

Abstract
Dipeptidyl peptidase 4 (DPP4) is an exopeptidase found either on cell surfaces where it is highly regulated in terms of its expression and surface availability (CD26) or in a free/circulating soluble constitutively available and intrinsically active form. It is responsible for proteolytic cleavage of many peptide substrates. In this review we discuss the idea that DPP4-cleaved peptides are not necessarily inactivated, but rather can possess either a modified receptor selectivity, modified bioactivity, new antagonistic activity, or even a novel activity relative to the intact parent ligand.
We examine in detail five different major DPP4 substrates: glucagon-like peptide 1 (GLP-1), glucose-dependent insulinotropic polypeptide (GIP), peptide tyrosine-tyrosine (PYY), and neuropeptide Y (NPY), and stromal derived factor 1 (SDF-1 aka CXCL12). We note that discussion of the cleaved forms of these five peptides are underrepresented in the research literature, and are both poorly investigated and poorly understood, representing a serious research literature gap. We believe they are understudied and misinterpreted as inactive due to several factors. This includes lack of accurate and specific quantification methods, sample collection techniques that are inherently inaccurate and inappropriate, and a general perception that DPP4 cleavage inactivates its ligand substrates.
Increasing evidence points towards many DPP4-cleaved ligands having their own bioactivity. For example, GLP-1 can work through a different receptor than GLP-1R, DPP4-cleaved GIP can function as a GIP receptor antagonist at high doses, and DPP4-cleaved PYY, NPY, and CXCL12 can have different receptor selectivity, or can bind novel, previously unrecognized receptors to their intact ligands, resulting in altered signaling and functionality. We believe that more rigorous research in this area could lead to a better understanding of DPP4’s role and the biological importance of the generation of novel cryptic ligands. This will also significantly impact our understanding of the clinical effects and side effects of DPP4-inhibitors as a class of anti-diabetic drugs that potentially have an expanding clinical relevance. This will be specifically relevant in targeting DPP4 substrate ligands involved in a variety of other major clinical acute and chronic injury/disease areas including inflammation, immunology, cardiology, stroke, musculoskeletal disease and injury, as well as cancer biology and tissue maintenance in aging.
Keywords: Dipeptidyl peptidase 4, glucagon-like peptide 1, glucose-dependent insulinotropic polypeptide, peptide tyrosine-tyrosine, neuropeptide Y, CXCL12
1. Introduction
Dipeptidyl peptidase 4 (DPP4) is a highly conserved exopeptidase found widely on the surface of epithelial, endothelial, stromal, stem and immune cells (Mulvihill & Drucker, 2014). In its cell membrane associated form it is referred to as cluster of differentiation 26 (CD26). DPP4 is a ubiquitous enzyme that is expressed in different compartments, including liver, kidney, gastrointestinal tract (GIT) and bone marrow (Ambrosi et al., 2017; Mentlein, 1999; Ou, O’Leary, & Broxmeyer, 2013). It also has a soluble form (sCD26/DPP4) that can be found in the circulation and in various tissue compartments. DPP4 regulates the activity of many different chemokines and peptide hormones, and it acts by cleaving off N-terminus dipeptides typically with a penultimate alanine or proline, although under some conditions other amino acids may be targeted (Broxmeyer, 2013; Drucker, 2007; Mortier, Gouwy, Van Damme, Proost, & Struyf, 2016).
CD26 is found as a single pass transmembrane protein that forms homodimers on most cells and in the majority of tissues (Mulvihill & Drucker, 2014). Regulation of trans-membrane CD26 activity occurs at multiple levels, including control of gene and protein expression, regulation of the transfer of CD26 to the cell surface, interaction with binding partners, and modulation of enzyme activity (Mulvihill & Drucker, 2014). Expression of cell surface trans-membrane, as well as intercellular membrane pools of trans-membrane CD26, is tightly controlled. Transition of CD26 to the surface of cells is highly regulated and can be initiated rapidly resulting in cell surface levels going from no CD26, or minimal basal levels, to very high levels in response to a large number of cell and tissue targeting factors such as interleukines, hypoxia-inducible factor 1 (HIF-1), interferons, retinoic acid, hepatocyte nuclear factor (HNF), granulocyte-colony stimulating factor (G-CSF), angiotensinogen II, classical leukocyte cellular activation (e.g. of T and B Cells), as well as modification of activity by post-translational glycosylation and subunit dimerization (Aroor et al., 2016; Bauvois, Djavaheri-Mergny, Rouillard, Dumont, & Wietzerbin, 2000; Christopherson, Cooper, & Broxmeyer, 2003; Chung et al., 2010; Dang et al., 2008; De Meester, Korom, Van Damme, & Scharpe, 1999; Gu et al., 2008; Klemann, Wagner, Stephan, & von Hörsten, 2016; Metzemaekers, Van Damme, Mortier, & Proost, 2016; Mortier et al., 2016; Mulvihill & Drucker, 2014; Shin et al., 2017). Functionally, DPP4 is involved in numerous cellular processes including cell-cell interaction, cell mobilization, cell migration, and membrane-associated activation of intracellular signal transduction pathways (De Meester et al., 1999; Lambeir, Durinx, Scharpé, & De Meester, 2003). CD26 is a relatively large protein consisting of 766 amino acids, most of which are extracellular, with a hydrophobic transmembrane anchor from amino acids 7 to 28, and a very short intracellular sequence consisting of 6 amino acids (Abbott, Baker, Sutherland, & McCaughan, 1994). The soluble form of the peptidase (sCD26/DPP4), composed of the extracellular amino acids 39–766, is shed into the extracellular environment by a number of matrix metalloproteinases (Havre et al., 2008; Varin et al., 2018). Unlike the tightly regulated expression/activity of the trans-membrane parent form (CD26) on cells and within tissues, the soluble DPP4 (sCD26) is widely present in plasma and other tissue compartments and is constitutively fully enzymatically active (Klemann et al., 2016; Mortier et al., 2016). Indeed endogenous levels of soluble DPP4 may not represent a rate-limiting factor, allowing cleavage of most of its substrate pools in those compartments (Carbone, Buzkova, Fink, Robbins, Bethel, Hamrick, et al., 2017; Carbone, Buzkova, Fink, Robbins, Bethel, Isales, et al., 2017).
Only a limited number of review and primary experimental papers have highlighted the complex role that DPP4 plays in the generation of cryptic substrate ligands and the need to consider how these changes to the nature of the truncated substrate’s signaling and functional activity impacts experimental data interpretation (Mortier, Van Damme, & Proost, 2008; Mulvihill & Drucker, 2014; O’Leary, Ou, & Broxmeyer, 2013). As a consequence, the idea that DPP4 proteolysis leads to partial or total inactivation of its substrates is still widely accepted and taken for granted. However, there is a growing literature that supports the idea that DPP4 metabolites can have selective, antagonistic or even novel signaling properties and functions (Cheng, Eby, LaPorte, Volkman, & Majetschak, 2017; Janssens et al., 2017; Kim, Yu, & Lee, 2014; Klemann et al., 2016; Mentlein, 1999; Mulvihill & Drucker, 2014; Ou et al., 2013; Szpakowska et al., 2018; Tarantola et al., 2012; Ziarek et al., 2017). We believe that the concept that in many cases DPP4 can alter substrate function, rather than just inactivating the ligand’s functionality is still an underrepresented view in the overall scientific literature; one that carries tremendous experimental, interpretative as well as clinical repercussions. In this review, we highlight five major DPP4 substrates that are not inactivated, but rather demonstrate new, or revealed cryptic, interactions and function. We show that DPP4 can modulate the function of the substrate not only by interfering with the substrate’s ability to either bind or activate its receptor, but truncation can antagonize the receptor or alter the ratio of binding and activation between primary and secondary receptors or lead to interactions with a new receptor by the cleaved substrate that is not a target of the intact parent ligand. In many cases, DPP4-truncated peptides have complex novel functions that are not adequately appreciated. Excitingly, understanding this unexpected cryptic bioactivity is an important new area for research as it is underappreciated and represents a major gap in the research literature. The cryptic ligand activities come with significant clinical implications and call for new interpretations of some previous research results (Metzemaekers et al., 2016; Mulvihill & Drucker, 2014; Ou et al., 2013).
This review will focus on DPP4 substrates that have bioactivity upon cleavage by DDP4. We are referring to these substrates in the title as cryptic bioactive ligands, which means that they develop a new previously hidden, bioactivity after post-translational modification (Brandt, Lambeir, Maes, Scharpe, & De Meester, 2006). These substrates include glucagon-like peptide 1 (GLP-1), glucose-dependent insulinotropic polypeptide (GIP), peptide tyrosine-tyrosine (PYY), neuropeptide Y (NPY), and the cytokine Stromal Cell-Derived Factor-1 (SDF-1, or CXCL12 in the revised cytokine nomenclature (Zlotnik & Yoshie, 2000)). These five substrates can be classified into three main classes: incretin hormones, pancreatic polypeptides, and chemokines. Both GLP-1 and GIP are incretin peptides released by gut, and are mainly involved in postprandial activation of insulin secretion and clearance of glucose (Drucker, 2003). PYY and NPY are both members of the pancreatic polypeptide family, which is involved in regulating a large array of biological functions including appetite control, behavioral stress responses, angiogenesis, and blood pressure (Groneberg, Folkerts, Peiser, Chung, & Fischer, 2004; Mentlein, 1999). A subset of chemokines and cytokines are starting to be recognized as DPP4 targets that may have biological functions following cleavage (Broxmeyer, Capitano, Campbell, Hangoc, & Cooper, 2016; Metzemaekers et al., 2016; Ou et al., 2013; Vanheule, Metzemaekers, Janssens, Struyf, & Proost, 2018). CXCL12 is the best studied of these. CXCL12 is a chemokine/cytokine involved in regulation of cell mobilization, migration and homing during tissue development, tissue maintenance and disease for numerous cell types, as well as being involved in the regulation of complex processes such as angiogenesis, hematopoiesis, and tissue repair (Brandt et al., 2006; Bromage, Davidson, & Yellon, 2014). While we have selected these five specifically due to their relatively accepted and maturing presence in the literature - they are not the only examples of DPP4 substrates with cryptic bioactivity. Additionally, not all DPP4 substrate metabolites will likely have cryptic bioactivity. We will introduce each one of these five substrates individually and compare bioactivity of the intact ligand and the DPP4-cleaved ligand, based on recent research. We will also discuss why some results in this area have been misleading or hard to accurately interpret, and demonstrate what we believe are essential tools for better and more accurate experimental results in the future for this field.
Problems related to the lack of understanding of these cryptic ligands include: 1. There are few tools to distinguish the ‘intact” ligand from the DPP4-cleaved ligand (or further proteolytically processed isoforms). Most antibodies to the ligands see both intact and N-terminal proteolytically cleaved isoforms equally well (Wang et al., 2014); 2. Consequently the short half-life, on the order of minutes, for most DPP4 ligand substrates leads to almost universal misinterpretation of the amount of intact ligands relative to the DPP4 cleaved ligands, which means some activities attributed to “intact” ligands are in fact due to the DPP4-cleaved isoforms; 3. Without the timely use of DPP4 inhibitors, or other proteolytic inhibitors, DPP4 activity continues during the time required to obtain and process the sample before stabilization and storage, with both in vivo and in vitro samples (e.g. blood draws followed by serum or plasma collection; or tissue culture media collection). This creates a number of problems when subsequently attempting to quantify ligands from such samples. For example, the actual amount of intact ligand is reduced by DPP4 activity, and once DPP4 initiates N-terminal cleavage, this allows other proteases to rapidly remove additional N-terminal amino acids accelerating the substrate’s degradation and clearance (Klemann et al., 2016) leading to a significant reduction in the “total” amount of detectable ligand within a sample (Baerts et al., 2015; Richter et al., 2014). Complicating the problem of mismeasurement of “total” ligand isoforms is increased variability when DPP4 and other protease inhibitors are not used at the point of initial sample collection, a significantly increased portion of the remaining ligand isoforms collected are in the DPP4-cleaved state, or even further processed. This results in increased variability in sample ligand measurement, as well as variability in the identification and quantification of the different biologically active cleaved ligand isoforms (Baerts et al., 2015). These challenges will be discussed in more detail below.
In this review, we look at the effect that DPP4 proteolytic cleavage has on the activity and biological functions of five major DPP4 substrate peptide hormones. This review establishes a better understanding of the clinical relevance of DPP4 inhibitors, by focusing on the biological actions of the cleaved isoforms, which are significantly understudied and usually overlooked when compared to the biological actions of intact isoforms of different DPP4 substrates. We report the most recent findings in this field, and present evidence that DPP4-cleaved peptides are far more interesting, as well as translationally and clinically relevant, than initially thought.
2. DPP4
2.1. DPP4 activity
Membrane associated and soluble DPP4 enzymes are responsible for cleaving several circulating substrates including peptide hormones and cytokines. This functional cleaving of dipeptides occurs at the N-terminal of the target substrate, by specifically cleaving off X-Proline and X-Alanine dipeptides from its polypeptide substrates (Lone, Nolte, Tinoco, & Saghatelian, 2010; Mabilleau, Mieczkowska, & Chappard, 2013). Recently CD26/DPPIV is also recognized to cleave some substrates that include hydroxyproline, serine, glycine, valine, threonine, and leucine in the second amino acid position (Havre et al., 2008). CD26 forms homodimer complexes with the enzymatic activity residing in the extracellular portion and with each subunit containing an alpha/beta hydrolase domain together with an eight-bladed beta-propeller domain to form the substrate binding pocket (Rasmussen, Branner, Wiberg, & Wagtmann, 2003). DPP4 is a member of a complex gene family that contains other enzymes responsible for cleaving other structurally related peptides. The DPP4-related enzymes include seprase, fibroblast activation protein α, DPP-6, -8, and -9, Attractin, and N-acetylated-α-linked acidic dipeptidases I and II (Busek, Malik, & Sedo, 2004; Frerker et al., 2007; Gorrell, 2005). Over the last few years, DPP4 has been thoroughly investigated because of its impact on several crucial cytokines and peptides implicated in different medical conditions. One of the most studied DPP4 substrates is GLP-1. GLP-1 is a 30 amino acid long incretin peptide hormone essential for glucose metabolism. GLP-1 enhances insulin secretion, which makes its role critical in the physiology and pathophysiology of diabetes. Normally, GLP-1 gets cleaved rapidly by DPP4 (Cantini, Di Franco, Mannucci, & Luconi, 2017; Xu et al., 2007). GLP-1–based drugs, including GLP-1 agonists and DPP4 inhibitors have been recently introduced into clinical practice with much enthusiasm (Carr et al., 2010; Mannucci et al., 2005).
In addition to its dipeptidase enzymatic activity, CD26 is also reported to have non-enzymatic functions mediated by molecular interactions with multiple binding sites in the beta-propeller domain (Havre et al., 2008). CD26 can form heterodimers with fibroblast activation protein α, and to associate with plasminogen 2, adenosine deaminase (ADA), CD45, C-X-C chemokine receptor type 4 (CXCR4), and mannose 6-phosphate/insulin-like growth factor II receptor, as well as binding to extracellular matrix (ECM) glycoproteins such as fibronectin and collagen proteins (Havre et al., 2008). DPP4 binding to ECM components may mediate turnover or modification of the ECM and DPP4 substrates near the cell surface. It permits soluble DPP4 to reside in specific ECM locations for either substrate interactions or DPP4 enzyme storage in the ECM reservoir for subsequent release from this pool, and attack of its substrates, upon ECM degradation during development, tissue maintenance or injury repair. A number of ECM molecules, including collagens and glycoproteins are modified by proteases to yield novel matrikines, which are ECM molecule fragments that act as trophic and growth factors. Proteolytic modification of ECM molecules can also yield novel binding sites for integrins and other EM receptors called matricryptins (Ricard-Blum & Salza, 2014; Su et al., 2016). If CD26 itself is not cleaving these ECM molecules directly it may be interacting with other proteases in regulating the creation of these molecules. The consequences of these interactions are unclear although they may serve to help rapidly modify ligands or binding partners of these molecules regulating signaling and adhesive interactions. Additionally, CD26 can internalize with some of these molecules, notably CD26 has been shown to bind CXCR4, the primary receptor for the cytokine/chemokine, and CD26 substrate, CXCL12 (Havre et al., 2008; Herrera et al., 2001). When CXCL12 binds CXCR4 initiating signaling, the CXCR4/CXCL12/CD26 complex can be internalized as part of the endosomal/lysosomal network. The exact function is not fully understood, but it may be part of the negative feedback regulation of the CXCL12/CXCR4 signaling pathway to temporally sharpen the end of the signal. The interaction of CD26 and CXCR4 will be further discussed in Section 7.
A naturally occurring loss of function mutation in the DPP4 gene helped identify the importance of the DPP4 enzyme. Fischer 344 (F344) rats have a Gly633-Arg mutation in the DPP4 gene. Although the mutant enzyme is synthesized normally, it is unable to be exported out of the endoplasmic reticulum, resulting in its rapid degradation. F344 rats showed a better glucose tolerance with higher GLP-1, GIP and insulin levels following an oral glucose challenge. The same results were demonstrated again in a 7-week high-fat feeding F344 rat model, in addition to reduced weight gain. Overall, it was shown that loss of DPP4 activity in rats is associated with an increase in GLP-1 action (Erickson, Suzuki, Sedlmayer, & Kim, 1992; Thompson et al., 1991; Tsuji et al., 1992).
