 μM concentration (Figure 4b).
Figure 4.
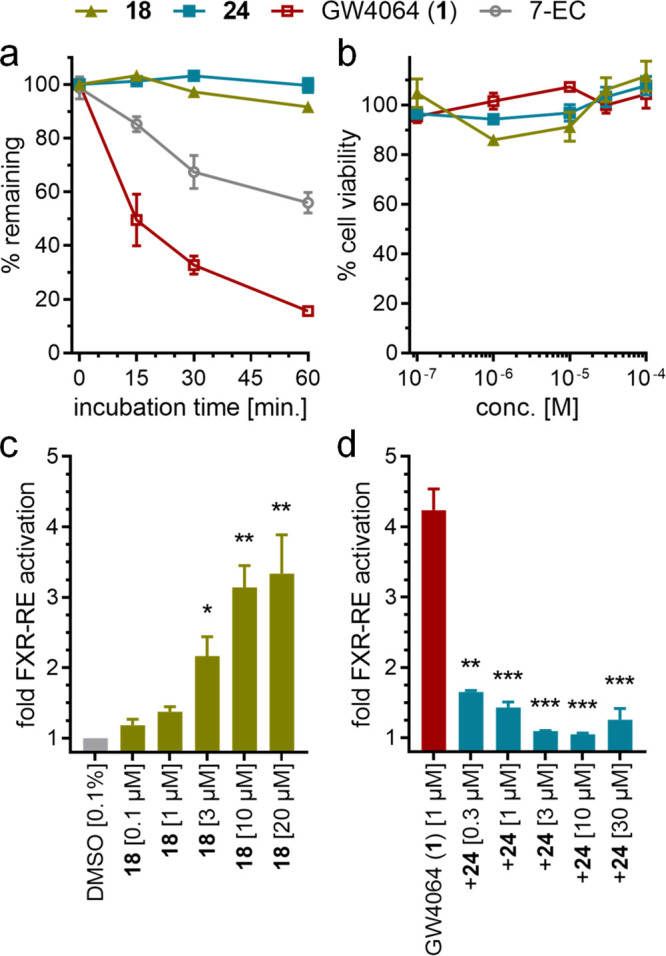
Microsomal stability, cytotoxicity, and FXR:RXR dimer modulatory activity of the new FXR ligands 18 and 24. (a) Stability of 18 and 24 against degradation by rat liver microsomes. Data for 1 are shown for comparison. 7-Ethoxycoumarin (7-EC) was used as the control. Data are mean ± SEM percent remaining compound, n = 3. (b) Cytotoxicity of 18 and 24 in human hepatocytes (HepG2) determined in a WST-1 assay. Data are mean ± SEM relative cell viability vs 0.1% DMSO, n = 4. (c, d) Activity of 18 and 24 on the human FXR:RXR heterodimer using a reporter gene under the control of the FXR response element from the promoter region of BSEP. Data are mean ± SEM fold FXR:RXR activation, n = 3. *, p < 0.05; **, p < 0.01; ***, p < 0.001 (t test vs (c) DMSO or (d) 1 μM 1).
To further validate the FXR modulator activity of 18 and 24 in a more physiological setting than the hybrid assay, we probed their effects on the human FXR response element from the promoter region of BSEP, which is regulated by the human full-length FXR:RXR heterodimer (Figure 4c). FXR agonist 18 robustly activated FXR:RXR (EC50 = 3 μM), whereas 24 acted as an FXR antagonist in competition with 1 on the FXR:RXR heterodimer as well.
In conclusion, from the nonselective virtual screening hit 5 as a lead, we have systematically probed the SAR of a novel FXR ligand chemotype that turned out to be tunable in its activity type between agonism and antagonism. With the central heterocycle and side chain saturation as switches, FXR activators and antagonists are accessible that comprise preferable characteristics compared with the reference FXR ligand 1. In particular, FXR agonist 18 and antagonist 24 evolved as optimized descendants of 5 with considerable potency and high selectivity. Both compounds modulated FXR activity in two cellular settings and bound to the recombinant FXR LBD, which orthogonally validated their effects as FXR-mediated. The compounds establish a valuable new FXR modulator scaffold to expand the collection of FXR ligand chemotypes as tools and potentially experimental drugs.
