

Abstract

Antitubercular 7-substituted 2-nitroimidazo[2,1-b][1,3]oxazines were previously shown to exhibit potent antileishmanial and antitrypanosomal activities, culminating in a new clinical investigational drug for visceral leishmaniasis (DNDI-0690). To offset development risks, we continued to seek further leads with divergent candidate profiles, especially analogues possessing greater aqueous solubility. Starting from an efficacious monoaryl derivative, replacement of the side chain ether linkage by novel amine, amide, and urea functionality was first explored; the former substitution was well-tolerated in vitro and in vivo but elicited marginal alterations to solubility (except through a less stable benzylamine), whereas the latter groups resulted in significant solubility improvements (up to 53-fold) but an antileishmanial potency reduction of at least 10-fold. Ultimately, we discovered that O-carbamate 66 offered a more optimal balance of increased solubility, suitable metabolic stability, excellent oral bioavailability (100%), and strong in vivo efficacy in a visceral leishmaniasis mouse model (97% parasite load reduction at 25 mg/kg).
Keywords: pretomanid, leishmaniasis, tuberculosis, Chagas disease, pharmacokinetics, in vivo efficacy
Leishmaniasis comprises a family of four diseases that are caused by Leishmania parasites and spread by infected sandflies.1 The most prevalent malady, cutaneous leishmaniasis (CL), leads to deep skin ulcers, whereas visceral leishmaniasis (VL) describes a condition where L. donovani (L. don) or L. infantum (L. inf) engulf critical organs, e.g., the spleen and liver, eventually causing death (without chemotherapy). Current options to treat VL include just four drugs (only one oral; miltefosine, 1; Figure 1) whose success varies greatly inside and among different regions.2 The World Health Organization recognizes that achievement of its 2030 target to “eliminate VL as a public health problem in 85% of countries” will need “critical action to develop more effective and user-friendly treatment and diagnostics, especially for East Africa”.3
Figure 1.

Various oral agents against VL (and/or tuberculosis).
Historically, drug discovery for leishmaniasis has relied heavily upon the repurposing of approved medicines.4 Today, there are still few well-validated drug targets for VL and CL, as the “biological pathways essential for survival and disease progression” are still being unraveled.5 Nevertheless, recent extensive investment in phenotypic screening of large compound collections and lead optimization activities4 has now produced five new VL drug candidates in phase I clinical trials (2-6) and another (DNDI-6174) in preclinical studies.5−8 Importantly, these agents possess diverse modes of action, providing the opportunity for future combination therapy; 2 (GSK3186899)9 is a CRK-12 inhibitor,2 both 3 (LXE408)8 and 6 (GSK3494245)10 are proteasome inhibitors, 4 (DNDI-6148)5 may inhibit mRNA maturation by binding to the enzyme CPSF3 (based on studies of other benzoxaboroles in T. brucei and target site identity in Leishmania(11)), and 5 (DNDI-0690)12 is activated by a novel nitroreductase, NTR2.13 Compound 5 originated from our VL lead optimization program with DNDi around analogues of the newly approved14 tuberculosis (TB) drug pretomanid (PA-824, 7). It exhibits very rapid leishmanicidal activity against L. don and L. inf without cytotoxicity and displays remarkable efficacy in mouse and hamster models of VL.12,15 Candidates 4 and 5 also demonstrate excellent oral activity in mouse models of CL,16,17 while 5 additionally retains the strong antitubercular properties of 7.12
Despite such progress, the failures of sitamaquine and fexinidazole in previous VL clinical trials2,12 and an unexpected recent suspension to the phase I study of 2(18) provide a timely reminder regarding the high attrition rate (∼90%) for small molecules in clinical development.19 To offset this risk, we aimed to discover novel backup candidates to 5 having greater aqueous solubility.20 An excellent starting point for new SAR exploration was analogue 8 (L. inf IC50 0.13 μM), which displayed in vivo activity comparable to that of 5 in a VL mouse model but had inferior safety.12 In previous SAR investigations on pretomanid (7), we demonstrated that replacement of the oxygen atom at C-6 by nitrogen-based linkers (viz. amines, amides, ureas, and N-carbamates) or extension of the O-linkage with additional polar functionality (e.g., acetamide, O-carbamate) were effective strategies to reduce compound lipophilicity, improve aqueous solubility, and modulate PK properties.20,21 An NH- (rather than O−)linkage to the phenyl ring of rac-9 also secured good potency and enhanced solubility (6-fold) in the 6-nitroimidazooxazole class.22
Our first targets encompassed several novel amino- and methylamino-linked analogues of 8. Reaction of known12 epoxides 13 and 16 with various anilines (catalyzed by anhydrous cobaltous chloride22) gave the uncyclized β-anilino alcohols, which were ring closed in low to moderate yield by careful treatment with sodium hydride22 (1.4–1.6 equiv; Scheme 1A, B). One pyridine congener (35) was also prepared, although here the epoxide-opening step was inefficient (17%) due to the poor nucleophilicity of methylaminopyridine 33 (Scheme 1C).