DPP4 inhibitors have been intensely investigated gaining wide popularity as a therapeutic approach for diabetes. Until just few year ago, the majority of scientists believed that DPP4 inactivates peptide hormones by cleaving them. However, recent data showed that this is not always the case. A study by Ban et al., reported that DDP4-truncated peptide hormones may still retain functional activity similar to the non-truncated peptides (Ban et al., 2010). More interestingly, other studies demonstrated that the DPP4-truncated peptides have different receptor specificities leading to a modified activity different from the parent peptide (Gautier-Stein & Mithieux, 2013; Sparre-Ulrich et al., 2017). Importantly, emerging research suggests the activity of some peptides is mainly attributed to the cleaved form instead of the intact or non-cleaved parent form (Deacon, Plamboeck, Møller, & Holst, 2002). Understanding this crucial stem-point would explain the heterogeneity of the results of many, previously performed studies. This review shows it is common, yet inaccurate, to simply attribute the experimental results to administrating the intact “non-cleaved” form of a DPP4 enzyme substrate. Rather, further investigation can reveal that the results are in fact due to a proteolytically modified form of the administered ligand substrate.
2.2. DPP4 substrates
The vast majority of research performed on DPP4 substrates is focused on the intact isoform of the substrate, as they are generally treated as being the sole bioactive form. This common assumption has resulted in a limited number of DPP4 substrates that have been well studied in terms of their proteolytic metabolic isoforms following DPP4 truncation, which is often the first proteolytic N-terminal step for DPP4 substrates (Klemann et al., 2016). Since DPP4 inhibitors used in the clinic are for treating diabetes, most DPP4-related research focuses on the incretin hormones GLP-1 or GIP as the target ligand in diabetic models.
We identified five major DPP4 substrates where research has focused on both their cleaved, as well as the intact, isoform (Table 1). Those ligand substrates include GLP-1, GIP, PYY, NPY, and CXCL12. Both GLP-1 and GIP are incretin hormones that are mainly involved in postprandial glucose level regulation (Nauck, 2013; Elahi, 1994). PYY and NPY belong to the polypeptide family of peptide hormones, and they both regulate appetite and satiety centers in brain (Flynn, Plata-Salamán, & Ffrench-Mullen, 1998; Unniappan et al., 2006). CXCL12 is a cytokine/chemokine and chemoattractant involved in regulating cell migration, homing of stem cells to injury sites, as well as the mobilization and recruiting of lymphocytes to sites of inflammation, or activated endothelium initiating cell attachment and diapedesis (Ghadge, Muhlstedt, Ozcelik, & Bader, 2011; Guo et al., 2015; Xue et al., 2014; Yu et al., 2016; Zhang et al., 2017). CXCL12 mediates the movement, differentiation and function of numerous other cells including various stem cells, neurons, angioblast/endothelial cells and tumor cells during tissue development, maintenance and disease, as well as complex processes such as angiogenesis, hematopoiesis, and tissue protection and repair following injury (e.g. cardioprotection, tissue response to ischeamic stroke, and bone repair) (Anderluh et al., 2016; Bromage et al., 2014; Cheng et al., 2017; Yang et al., 2018).
Table 1.
DPP4 intact substrates, and their amino acid sequences
| Substrate | Amino acid sequence | Ref. |
|---|---|---|
| GLP-1(7–36) | HA ↑ EGTFTSDVSSYLEGQAAKEFIAWLVKGR | Orskov, Bersani, Johnsen, Hojrup, and Holst (1989)) |
| GIP(1–42) | YA ↑ EGTFISDYSIAMDKIHQQDFVNWLLAQKGKKNDWKHNITQ | (Sparre-Ulrich et al. (2017)) |
| PYY(1–36) | YP ↑ IKPEAPGEDASPEELNRYYASLRHYLNLVTRQRY | Grandt et al. (1994)) |
| NPY(1–36) | YP ↑ SKPDNPGEDAPAEDMARYYSALRHYINLITRQRY | Tatemoto (1982) |
| CXCL12α(1–67) | KP ↑ VSLSYRCPCRFFESHVARANVKHLKILNTPNCALQIVARLKNNNRQVCIDPKLKWIQEYLEKALNK | Dong et al. (2012) |
↑ DPP4 proteolytic site
2.3. Troubleshooting the challenges in studying DPP4-cleaved substrates
While the listed DPP4 substrates are thoroughly studied in their intact form, more limited, but an increasing amount of research has been conducted to investigate dose effects, signaling pathways, and functions, related to their DPP4-cleaved forms. Furthermore, accurate interpretation of intact substrate research results is complicated by the common assumption by researchers that relates their outcome measures - any biological response or signaling event - to the intact form, without giving consideration to the idea that the cleaved form might at least partially be responsible for the observed effects.
One issue that complicates research of DPP4 metabolic substrates is the lack of commercially-available, reliable, sensitive, and accurate quantification methods to detect and measure the cleaved isoforms or even distinguish them from the intact substrates. Rigorous research requires the ability to accurately quantify both intact and DPP4-cleaved ligands in both in vivo biological and in vitro samples. Since the difference between intact and DPP4-cleaved ligands is only 2 amino acids, finding or designing a good antibody that only reacts with one of the two forms is problematic. Until last year, only GIP out of the five mentioned DPP4 substrates had an antibody that can differentiate between intact or full length GIP, and DPP4-cleaved GIP, this antibody is utilized in an ELISA assay to quantify GIP(1–42) (Troutt et al., 2011). In 2017, Bromage et al., managed to design a novel recombinant antibody that can be used in an ELISA assay to specifically quantify intact (non-DPP4 cleaved) CXCL12, with reportedly no cross-reactivity to DPP4-cleaved CXCL12 (Bromage, Taferner, Pillai, Yellon, & Davidson, 2017). Recently, our lab has generated a DPP4-cleaved CXCL12 specific polyclonal antibody that does not cross-react with the intact form and can detect physiological levels of DPP4-cleaved CXCL12 (Elmansi et al., 2018). Other commonly available antibodies that recognize DPP4 substrates are intended to detect the intact (non-DPP4 cleaved) form, but have not been characterized as to whether they also recognize proteolytic isoforms, or if they have been characterized, they see the intact and multiple N-terminal proteolytic variants (Antonsson, De Lys, Dechavanne, Chevalet, & Boschert, 2010; Baerts et al., 2015; Bromage et al., 2017; Richter et al., 2014; Wang et al., 2014). Quantification methods based on these antibodies misrepresent how much intact or cleaved ligand are in a biological sample and result in inaccurate or misleading results. A good example for misleading associations that resulted from not having the right quantification tools is the association between remote ischemic conditioning (RIC) and upregulated levels of CXCL12 (Fortunato et al., 2013; Jiang et al., 2013; Kamota et al., 2009). Using an inflatable cuff, RIC applied to either arms of healthy human volunteers, or to hind limps of Sprague-Dawley rats, and utilizing their new intact CXCL12-specific antibody; Bromage et al., demonstrated that although there is an upregulation in total CXCL12 levels as reported by previous literature, there is an unexpected significant decrease in intact CXCL12 levels after RIC, suggesting a dramatic increase in CXCL12 N-terminal cleavage following RIC and potentially flipping the interpretation of study results while revealing the presence of DPP4 isoforms that may themselves be influencing the results. Baerts et al, found similar results in ischemic heart disease (Baerts et al., 2015). These cautionary findings are applicable to all studies of DPP4 substrate ligands, but most especially where the metabolite products may have their own biological activity (Mulvihill & Drucker, 2014).
To avoid the daunting challenge of designing an antibody that is specific for either the intact or DPP4-cleaved ligand researchers turned to using different mass spectrometric techniques. Mass spectrometry (MS) characterizes and quantifies different peptides depending on their mass, making a two-amino acids difference easier to detect. The problem, however, is that in a complex biological sample, there is a vast pool of peptides that might share similar molecular weights. A second problem is the very low physiological concentration of some of the peptides in question are below the MS detection limits, resulting in the need to artificially add superphysiological concentrations of the ligands to biological samples to produce enough of the intact and cleaved variants to be detected using standard MS (Busso et al., 2005; De La Luz Sierra et al., 2004). These two challenges in MS analytical methods have confined it for a long time to qualitative analysis, which is identification and characterization of different cleaved variants of specific peptides under very limited conditions (Richter et al., 2014). To increase selectivity and obtain a lower physiological limit of detection, Chappell et al., utilized immunoaffinity enrichment MS with liquid chromatography sample purification to be able to quantify both GLP-1(7–36) and GLP-1(9–36) in human plasma (Chappell et al., 2014). Another pioneering paper is CXCL12 quantification by Wang et al., followed the same general scheme to accurately quantify intact and DPP4-cleaved CXCL12 in circulation of mice and rhesus monkeys (Wang et al., 2014). Wang et al., found that a DPP4 inhibitor while decreasing total levels of CXCL12, measured through other commercially-available quantification methods, actually significantly increased levels of the intact CXCL12; a major unexpected finding that demonstrates the importance of measuring intact and cleaved peptides in interpreting experimental data. This effect was speculated by the authors to represent a reduction in the overall amount of proteolytic isoforms while retaining or slightly increasing the amount of intact CXCL12. The inhibition of CXCL12 turnover results in negative homeostatic feedback regulation of the synthesis and release of CXCL12 by CXCR4+ cells that would normally increase CXCL12 synthesis when there is a reduction in intact CXCL12/CXCR4 signaling following proteolysis. The overall effect would be a decrease in total CXCL12 levels (Wang et al., 2014).
Even with having the right specific antibody, or the right immunoaffinity enrichment mass spectrometry protocol with a low enough limit of detection that works for biological samples, there is still the problem of very short half-life for some of these DPP4 substrates, and the importance of inhibition of DPP4 proteolysis during sample handling to allow accurate measurement of the substrates and their metabolic products. Utilizing labeled peptides, Kieffer et al., demonstrated that more than 50% of GLP-1 is metabolized in rats to DPP4-cleaved form within 2 minutes (Kieffer, McIntosh, & Pederson, 1995). Antonsson et al., reported 80% of CXCL12 was cleaved in plasma by DPP4 within 5 minutes. Interestingly, they also showed that the DPP4 cleaved isoform has a significantly longer half-life than full length CXCL12 and consequently may have a longer time for its own biological actions to be induced than the intact ligand (Antonsson et al., 2010). Baerts et al., analyzed immunoreactivity of CXCL12α in human plasma samples immediately after sample collection with, or without, DPP4 inhibitors in the collection tube. They showed a 25% decrease in the total amount of detectable CXCL12 when DPP4 was not inhibited at the time of collection. This would include not only intact CXCL12, but the DPP4-cleaved isoform and other further proteolytic isoforms. Importantly with further incubation (one hour) the total detectable CXCL12 dropped by 75% (Baerts et al., 2015). The results of this experiment and others show that ex vivo DPP4 proteolysis of CXCL12, and by extension other DPP4 substrates, can lead to misleading experimental data, both immediately at the time of collection or following the time needed for processing samples prior to analysis or storage (Antonsson et al., 2010; Baerts et al., 2015; Bromage et al., 2017; Hall, 2014; Mortier, Gouwy, Van Damme, & Proost, 2011; Richter et al., 2014; Wang et al., 2014). This raises an important and generally overlooked point; when it comes to DPP4 substrates quantification, sample collection and handling can greatly affect results and lead to data misinterpretation. We recommend that specific DPP4 inhibitors such as sitagliptin or linagliptin should be used not only for sample collection and preservation but also used as an important control treatment group when analyzing results. We also recommend the potential addition of broad spectrum proteolytic cocktails in combination with DPP4 inhibitors to block further degradation of extant DPP4-cleaved substrate metabolites in samples. This will help dissect out the impact of the metabolites and see the “specific” effect of intact or cleaved ligands in animals and cell culture studies. Without the use of DPP4 inhibitors in an experiment, researchers are looking at the effect of mixed populations of intact and cleaved ligands on their outcome measures. If DPP4 substrates are used as treatments in experiments where the DPP4 metabolites may have separate bioactivity, the generation of those isoforms and their impact on outcome measures should be considered.
All of these factors make the task of quantifying the cleaved or intact peptide challenging, and hence it is harder to establish an association between a certain biological function or trait, and appropriately assigning it to the intact ligand, the DPP4-cleaved ligand or a summing of effects from a combination of isoforms.
Another factor that interferes with both quantification of ligands, and interpretation of results; is binding of intact or cleaved forms to ECM molecules like glycosaminoglycans (GAGs). It was found that both GLP-1 and GLP-2 can be stored in aggregates with GAGs (Maji et al., 2009). Different GAGs have also been shown to bind CXCL12 and inhibit its cardioprotectitive effect (Peterson et al., 2004; Ziarek et al., 2013). This indicates that levels of other circulating factors like GAGs as well as GAGs in the extracellular microenvironment can affect the concentration and function of different cytokines and peptide hormones (Fig. 1). Importantly this may impact the amount of DPP4 substrate ligands that are available to be measured both due to GAG-ligand binding removing them from the soluble pool to be quantified, as well as masking them from antibody-based assays by blocking antigenic sites targeted by the antibodies. Some investigators are now also trying to liberate GAG-bound DPP4-substrate ligands in samples prior to quantification to be able to assess the different ligand pools (Irhimeh, Fitton, & Lowenthal, 2007; Sweeney & Papayannopoulou, 2001).
Fig. 1.
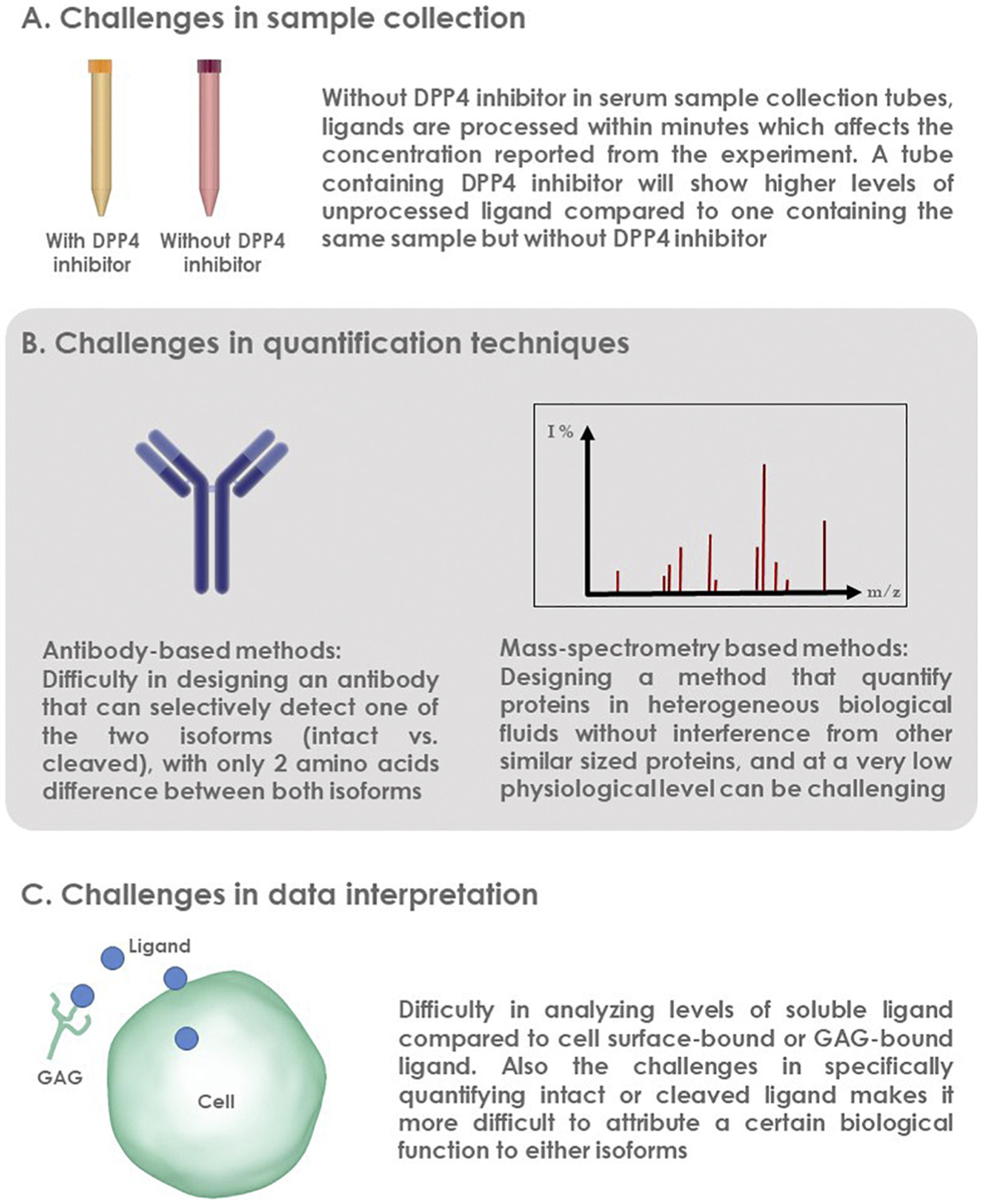
Challenges in studying DPP4-cleaved substrates can be classified into challenges related to sample collection, quantification of cleaved substrate, or data interpretation
All of the previous factors demonstrate how complicated and challenging the study of DPP4 substrates can be. However, with the right experimental tools, study design, and data interpretation based on critical thinking, researchers have been able to start to show that many DPP4-cleaved peptides possess separate bioactivity from their parent peptides. In the next section, we will present their data and try to connect the dots between different results to form a better understanding of how DPP4 changes its substrates’ bioactivity.