Acknowledgments
The authors thank Edith Alig, Emre Duman, and Dr. Lothar Fink (Goethe University Frankfurt) for performing X-ray powder diffraction and structure solution for compounds 10, 13, and 17. D.M. is grateful for financial support by the Aventis Foundation.
Glossary
Abbreviations
- BSEP
bile salt export protein
- CDCA
chenodeoxycholic acid
- FXR
farnesoid X receptor
- ITC
isothermal titration calorimetry
- LBD
ligand binding domain
- LXR
liver X receptor
- PPAR
peroxisome proliferator-activated receptor
- RAR
retinoic acid receptor
- RXR
retinoid X receptor
- SAR
structure–activity relationship
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00647.
Figures S1–S3, synthetic procedures and analytical characterization data for compounds 6–26 and intermediates, methods for in vitro characterization, and computational procedures (PDF)
Author Contributions
† M.H. and J.V. contributed equally to this study.
The authors declare no competing financial interest.
Notes
X-ray powder diffraction data for compounds 10 (CCDC 2058900), 13 (CCDC 2058898), and 17 (CCDC 2058882) are available from the Cambridge Crystallographic Data Centre (CCDC).
Supplementary Material
References
- Moore D. D.; Kato S.; Xie W.; Mangelsdorf D. J.; Schmidt D. R.; Xiao R.; Kliewer S. A. International Union of Pharmacology. LXII. The NR1H and NR1I Receptors: Constitutive Androstane Receptor, Pregnene X Receptor, Farnesoid X Receptor Alpha, Farnesoid X Receptor Beta, Liver X Receptor Alpha, Liver X Receptor Beta, and Vitamin D Receptor. Pharmacol. Rev. 2006, 58 (4), 742–759. 10.1124/pr.58.4.6. [DOI] [PubMed] [Google Scholar]
- Merk D.; Steinhilber D.; Schubert-Zsilavecz M. Medicinal Chemistry of Farnesoid X Receptor Ligands: From Agonists and Antagonists to Modulators. Future Med. Chem. 2012, 4 (8), 1015–1036. 10.4155/fmc.12.47. [DOI] [PubMed] [Google Scholar]
- Shin D. J.; Wang L. Bile Acid-Activated Receptors: A Review on FXR and Other Nuclear Receptors. Handb. Exp. Pharmacol. 2019, 256, 51–72. 10.1007/164_2019_236. [DOI] [PubMed] [Google Scholar]
- Adorini L.; Pruzanski M.; Shapiro D. Farnesoid X Receptor Targeting to Treat Nonalcoholic Steatohepatitis. Drug Discovery Today 2012, 17 (17–18), 988–997. 10.1016/j.drudis.2012.05.012. [DOI] [PubMed] [Google Scholar]
- Arab J. P.; Karpen S. J.; Dawson P. A.; Arrese M.; Trauner M. Bile Acids and Nonalcoholic Fatty Liver Disease: Molecular Insights and Therapeutic Perspectives. Hepatology 2017, 65 (1), 350–362. 10.1002/hep.28709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuipers F.; Bloks V. W.; Groen A. K. Beyond Intestinal Soap-Bile Acids in Metabolic Control. Nat. Rev. Endocrinol. 2014, 10 (8), 488–498. 10.1038/nrendo.2014.60. [DOI] [PubMed] [Google Scholar]
- Maloney P. R.; Parks D. J.; Haffner C. D.; Fivush A. M.; Chandra G.; Plunket K. D.; Creech K. L.; Moore L. B.; Wilson J. G.; Lewis M. C.; Jones S. A.; Willson T. M. Identification of a Chemical Tool for the Orphan Nuclear Receptor FXR. J. Med. Chem. 2000, 43 (16), 2971–2974. 10.1021/jm0002127. [DOI] [PubMed] [Google Scholar]