Scheme 1. Synthesis of Novel Arylamino Analogues of 8.

Reagents and conditions: (i) CoCl2, CH3CN, 65–75 °C, 1–3 d (17–97%); (ii) NaH, DMF, 0–20 °C, 2–4.3 h (or 50–70 °C, 2–3 h) (9–63%); (iii) HCOOH/Ac2O, THF, 20 °C, 23 h (97%); (iv) Me2S·BH3, THF, 0–20 °C, 0.5 h, then 65 °C, 3.5 h (72%).
Additional N-linked targets were obtained through azide intermediates 37 and 42, generated from epoxide 16 (via ring opening with sodium azide23 and ring closure as above) and alcohol 41(12) (via Mitsunobu reaction with diphenylphosphoryl azide), respectively (Scheme 2A, B). Reduction of 37 to amine 38 using 1:1 propane-1,3-dithiol/triethylamine20 was complicated by low solubility and side product formation (acylation then gave amide 45 in just 38% yield; Scheme 2B). Therefore, we turned to a one-pot amidation method involving modified Staudinger conditions (triphenylphosphine added to a mixture of the azide and acid chloride),24 which furnished the carboxamides (44 and 46–48) directly and in high yield (73–84%; Scheme 2B). Carbamoylation (urea 51) was also achieved through another Staudinger approach25 (triphenylphosphine added to a mixture of 37 and the appropriate aniline in the presence of 2 M triethylammonium bicarbonate; Scheme 2C), albeit, these conditions seemed to favor the formation of symmetrical ureas (52). The same chemistry was applied to 7H azide 42 to give amide 43 and urea 49 (Scheme 2B, C).
Scheme 2. Synthesis of Further N-Linked Analogues of 8.

Reagents and conditions: (i) NaN3, CTAB, MeOH, 20 °C, 45 min, then 40 °C, 17 h (73%); (ii) NaH, DMF, 0–20 °C, 2.5 h (70%); (iii) PPh3, aq dioxane, 12–20 °C, 1 d (38: 48%, 39: 32%); (iv) 4-OCF3PhCHO, NaBH3CN, AcOH, DMF, 0–20 °C, 21 h (50%); (v) PPh3, DEAD, DPPA, DMF, 0–20 °C, 45 h (83%); (vi) PPh3, ArCOCl (or 4-OCF3PhOCH2COCl), CH2Cl2, 20 °C, 1.5–2.2 h (51–84%); (vii) HS(CH2)3SH, Et3N, MeOH, CH2Cl2, 15–20 °C, 12 h (83%); (viii) 3-OCF3PhCOCl or RPhSO2Cl, DIPEA, DMF, 0–20 °C, 3–19 h (46–81%); (ix) PPh3, 4-OCF3PhNH2, 2 M TEAB, dioxane, 12–20 °C, 35 h (14–17%); (x) 4-OCF3BnNCO, DIPEA, Bu2Sn(OAc)2, DMF, 20 °C, 16 h (93%); (xi) 4-NO2PhOCOCl, pyridine, CH2Cl2, 0–20 °C, 20 h (98%); (xii) 38, DMAP, DIPEA, DMF, 20 °C, 44 h (71%); (xiii) PPh3, Boc-ON, CH2Cl2, 0–20 °C, 1 d (67%); (xiv) TFA, CH2Cl2, 20 °C, 7 h (100%).
In related methodology, the Boc derivative 56 was obtained from azide 37 using triphenylphosphine and Boc-ON26 [2-(tert-butoxycarbonyloxyimino)-2-phenylacetonitrile] (Scheme 2E), but purification of this compound was not straightforward. Removal of the Boc group (TFA/CH2Cl2) and basification gave hygroscopic material (after chromatography), so this was elaborated immediately to sulfonamides 57 and 58. Alternatively, treatment of 37 with triphenylphosphine alone (in aqueous dioxane) generated a ca. 4:3 mixture of amine 38 and iminophosphorane 39. Although 39 resisted mild hydrolysis, some pure 38 could be separated out as a stable, nonhygroscopic solid, enabling the syntheses of benzylamine 40 (Scheme 2A) and urea 53 (Scheme 2D) via standard methods,20 and N-carbamate 55 (via aminolysis of the novel 4-nitrophenyl carbonate 54; Scheme 2D). One ether-linked amide (64) was also accessed in meager yield (4%) by coupling alcohol 63(12) and iodoacetamide 62 (Scheme 3A). Finally, several O-carbamate derivatives (65, 66, and 68–74) were formed by Cu(I)-induced reaction20 of alcohols 41, 63, 67, and ent-67(12,27) with aryl isocyanates (Scheme 3B).
Scheme 3. Synthesis of New O-Linked Analogues of 8.