3. GLP-1
GLP-1(7–36) is a peptide hormone released from the L cells of the small intestine in response to food ingestion. GLP-1 hormone binds to the GLP-1 receptor (GLP-1R) and results in stimulating insulin secretion as well as inhibiting glucagon release. As a result, the blood glucose level usually decreases following food ingestion (Nauck, 2013). GLP-1 possesses additional, non-gastrointestinal related effects. These effects include protection against post-myocardial infarction remodeling and inhibition of adipose stem cells (ASC) differentiation. A higher dose of GLP-1 leads to significant inhibition of both ASC cell count and cell growth (Chen, Angeli, Shen, & Shannon, 2014; Goodwill et al., 2014; Sauve et al., 2010; Sokos, Nikolaidis, Mankad, Elahi, & Shannon, 2006).
Full length GLP-1 has a limited half-life prior to DPP4 cleavage. GLP-1 injected subcutaneously in beagle dogs has a short half-life of less than 60 minutes prior to being cleaved by DPP4 into GLP-1(9–36). In rats the half-life prior to DPP4 cleavage has been reported to be as short as 2 minutes (Kieffer et al., 1995). For a long time, GLP-1(9–36) was considered to be inactive (Knudsen & Pridal, 1996). However, a recent study by Chen et al., showed that it possesses some of the functional activity of intact GLP-1 (Chen et al., 2014).
3.1. GLP-1 effect on postprandial glycemia:
Studies have shown that exogenous administration of either intact or DPP4-cleaved GLP-1 to human subjects leads to reduction of both fasting and postprandial glucose levels. However, the effect of intact GLP-1 was different from the cleaved. It has been found that the cleaved form had minimal effect on reducing blood glucose levels, in addition that effect was only seen with the use of supraphysiological levels of cleaved GLP-1. In contrast, intact GLP-1 reduced blood glucose level much more efficiently and that effect was attributed to delaying gastric emptying and lowering glucagon levels. Cleaved GLP-1 had no significant effect on either gastric emptying or glucagon levels (Meier et al., 2006; Vahl, Paty, Fuller, Prigeon, & D’Alessio, 2003).
These findings were further confirmed by another study in pigs conducted by Deacon et al. The aim of the study was to assess the effect of cleaved GLP-1 on blood glucose levels. The study found that both intact and cleaved GLP-1 reduced blood glucose. They also suggested that cleaved GLP-1 works through enhancing the glucose disposal rate with no effect on insulin secretion (Deacon et al., 2002). Another study by Cantini et al., was conducted to evaluate the effect of intact and cleaved GLP-1 on glucose uptake. To do that, they treated ASCs for 1 hour with 10 nM insulin in the presence or absence of 10 nM of either intact or cleaved GLP-1. They found that both intact and cleaved GLP-1 significantly inhibited glucose uptake to a similar extent (Cantini et al., 2017).
Altogether, it has been strongly suggested that the cleaved GLP-1 form shares some effects with intact GLP-1, but has significant differences in how it achieves those effects. Interestingly, it has been reported that the cleaved form might be signaling and acting through receptors other than GLP-1R, which would explain the different mechanisms of action (Cantini, Mannucci, & Luconi, 2016).
3.2. GLP-1 effect on adipogenesis
Intact GLP-1 is known to inhibit ASC adipogenesis, and to downregulate the expression of adipocyte-specific markers like adipocyte protein 2, lipoprotein lipase and peroxisome proliferator-activated receptor gamma (El Bekay et al., 2016; Lee et al., 2015).
Cantini et al., evaluated the effect of intact and cleaved GLP-1 on ASC adipogenesis in vitro using Oil Red O and AdipoRed stains. Their results showed that treating ASCs with either intact or cleaved GLP-1 reduced the number of differentiated adipocytes as well as cytoplasmic lipid droplets. Also, both intact and cleaved GLP-1 reduced mRNA expression of FABP4, an adipose differentiation marker. They found that exendin (9–39), a competitive antagonist of GLP-1R, was able to reverse only the intact GLP-1 effect, but not that of cleaved GLP-1. This suggests that both intact and cleaved GLP-1 are working through different receptors (Cantini et al., 2017) (Table 2).
Table 2.
Biological functions of intact and cleaved GLP-1 alone and with exendin(9–39) (GLP-1R blocker)
| Effect | Intact GLP-1(7–36) | Cleaved GLP-1(9–36) | Ref. | ||
|---|---|---|---|---|---|
| Alone | With exendin(9–39) | Alone | With exendin(9–39) | ||
| Cell count | Decrease | No decrease | Decrease | Decrease | Cantini et al. (2017) |
| Cell proliferation | Decrease | ND | Decrease | ND | Cantini et al. (2017) |
| Apoptosis | Increase | ND | Increase | ND | Cantini et al. (2017) |
| Adipogenesis | Decrease | No decrease | Decrease | Decrease | Cantini et al. (2017), El Bekay et al. (2016) |
| Glucose uptake | Decrease | ND | Decrease | ND | Cantini et al. (2017) |
| Myocardial protective effects | Increase | Increase | Increase | Increase | Robinson et al. (2016), Sonne et al. (2008) |
ND: not determined
3.3. GLP-1 effect on apoptosis and cell proliferation
Both intact and cleaved GLP-1 significantly inhibited cell growth and decreased cell count. Exendin (9–39) itself did not affect cell numbers. However, it was able to reverse the decrease in cell numbers that was caused by intact GLP-1, but not cleaved GLP-1. Cytofluorimetric assays of annexin-V exposure on cell surface showed that both intact and cleaved GLP-1 significantly induced cellular apoptosis and caused a significant increase in apoptotic ASCs. In addition, both forms inhibited cell proliferation as shown by results of MTT assays. Similarly, exendin (9–39) was able to reverse the apoptotic effect of the intact GLP-1 with no significant action on cleaved GLP-1’s effect (Cantini et al., 2017).
3.4. GLP-1 effect on the cardiovascular system
The effects of intact and cleaved GLP-1 on the cardiovascular system have been assessed in different models. Ex vivo, rat thoracic aortas were excised and cut into 3 mm rings to assess cumulative relaxation responses for both intact and cleaved GLP-1. Both intact and cleaved GLP-1 significantly relaxed the rat aorta vasculature in a dose-dependent manner (Green et al., 2008).
In vivo, intact GLP-1 is known to protect myocardial tissue from ischemic events, an effect that GLP-1R antagonist exendin (9–39) was not able to reverse. The myocardial protective effect of GLP-1 even persisted in GLP-1R knockout mice (Ban et al., 2008; Sonne, Engstrøm, & Treiman, 2008). Cleaved GLP-1, on the other hand, was shown to be specifically protective against the development of diastolic dysfunction after myocardial infarction. It also reduced myocardial inflammation by affecting infiltrating macrophages. In addition, cleaved GLP-1 increased mitral valve E/A ratios, and decreased E wave deceleration rate in echocardiography in mice with myocardial infarction. These protective effects of cleaved GLP-1 were not reversed by exendin (9–39) (Robinson et al., 2016). Accordingly, it is now starting to be thought that this protective effect against cardiac ischemia can be attributed exclusively to cleaved GLP-1, and that previously it was mistakenly attributed to intact GLP-1. An experiment that proved this point was conducted by Ban et al., where sitagliptin, a DPP4 inhibitor, was used to prevent the truncation of the intact GLP-1 resulting in diminished intact GLP-1 protective effects against ischemia (Ban et al., 2008; Sonne et al., 2008).
3.5. GLP-1R Receptor binding
Based on findings of the studies mentioned above, cleaved GLP-1 possesses physiological actions that are not inhibited by blocking the GLP-1R or knocking down its expression. This suggests that cleaved GLP-1’s actions may be through another pathway independent of the classical GLP-1R pathway (Fig. 2). This provides a strong rationale to perform receptor binding assays of both intact and cleaved GLP-1 to GLP-1R. These studies will be essential in the interpretation of various experimental results.
Fig. 2.
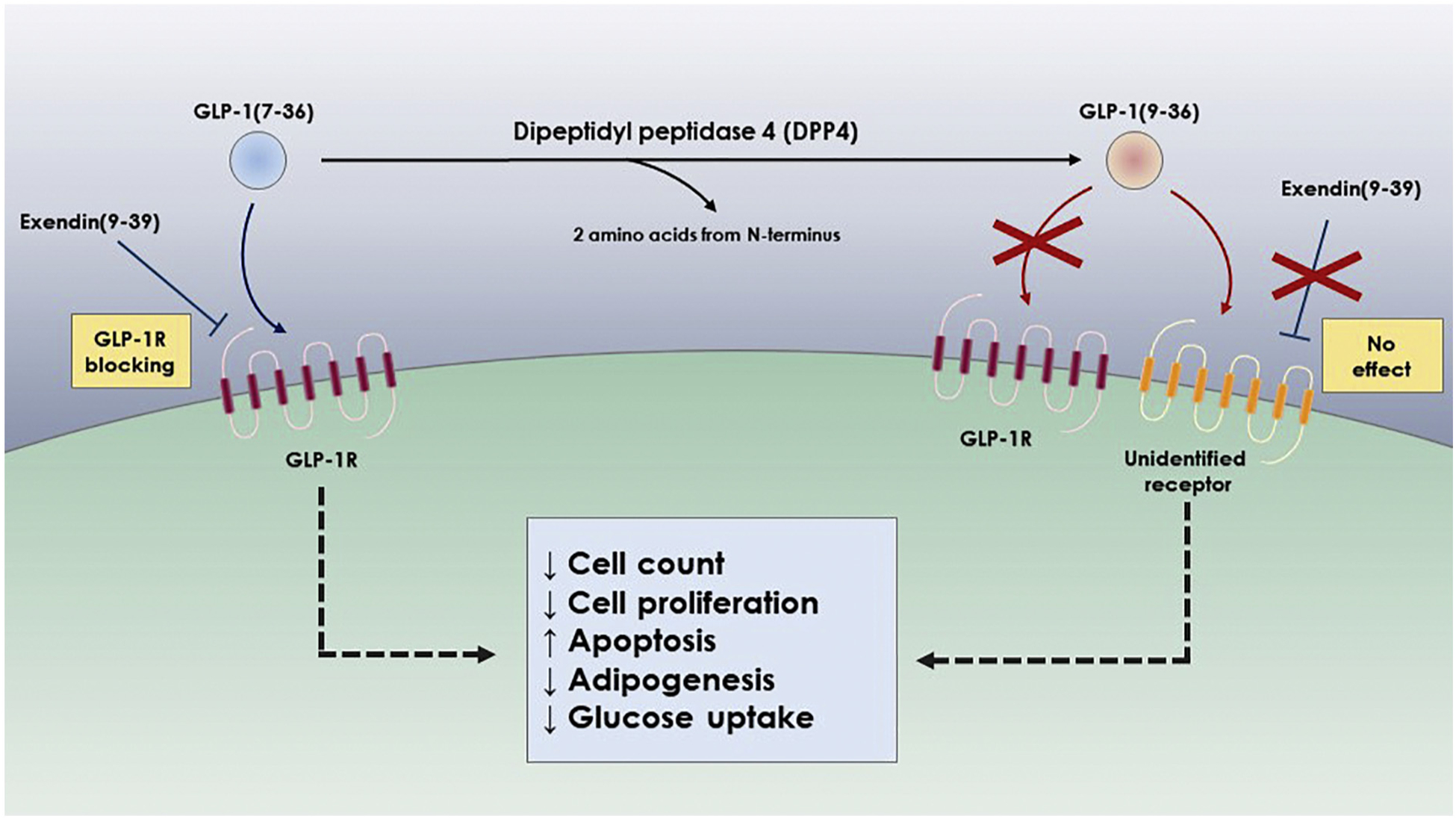
Effect of DPP4 proteolysis on receptor activation and biological functions of GLP-1
To determine affinity and binding density of both intact and cleaved GLP-1 to GLP-1R, Kuc et al., performed radioligand binding assays on coronal sections of mouse brain. These sections were selected since the tissues have high expression levels of GLP-1R as indicated by the localized in situ hybridization expression of its mRNA. Their results showed that both binding affinity (KD) and binding density (BMAX) of cleaved GLP-1(9–36) to GLP-1R in mouse brain coronal sections were significantly lower than intact GLP-1(3–36). Moreover, scintillation proximity assays (SPA) showed differential binding of intact and cleaved GLP-1 to GLP-1R. Based on SPA, the affinity of cleaved GLP-1 to GLP-1R receptor (Ki) was 70,000-fold less than intact GLP-1. Finally, cAMP assays were performed in CHO-K1 cell line expressing wild type GLP-1R receptors to evaluate receptor activation. In line with minimal association and binding of cleaved GLP-1 to the GLP-1R receptor, the cAMP assay results showed minimal signaling activation of GLP-1R by cleaved GLP-1. The maximum intracellular signaling of cleaved GLP-1 (EMAX) was 10% of the intact form, and an agonist response curve showed that almost 105 fold increase in cleaved GLP-1(9–36) concentration was required to reach this EMAX as shown by EC50 values (Kuc et al., 2014) (Table 3). This supports the idea that most - if not all - of cleaved GLP-1’s biological effects are mediated through a different receptor than GLP-1R.
Table 3.
Receptor binding assays of intact and cleaved GLP-1 to GLP-1R
| Intact GLP-1(7–36) | Cleaved GLP-1(9–36) | Ref. | |
|---|---|---|---|
| Saturation binding assay KD | 1.29 ± 0.26 AU | 0.214 ± 0.08 AU | Kuc et al. (2014) |
| Saturation binding assay BMAX | 57.0 ± 14.5 AU | 2.69 ± 0.74 AU | Kuc et al. (2014) |
| SPA binding assay Ki | 0.102 nM | 156 nM | Kuc et al. (2014) |
| cAMP assay EC50 | 0.072 nM | 7270 nM | Kuc et al. (2014) |
| cAMP assay EMAX | 104% | 8.93% | Kuc et al. (2014) |
3.6. GLP-1 summary
While DPP4-cleaved GLP-1(9–36) displays some of the intact ligand’s biological functions, we believe that the cleaved form is acting through a different mechanism than intact GLP-1(7–36) since the effects are not blocked by GLP-1R inhibitors like exendin or by knock-down of the receptor. Of all the important biological functions that intact GLP-1 influences, such as decreasing ASCs count and proliferation, increasing apoptosis, and downregulating glucose uptake, we think there is evidence that GLP-1’s myocardial protective function against ischemic events and diastolic dysfunction is in fact due to DPP4-cleaved GLP-1(9–36) mediated through a receptor other than GLP-1R.
4. GIP
GIP is a 42 amino acid long peptide hormone. It is synthesized by enteroendocrine K-cells of the stomach and duodenum. Similar to GLP-1, GIP is responsible for regulation of postprandial glycemia (Elahi, 1994). Structurally, the C-terminal domain is dispensable for GIP effects. As a matter of fact, the functional activity of GIP wasn’t reduced after removing three to four amino acids from the C-terminal. Moreover, the GIP(1–31) fragment, missing 11 amino acids from the C-terminal, was still able to retain cAMP stimulatory effect equal to that of intact GIP (Fujita et al., 2010; Fujita, Asadi, Yang, Kwok, & Kieffer, 2010). On the contrary, the N-terminal is critical for the activity of GIP-1. It has been found that while recombinant GIP missing 4 amino acids from the N-terminal domain was able to bind to the receptor with a similar affinity to the intact form, its insulintropic activity was significantly reduced (Hinke et al., 2001). As a DPP4 substrate, DPP4 truncates GIP peptide at its N-terminal by removing the two terminal amino acids. Lacking the N-terminal essential for its signaling activity, cleaved GIP was believed to be biologically inactive (Deacon, 2004; Kieffer et al., 1995). However, its ability to still bind the receptor suggests it may act as a competitive inhibitor.
Normally GIP is cleaved on both the N and C terminals though depending on the cleavage site the activity of the resulting fragment significantly varies. We are going to be discussing how DPP4 N-terminal cleavage affects both intact GIP(1–42) and C-terminal cleaved GIP(1–30), converting them into GIP(3–42) and GIP(3–30), respectively.
4.1. GIP effect on glucose regulation
In ob/ob mice, intact GIP(1–42) results in decreased plasma glucose after glucose administration. In contrast GIP(3–42) leads to an initial increase in glucose reaching its peak after 15 minutes, before it starts to decrease. Administration of other N-terminal cleaved GIP isoforms to ob/ob mice, like GIP(8–42), also showed a significant inhibition to intact GIP glucose lowering effect (Kerr, Flatt, Flatt, & Gault, 2011).
4.2. GIP effect on Insulin secretion
In pancreatic BRIN-BD11 cells, intact GIP stimulated insulin secretion in a dose-dependent manner. GIP (3–42) on the other hand significantly decreased insulin secretion along with other N-terminal truncated variants including GIP(4–42) and GIP(8–42). When intact GIP was co-administered with GIP(8–42), another N-terminal truncated variant, GIP(8–42) significantly decreased intact GIP-induced insulin release at 15 and 30 minutes after administration. Also, when cells were incubated with either GIP(3–42) or GIP(8–42), together with intact GIP, insulin secretion stimulated by intact GIP was significantly inhibited (Kerr et al., 2011). This could mean either that N-terminal truncated GIP variants possess an antagonistic effect to intact GIP through the same receptor by competitive inhibition, or via another unidentified receptor.