- Downes M.; Verdecia M. A.; Roecker A. J.; Hughes R.; Hogenesch J. B.; Kast-Woelbern H. R.; Bowman M. E.; Ferrer J.-L.; Anisfeld A. M.; Edwards P. A.; Rosenfeld J. M.; Alvarez J. G. A.; Noel J. P.; Nicolaou K. C.; Evans R. M. A Chemical, Genetic, and Structural Analysis of the Nuclear Bile Acid Receptor FXR. Mol. Cell 2003, 11 (4), 1079–1092. 10.1016/S1097-2765(03)00104-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flatt B.; Martin R.; Wang T. L.; Mahaney P.; Murphy B.; Gu X. H.; Foster P.; Li J.; Pircher P.; Petrowski M.; Schulman I.; Westin S.; Wrobel J.; Yan G.; Bischoff E.; Daige C.; Mohan R. Discovery of XL335 (WAY-362450), a Highly Potent, Selective, and Orally Active Agonist of the Farnesoid X Receptor (FXR). J. Med. Chem. 2009, 52 (4), 904–907. 10.1021/jm8014124. [DOI] [PubMed] [Google Scholar]
- Pellicciari R.; Fiorucci S.; Camaioni E.; Clerici C.; Costantino G.; Maloney P. R.; Morelli A.; Parks D. J.; Willson T. M. 6α-Ethyl-Chenodeoxycholic Acid (6-ECDCA), a Potent and Selective FXR Agonist Endowed with Anticholestatic Activity. J. Med. Chem. 2002, 45 (17), 3569–3572. 10.1021/jm025529g. [DOI] [PubMed] [Google Scholar]
- Xu Y. Recent Progress on Bile Acid Receptor Modulators for Treatment of Metabolic Diseases. J. Med. Chem. 2016, 59 (14), 6553–6579. 10.1021/acs.jmedchem.5b00342. [DOI] [PubMed] [Google Scholar]
- Mullard A. FDA Rejects NASH Drug. Nat. Rev. Drug Discovery 2020, 19 (8), 501. 10.1038/d41573-020-00126-9. [DOI] [PubMed] [Google Scholar]
- Neuschwander-Tetri B. A.; Loomba R.; Sanyal A. J.; Lavine J. E.; Van Natta M. L.; Abdelmalek M. F.; Chalasani N.; Dasarathy S.; Diehl A. M.; Hameed B.; Kowdley K. V.; McCullough A.; Terrault N.; Clark J. M.; Tonascia J.; Brunt E. M.; Kleiner D. E.; Doo E. Farnesoid X Nuclear Receptor Ligand Obeticholic Acid for Non-Cirrhotic, Non-Alcoholic Steatohepatitis (FLINT): A Multicentre, Randomised, Placebo-Controlled Trial. Lancet 2015, 385 (9972), 956–965. 10.1016/S0140-6736(14)61933-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younossi Z. M.; Ratziu V.; Loomba R.; Rinella M.; Anstee Q. M.; Goodman Z.; Bedossa P.; Geier A.; Beckebaum S.; Newsome P. N.; Sheridan D.; Sheikh M. Y.; Trotter J.; Knapple W.; Lawitz E.; Abdelmalek M. F.; Kowdley K. V.; Montano-Loza A. J.; Boursier J.; et al. Obeticholic Acid for the Treatment of Non-Alcoholic Steatohepatitis: Interim Analysis from a Multicentre, Randomised, Placebo-Controlled Phase 3 Trial. Lancet 2019, 394 (10215), 2184–2196. 10.1016/S0140-6736(19)33041-7. [DOI] [PubMed] [Google Scholar]
- Musso G.; Cassader M.; Gambino R. Non-Alcoholic Steatohepatitis: Emerging Molecular Targets and Therapeutic Strategies. Nat. Rev. Drug Discovery 2016, 15 (4), 249–274. 10.1038/nrd.2015.3. [DOI] [PubMed] [Google Scholar]
- Steri R.; Schneider P.; Klenner A.; Rupp M.; Kriegl J. M.; Schubert-Zsilavecz M.; Schneider G. Target Profile Prediction: Cross-Activation of Peroxisome Proliferator-Activated Receptor (PPAR) and Farnesoid X Receptor (FXR). Mol. Inf. 2010, 29 (4), 287–292. 10.1002/minf.200900009. [DOI] [PubMed] [Google Scholar]
- Imai S.; Kikui H.; Moriyama K.; Togo H. One-Pot Preparation of 2,5-Disubstituted and 2,4,5-Trisubstituted Oxazoles from Aromatic Ketones with Molecular Iodine, Oxone, and Trifluoromethanesulfonic Acid in Nitriles. Tetrahedron 2015, 71 (33), 5267–5274. 10.1016/j.tet.2015.06.022. [DOI] [Google Scholar]