Reagents and conditions: (i) NaI, acetone, 56 °C, 2 h, then 20 °C, 15 h (96%); (ii) 62, NaH, DMF, 0–20 °C, 80 min (4%); (iii) ArNCO, CuCl, DMF, 20 °C, 32–52 h (44–98%).
The 32 new analogues were screened for activity against L. inf (Table 1), two Trypanosoma parasites, and Mycobacterium tuberculosis as well as for cytotoxicity toward MRC-5 cells (see Table S1 in the Supporting Information for additional data). All compounds were classified as nontoxic (MRC-5 IC50s ≥ 40 μM) with many providing VL selectivity indices of >100. For substituted phenyl side chains, good anti-VL activity was maintained across both the amino (NH) and methylamino (NMe) linker series (15–30). Compared to known ether-linked counterparts (8, 10–12), potency was similar or ∼2-fold lower for the new 7-Me analogues (L. inf IC50s 0.18 and 0.27 μM for 18 and 26), and 3- to 5-fold lower for 7H congeners 15 and 24 (L. inf IC50s 0.15 and 0.22 μM). In both NH and NMe linker series, the best phenyl substituent was 4-trifluoromethoxy, followed by 4-chloro, whereas the more hydrophilic 4-fluoro derivatives displayed weaker inhibition. The trifluoromethylpyridine analogue 35 was ∼8-fold less potent than 26.
Table 1. Structures, Calculated Lipophilicities, and Antileishmanial Activities of New Linker Analogues.
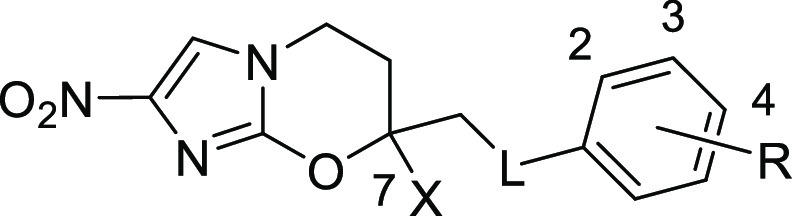
| compound | X | linker (L) | R | CLogPa | IC50 (μM)b | compound | X | linker (L) | R | CLogPa | IC50 (μM)b |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 10c | H | O | 4-OCF3 | 3.21 | 0.047 | 48 | Me | NHCOCH2O | 4-OCF3 | 2.85 | 1.7 |
| 8c | Me | O | 4-OCF3 | 3.73 | 0.13 | 49 | H | NHCONH | 4-OCF3 | 2.36 | 3.3 |
| 11d | Me | O | 4-Cl | 3.38 | 0.13 | 51 | Me | NHCONH | 4-OCF3 | 2.88 | 1.4 |
| 12d | Me | O | 4-F | 2.81 | 0.45 | 53 | Me | NHCONHCH2 | 4-OCF3 | 2.91 | 1.8 |
| 15 | H | NH | 4-OCF3 | 2.80 | 0.15 | 55 | Me | NHCOOCH2 | 4-OCF3 | 3.21 | 0.16 |
| 18 | Me | NH | 4-OCF3 | 3.32 | 0.18 | 57 | Me | NHSO2 | 4-OCF3 | 2.92 | 2.2 |
| 20 | Me | NH | 4-Cl | 3.12 | 0.29 | 58 | Me | NHSO2 | 3-OCF3 | 2.92 | 2.0 |
| 22 | Me | NH | 4-F | 2.55 | 0.93 | 59c | H | OCH2 | 4-OCF3 | 2.84 | 0.12 |
| 24 | H | NMe | 4-OCF3 | 3.39 | 0.22 | 60c | Me | OCH2 | 4-OCF3 | 3.36 | 0.30 |
| 26 | Me | NMe | 4-OCF3 | 3.91 | 0.27 | 64 | Me | OCH2CONH | 4-OCF3 | 2.63 | 0.49 |
| 28 | Me | NMe | 4-Cl | 3.66 | 0.31 | 65 | H | OCONH | 4-OCF3 | 2.71 | 0.36 |
| 30 | Me | NMe | 4-F | 3.09 | 0.48 | 66 | Me | OCONH | 4-OCF3 | 3.23 | 0.36 |
| 35 | Me | NMe | 3-aza, 4-CF3 | 2.86 | 2.2 | 68 | Mee | OCONH | 4-OCF3 | 3.23 | 0.14 |
| 40 | Me | NHCH2 | 4-OCF3 | 3.13 | 0.13 | 69 | Mef | OCONH | 4-OCF3 | 3.23 | 0.20 |
| 43 | H | NHCO | 4-OCF3 | 2.28 | 1.5 | 70 | Me | OCONH | 2-OCF3 | 3.23 | 0.64 |
| 44 | Me | NHCO | 4-OCF3 | 2.80 | 1.4 | 71 | H | OCONHCH2 | 4-OCF3 | 2.69 | 0.18 |
| 45 | Me | NHCO | 3-OCF3 | 2.80 | 1.3 | 72 | Me | OCONHCH2 | 4-OCF3 | 3.21 | 0.32 |
| 46 | Me | NHCO | 2-OCF3 | 2.73 | 9.9 | 73 | H | OCONZCH2g | 4-OCF3 | 6.09 | 0.79 |
| 47 | Me | NHCOCH2 | 4-OCF3 | 2.50 | 2.0 | 74 | Me | OCONZCH2g | 4-OCF3 | 6.61 | 1.3 |
Calculated log P values from ChemDraw v19.1.