In vivo, treatment with different variants of N-terminal truncated GIP did not significantly alter plasma insulin response after glucose injection in ob/ob mice, while intact GIP significantly increased plasma insulin level (Kerr et al., 2011). In isolated perfused rat pancreas, GIP (3–30) competitively inhibited GIP(1–42) insulin, glucagon, and somatostatin secretion (Sparre-Ulrich et al., 2017) (Table 4). The reason for different results in isolated perfused rat pancreas versus the whole mice is not explained, but it could be due to the fact that in vivo serum insulin level is more tightly controlled than secreted insulin level in an isolated pancreas.
Table 4.
Effect of C-terminal and N-terminal cleavage on receptor binding and biological functions of GIP
| Effect | GIP(1–42) | GIP(3–42) | GIP(1–30) | GIP(3–30) | Ref. |
|---|---|---|---|---|---|
| GIP Receptor binding | High | Low | High | High | Hansen et al. (2016) |
| Receptor activity | Agonist | Weak antagonist | Agonist | Strong antagonist | Hansen et al. (2016) |
| cAMP production | Increase | Decrease | Increase | Decrease | Kerr et al. (2011) |
| Insulin secretion | Increase | Decrease | Increase | Decrease | Kerr et al. (2011), Sparre-Ulrich et al. (2017) |
4.3. GIP effect on cAMP response
In terms of receptor activation, Hansen et al showed that cAMP activation in response to GIP receptor activation was almost the same for both GIP (1–42) and GIP(1–30) in COS-7 cells, which are fibroblast-like cell lines derived from monkey kidney tissue. This suggests that the C-terminal of GIP is not essential for GIP receptor activation and down-stream responses. However, Kerr et al., found that DPP4 N-terminal truncated GIP (3–42) had a significantly lower cAMP expression, when compared to intact GIP in BRIN-BD11 cells (Hansen et al., 2016; Kerr et al., 2011). This shows again that the N-terminal is necessary for receptor activation and subsequent cAMP activation, and that cleavage of N-terminal leads to loss of signaling events downstream to GIP receptor activation. However, the mechanism of ligand inactivation versus antagonism is important to distinguish.
4.4. GIP receptor binding
Hansen et al., demonstrated that the N-terminal was critical for high-affinity binding to GIP receptor (GIP-R). The C-terminal on the other hand was a modulator for agonistic versus antagonistic actions (Fig. 3). It was found that GIP(3–30) acted as a GIP-R antagonist in both human and rat cells. GIP(3–30) competitively inhibited GIP receptor activation by the intact form GIP(1–42). A very interesting finding was that although GIP(3–42) did not possess any agonistic activity, it was a significantly less potent antagonist than GIP(3–30) both in human and rat systems, suggesting that the C-terminal can be acting as a negative regulator of the antagonist action. Competitive binding assays for GIP(1–30) and GIP(3–30) were performed to compare affinity of both ligands to the GIP-R. It was found that DPP4-cleaved GIP(3–30) had a lower log(IC50) and higher Ki, suggesting it had a higher affinity to bind to the GIP-R than GIP(1–30) (Hansen et al., 2016) (Table 5).
Fig. 3.
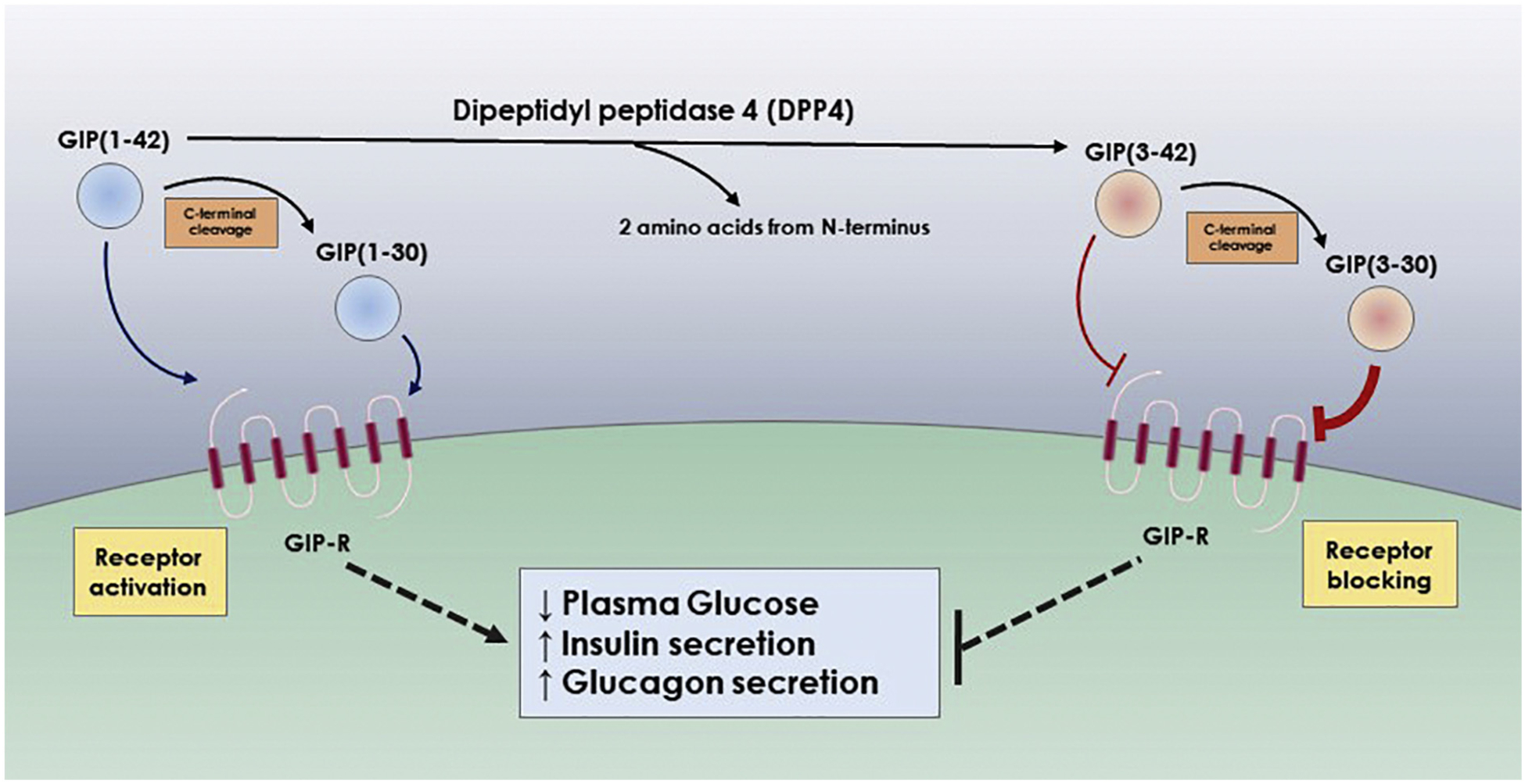
Effect of DPP4 proteolysis on receptor activation and biological functions of GIP
Table 5.
Receptor binding assays of GIP(1–30) and GIP(3–30) to GIP receptor
| GIP(1–30) | GIP(3–30) | Ref. | |
|---|---|---|---|
| Competitive binding assay logIC50 | −9.05 ± 0.02 AU | −8.63 ± 0.04 AU | Hansen et al. (2016)) |
| Competitive binding assay Ki | 0.89 nM | 2.3 nM | Hansen et al. (2016) |
4.5. GIP summary
GIP is one of the most intriguing DPP4 substrates. While intact GIP(1–42) is a critical incretin hormone that increases insulin and glucagon secretion, N- and C-terminals cleaved GIP(3–30) results in strong antagonism to GIP-R mediated functions. It is believed that the N-terminus, cleaved by DPP4, controls whether GIP variants are agonistic or antagonistic to GIP-R signaling, while the C-terminus controls the strength of the antagonistic effect of the N-terminal DPP4 cleaved GIP on the GIP-R.
5. PYY
PYY is a 36 amino acid member of the PP-fold family of peptide hormones. It is released from the distal intestine along with GLP-1 during digestion (Adrian et al., 1985). This family of peptides act preferentially through binding specific receptors known as Y1, Y2, Y4, Y5, and Y6. Similar to the intestine, PYY is also expressed in the brain, where it binds at least three PYY receptors (Y1, Y2, and Y5) with different affinities (Blomqvist & Herzog, 1997). PYY exerts a wide variety of physiological actions, the most distinct being its ability to decrease appetite and inhibit food intake. In addition, it also inhibits gastrointestinal motility, gastric and pancreatic secretions, decreases glomerular filtration rate and promotes vasoconstriction (Ferrier, 2000; Playford et al., 1992; Playford et al., 1995; Yang, 2002).
5.1. Y receptors subtypes
The different actions of PYY are achieved through binding to different receptors. Y1 and Y5 activation is responsible for triggering hunger and increasing appetite. Interestingly, Y2 receptor activation decreases appetite and consequently decreases body weight in mice (Gerald et al., 1996). Another main functional contrast between Y1 and Y2, is that Y1 specific agonists decrease anxiety, while Y2 specific agonists are anxiogenic. Receptors also vary in their distribution pattern within different tissues. Y1 receptor is expressed in adipocytes, colon, vascular smooth muscles, and the cerebral cortex (Castan et al., 1993; Dumont, Fournier, St-Pierre, Schwartz, & Quirion, 1990; Grundemar et al., 1992; Mannon, Mervin, & Sheriff-Carter, 1994). It is involved in the regulation of decreased anxiety and depression (Caberlotto et al., 1999; Wahlestedt, Pich, Koob, Yee, & Heilig, 1993). The Y2 receptor is expressed in nerve fibers, the hippocampus, and intestine (Gehlert, Gackenheimer, & Schober, 1992; Rettenbacher & Reubi, 2001; Stjernquist & Owman, 1990), and it is associated with memory retention and angiogenesis (Flood & Morley, 1989; Zukowska-Grojec et al., 1998). The Y5 receptor is expressed in the hypothalamus, intestine, ovary, testis, pancreas, and skeletal muscle (Gerald et al., 1996; Herzog et al., 1997). It is involved with stimulation of appetite, and regulation of brain seizures and circadian rhythm (Gerald et al., 1996; Gribkoff, Pieschl, Wisialowski, van den Pol, & Yocca, 1998; Guo, Castro, Palmiter, & Baraban, 2002; Matsumoto, Basil, Jetton, Lehman, & Bittman, 1996).
DPP4 cleaves PYY (1–36) and yields PYY (3–36). PYY (3–36) has been proven to possess a distinct biological activity. Grandt et al., showed that DPP4-cleaved PYY(3–36) is a selective Y2 receptor agonist, as compared to intact PYY(1–36) that binds to both Y1 and Y2 receptors (Grandt et al., 1994). This shows a very interesting effect for DPP4 on PYY, where it shifts it from a limited-selective multi-receptor agonist that binds to three different Y receptors, to a highly selective agonist that binds only to one of these receptors. Based on that, there is a great chance that cleaved PYY (3–36) would be able to show a more prominent pattern of Y2 receptor activation compared to PYY (1–36). Therefore, DPP4 may be implicated in physiological control of appetite, by decreasing hunger-triggering Y1 and Y5 receptor signals, and upregulating anxiogenic Y2 receptor signal. Further studies, however, need to be conducted to support this hypothesis, and to test if pharmacological DPP4 inhibitors lead to increased food intake by downregulating PYY(3–36) protein levels and how this might interact with DPP4 mediated insulin and glucose regulation.
5.2. PYY effect on food intake
According to a study conducted by Unniappan et al., they showed that in normal rats, both PYY (1–36) and PYY (3–36) significantly reduced 24-h food intake. In contrast, in DPP4 deficient rats, PYY(3–36) significantly reduced 24-h food intake, while PYY(1–36) had no notable effect (Unniappan et al., 2006) (Table 6). This result suggests another instance where the effect of the DPP4-cleaved substrate may be mistakenly attributed to an intact form of the parent ligand. Without proper inhibition of the degrading enzyme, effects of both intact and cleaved form may be confused (Fig. 4).
Table 6.
Receptor selectivity and effect on food intake of intact and cleaved PYY
| Intact PYY(1–36) | Cleaved PYY(3–36) | Ref. | |
|---|---|---|---|
| Receptor selectivity | Y1, Y2, Y4, and Y5 agonist | Y2 agonist | Grandt, Schimiczek, Beglinger, et al. (1994) |
| Food intake | Decrease (no effect on DPP4-deficient mice) | Decrease (acts on DPP4-deficient mice) | Batterham et al. (2003), Chelikani et al. (2005), Moran et al. (2005), Pittner et al. (2004), Tschop et al. (2004) |
Fig. 4.
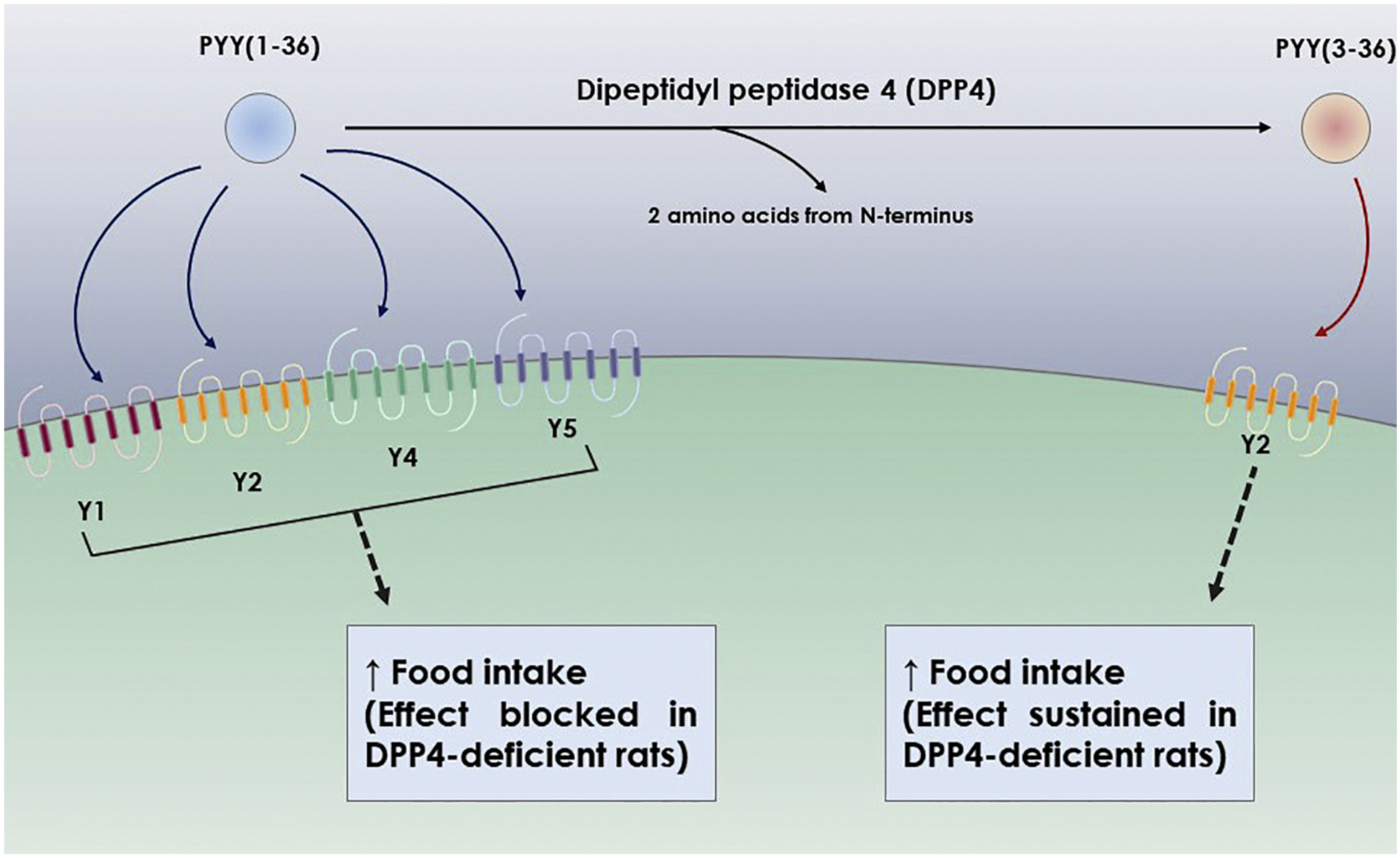
Effect of DPP4 proteolysis on receptor activation and biological functions of PYY
After fasting, DPP4 deficient rats also showed a higher 24-h food intake and weight gain. They also showed higher cumulative food intake during light phase and dark phase (Unniappan et al., 2006). It has been also shown that treatment with PYY(3–36) through various routes of administration in different rat, mouse, and rhesus monkey models, in addition to obese humans, lead to reduction of food intake (Batterham et al., 2003; Chelikani, Haver, & Reidelberger, 2005; Moran et al., 2005; Pittner et al., 2004; Tschop et al., 2004). Taken together, these results support the idea that inhibition of food intake may in part be caused by the DPP4-cleaved form of PYY (3–36).