- Merk D.; Sreeramulu S.; Kudlinzki D.; Saxena K.; Linhard V.; Gande S. L.; Hiller F.; Lamers C.; Nilsson E.; Aagaard A.; Wissler L.; Dekker N.; Bamberg K.; Schubert-Zsilavecz M.; Schwalbe H. Molecular Tuning of Farnesoid X Receptor Partial Agonism. Nat. Commun. 2019, 10 (1), 2915. 10.1038/s41467-019-10853-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merk D.; Lamers C.; Ahmad K.; Carrasco Gomez R.; Schneider G.; Steinhilber D.; Schubert-Zsilavecz M. Extending the Structure-Activity Relationship of Anthranilic Acid Derivatives as Farnesoid x Receptor Modulators: Development of a Highly Potent Partial Farnesoid x Receptor Agonist. J. Med. Chem. 2014, 57 (19), 8035–8055. 10.1021/jm500937v. [DOI] [PubMed] [Google Scholar]
- Schmidt J.; Rotter M.; Weiser T.; Wittmann S.; Weizel L.; Kaiser A.; Heering J.; Goebel T.; Angioni C.; Wurglics M.; Paulke A.; Geisslinger G.; Kahnt A.; Steinhilber D.; Proschak E.; Merk D. A Dual Modulator of Farnesoid X Receptor and Soluble Epoxide Hydrolase to Counter Nonalcoholic Steatohepatitis. J. Med. Chem. 2017, 60 (18), 7703–7724. 10.1021/acs.jmedchem.7b00398. [DOI] [PubMed] [Google Scholar]
- Jang J. Y.; Bae H.; Lee Y. J.; Choi Y. Il; Kim H.-J.; Park S. B.; Suh S. W.; Kim S. W.; Han B. W. Structural Basis for the Enhanced Anti-Diabetic Efficacy of Lobeglitazone on PPARγ. Sci. Rep. 2018, 8 (1), 31. 10.1038/s41598-017-18274-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanke T.; Cheung S. Y.; Kilu W.; Heering J.; Ni X.; Planz V.; Schierle S.; Faudone G.; Friedrich M.; Wanior M.; Werz O.; Windbergs M.; Proschak E.; Schubert-Zsilavecz M.; Chaikuad A.; Knapp S.; Merk D. A Selective Modulator of Peroxisome Proliferator-Activated Receptor Γwith an Unprecedented Binding Mode. J. Med. Chem. 2020, 63 (9), 4555–4561. 10.1021/acs.jmedchem.9b01786. [DOI] [PubMed] [Google Scholar]
- Lamers C.; Schubert-Zsilavecz M.; Merk D. Therapeutic Modulators of Peroxisome Proliferator-Activated Receptors (PPAR): A Patent Review (2008-Present). Expert Opin. Ther. Pat. 2012, 22 (7), 803–841. 10.1517/13543776.2012.699042. [DOI] [PubMed] [Google Scholar]
- Hopkins A. L.; Keserü G. M.; Leeson P. D.; Rees D. C.; Reynolds C. H. The Role of Ligand Efficiency Metrics in Drug Discovery. Nat. Rev. Drug Discovery 2014, 13 (2), 105–121. 10.1038/nrd4163. [DOI] [PubMed] [Google Scholar]
- Akwabi-Ameyaw A.; Bass J. Y.; Caldwell R. D.; Caravella J. A.; Chen L.; Creech K. L.; Deaton D. N.; Jones S. A.; Kaldor I.; Liu Y.; Madauss K. P.; Marr H. B.; McFadyen R. B.; Miller A. B.; Navas F. III; Parks D. J.; Spearing P. K.; Todd D.; Williams S. P.; Wisely G. B. Conformationally Constrained Farnesoid X Receptor (FXR) Agonists: Naphthoic Acid-Based Analogs of GW 4064. Bioorg. Med. Chem. Lett. 2008, 18 (15), 4339–4343. 10.1016/j.bmcl.2008.06.073. [DOI] [PubMed] [Google Scholar]
- Tetko I. V.; Tanchuk V. Y. Application of Associative Neural Networks for Prediction of Lipophilicity in ALOGPS 2.1 Program. J. Chem. Inf. Comput. Sci. 2002, 42 (5), 1136–1145. 10.1021/ci025515j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.