Values for 50% growth inhibition of L. inf (in mouse macrophages); all data are averages from two or more independent experiments (for standard deviations, see the Supporting Information).
Ref (12).
Ref (27).
(7R)-Enantiomer.
(7S)-Enantiomer.
Z = CONHCH2(4-OCF3Ph).
Two promising examples (18 and 26) showed modest (2- to 4-fold) solubility improvements over 8 at low pH only (Table 2), consistent with small lipophilicity changes (ΔCLogP −0.4 and +0.2 units, respectively). The NH-linked compound (18) also demonstrated reasonable stability toward mouse liver microsomes (MLM) (45% parent remaining vs 50% for 8; Table 2) but its NMe-linked derivative 26 was metabolized more quickly (an MLM half-life of 17 min). However, because 26 was considerably easier to synthesize than 18, we elected to evaluate both compounds in the L. don mouse model.12 Here, once daily oral dosing of 18 for 5 days gave essentially complete parasite clearance at 50 mg/kg (99.7%; Tables 2 and S3), but 26 was only moderately effective (58% at 50 mg/kg). Further testing of 18 at 6.25 mg/kg suggested that it was ∼2-fold less dose-potent than 8.
Table 2. Aqueous Solubility, Microsomal Stability, and In Vivo Efficacy Data for Selected Compounds.
| aqueous
solubility (μg/mL)a |
stability
in liver microsomes (% parent remaining at 1 h) |
efficacy
in L. don mouse model (% parasite burden reduction
at oral dose in mg/kg)b |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| compound | pH 7 | pH 1 | human | mouse | 50 | 25 | 12.5 | 6.25 | 3.13 |
| 8c | 2.3 | 58 | 50 | 100 | 100 | 83 | 25 | ||
| 10c | 4.0 | 85 | 57 | 87 | |||||
| 18 | 3.0 | 10 | 47 | 45 | >99 | 29 | |||
| 26 | 1.5 | 4.6 | 8.0 | 9.2 | 58 | ||||
| 40 | 124 | 33 000 | 56 | 36 | |||||
| 44 | 78 | 67 | 65 | 71 | |||||
| 45 | 14 | 58 | 36 | ||||||
| 51 | 121 | 54 | 39 | 36 | |||||
| 55 | 28 | 44 | 22 | ||||||
| 65 | 4.5 | 93 | 58 | ||||||
| 66 | 11 | 68 | 46 | >99 | 97 | 7 | |||
| 68 | 6.3 | 95 | 51 | 89 | 40 | ||||
| 69 | 7.9 | 78 | 55 | 51 | 31 | ||||
| 72 | 117 | 45 | 8.7 | ||||||
Solubility in water (pH 7) or 0.1 M HCl (pH 1).
Dosing was once daily for 5 days (see the Supporting Information for all protocols).
Ref (12).
In subsequent work, we prepared a methylene homologue of 18, benzylamine 40, which retained the excellent potency of 8 (L. inf IC50 0.13 μM) and exhibited markedly better aqueous solubility (54- to 14 300-fold at pH 7 and pH 1, respectively). However, inferior MLM stability (36%) discouraged any in vivo appraisal.
Incorporation of an amide (43–48), urea (49, 51, 53) or sulfonamide (57, 58) linkage led to much larger lipophilicity changes over 8 and 10 (ΔCLogP −0.8 to −1.2 units) and greater aqueous solubility (e.g., 44 and 51: 78 and 121 μg/mL, respectively). Nevertheless, much weaker potencies were observed in all cases (L. inf IC50s generally 1–3 μM). The most useful compounds were benzamides 44 and 45 and phenylurea 51 (L. inf IC50s 1.3–1.4 μM). Linker extension (47, 48, 53) led to reduced potency, while aryl sulfonamides 57 and 58 were also less impressive (L. inf IC50s ∼ 2 μM). Stability testing of three molecules in MLM (44, 45, 51) revealed that benzamide 44 had the best profile (65 vs 39% for urea 51). This translated into stronger efficacy for 44 over 51 in the L. don mouse model (71 vs 36% parasite burden reduction at 50 mg/kg), albeit, this level of activity was considered only borderline according to published lead selection criteria.28
To complete our investigation of acylamino linkers, N-linked benzyl carbamate 55 was prepared (ΔCLogP −0.5 units vs 8). Unexpectedly, this compound afforded excellent potency (L. inf IC50 0.16 μM) and 12-fold better aqueous solubility than 8, but was much less stable toward MLM (22 vs 50%, respectively). These findings reinforced the difficulty of simultaneously attaining high potency, solubility, and stability in one molecule.