5.3. PYY effect on GIT motility
Moriya et al., showed that PYY (3–36) - in addition to other Y2 receptor agonists – significantly inhibited castor oil-induced diarrhea in a dose-dependent manner in mice. PYY (3–36) also significantly blocked 16,16-dimethyl Prostaglandin E2-induced intestinal fluid accumulation. It also significantly and dose-dependently reduced wet fecal weight and bead expulsion time by slowing colonic transit (Moriya et al., 2010). This is an effect that was not reported for PYY(1–36). This could be because PYY(3–36) is a pure Y2 receptor agonist, while the effects of PYY (1–36) is the result of Y1, Y2, and Y5 activation, which dilutes the overall effect producing a less distinct phenotype.
5.4. PYY effect on locomotor and exploratory behavior
PYY (3–36) effects are not only restricted to food intake and GIT motility. Stadlbauer et al., showed that mice treated with PYY (3–36) show increased novel object exploration compared to vehicle treated mice. Also, mice treated with PYY (3–36) showed a higher locomotor habituation response to novel environments than mice treated with vehicle alone. PYY (3–36) potentiated the increase in locomotor response to amphetamine, as measured by the distance moved in an open field. Apomorphine treatment induces stereotyped behavior in mice, which is evidenced by increased leaning and climbing. PYY(3–36) also increased this stereotyped apomorphine behavior (Stadlbauer, Weber, Langhans, & Meyer, 2013). These effects are also believed to be a result of Y2 receptor activation in the brain.
5.5. PYY summary
Intact PYY(1–36) activates 4 different receptors (Y1, Y2, Y4, and Y5), and as expected, the biological effects from activating these 4 different receptors is complicated and hard to accurately measure compared to PYY(3–36) which appears to work as a Y2-specific receptor agonist. The only well documented biological function where PYY(1–36) is compared to PYY(3–36) is the effect on food intake. Both intact and cleaved variants decrease 24-h food intake in rats, and only PYY(3–36) decreases 24-h food intake in DPP4-deficient rats. This suggests that the decrease in food intake following intact PYY(1–36) delivery is actually due to the DPP4 cleavage yielding PYY(3–36) after treatment in rats. Cleaved PYY also possesses other important biological activities on GIT mobility, and CNS mediated locomotor and exploratory behavior in mice. A better understanding of these functions as well as other possible effects of cleaved and intact PYY and interaction with other DPP4 cleaved substrate signaling pathways is necessary to assess benefits and drawbacks of using clinical DPP4 inhibitors for treatment of diabetic patients or in other populations.
6. NPY
NPY is another 36 amino acid peptide hormone that is a Y receptor family agonist. Similar to PYY, it is expressed in the hypothalamus and is truncated by DPP4 to also produce a selective Y2 receptor agonist. NPY (1–36) increases short term 2-h food intake. In addition, NPY inhibits osteoblast activity centrally by binding to the Y2 receptor and peripherally by binding to Y1 receptor. NPY also affects adiposity both centrally in the hypothalamus and peripherally. Centrally, NPY increases fat accretion through Y1 and Y5 receptors leading to increased bone marrow adiposity, and decreases bone marrow fat accretion through the Y2 receptor. Bone marrow and peripheral adiposity are differentially regulated, therefore it is interesting that in peripheral tissues, NPY leads to increased fat accretion through activation of both Y1 and Y2 receptors. NPY also plays a role in increasing adhesion and chemotaxis of macrophages. It also contributes in synaptic transmission and modulates neuroplasticity. NPY affects a complex interdigitating network of biological functions through four different receptors, and a better understanding of how DPP4-cleaved PYY is contributing to this complex network is essential to grasp the extents to which DPP4 inhibitors are affecting neuropeptide biology.
6.1. NPY receptor binding
In 1996, Grandt et al., found that both NPY(1–36) and NPY(3–36) compete in binding to CHP 234 human neuroblastoma cells; a type of cell that expresses the Y2, but not the Y1 receptor, on their surface. However, SK-N-MC human neuroblastoma cells, which in contrast express Y1 receptor, but not the Y2 receptor, only bind NPY(1–36). These results suggest that similar to PYY; the intact form is a non-selective agonist for both Y1 and Y2 receptors, while the cleaved form is a selective Y2 agonist (Grandt et al., 1996) (Fig. 5).
Fig. 5.

Effect of DPP4 proteolysis on receptor activation and biological functions of NPY
6.2. NPY effect on cell migration
It has been found by Ghersi et al., that both NPY(1–36) and NPY (3–36) cause human umbilical vein endothelial cells (HUVECs) to migrate in a wound healing model. However, when DPP4 activity was inhibited using a DPP4 neutralizing mAb the anti-migratory effect of NPY(1–36) was significantly decreased. This suggests that increasing cell migration in wound healing model may be due to DDP4 cleaved NPY(3–36) rather than the intact form (Ghersi, Chen, Lee, & Zukowska, 2001) (Table 7).
Table 7.
Receptor selectivity and effect on cell migration and food intake of intact and cleaved NPY
| Intact NPY(1–36) | Cleaved NPY(3–36) | Ref. | |
|---|---|---|---|
| Receptor selectivity | Y1, Y2, Y4, and Y5 agonist | Y2 agonist | Grandt et al. (1996) |
| Cell migration | + (effect blocked with DPP4 inhibition) | +(effect retained with DPP4 inhibition) | Ghersi et al. (2001) |
| 2 Hour food intake | + | ++ | Flynn et al. (1998) |
Moreover, Singh et al reported that in the hematopoietic microenvironment, DPP4-cleaved NPY(3–36) reduced VE-cadherin and CD31 expression in endothelial cell junctions, and mobilized hematopoietic stem and progenitor cells (HSPCs) through Y2 and Y5 receptor activation. HSPCs trafficking was impaired in mice lacking either DPP4 or NPY, and was restored upon treatment of mice with DPP4-cleaved NPY(3–36) (Singh et al., 2017).
6.3. NPY(3–36) effect on food intake
The effect of NPY(3–36) on food intake has not been as thoroughly studied as PYY(3–36). One publication comparing NPY(1–36) to NPY(3–36) in rats in the context of food intake showed that NPY(3–36) leads to a greater increase in 2-hour food intake compared with NPY(1–36). The increase in food intake was found to be dose-dependent and it plateaued as the dose increased beyond 2.5 μg/rat (Flynn et al., 1998) (Table 7). This is an unexpected result, and is in contrast to the effects of PYY(3–36) on food intake. An explanation for why NPY(3–36), a Y2 selective agonist, leads to increased food intake is unclear, and further work needs to be done to assess this effect of NPY(3–36) on a longer timeline. This suggests DPP4-cleaved PYY and NPY can differentially act on the Y2 receptor.
6.4. NPY summary
NPY is not as thoroughly studied as GLP-1 or GIP, but it has been shown that intact NPY(1–36) can activate 4 different Y receptors (1, 2, 4, and 5) while DPP4-cleaved NPY(3–36) only activates the Y2 receptor. Both NPY(1–36) and (3–36) induce HUEVCs migration, but with DPP4 inhibition, only NPY(3–36) retains this effect, which suggests that NPY(1–36) can only induce cell migration after being cleaved by DPP4. Also, the effect of NPY on food intake is limited, however, in 2-h food intake studies DPP4-cleaved NPY(3–36) shows a greater inhibition of intake in rats compared to intact NPY(1–36).
7. CXCL12, stromal cell-derived factor 1 (SDF-1)
SDF-1/CXCL12 is a highly conserved pleiotropic chemokine/cytokine that is considered one of the earliest cytokines evolutionarily (Shirozu et al., 1995). It is produced by numerous cell types and tissues constitutively and its expression is induced by most tissues in response to injury (Herberg et al., 2013). Common sources are tissue stromal/support cells such as mesenchymal derived cells involved in stem cell niche organization, cardiomyocytes, neural stem cells (NSCs), osteogenic cells, and endothelial cells (Carbone et al., 2017b; Herberg, Fulzele, et al., 2013; Mortensen & Hill, 2015). For example, we and others have shown that in the brain astrocytes are the primary constitutive source of CXCL12. Astrocytes increase CXCL12 secretion during development regulating neuronal cell and process migration, as well as in maintenance of the organization of the adult neurogenic zone, and critically in tissue repair following injury (Hill et al., 2004; Miller et al., 2005; Shin et al., 2014). CXCL12 is also highly secreted by lung and bone marrow stromal cells, as well as stromal and endothelial cells in other tissues being involved in a large spectrum of biological and developmental processes (Bleul, 1996; Nagasawa et al., 1996). CXCL12 is involved with cardioprotection, bone repair, neuronal regeneration, tumor cell mobilization, migration and homing/metastasis (Bromage et al., 2014; Broxmeyer et al., 2005; Cheng, Wang, et al., 2017; Li, Basu, Han, Kim, & Broxmeyer, 2007; Yang et al., 2018). On the cellular level, CXCL12 affects migration, survival, and proliferation (Herberg et al., 2013; Leite Pereira et al., 2018; Reid et al., 2018). In humans CXCL12 has six alternative splice variants (α to φ) differing only in the exon related to the C-terminus. Out of these six, the most common splice variants are CXCL12α (1–68) and CXCL12β (1–72) (Yu et al., 2006). All CXCL12 variants, including CXCL12α and CXCL12β, are cleaved by DPP4 removing their N-terminus KP dipeptide amino acids (Kato et al., 1998; Lambeir et al., 2001). Importantly, the ability to initiate the DPP4 attack is regulated by the C-terminus. CXCL12α is rapidly cleaved when its C-terminal lysine is removed by Carboxypeptidase M or N, in contrast the half-life of the N-terminal intact CXCL12β is longer due to the protection of lysine 68 by the four additional C-terminal amino acids blocking the binding of the carboxypeptidases (Herberg, Fulzele, et al., 2013; Herberg, Shi, et al., 2013). Other than difference in half-life, there seems to be no significant difference in receptor selectivity or functions between CXCL12α and CXCL12β (De La Luz Sierra et al., 2004; Herberg, Fulzele, et al., 2013; Herberg, Shi, et al., 2013; McQuibban et al., 2001; Proost et al., 1998; Richter et al., 2014; Vergote et al., 2006). Interestingly for CXCL12, not only does DPP4 cleave two amino acids from its polypeptide structure, but it sets CXCL12 up for further proteolysis by other peptidases that lead to even more circulating proteolytic metabolites of CXCL12 (Richter et al., 2014). Until recently DPP4-cleaved CXCL12 has generally been thought to be inactive, or at least much less active than intact CXCL12 (Anderluh et al., 2016; Crump et al., 1997; Kato et al., 1998).
The two main receptors for CXCL12 are CXCR4, a ligand concentration biased G-protein coupled receptor (GPCR), and CXCR7 (AKA - Atypical Chemokine Receptor 3 or ACKR3), a β-arrestin associated receptor that has a 1000-fold higher affinity for CXCL12 than CXCR4 (Quinn, Mackie, & Caron, 2018; Reid et al., 2018; Sanchez-Martin, Sanchez-Mateos, & Cabanas, 2012). Data suggests that both receptors can homodimerize, as well as heterodimerize with each other, or with other receptors including α1A/B-adrenoceptors (Decaillot et al., 2011; Levoye, Balabanian, Baleux, Bachelerie, & Lagane, 2009; Tripathi et al., 2015). This ability to dimerize regulates receptor functions and affects their downstream signaling events (Ferre et al., 2014; Milligan, Canals, Pediani, Ellis, & Lopez-Gimenez, 2007; Rozenfeld & Devi, 2010; Tripathi et al., 2015). ACKR3 may have additional heterodimerization partners including epidermal growth factor receptor (Salazar et al., 2014). The potential for multiple heterodimeric complexes with CXCL12 receptors opens the door for possible combinations that respond to DPP4, and other N-terminal, cleaved CXCL12 isoforms. The ligand, CXCL12, can itself homodimerize, and it can activate the CXCR4 receptor either in monomeric or a dimeric form (Drury et al., 2011; Roy et al., 2015). There is, however, a ligand bias associated with CXCR4 response to monomeric or dimeric CXCL12 activation. In this case, monomeric CXCL12 activation of CXCR4 leads to G-protein-dependent signaling. However, as the CXCL12 ligand concentrations increase, the likelihood of both ligand binding pockets of the CXCR4 receptor dimer being occupied increases as well. This is accompanied with a switch from GPCR-mediated signaling to β-arrestin-dependent signaling, which blocks actin remodeling and inhibits cell migration (Drury et al., 2011; Ziarek et al., 2017).
7.1. Receptor activation by DPP4-cleaved CXCL12
Crump et al., assessed the ability of N-terminal modified, and other CXCL12 variants, to bind to and activate CXCR4 demonstrating that removal of the first 2 amino acids or beyond dramatically inhibited or eliminated binding to and activation of CXCR4 (Crump et al., 1997). More recently, Janssens et al., used CXCR4-transfected, ACKR3-negative CHO and COS-7 cells to test receptor activation and signaling specifically through the CXCR4 receptor. It was found that starting at 100 pM, intact CXCL12(1–68) induced IP3 accumulation in the cytosol, and triggered phosphorylation of the second messengers ERK and Akt, in a dose-dependent manner. In contrast DPP4-cleaved CXCL12(3–68) even at concentrations up to 100 nM, did not induce IP3 accumulation, or ERK and Akt phosphorylation (Janssens et al., 2017). Janssens et al., also demonstrated that while DPP4-cleaved CXCL12α cannot activate CXCR4, it can still recruit β-arrestin through activation of the ACKR3 receptor. However, the activation of ACKR3 is about 10-fold less than that induced by intact CXCL12 (Janssens et al., 2017) (Table 8). Cheng et al., showed very similar results using the Presto-Tango β-arrestin 2 recruitment assays for CXCL12(1–68) and CXCL12(3–68). The assays showed that while 1 μM CXCL12(1–68) recruited β-arrestin 2 through both CXCR4 and ACKR3, CXCL12(3–68) only managed to recruit β-arrestin 2 through the CXCR4 receptor at extremely high doses in vitro [EC50 (nM) N103) with no detectable efficiency% while the ACKR3 receptor binding of CXCL12 (3–68) lead to ACKR3 recruitment of β-arrestin 2 that was significantly greater [EC50 (nM) N13.3±13.5), but reduced to about 20% efficiency compared to intact CXCL12(1–68) (Cheng, Eby, et al., 2017). Ziarek et al., 2017 also reported that DPP4-truncated CXCL12(3–68) shows a dramatic drop in CXCR4 binding affinity, in addition to complete loss of G protein agonist activity. They also described that cleaved CXCL12 possesses a very weak CXCR4 GPCR antagonistic activity (Ziarek et al., 2017).
Table 8.
Effect of N-terminal cleavage on receptor activation and biological functions of CXCL12α and β
| Intact CXCL12 | Cleaved CXCL12 | Ref. | |
|---|---|---|---|
| CXCR4 receptor (G-protein agonist) | High | No activation | Cheng, Eby, et al. (2017), Janssens et al. (2017), Ziarek et al. (2017) |
| CXCR4 receptor (β-arrestin recruitment) | High (concentration biased) | Very low with EC50 >103 | Cheng, Eby, et al. (2017) |
| ACKR3 receptor (β-arrestin recruitment) | High | Decreased by 80–90% | Cheng, Eby, et al. (2017), Janssens et al. (2017) |
| GAG binding | High | Slightly decreased | Janssens et al. (2017) |
| BMSCs osteogenic differentiation | increases | inhibits | Elmansi et al. (2018) |
| Cell migration | chemotactic | inhibits | Elmansi et al. (2018), Janssens et al. (2017), Kato et al. (1998) |
| Anti-HIV-1 activity | Present | Absent | Kato et al. (1998) |
Research done by Szpakowska et al., on CXCL11, another cytokine that activates CXCR3 and ACKR3, and is cleaved by DPP4 in a fashion similar to CXCL12, showed that DPP4-cleaved CXCL11 loses its ability to activate the CXCR3 receptor, but retains the ability to bind and activate ACKR3 (Szpakowska et al., 2018). This result, along with the previously mentioned CXCL12 receptor binding and activation assays, shows that removal of N-terminal lysine/proline by DPP4 is not able to initiate GPCR-biased activity through CXCR4, but can still induce β-arrestin mediated activity through ACKR3 and possibly to a lesser extent through CXCR4 β-arrestin mediated activity.
7.2. CXCL12 GAG binding affinity
As mentioned previously, CXCL12 concentration and biological activity are affected by binding to GAGs. Janssens et al., utilized heparan sulfate and dermatan sulfate coated plates to compare the binding affinity of intact and cleaved CXCL12. The results showed a moderate, but significant, decrease in affinity of cleaved CXCL12 to dermatan sulfate compared to intact CXCL12. However, there was no significant difference in affinity of both intact and cleaved CXCL12 to heparan sulfate, which is thought to be a major sink for extracellular CXCL12 (Janssens et al., 2017).
7.3. CXCL12 effect on bone marrow mesenchymal stem cells differentiation
The role of CXCL12 in bone health is well established. CXCL12β was previously shown to potentiate the osteoinductive effect of bone morphogenetic protein 2. This effect was shown in vitro in genetically engineered bone marrow-derived mesenchymal stem cells (BMSCs), and in vivo in bone regeneration in critical-size rat calvarial defects (Herberg et al., 2014; Herberg et al., 2015). Our lab recently found that while intact CXCL12β(1–72) induces osteogenic differentiation in BMSCs, cleaved CXCL12β(3–72) inhibits osteogenic differentiation (Elmansi et al., 2018) (Table 8). This result has major implications, as the balance between osteogenic and adipogenic differentiation in BMSCs is a crucial aspect in bone healing and healthy bone aging (Infante & Rodriguez, 2018).
7.4. CXCL12 effect on cell migration
CXCR4 is required for CXCL12-mediated chemotaxis, and is a co-receptor for HIV cell binding and transfection. A paper by Shioda et al., in 1998 demonstrated that DPP4 abolished both chemotactic and anti-HIV-1 activities for CXCL12α and CXCL12β. Shioda et al., infected two different cell lines with a recombinant CXCL12α-expressing vector. The two cell lines were H9, a DPP4-expressing T-cell line, and MT4, a T-cell line that does not express DPP4. After 3 days, the results they found were intriguing; western blot analysis showed while there were similar total levels of CXCL12α in the supernatant of both cell lines, the chemotactic and anti-HIV-1 activities widely varied from remarkably high levels in non-DPP4 expressing MT4 cells to very low activity in DPP4 expressing H9 cells (Kato et al., 1998). Although this experiment did not directly test the biological activity of DPP4-cleaved CXCL12, it hints that there is an antagonistic action resulting from having DPP4 in close proximity to released CXCL12.
Another experiment by Janssens et al., showed that DPP4-cleaved CXCL12α does not induce endothelial cell migration compared to intact CXCL12α. However, there was no control group to test the possibility that cleaved CXCL12α inhibits migration, instead of simply being inactive (Janssens et al., 2017). Christopherson et al, reported that DPP4-cleaved CXCL12α(3–68) does not induced the migration of CD34+ HSCPs and even inhibits cellular migratory activity induced by intact CXCL12α (1–68). They suggested that DPP4 truncation of CXCL12α could represent a novel mechanism that regulates migration, mobilization, and homing of HSPCs (Christopherson, Cooper, & Broxmeyer, 2003; Christopherson II, Hangoc, & Broxmeyer, 2002; Christopherson II, Hangoc, Mantel, & Broxmeyer, 2004). Moreover, our lab recently found that pretreating BMSCs with cleaved CXCL12β(3–72) for 16 hours, diminishes their ability to migrate towards media containing intact CXCL12β(1–72) in transwell assays. This result is important in understanding stem cell migration and cancer cell metastasis. It also means that DPP4-cleaved CXCL12 is not simply inactive, but possesses a separate bioactivity that might be antagonistic to intact CXCL12 (Elmansi et al., 2018) (Table 8) (See Fig. 6). However, since DPP4 cleaved CXCL12 does not appear to effectively bind to CXCR4 based on competitive displacement of radiolabeled CXCL12 (1–68) by cold CXCL12 (3–68) [CXCL12(3–68) kd = 464 ± 2nM compared to Kd = 1.44 ± 1.5 nM for CXCL12(1–68) (Ziarek et al., 2017) the reports of apparent antagonistic activity actually may be mediated by ACKR3 or alternative mechanisms via interactions with other receptors (Cheng, Eby, et al., 2017; Janssens et al., 2017). For example, ACKR3 activation may down-regulate CXCR4 expression and therefore G-protein coupled signaling directly, or as a consequence of CXCR4/ACKR3 heterodimerization, which results in β-arrestin signaling while abrogating G-protein coupled signaling through CXCR4. Therefore, ACKR3 activation leads to decreased CXCR4 signaling and inhibition of CXCR4 mediated migration and other actions (Decaillot et al., 2011; Janssens, Struyf, & Proost, 2018; Levoye et al., 2009; Zabel et al., 2009). (See Fig. 6.)
Fig. 6.

Effect of DPP4 proteolysis on receptor activation and biological functions of CXCL12
7.5. CXCL12 effect on cell viability
We previously showed that intact CXCL12β mediates cell survival via upregulation of the autophagic pathway and inhibition of apoptosis in BMSCs (Herberg, Shi, et al., 2013). However, another form of cleaved CXCL12(5–67) produced by the action of matrix metalloproteinase-2, was shown to induce NSC death through activating the intrinsic apoptotic pathway. This was evident by increased caspase 3/7 activation in NSCs treated with cleaved CXCL12(5–67) compared to control. Intact CXCL12 on the other hand was shown to inhibit apoptosis and decrease caspase 3/7 activation in NSCs compared to control (Adelita, Stilhano, Han, Justo, & Porcionatto, 2017).
7.6. CXCL12 effect on brain and nervous system
While there is no available data on DPP4-cleaved CXCL12(3–67) activity in the brain, studies showed that the previously-mentioned cleaved CXCL12 (5–67) is neurotoxic to human and mouse neurons with no chemotactic properties. It was also found that higher levels of CXCL12(5–67) in brain were associated with HIV-1 infection (McQuibban et al., 2001; Zhang et al., 2003). The interesting and relevant part, is that this form of cleaved CXCL12(5–67) did not act through CXCR4, the regular receptor for intact CXCL12, but exerted its neurotoxic and immunogenic actions through the chemokine receptor CXCR3, which also shares the ligand CXCL11 with ACKR3, which is promiscuous in being activation-prone by N-terminal modified CXCL11 and CXCL12 (Benhadjeba, Edjekouane, Sauve, Carmona, & Tremblay, 2018; Vergote et al., 2006). This does not mean that DPP4-cleaved CXCL12 acts through CXCR3, but it reinforces the idea that a proteolytic enzyme can generate a new cryptic ligand to a receptor that was not activated by the intact parent peptide.
7.7. CXCR4 and DPP4 interactions
Among the non-substrate proteins that bind CD26 are a number of receptors including the CXCL12 receptor CXCR4 (Havre et al., 2008). Binding of CXCL12 initiates the internalization of both CXCR4 and CD26 in the T-cell line, Jurkat J32, the B-cell line, SKW6.4, and peripheral blood lymphocytes, suggesting that these two proteins may function together and emphasizing the importance of understanding the actions of DPP4 cleaved ligands (Herrera et al., 2001).
7.8. CXCL12 summary
CXCL12 is one of the most thoroughly studied cytokines in the literature. However, data regarding biological functions of DPP4-cleaved CXCL12 are just beginning to emerge. There have been a number of papers measuring DPP4-cleaved CXCL12(3–68) affinity, binding, and potential to activate CXCR4 or ACKR3 receptors (Cheng, Eby, et al., 2017; Janssens et al., 2017). It has been shown that DPP4-cleaved CXCL12 (3–68) can bind and activate ACKR3 with reduced affinity, while it cannot activate CXCR4 at all, at least via GPCR signaling. There is however a suggestion that CXCL12(3–68) may signal via β-Arrestin 2 pathways for both ACKR3, and possibly CXCR4 (Cheng, Eby, et al., 2017; Janssens et al., 2017; Ziarek et al., 2017). In terms of binding to GAGs in extracellular matrix, cleaved CXCL12(3–68) binds a subset of GAGs with a slightly decreased affinity compared to intact CXCL12. The main biological function measured for cleaved CXCL12 in the literature has been its inability to provide the anti-HIV activity of the intact isoform (Crump et al., 1997; Proost et al., 1998). Our lab recently found that DPP4-cleaved CXCL12β(3–72) has an antagonistic-like action to intact CXCL12β(1–72) in terms of BMSCs cell migration and osteogenic differentiation (Elmansi et al., 2018), while intact CXCL12β(1–72) induces BMSCs migration and osteogenic differentiation, cleaved CXCL12β(3–72) inhibits both of those CXCR4 G-protein mediated functions (Elmansi et al., 2018). Again, this likely does not represent direct competitive inhibition of the intact isoform with the CXCR4 receptor, certainly not through the CXCR4 GPCR mechanism, and it is unclear if CXCL12α(3–68) and CXCL12β(3–72) can bind effectively at physiological levels to CXCR4 (Cheng, Eby, et al., 2017; Crump et al., 1997; Janssens et al., 2017; Ziarek et al., 2017). Further work needs to be done to assess other biological functions and potential mechanistic pathways through which DPP4-cleaved CXCL12 is acting.
8. Conclusion
DPP4 proteolytic cleavage of different peptide hormones does not simply deactivate them. Rather, many studies have shown that DPP4 cleavage leads to changes in the intact ligand/substrate’s biological function mediated through alterations in the normal receptor(s) interactions or revealed cryptic interactions with receptors that do not recognize the intact ligand. Additionally, biological functions and receptor interactions usually attributed to the intact ligand may actually be due to the DPP4-cleaved ligand. While this review focuses on the consequences of DPP4 enzymatic cleavage of five different substrates that belong to three different classes of biological molecules, it is important to mention that these constitute only a small number of DPP4 substrates. A large array of other proteins –including cytokines, modulatory factors, colony-stimulating factors, etc.- have been reported to be cleaved by DPP4 in different tissues and organs (Broxmeyer et al., 2012; Christopherson, Cooper, Hangoc, & Broxmeyer, 2003; Ou et al., 2013).
In the case of GLP-1, it was found that some of the actions attributed to intact GLP-1 can be attenuated or even completely blocked upon inhibition of DPP4 proteolytic activity. This, in addition to the previously mentioned findings, suggests that cleaved GLP-1 is itself responsible for some of the physiological actions that has been attributed to intact GLP-1. Blocking of GLP-1R reverses intact GLP-1 activity, with no effect on cleaved GLP-1 activity. This observation indicates that cleaved GLP-1 is also acting through an unidentified receptor that is distinct from GLP-1R. This surprising conclusion about DPP4-cleaved ligands is paradigm shift that reveals a critical gap in our understanding and interpretation of research and clinical data related to vital signaling pathways. In the case of GLP-1 the clinical implications are significant, since it affects our understanding of insulin regulation in general and specifically through new implications of how anti-diabetic DPP4 inhibitors work. Further research in this area is essential, to better understand the potentially beneficial biological actions of cleaved GLP-1 that can be suppressed by the use of DPP4 inhibitors, as well as better understanding the physiology of extending the functional half-life of intact GLP-1.
For GIP, DPP4 cleavage introduces a GIP receptor antagonistic feature that is strengthened by C-terminal cleavage by other proteolytic enzymes. While for the two neuropeptides PYY and NPY, DPP4 cleavage means the production of a more selective Y2 receptor agonist, instead of the non-selective intact form.
For CXCL12, it was found that DPP4 cleavage completely abolishes its ability to initiate CXCR4 GPCR activation, while it retains some of its ability to activate ACKR3, making it more selective to β-arrestin signal transduction pathways instead of GPCR pathways. Further, the ability of DPP4-cleaved CXCL12 to activate ACKR3 inhibiting CXCR4 signaling might explain DPP4-cleaved CXCL12’s apparent CXCR4 antagonistic action in light of its apparent inability to bind effectively to CXCR4 (Ziarek et al., 2017). Although this last point is needs further clarification. Importantly, there are hints that DPP4-cleaved CXCL12 may also act through alternative receptors and signaling pathways. This underscores the need to explore DPP4 cryptic ligands bioactivity not only for CXCL12, but more broadly among DPP4 substrates. In addition to the clinical significance these findings carry, they can also help us predict or understand how DPP4 and DPP4 inhibitors may affect cellular functions and biological processes of other important chemokines and polypeptides.
Further, this review highlights the crucial need to standardize the collection and processing of samples to preserve the true in vivo molecular profiles. This is both in terms of the total quantities of specific molecules as well as the ratio or individual profiles of different proteolytic metabolites. Additionally, there is a critical need to develop new tools to measure these ligand isoforms for accurate quantitation and interpretation of experimental results. Importantly, we are also proposing that plasma, serum, urine, and other biological samples collected in clinical trials and for biorepositories need to have the samples protected at the point of collection from DPP4 and other proteolytic or enzymatic degradation to allow for not only accurate prospective analysis, but for retrospective use of these tissue banks and biorepositories. This has real world implications in understanding normal and pathological systems and quality of life and even life-or-death repercussions in the development of therapeutics for major diseases such as diabetes, osteoporosis, inflammation, stroke, myocardial infarction, wound repair and cancer.
Acknowledgements
This publication is based upon work supported in part by the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Clinical Science Research and Development Program (VA Merit Award 1I01CX000930–01, WDH) and the National Institutes of Health (NIA-AG036675 SF, MWH, CMI, MEML, and WDH), United States. The contents of this publication do not represent the views of the Department of Veterans Affairs, or the United States Government.
Abbreviations:
- ACKR3
Atypical chemokine receptor 3
- ADA
Adenosine deaminase
- ASCs
Adipose stem cells
- BMSCs
Bone marrow mesenchymal stem cells
- CD26
Cluster of differentiation 26
- CXCR4
C-X-C chemokine receptor type 4
- DPP4
Dipeptidyl peptidase 4
- ECM
Extracellular matrix
- F344
Fischer 344 rats
- GAGs
Glycosaminoglycans
- G-CSF
Granulocyte-colony stimulating factor
- GIP
Glucose-dependent insulinotropic polypeptide
- GIP-R
Glucose-dependent insulinotropic polypeptide receptor
- GIT
Gastrointestinal tract
- GLP-1
Glucagon-like peptide 1
- GLP-1R
Glucagon-like peptide 1 receptor
- GPCR
G-protein coupled receptor
- HIF-1
Hypoxia-inducible factor
- HNF
Hepatocyte nuclear factor
- HSPCs
Hematopoietic stem and progenitor cells
- HUVECs
Human umbilical vein endothelial cells
- MS
Mass spectrometry
- NPY
Neuropeptide Y
- NSC
Neural stem cells
- PYY
Peptide tyrosine-tyrosine
- RIC
Remote ischemic conditioning
- sCD26
Soluble cluster of differentiation 26
- SDF-1
Stromal Cell-Derived Factor-1
- SPA
Scintillation proximity assays
Footnotes
Conflict of interest statement
The above mentioned funding did not lead to any conflict of interests regarding the publication of this manuscript. The authors also declare that there is no other conflict of interest regarding the publication of this manuscript.