Extension of the ether linkage of 8 by including an amide functionality (64) also gave a considerable lipophilicity reduction (ΔCLogP −1.1 units) but correspondingly weaker potency (4-fold vs 8), so we switched our focus to O-carbamates (65–72). These new analogues provided more moderate lipophilicity changes over 8 and 10 (ΔCLogP −0.5 units) and generally displayed potencies in a more acceptable range (L. inf IC50s 0.18–0.36 μM). In terms of both solubility and microsomal stability, the 7H phenyl carbamate 65 was not significantly superior to its more active ether-linked counterpart 10, whereas the 7-Me congener 66 was 5-fold more soluble than 8 and provided better stability toward human liver microsomes (HLM; 68% parent for 66 vs 58% for 8). Benzyl homologue 72 demonstrated ∼11-fold greater solubility than 66 (117 μg/mL) but, like 26, this molecule was rapidly metabolized by MLM (a half-life of 17 min).
Previous VL mouse model efficacy studies for this 7-substituted 2-nitroimidazooxazine class have identified different preferences for 7H or 7-Me derivatives, depending upon the length of the side chain.12,27 With PK effects being largely responsible, we elected to measure mouse PK data on both 65 and 66. Compared to their ether-linked counterparts (10, 8), both carbamate derivatives afforded slower clearance rates and much greater oral absorption, leading to improved exposure levels and oral bioavailability values of 100% (Table 3 and Supporting Information, Figures S2 and S3; no in vitro or in vivo metabolite identification studies were performed on any compounds).
Table 3. Mouse Pharmacokinetic Parameters for Selected Compounds.
| intravenous (1 mg/kg) |
oral
(25 mg/kg) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| compound | C0 (μg/mL) | CL (mL/min/kg) | Vdss (L/kg) | t1/2 (h) | AUClasta (μg·h/mL) | Cmax (μg/mL) | Tmax (h) | t1/2 (h) | AUClasta (μg·h/mL) | Fb (%) |
| 10c | 0.36 | 48 | 3.2 | 1.1 | 0.341 | 1.3 | 0.50 | 3.86 | 45 | |
| 8c | 0.79 | 12 | 2.5 | 2.8 | 1.31 | 1.4 | 3.0 | 11.5 | 35 | |
| 65 | 0.74 | 14 | 1.6 | 1.2 | 1.17 | 9.6 | 1.0 | 1.6 | 35.4 | 100 |
| 66 | 0.80 | 8.5 | 1.2 | 1.3 | 2.07 | 13.5 | 0.75 | 1.7 | 57.8 | 100 |
Area under the curve calculated to the last time point.
Oral bioavailability, determined using dose normalized AUClast values.
Ref (12).
Overall, 7-Me congener 66 delivered better oral exposure (Figure 2) and was advanced to efficacy testing. At 50 mg/kg, 66 gave essentially complete clearance of parasites (liver: >99.9%, spleen: 100%); it also showed strong efficacy at 25 mg/kg (liver: 97%, spleen: 96%). The ED50 (liver) was 16 mg/kg, ∼4-fold lower than the value obtained for 8 but still very promising; hence, the enantiomers of 66 (68, 69) were prepared and assessed. Compared to the reported enantiomers of 8 (75 and 76 in Table S2),27 these chiral carbamates were only marginally less effective in vitro (1.2- to 1.4-fold) with the 7R form (68) being slightly more potent than the 7S form (69) (L. inf IC50s 0.14 and 0.20 μM, respectively). Moreover, 68 exhibited strikingly improved HLM stability (95% parent, cf. 68% for 66, 78% for 69, 58% for 5,12 and 63% for the 7R enantiomer of 8(27)). Finally, 68 delivered significantly greater efficacy than 69 in the L. don mouse model (89% parasite burden reduction at 25 mg/kg for 68 vs 51% for 69). Thus, the present investigation of novel linker analogues of 8 has identified 7R carbamate 68 as a preferred new lead for VL.
Figure 2.
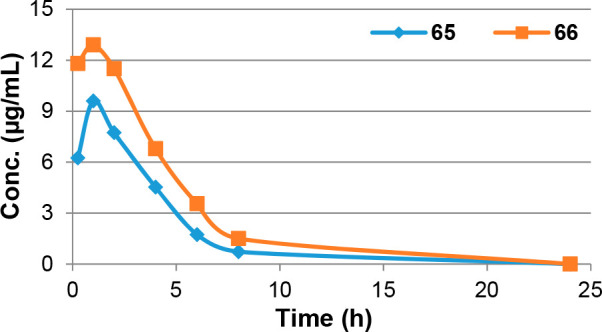
Plasma concentration–time profiles for 65 and 66, dosed orally at 25 mg/kg in BALB/c mice.