References
- Abbott CA, Baker E, Sutherland GR, & McCaughan GW (1994). Genomic organization, exact localization, and tissue expression of the human CD26 (dipeptidyl peptidase IV) gene. Immunogenetics 40, 331–338. [DOI] [PubMed] [Google Scholar]
- Adelita T, Stilhano RS, Han SW, Justo GZ, & Porcionatto M (2017). Proteolytic processed form of CXCL12 abolishes migration and induces apoptosis in neural stem cells in vitro. Stem Cell Research 22, 61–69. [DOI] [PubMed] [Google Scholar]
- Adrian TE, Ferri GL, Bacarese-Hamilton AJ, Fuessl HS, Polak JM, & Bloom SR (1985). Human distribution and release of a putative new gut hormone, peptide YY. Gastroenterology 89, 1070–1077. [DOI] [PubMed] [Google Scholar]
- Ambrosi TH, Scialdone A, Graja A, Gohlke S, Jank AM, Bocian C, et al. (2017). Adipocyte accumulation in the bone marrow during obesity and aging impairs stem cell-based hematopoietic and bone regeneration. Cell Stem Cell 20 (771–784.e776). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderluh M, Kocic G, Tomovic K, Kocic R, Deljanin-Ilic M, & Smelcerovic A (2016). Cross-talk between the dipeptidyl peptidase-4 and stromal cell-derived factor-1 in stem cell homing and myocardial repair: Potential impact of dipeptidyl peptidase-4 inhibitors. Pharmacology & Therapeutics 167, 100–107. [DOI] [PubMed] [Google Scholar]
- Antonsson B, De Lys P, Dechavanne V, Chevalet L, & Boschert U (2010). In vivo processing of CXCL12 alpha/SDF-1 alpha after intravenous and subcutaneous administration to mice. Proteomics 10, 4342–4351. [DOI] [PubMed] [Google Scholar]
- Aroor A, Zuberek M, Duta C, Meuth A, Sowers JR, Whaley-Connell A, et al. (2016). Angiotensin II stimulation of DPP4 activity regulates megalin in the proximal tubules. International Journal of Molecular Sciences 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baerts L, Waumans Y, Brandt I, Jungraithmayr W, Van der Veken P, Vanderheyden M, et al. (2015). Circulating stromal cell-derived factor 1alpha levels in heart failure: A matter of proper sampling. PLoS One 10 e0141408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban K, Kim K-H, Cho C-K, Sauvé M, Diamandis EP, Backx PH, et al. (2010). Glucagon-like peptide (GLP)-1(9–36)amide-mediated cytoprotection is blocked by exendin(9–39) yet does not require the known GLP-1 receptor. Endocrinology 151, 1520–1531. [DOI] [PubMed] [Google Scholar]
- Ban K, Noyan-Ashraf MH, Hoefer J, Bolz SS, Drucker DJ, & Husain M (2008). Cardioprotective and vasodilatory actions of glucagon-like peptide 1 receptor are mediated through both glucagon-like peptide 1 receptor-dependent and -independent pathways. Circulation 117, 2340–2350. [DOI] [PubMed] [Google Scholar]
- Batterham RL, Cohen MA, Ellis SM, Le Roux CW, Withers DJ, Frost GS, et al. (2003). Inhibition of food intake in obese subjects by peptide YY3–36. New England Journal of Medicine 349, 941–948. [DOI] [PubMed] [Google Scholar]
- Bauvois B, Djavaheri-Mergny M, Rouillard D, Dumont J, & Wietzerbin J (2000). Regulation of CD26/DPPIV gene expression by interferons and retinoic acid in tumor B cells. Oncogene 19, 265. [DOI] [PubMed] [Google Scholar]
- Benhadjeba S, Edjekouane L, Sauve K, Carmona E, & Tremblay A (2018). Feedback control of the CXCR7/CXCL11 chemokine axis by estrogen receptor alpha in ovarian cancer. Molecular Oncology 12, 1689–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleul CC (1996). A highly efficacious lymphocyte chemoattractant, stromal cell-derived factor 1 (SDF-1). Journal of Experimental Medicine 184, 1101–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomqvist AG, & Herzog H (1997). Y-receptor subtypes—how many more? Trends in Neurosciences 20, 294–298. [DOI] [PubMed] [Google Scholar]
- Brandt I, Lambeir AM, Maes MB, Scharpe S, & De Meester I (2006). Peptide substrates of dipeptidyl peptidases. Advances in Experimental Medicine and Biology 575, 3–18. [DOI] [PubMed] [Google Scholar]
- Bromage DI, Davidson SM, & Yellon DM (2014). Stromal derived factor 1alpha: a chemokine that delivers a two-pronged defence of the myocardium. Pharmacology & Therapeutics 143, 305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromage DI, Taferner S, Pillai M, Yellon DM, & Davidson SM (2017). A novel recombinant antibody specific to full-length stromal derived factor-1 for potential application in biomarker studies. PLoS One 12 e0174447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broxmeyer HE (2013). Counteracting the enzymatic activity of dipeptidylpeptidase 4 for potential therapeutic advantage, with an emphasis on cord blood transplantation. The Korean Journal of Internal Medicine 28, 639–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broxmeyer HE, Capitano M, Campbell TB, Hangoc G, & Cooper S (2016). Modulation of hematopoietic chemokine effects in vitro and in vivo by DPP-4/CD26. Stem Cells and Development 25, 575–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broxmeyer HE, Hoggatt J, O’Leary HA, Mantel C, Chitteti BR, Cooper S, et al. (2012). Dipeptidylpeptidase 4 negatively regulates colony-stimulating factor activity and stress hematopoiesis. Nature Medicine 18, 1786–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broxmeyer HE, Orschell CM, Clapp DW, Hangoc G, Cooper S, Plett PA, et al. (2005). Rapid mobilization of murine and human hematopoietic stem and progenitor cells with AMD3100, a CXCR4 antagonist. The Journal of Experimental Medicine 201, 1307–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busek P, Malik R, & Sedo A (2004). Dipeptidyl peptidase IV activity and/or structure homologues (DASH) and their substrates in cancer. The International Journal of Biochemistry & Cell Biology 36, 408–421. [DOI] [PubMed] [Google Scholar]
- Busso N, Wagtmann N, Herling C, Chobaz-Peclat V, Bischof-Delaloye A, So A, et al. (2005). Circulating CD26 is negatively associated with inflammation in human and experimental arthritis. The American Journal of Pathology 166, 433–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caberlotto L, Jimenez P, Overstreet DH, Hurd YL, Mathé AA, & Fuxe K (1999). Alterations in neuropeptide Y levels and Y1 binding sites in the Flinders Sensitive Line rats, a genetic animal model of depression. Neuroscience Letters 265, 191–194. [DOI] [PubMed] [Google Scholar]
- Cantini G, Di Franco A, Mannucci E, & Luconi M (2017). Is cleaved glucagon-like peptide 1 really inactive? Effects of GLP-1(9–36) on human adipose stem cells. Molecular and Cellular Endocrinology 439, 10–15. [DOI] [PubMed] [Google Scholar]
- Cantini G, Mannucci E, & Luconi M (2016). Perspectives in GLP-1 Research: New Targets, New Receptors. Trends in Endocrinology and Metabolism 27, 427–438. [DOI] [PubMed] [Google Scholar]
- Carbone LD, Buzkova P, Fink HA, Robbins JA, Bethel M, Hamrick MW, et al. (2017b). Association of plasma SDF-1 with bone mineral density, body composition, and hip fractures in older adults: The cardiovascular health study. Calcified Tissue International 100, 599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbone LD, Buzkova P, Fink HA, Robbins JA, Bethel M, Isales CM, et al. (2017a). Association of DPP-4 activity with BMD, body composition, and incident hip fracture: the Cardiovascular Health Study. Osteoporosis International 28, 1631–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr RD, Larsen MO, Jelic K, Lindgren O, Vikman J, Holst JJ, et al. (2010). Secretion and dipeptidyl peptidase-4-mediated metabolism of incretin hormones after a mixed meal or glucose ingestion in obese compared to lean, nondiabetic men. The Journal of Clinical Endocrinology & Metabolism 95, 872–878. [DOI] [PubMed] [Google Scholar]
- Castan I, Valet P, Larrouy D, Voisin T, Remaury A, Daviaud D, et al. (1993). Distribution of PYY receptors in human fat cells: an antilipolytic system alongside the alpha 2-adrenergic system. American Journal of Physiology. Endocrinology and Metabolism 265, E74–E80. [DOI] [PubMed] [Google Scholar]
- Chappell DL, Lee AY, Castro-Perez J, Zhou H, Roddy TP, Lassman ME, et al. (2014). An ultrasensitive method for the quantitation of active and inactive GLP-1 in human plasma via immunoaffinity LC–MS/MS. Bioanalysis 6, 33–42. [DOI] [PubMed] [Google Scholar]
- Chelikani PK, Haver AC, & Reidelberger RD (2005). Intravenous infusion of peptide YY(3–36) potently inhibits food intake in rats. Endocrinology 146, 879–888. [DOI] [PubMed] [Google Scholar]
- Chen M, Angeli FS, Shen Y-T, & Shannon RP (2014). GLP-1 (7–36) amide restores myocardial insulin sensitivity and prevents the progression of heart failure in senescent beagles. Cardiovascular Diabetology 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X, Wang H, Zhang X, Zhao S, Zhou Z, Mu X, et al. (2017). The role of SDF-1/CXCR4/CXCR7 in neuronal regeneration after cerebral ischemia. Frontiers in Neuroscience 11, 590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y-H, Eby JM, LaPorte HM, Volkman BF, & Majetschak M (2017). Effects of cognate, non-cognate and synthetic CXCR4 and ACKR3 ligands on human lung endothelial cell barrier function. PLoS One 12 e0187949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopherson KW II, Hangoc G, & Broxmeyer HE (2002). Cell surface peptidase CD26/dipeptidylpeptidase IV regulates CXCL12/stromal cell-derived factor-1 alpha-mediated chemotaxis of human cord blood CD34+ progenitor cells. Journal of Immunology 169, 7000–7008. [DOI] [PubMed] [Google Scholar]
- Christopherson KW II, Hangoc G, Mantel CR, & Broxmeyer HE (2004). Modulation of hematopoietic stem cell homing and engraftment by CD26. Science 305, 1000–1003. [DOI] [PubMed] [Google Scholar]
- Christopherson KW, Cooper S, & Broxmeyer HE (2003). Cell surface peptidase CD26/DPPIV mediates G-CSF mobilization of mouse progenitor cells. Blood 101, 4680. [DOI] [PubMed] [Google Scholar]
- Christopherson KW, Cooper S, Hangoc G, & Broxmeyer HE (2003). CD26 is essential for normal G-CSF-induced progenitor cell mobilization as determined by CD26−/− mice. Experimental Hematology 31, 1126–1134. [DOI] [PubMed] [Google Scholar]
- Chung K-M, Cheng J-H, Suen C-S, Huang C-H, Tsai C-H, Huang L-H, et al. (2010). The dimeric transmembrane domain of prolyl dipeptidase DPP-IV contributes to its quaternary structure and enzymatic activities. Protein Science: A Publication of the Protein Society 19, 1627–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crump MP, Gong JH, Loetscher P, Rajarathnam K, Amara A, Arenzana-Seisdedos F, et al. (1997). Solution structure and basis for functional activity of stromal cell derived factor-1; dissociation of CXCR4 activation from binding and inhibition of HIV-1. The EMBO Journal 16, 6996–7007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang DT, Chun SY, Burkitt K, Abe M, Chen S, Havre P, et al. (2008). Hypoxia-inducible factor-1 target genes as indicators of tumor vessel response to vascular endothelial growth factor inhibition. Cancer Research 68, 1872–1880. [DOI] [PubMed] [Google Scholar]
- De La Luz Sierra M, Yang F, Narazaki M, Salvucci O, Davis D, Yarchoan R, et al. (2004). Differential processing of stromal-derived factor-1alpha and stromal-derived factor-1beta explains functional diversity. Blood 103, 2452–2459. [DOI] [PubMed] [Google Scholar]
- De Meester I, Korom S, Van Damme J, & Scharpe S (1999). CD26, let it cut or cut it down. Immunology Today 20, 367–375. [DOI] [PubMed] [Google Scholar]
- Deacon CF (2004). Circulation and degradation of GIP and GLP-1. Hormone and Metabolic Research 36, 761–765. [DOI] [PubMed] [Google Scholar]
- Deacon CF, Plamboeck A, Møller S, & Holst JJ (2002). GLP-1-(9–36) amide reduces blood glucose in anesthetized pigs by a mechanism that does not involve insulin secretion. American Journal of Physiology. Endocrinology and Metabolism 282, E873–E879. [DOI] [PubMed] [Google Scholar]
- Decaillot FM, Kazmi MA, Lin Y, Ray-Saha S, Sakmar TP, & Sachdev P (2011). CXCR7/CXCR4 heterodimer constitutively recruits beta-arrestin to enhance cell migration. The Journal of Biological Chemistry 286, 32188–32197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C-Z, Tian S, Choi W-T, Kumar S, Liu D, Xu Y, et al. (2012). Critical role in CXCR4 signaling and internalization of the polypeptide main chain in the amino terminus of SDF-1α probed by novel N-methylated synthetically and modularly modified chemokine analogues. Biochemistry 51, 5951–5957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drucker DJ (2003). Enhancing incretin action for the treatment of type 2 diabetes. Diabetes Care 26, 2929–2940. [DOI] [PubMed] [Google Scholar]
- Drucker DJ (2007). Dipeptidyl peptidase-4 inhibition and the treatment of type 2 diabetes. Preclinical Biology and Mechanisms of Action 30, 1335–1343. [DOI] [PubMed] [Google Scholar]
- Drury LJ, Ziarek JJ, Gravel S, Veldkamp CT, Takekoshi T, Hwang ST, et al. (2011). Monomeric and dimeric CXCL12 inhibit metastasis through distinct CXCR4 interactions and signaling pathways. Proceedings of the National Academy of Sciences 108, 17655–17660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumont Y, Fournier A, St-Pierre S, Schwartz TW, & Quirion R (1990). Differential distribution of neuropeptide Y1 and Y2 receptors in the rat brain. European Journal of Pharmacology 191, 501–503. [DOI] [PubMed] [Google Scholar]
- El Bekay R, Coín-Aragüez L, Fernández-García D, Oliva-Olivera W, Bernal-López R, Clemente-Postigo M, et al. (2016). Effects of glucagon-like peptide-1 on the differentiation and metabolism of human adipocytes. British Journal of Pharmacology 173, 1820–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elahi D, McAloon-Dyke M, Fukagawa NK, Meneilly GS, Sclater AL, Minaker KL, Habener JF, & Andersen DK (1994). The insulinotropic actions of glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (7–37) in normal and diabetic subjects. Regulatory Peptides 51, 63–74. [DOI] [PubMed] [Google Scholar]
- Elmansi AM, Hussein KA, Volkman BF, Kondrikova G, Bollag W, Fulzele S, et al. (2018). DPP-4-cleaved SDF-1β diminishes migration and osteogenic differentiation capacities of bone marrow mesenchymal stem cells. Journal of Bone and Mineral Research 32(Suppl. 1) Montréal, QC, Canada: ASBMR 2018 Annual Meeting. [Google Scholar]
- Erickson RH, Suzuki Y, Sedlmayer A, & Kim YS (1992). Biosynthesis and degradation of altered immature forms of intestinal dipeptidyl peptidase IV in a rat strain lacking the enzyme. The Journal of Biological Chemistry 267, 21623–21629. [PubMed] [Google Scholar]
- Ferre S, Casado V, Devi LA, Filizola M, Jockers R, Lohse MJ, et al. (2014). G protein-coupled receptor oligomerization revisited: Functional and pharmacological perspectives. Pharmacological Reviews 66, 413–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrier L (2000). Pathways and receptors involved in peptide YY induced contraction of rat proximal colonic muscle in vitro. Gut 46, 370–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flood JF, & Morley JE (1989). Dissociation of the effects of neuropeptide Y on feeding and memory: Evidence for pre- and postsynaptic mediation. Peptides 10, 963–966. [DOI] [PubMed] [Google Scholar]
- Flynn MC, Plata-Salamán CR, & Ffrench-Mullen JMH (1998). Neuropeptide Y-Related Compounds and Feeding. Physiology & Behavior 65, 901–905. [DOI] [PubMed] [Google Scholar]
- Fortunato O, Spinetti G, Specchia C, Cangiano E, Valgimigli M, & Madeddu P (2013). Migratory activity of circulating progenitor cells and serum SDF-1α predict adverse events in patients with myocardial infarction. Cardiovascular Research 100, 192–200. [DOI] [PubMed] [Google Scholar]
- Frerker N, Wagner L, Wolf R, Heiser U, Hoffmann T, Rahfeld JU, et al. (2007). Neuropeptide Y (NPY) cleaving enzymes: structural and functional homologues of dipeptidyl peptidase 4. Peptides 28, 257–268. [DOI] [PubMed] [Google Scholar]
- Fujita Y, Asadi A, Yang GK, Kwok YN, & Kieffer TJ (2010). Differential processing of pro-glucose-dependent insulinotropic polypeptide in gut. American Journal of Physiology. Gastrointestinal and Liver Physiology 298, G608–G614. [DOI] [PubMed] [Google Scholar]
- Fujita Y, Wideman RD, Asadi A, Yang GK, Baker R, Webber T, et al. (2010). Glucose-dependent insulinotropic polypeptide Is expressed in pancreatic islet α-cells and promotes insulin secretion. Gastroenterology 138 1966–1975.e1961. [DOI] [PubMed] [Google Scholar]
- Gautier-Stein A, & Mithieux G (2013). A role for PYY3–36 in GLP1-induced insulin secretion. Molecular Metabolism 2, 123–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehlert DR, Gackenheimer SL, & Schober DA (1992). [Leu31-Pro34] neuropeptide Y identifies a subtype of 125I-labeled peptide YY binding sites in the rat brain. Neurochemistry International 21, 45–67. [DOI] [PubMed] [Google Scholar]
- Gerald C, Walker MW, Criscione L, Gustafson EL, Batzl-Hartmann C, Smith KE, et al. (1996). A receptor subtype involved in neuropeptide-Y-induced food intake. Nature 382, 168–171. [DOI] [PubMed] [Google Scholar]
- Ghadge SK, Muhlstedt S, Ozcelik C, & Bader M (2011). SDF-1alpha as a therapeutic stem cell homing factor in myocardial infarction. Pharmacology & Therapeutics 129, 97–108. [DOI] [PubMed] [Google Scholar]
- Ghersi G, Chen W-T, Lee EW, & Zukowska Z (2001). Critical role of dipeptidyl peptidase IV in neuropeptide Y-mediated endothelial cell migration in response to wounding. Peptides 22, 453–458. [DOI] [PubMed] [Google Scholar]
- Goodwill AG, Tune JD, Noblet JN, Conteh AM, Sassoon D, Casalini ED, et al. (2014). Glucagon-like peptide-1 (7–36) but not (9–36) augments cardiac output during myocardial ischemia via a Frank–Starling mechanism. Basic Research in Cardiology 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorrell MD (2005). Dipeptidyl peptidase IV and related enzymes in cell biology and liver disorders. Clinical Science (London, England) 108, 277–292. [DOI] [PubMed] [Google Scholar]