In addition to their antileishmanial effects, the new compounds of Table 1 also displayed low- or submicromolar activities against TB and Chagas disease (see the Supporting Information, Table S1). In both cases, amino-linked analogues were favored, but a broader range of linking groups was tolerated for TB. The best TB lead was 7R carbamate 68 (MIC90s 0.085 and 1.0 μM under aerobic and hypoxic conditions, respectively). However, across all 7-Me linker derivatives, the association between VL potency (as pIC50) and effectiveness against TB (as pMIC in the aerobic MABA assay) was only moderate (R2 = 0.54; Figure 3), and neither activity correlated well with CLogP data (see the Supporting Information, Figure S4). These results were consistent with the involvement of unrelated nitroreductases in the activation mechanisms of 2-nitroimidazooxazines against VL and TB.13,29
Figure 3.

Similarity of SAR against VL and TB for 7-Me linker analogues.
In summary, we prepared and evaluated 32 novel linker analogues of 8 with the objective of identifying efficacious backup leads to clinical candidate 5 that were more soluble. While amino, amide, urea and N-carbamate linkages all provided some solubility improvements (up to 54-fold over 8), O-carbamate 66 offered the best overall balance of drug-like properties, displaying 100% oral bioavailability in mice and excellent in vivo efficacy in a VL mouse model. Appraisal of its enantiomers pinpointed 7R carbamate 68 as superior, having notably improved HLM stability and 14-fold better solubility than 5.12 This molecule was also a potent lead against TB, suggesting that further assessments are warranted. Overall, these results have illustrated the utility of linker replacement as a lipophilicity reduction strategy (to enhance aqueous solubility), facilitating the discovery of a potential new drug candidate from this very promising 7-substituted 2-nitroimidazooxazine class.
Acknowledgments
The authors thank the Drugs for Neglected Diseases initiative for financial support through a collaborative research agreement. For this project, DNDi received financial support from the following donors: UK aid, UK; Federal Ministry of Education and Research (BMBF), through KfW, Germany; Dutch Ministry of Foreign Affairs (DGIS), The Netherlands; French Ministry for Europe and Foreign Affairs (MEAE), France; Swiss Agency for Development and Cooperation (SDC), Switzerland; Médecins Sans Frontières (MSF), International. The donors had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The authors also thank Fanny Escudié (DNDi) for coordination support and Sisira Kumara (ACSRC) for the solubility measurements.
Glossary
Abbreviations
- CL
cutaneous leishmaniasis;
- VL
visceral leishmaniasis
- L. don
Leishmania donovani
- L. inf
Leishmania infantum
- CTAB
cetyltrimethylammonium bromide
- TEAB
triethylammonium bicarbonate
- DPPA
diphenylphosphoryl azide
- Boc-ON
2-(tert-butoxycarbonyloxyimino)-2-phenylacetonitrile
- DIPEA
N,N-diisopropylethylamine
- MLM
mouse liver microsomes
- HLM
human liver microsomes.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00649.
Additional biological and pharmacokinetic data and methods, experimental section, combustion analytical data, and NMR spectra for key compounds (PDF)
Author Present Address
□ P.D.O.: Helmholtz Zentrum München, Ingolstädter Landstrasse 1, 85764 Neuherberg, Germany
Author Present Address
○ Z.M.: TenNor Therapeutics Ltd., 218 Xinghu Street, Suzhou Industrial Park, Suzhou 215123, China
The authors declare no competing financial interest.
Supplementary Material
References
- Burza S.; Croft S. L.; Boelaert M. Leishmaniasis. Lancet 2018, 392, 951–970. 10.1016/S0140-6736(18)31204-2. [DOI] [PubMed] [Google Scholar]
- Alves F.; Bilbe G.; Blesson S.; Goyal V.; Monnerat S.; Mowbray C.; Muthoni Ouattara G.; Pécoul B.; Rijal S.; Rode J.; Solomos A.; Strub-Wourgaft N.; Wasunna M.; Wells S.; Zijlstra E. E.; Arana B.; Alvar J. Recent development of visceral leishmaniasis treatments: successes, pitfalls, and perspectives. Clin. Microbiol. Rev. 2018, 31, e00048–18. 10.1128/CMR.00048-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ending the Neglect to Attain the Sustainable Development Goals: A Road Map for Neglected Tropical Diseases 2021–2030; World Health Organization: Geneva, Switzerland, 2020.
- Hendrickx S.; Caljon G.; Maes L. Need for sustainable approaches in antileishmanial drug discovery. Parasitol. Res. 2019, 118, 2743–2752. 10.1007/s00436-019-06443-2. [DOI] [PubMed] [Google Scholar]
- Balaña-Fouce R.; Pérez Pertejo M. Y.; Domínguez-Asenjo B.; Gutiérrez-Corbo C.; Reguera R. M. Walking a tightrope: drug discovery in visceral leishmaniasis. Drug Discovery Today 2019, 24, 1209–1216. 10.1016/j.drudis.2019.03.007. [DOI] [PubMed] [Google Scholar]
- 2019 R&D portfolio in review: leishmaniasis. DNDi, 26 February 2020. https://www.dndi.org/2020/media-centre/news-views-stories/news/leishmaniasis-rnd-portfolio-update/ (accessed October 9, 2020).
- NCT04504435: Safety, tolerability and pharmacokinetics (PK) investigation of GSK3494245 in healthy participants. https://clinicaltrials.gov/ct2/show/NCT04504435 (accessed October 9, 2020).
- Nagle A.; Biggart A.; Be C.; Srinivas H.; Hein A.; Caridha D.; Sciotti R. J.; Pybus B.; Kreishman-Deitrick M.; Bursulaya B.; Lai Y. H.; Gao M.-Y.; Liang F.; Mathison C. J. N.; Liu X.; Yeh V.; Smith J.; Lerario I.; Xie Y.; Chianelli D.; Gibney M.; Berman A.; Chen Y.-L.; Jiricek J.; Davis L. C.; Liu X.; Ballard J.; Khare S.; Eggimann F. K.; Luneau A.; Groessl T.; Shapiro M.; Richmond W.; Johnson K.; Rudewicz P. J.; Rao S. P. S.; Thompson C.; Tuntland T.; Spraggon G.; Glynne R. J.; Supek F.; Wiesmann C.; Molteni V. Discovery and characterization of clinical candidate LXE408 as a kinetoplastid-selective proteasome inhibitor for the treatment of leishmaniases. J. Med. Chem. 2020, 63, 10773–10781. 10.1021/acs.jmedchem.0c00499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasco R. F.; Guerrero C.; Fra G.; Moure A.; Miguel-Siles J.; Quesada-Campos M. T.; Ruiz-Gomez J. R.; Gilbert I. H.; Thomas M. G.; Miles T. J. Optimisation of a key cross-coupling reaction toward the synthesis of a promising antileishmanial compound. Tetrahedron Lett. 2019, 60, 1243–1247. 10.1016/j.tetlet.2019.03.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie S. C.; Dick L. R.; Gould A.; Brand S.; Tilley L. The proteasome as a target for protozoan parasites. Expert Opin. Ther. Targets 2019, 23, 903–914. 10.1080/14728222.2019.1685981. [DOI] [PubMed] [Google Scholar]
- Wall R. J.; Rico E.; Lukac I.; Zuccotto F.; Elg S.; Gilbert I. H.; Freund Y.; Alley M. R. K.; Field M. C.; Wyllie S.; Horn D. Clinical and veterinary trypanocidal benzoxaboroles target CPSF3. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 9616–9621. 10.1073/pnas.1807915115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson A. M.; O’Connor P. D.; Marshall A. J.; Yardley V.; Maes L.; Gupta S.; Launay D.; Braillard S.; Chatelain E.; Franzblau S. G.; Wan B.; Wang Y.; Ma Z.; Cooper C. B.; Denny W. A. 7-Substituted 2-nitro-5,6-dihydroimidazo[2,1-b][1,3]oxazines: novel antitubercular agents lead to a new preclinical candidate for visceral leishmaniasis. J. Med. Chem. 2017, 60, 4212–4233. 10.1021/acs.jmedchem.7b00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyllie S.; Roberts A. J.; Norval S.; Patterson S.; Foth B. J.; Berriman M.; Read K. D.; Fairlamb A. H. Activation of bicyclic nitro-drugs by a novel nitroreductase (NTR2) in Leishmania. PLoS Pathog. 2016, 12 (11), e1005971. 10.1371/journal.ppat.1005971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keam S. J. Pretomanid: first approval. Drugs 2019, 79, 1797–1803. 10.1007/s40265-019-01207-9. [DOI] [PubMed] [Google Scholar]
- Van den Kerkhof M.; Mabille D.; Chatelain E.; Mowbray C. E.; Braillard S.; Hendrickx S.; Maes L.; Caljon G. In vitro and in vivo pharmacodynamics of three novel antileishmanial lead series. Int. J. Parasitol.: Drugs Drug Resist. 2018, 8, 81–86. 10.1016/j.ijpddr.2018.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Bocxlaer K.; Caridha D.; Black C.; Vesely B.; Leed S.; Sciotti R. J.; Wijnant G.-J.; Yardley V.; Braillard S.; Mowbray C. E.; Ioset J.-R.; Croft S. L. Novel benzoxaborole, nitroimidazole and aminopyrazoles with activity against experimental cutaneous leishmaniasis. Int. J. Parasitol.: Drugs Drug Resist. 2019, 11, 129–138. 10.1016/j.ijpddr.2019.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijnant G.-J.; Croft S. L.; de la Flor R.; Alavijeh M.; Yardley V.; Braillard S.; Mowbray C.; Van Bocxlaer K. Pharmacokinetics and pharmacodynamics of the nitroimidazole DNDI-0690 in mouse models of cutaneous leishmaniasis. Antimicrob. Agents Chemother. 2019, 63, e00829–19. 10.1128/AAC.00829-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT03874234: Safety, tolerability and pharmacokinetics (PKs) investigation of GSK3186899 in healthy subjects. https://clinicaltrials.gov/ct2/show/study/NCT03874234 (accessed December 12, 2020).
- Smietana K.; Siatkowski M.; Møller M. Trends in clinical success rates. Nat. Rev. Drug Discovery 2016, 15, 379–380. 10.1038/nrd.2016.85. [DOI] [PubMed] [Google Scholar]
- Thompson A. M.; O’Connor P. D.; Marshall A. J.; Blaser A.; Yardley V.; Maes L.; Gupta S.; Launay D.; Braillard S.; Chatelain E.; Wan B.; Franzblau S. G.; Ma Z.; Cooper C. B.; Denny W. A. Development of (6R)-2-nitro-6-[4-(trifluoromethoxy)phenoxy]-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazine (DNDI-8219): a new lead for visceral leishmaniasis. J. Med. Chem. 2018, 61, 2329–2352. 10.1021/acs.jmedchem.7b01581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaser A.; Palmer B. D.; Sutherland H. S.; Kmentova I.; Franzblau S. G.; Wan B.; Wang Y.; Ma Z.; Thompson A. M.; Denny W. A. Structure-activity relationships for amide-, carbamate-, and urea-linked analogues of the tuberculosis drug (6S)-2-nitro-6-{[4-(trifluoromethoxy)benzyl]oxy}-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazine (PA-824). J. Med. Chem. 2012, 55, 312–326. 10.1021/jm2012276. [DOI] [PubMed] [Google Scholar]
- Thompson A. M.; O’Connor P. D.; Blaser A.; Yardley V.; Maes L.; Gupta S.; Launay D.; Martin D.; Franzblau S. G.; Wan B.; Wang Y.; Ma Z.; Denny W. A. Repositioning antitubercular 6-nitro-2,3-dihydroimidazo[2,1-b][1,3]oxazoles for neglected tropical diseases: structure-activity studies on a preclinical candidate for visceral leishmaniasis. J. Med. Chem. 2016, 59, 2530–2550. 10.1021/acs.jmedchem.5b01699. [DOI] [PubMed] [Google Scholar]
- Fringuelli F.; Piermatti O.; Pizzo F.; Vaccaro L. Ring opening of epoxides with sodium azide in water. A regioselective pH-controlled reaction. J. Org. Chem. 1999, 64, 6094–6096. 10.1021/jo990368i. [DOI] [Google Scholar]
- Maunier V.; Boullanger P.; Lafont D. A one-pot synthesis of glycosyl amides from glycosyl azides using a modified Staudinger reaction. J. Carbohydr. Chem. 1997, 16, 231–235. 10.1080/07328309708006523. [DOI] [Google Scholar]
- Yagodkin A.; Löschcke K.; Weisell J.; Azhayev A. Straightforward carbamoylation of nucleophilic compounds employing organic azides, phosphines, and aqueous trialkylammonium hydrogen carbonate. Tetrahedron 2010, 66, 2210–2221. 10.1016/j.tet.2010.01.017. [DOI] [Google Scholar]
- Ariza X.; Urpí F.; Viladomat C.; Vilarrasa J. One-pot conversion of azides to Boc-protected amines with trimethylphosphine and Boc-ON. Tetrahedron Lett. 1998, 39, 9101–9102. 10.1016/S0040-4039(98)02006-1. [DOI] [Google Scholar]
- Thompson A. M.; O’Connor P. D.; Marshall A. J.; Yardley V.; Maes L.; Gupta S.; Launay D.; Braillard S.; Chatelain E.; Wan B.; Franzblau S. G.; Ma Z.; Cooper C. B.; Denny W. A. Heteroaryl ether analogues of an antileishmanial 7-substituted 2-nitroimidazooxazine lead afford attenuated hERG risk: In vitro and in vivo appraisal. Eur. J. Med. Chem. 2021, 209, 112914. 10.1016/j.ejmech.2020.112914. [DOI] [PubMed] [Google Scholar]
- Katsuno K.; Burrows J. N.; Duncan K.; Hooft van Huijsduijnen R.; Kaneko T.; Kita K.; Mowbray C. E.; Schmatz D.; Warner P.; Slingsby B. T. Hit and lead criteria in drug discovery for infectious diseases of the developing world. Nat. Rev. Drug Discovery 2015, 14, 751–758. 10.1038/nrd4683. [DOI] [PubMed] [Google Scholar]
- Manjunatha U.; Boshoff H. I. M.; Barry C. E. The mechanism of action of PA-824: Novel insights from transcriptional profiling. Commun. Integr. Biol. 2009, 2, 215–218. 10.4161/cib.2.3.7926. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.