- Grandt D, Schimiczek M, Beglinger C, Layer P, Goebell H, Eysselein VE, et al. (1994). Two molecular forms of Peptide YY (PYY) are abundant in human blood: characterization of a radioimmunoassay recognizing PYY 1–36 and PYY 3–36. Regulatory Peptides 51, 151–159. [DOI] [PubMed] [Google Scholar]
- Grandt D, Schimiczek M, Rascher W, Feth F, Shively J, Lee TD, et al. (1996). Neuropeptide Y 3–36 is an endogenous ligand selective for Y2 receptors. Regulatory Peptides 67, 33–37. [DOI] [PubMed] [Google Scholar]
- Grandt D, Schimiczek M, Struk K, Shively J, Eysselein VE, Goebell H, et al. (1994). Characterization of two forms of peptide YY, PYY(1–36) and PYY(3–36), in the rabbit. Peptides 15, 815–820. [DOI] [PubMed] [Google Scholar]
- Green BD, Hand KV, Dougan JE, McDonnell BM, Cassidy RS, & Grieve DJ (2008). GLP-1 and related peptides cause concentration-dependent relaxation of rat aorta through a pathway involving KATP and cAMP. Archives of Biochemistry and Biophysics 478, 136–142. [DOI] [PubMed] [Google Scholar]
- Gribkoff VK, Pieschl RL, Wisialowski TA, van den Pol AN, & Yocca FD (1998). Phase shifting of circadian rhythms and depression of neuronal activity in the rat suprachiasmatic nucleus by neuropeptide Y: Mediation by different receptor subtypes. The Journal of Neuroscience 18, 3014–3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groneberg DA, Folkerts G, Peiser C, Chung KF, & Fischer A (2004). Neuropeptide Y (NPY). Pulmonary Pharmacology & Therapeutics 17, 173–180. [DOI] [PubMed] [Google Scholar]
- Grundemar L, Jonas SE, Mörner N, Högestätt ED, Wahlestedt C, & Håkanson R (1992). Characterization of vascular neuropeptide Y receptors. British Journal of Pharmacology 105, 45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu N, Tsuda M, Matsunaga T, Adachi T, Yasuda K, Ishihara A, et al. (2008). Glucose regulation of dipeptidyl peptidase IV gene expression is mediated by hepatocyte nuclear factor-1alpha in epithelial intestinal cells. Clinical and Experimental Pharmacology & Physiology 35, 1433–1439. [DOI] [PubMed] [Google Scholar]
- Guo H, Castro PA, Palmiter RD, & Baraban SC (2002). Y5 receptors mediate neuropeptide Y actions at excitatory synapses in area CA3 of the mouse hippocampus. Journal of Neurophysiology 87, 558–566. [DOI] [PubMed] [Google Scholar]
- Guo R, Chai L, Chen L, Chen W, Ge L, Li X, et al. (2015). Stromal cell-derived factor 1 (SDF-1) accelerated skin wound healing by promoting the migration and proliferation of epidermal stem cellsVol. 51.. [DOI] [PubMed] [Google Scholar]
- Hall MP (2014). Biotransformation and in vivo stability of protein biotherapeutics: impact on candidate selection and pharmacokinetic profiling. Drug Metabolism and Disposition 42, 1873–1880. [DOI] [PubMed] [Google Scholar]
- Hansen LS, Sparre-Ulrich AH, Christensen M, Knop FK, Hartmann B, Holst JJ, et al. (2016). N-terminally and C-terminally truncated forms of glucose-dependent insulinotropic polypeptide are high-affinity competitive antagonists of the human GIP receptor. British Journal of Pharmacology 173, 826–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havre P, Abe M, Urasaki Y, Ohnuma K, Morimoto C, & H Dang N (2008). The role ofCD26/dipeptidyl peptidase IV in cancerVol. 13.. [DOI] [PubMed] [Google Scholar]
- Herberg S, Fulzele S, Yang NL, Shi XM, Hess M, Periyasamy-Thandavan S, et al. (2013). Stromal cell-derived factor-1 beta potentiates bone morphogenetic protein-2-stimulated osteoinduction of genetically engineered bone marrow-derived mesenchymal stem cells in vitro. Tissue Engineering Part A 19, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herberg S, Kondrikova G, Hussein KA, Johnson MH, Elsalanty ME, Shi X, et al. (2015). Mesenchymal stem cell expression of stromal cell-derived factor-1β augments bone formation in a model of local regenerative therapy. Journal of Orthopaedic Research: Official Publication of the Orthopaedic Research Society 33, 174–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herberg S, Shi X, Johnson MH, Hamrick MW, Isales CM, & Hill WD (2013). Stromal cell-derived factor-1β mediates cell survival through enhancing autophagy in bone marrow-derived mesenchymal stem cells. PLoS One 8 e58207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herberg S, Susin C, Pelaez M, Howie RN, Moreno de Freitas R, Lee J, et al. (2014). Low-dose bone morphogenetic protein-2/stromal cell-derived factor-1beta cotherapy induces bone regeneration in critical-size rat calvarial defects. Tissue Engineering. Part A 20, 1444–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera C, Morimoto C, Blanco J, Mallol J, Arenzana F, Lluis C, et al. (2001). Comodulation of CXCR4 and CD26 in Human Lymphocytes. Journal of Biological Chemistry 276, 19532–19539. [DOI] [PubMed] [Google Scholar]
- Herzog H, Darby K, Ball H, Hort Y, Beck-Sickinger A, & Shine J (1997). Overlapping gene structure of the human neuropeptide Y receptor subtypes Y1 and Y5 suggests coordinate transcriptional regulation. Genomics 41, 315–319. [DOI] [PubMed] [Google Scholar]
- Hill WD, Hess DC, Martin-Studdard A, Carothers JJ, Zheng J, Hale D, et al. (2004). SDF-1 (CXCL12) is upregulated in the ischemic penumbra following stroke: association with bone marrow cell homing to injury. Journal of Neuropathology and Experimental Neurology 63, 84–96. [DOI] [PubMed] [Google Scholar]
- Hinke SA, Manhart S, Pamir N, Demuth H-U, Gelling RW, Pederson RA, et al. (2001). Identification of a bioactive domain in the amino-terminus of glucose-dependent insulinotropic polypeptide (GIP). Biochimica et Biophysica Acta (BBA) - Protein Structure and Molecular Enzymology, 1547, 143–155. [DOI] [PubMed] [Google Scholar]
- Infante A, & Rodriguez CI (2018). Osteogenesis and aging: lessons from mesenchymal stem cells. Stem Cell Research & Therapy 9, 244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irhimeh MR, Fitton JH, & Lowenthal RM (2007). Fucoidan ingestion increases the expression of CXCR4 on human CD34+ cells. Experimental Hematology 35, 989–994. [DOI] [PubMed] [Google Scholar]
- Janssens R, Mortier A, Boff D, Ruytinx P, Gouwy M, Vantilt B, et al. (2017). Truncation of CXCL12 by CD26 reduces its CXC chemokine receptor 4- and atypical chemokine receptor 3-dependent activity on endothelial cells and lymphocytes. Biochemical Pharmacology 132, 92–101. [DOI] [PubMed] [Google Scholar]
- Janssens R, Struyf S, & Proost P (2018). The unique structural and functional features ofCXCL12. Cellular & Molecular Immunology 15, 299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Q, Song P, Wang E, Li J, Hu S, & Zhang H (2013). Remote ischemic postconditioning enhances cell retention in the myocardium after intravenous administration of bone marrow mesenchymal stromal cells. Journal of Molecular and Cellular Cardiology 56, 1–7. [DOI] [PubMed] [Google Scholar]
- Kamota T, Li T-S, Morikage N, Murakami M, Ohshima M, Kubo M, et al. (2009). Ischemic Pre-Conditioning Enhances the Mobilization and Recruitment of Bone Marrow Stem Cells to Protect Against Ischemia/Reperfusion Injury in the Late Phase. Journal of the American College of Cardiology 53, 1814–1822. [DOI] [PubMed] [Google Scholar]
- Kerr BD, Flatt AJS, Flatt PR, & Gault VA (2011). Characterization and biological actions of N-terminal truncated forms of glucose-dependent insulinotropic polypeptide. Biochemical and Biophysical Research Communications 404, 870–876. [DOI] [PubMed] [Google Scholar]
- Kieffer TJ, McIntosh CH, & Pederson RA (1995). Degradation of glucose-dependent insulinotropic polypeptide and truncated glucagon-like peptide 1 in vitro and in vivo by dipeptidyl peptidase IV. Endocrinology 136, 3585–3596. [DOI] [PubMed] [Google Scholar]
- Kim NH, Yu T, & Lee DH (2014). The nonglycemic actions of dipeptidyl peptidase-4 inhibitors. BioMed Research International 2014, 368703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemann C, Wagner L, Stephan M, & von Hörsten S (2016). Cut to the chase: a review of CD26/dipeptidyl peptidase-4’s (DPP4) entanglement in the immune system. Clinical and Experimental Immunology 185, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen LB, & Pridal L (1996). Glucagon-like peptide-1-(9–36) amide is a major metabolite of glucagon-like peptide-1-(7–36) amide after in vivo administration to dogs, and it acts as an antagonist on the pancreatic receptor. European Journal of Pharmacology 318, 429–435. [DOI] [PubMed] [Google Scholar]
- Kuc RE, Maguire JJ, Siew K, Patel S, Derksen DR, Margaret Jackson V, et al. (2014). Characterization of [125I]GLP-1(9–36), a novel radiolabeled analog of the major metabolite of glucagon-like peptide 1 to a receptor distinct from GLP1-R and function of the peptide in murine aorta. Life Sciences 102, 134–138. [DOI] [PubMed] [Google Scholar]
- Lambeir A-M, Durinx C, Scharpé S, & De Meester I (2003). Dipeptidyl-Peptidase IV from Bench to Bedside: An Update on Structural Properties, Functions, and Clinical Aspects of the Enzyme DPP IV. Critical Reviews in Clinical Laboratory Sciences 40, 209–294. [DOI] [PubMed] [Google Scholar]
- Lambeir AM, Proost P, Durinx C, Bal G, Senten K, Augustyns K, et al. (2001). Kinetic investigation of chemokine truncation by CD26/dipeptidyl peptidase IV reveals a striking selectivity within the chemokine family. The Journal of Biological Chemistry 276, 29839–29845. [DOI] [PubMed] [Google Scholar]
- Lee HM, Joo BS, Lee CH, Kim HY, Ock JH, & Lee YS (2015). Effect of glucagon-like peptide-1 on the differentiation of adipose-derived stem cells into osteoblasts and adipocytes. Journal of Menopausal Medicine 21, 93–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leite Pereira C, Q. T. G, R. F. J, D’Este M, Eglin D, Alini MP, et al. (2018). SDF-1-mediated migration of MSCs enhances collagen type II expression in intervertebral disc. Tissue Engineering. Part A. 10.1089/ten.TEA.2018.0131. [DOI] [PubMed] [Google Scholar]
- Levoye A, Balabanian K, Baleux F, Bachelerie F, & Lagane B (2009). CXCR7 heterodimerizes with CXCR4 and regulates CXCL12-mediated G protein signaling. Blood 113, 6085–6093. [DOI] [PubMed] [Google Scholar]
- Li G, Basu S, Han M-K, Kim Y-J, & Broxmeyer HE (2007). Influence of ERK activation on decreased chemotaxis of mature human cord blood monocyte-derived dendritic cells to CCL19 and CXCL12. Blood 109, 3173–3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lone AM, Nolte WM, Tinoco AD, & Saghatelian A (2010). Peptidomics of the ProlylPeptidases. The AAPS Journal 12, 483–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabilleau G, Mieczkowska A, & Chappard D (2013). Use of glucagon-like peptide-1 receptor agonists and bone fractures: A meta-analysis of randomized clinical trials (胰高血糖素样肽-1受体激动剂的使用与骨折的关系:一项对随机临床试验的meta分析). Journal of Diabetes 6, 260–266. [DOI] [PubMed] [Google Scholar]
- Maji SK, Perrin MH, Sawaya MR, Jessberger S, Vadodaria K, Rissman RA, et al. (2009). Functional amyloids as natural storage of peptide hormones in pituitary secretory granules. Science 325, 328–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannon PJ, Mervin SJ, & Sheriff-Carter KD (1994). Characterization of a Y1-preferring NPY/PYY receptor in HT-29 cells. American Journal of Physiology. Gastrointestinal and Liver Physiology 267, G901–G907. [DOI] [PubMed] [Google Scholar]
- Mannucci E, Pala L, Ciani S, Bardini G, Pezzatini A, Sposato I, et al. (2005). Hyperglycaemia increases dipeptidyl peptidase IV activity in diabetes mellitus. Diabetologia 48, 1168–1172. [DOI] [PubMed] [Google Scholar]
- Matsumoto S-I, Basil J, Jetton AE, Lehman MN, & Bittman EL (1996). Regulation of the phase and period of circadian rhythms restored by suprachiasmatic transplants. Journal of Biological Rhythms 11, 145–162. [DOI] [PubMed] [Google Scholar]
- McQuibban GA, Butler GS, Gong J-H, Bendall L, Power C, Clark-Lewis I, et al. (2001). Matrix metalloproteinase activity inactivates the CXC chemokine stromal cell-derived Factor-1. Journal of Biological Chemistry 276, 43503–43508. [DOI] [PubMed] [Google Scholar]
- Meier JJ, Gethmann A, Nauck MA, Götze O, Schmitz F, Deacon CF, et al. (2006). The glucagon-like peptide-1 metabolite GLP-1-(9–36) amide reduces postprandial glycemia independently of gastric emptying and insulin secretion in humans. American Journal of Physiology. Endocrinology and Metabolism 290, E1118–E1123. [DOI] [PubMed] [Google Scholar]
- Mentlein R (1999). Dipeptidyl-peptidase IV (CD26)–role in the inactivation of regulatory peptides. Regulatory Peptides 85, 9–24. [DOI] [PubMed] [Google Scholar]
- Metzemaekers M, Van Damme J, Mortier A, & Proost P (2016). Regulation of chemokine activity – A focus on the role of dipeptidyl peptidase IV/CD26. Frontiers in Immunology 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JT, Bartley JH, Wimborne HJ, Walker AL, Hess DC, Hill WD, et al. (2005). The neuroblast and angioblast chemotaxic factor SDF-1 (CXCL12) expression is briefly up regulated by reactive astrocytes in brain following neonatal hypoxic-ischemic injury. BMC Neuroscience 6, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan G, Canals M, Pediani JD, Ellis J, & Lopez-Gimenez JF (2007). The role of GPCR dimerisation/oligomerisation in receptor signalling Ernst Schering Foundation Symposium Proceedings (pp. 145–162). Springer; Berlin Heidelberg. [DOI] [PubMed] [Google Scholar]
- Moran TH, Smedh U, Kinzig KP, Scott KA, Knipp S, & Ladenheim EE (2005). Peptide YY(3–36) inhibits gastric emptying and produces acute reductions in food intake in rhesus monkeys. American Journal of Physiology. Regulatory, Integrative and Comparative Physiology 288, R384–R388. [DOI] [PubMed] [Google Scholar]
- Moriya R, Shirakura T, Hirose H, Kanno T, Suzuki J, & Kanatani A (2010). NPY Y2 receptor agonist PYY(3–36) inhibits diarrhea by reducing intestinal fluid secretion and slowing colonic transit in mice. Peptides 31, 671–675. [DOI] [PubMed] [Google Scholar]
- Mortensen LJ, & Hill WD (2015). Skeletal stem cells for bone development, homeostasis and repair: one or many? Bonekey Reports 4, 769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortier A, Gouwy M, Van Damme J, & Proost P (2011). Effect of posttranslational processing on the in vitro and in vivo activity of chemokines. Experimental Cell Research 317, 642–654. [DOI] [PubMed] [Google Scholar]
- Mortier A, Gouwy M, Van Damme J, Proost P, & Struyf S (2016). CD26/dipeptidylpeptidase IV-chemokine interactions: double-edged regulation of inflammation and tumor biology. Journal of Leukocyte Biology 99, 955–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortier A, Van Damme J, & Proost P (2008). Regulation of chemokine activity by post-translational modification. Pharmacology & Therapeutics 120, 197–217. [DOI] [PubMed] [Google Scholar]
- Mulvihill EE, & Drucker DJ (2014). Pharmacology, physiology, and mechanisms of action of dipeptidyl peptidase-4 inhibitors. Endocrine Reviews 35, 992–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagasawa T, Hirota S, Tachibana K, Takakura N, Nishikawa S, Kitamura Y, et al. (1996). Defects of B-cell lymphopoiesis and bone-marrow myelopoiesis in mice lacking the CXC chemokine PBSF/SDF-1. Nature 382, 635–638. [DOI] [PubMed] [Google Scholar]
- Nauck MA (2013). A critical analysis of the clinical use of incretin-based therapies: The benefits by far outweigh the potential risks. Diabetes Care 36, 2126–2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Leary H, Ou X, & Broxmeyer HE (2013). The role of dipeptidyl peptidase 4 in hematopoiesis and transplantation. Current Opinion in Hematology 20, 314–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orskov C, Bersani M, Johnsen AH, Hojrup P, & Holst JJ (1989). Complete sequences of glucagon-like peptide-1 from human and pig small intestine. The Journal of Biological Chemistry 264, 12826–12829. [PubMed] [Google Scholar]
- Ou X, O’Leary HA, & Broxmeyer HE (2013). Implications of DPP4 modification of proteins that regulate stem/progenitor and more mature cell types. Blood 122, 161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson FC, Elgin ES, Nelson TJ, Zhang F, Hoeger TJ, Linhardt RJ, et al. (2004). Identification and characterization of a glycosaminoglycan recognition element of the C chemokine lymphotactin. The Journal of Biological Chemistry 279, 12598–12604. [DOI] [PubMed] [Google Scholar]
- Pittner RA, Moore CX, Bhavsar SP, Gedulin BR, Smith PA, Jodka CM, et al. (2004). Effects of PYY[3–36] in rodent models of diabetes and obesity. International Journal of Obesity 28, 963–971. [DOI] [PubMed] [Google Scholar]
- Playford RJ, Benito-Orfila MA, Nihoyannopoulos P, Nandha KA, Cockcroft J, Todd S, et al. (1992). Effects of peptide YY on the human cardiovascular system: reversal of responses to vasoactive intestinal peptide. American Journal of Physiology. Endocrinology and Metabolism 263, E740–E747. [DOI] [PubMed] [Google Scholar]
- Playford RJ, Mehta S, Upton P, Rentch R, Moss S, Calam J, et al. (1995). Effect of peptide YY on human renal function. American Journal of Physiology. Renal Physiology 268, F754–F759. [DOI] [PubMed] [Google Scholar]
- Proost P, Struyf S, Schols D, Durinx C, Wuyts A, Lenaerts J -P., et al. (1998). Processing by CD26/dipeptidyl-peptidase IV reduces the chemotactic and anti-HIV-1 activity of stromal-cell-derived factor-1α. FEBS Letters 432, 73–76. [DOI] [PubMed] [Google Scholar]
- Quinn KE, Mackie DI, & Caron KM (2018). Emerging roles of atypical chemokine receptor 3 (ACKR3) in normal development and physiology. Cytokine 109, 17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen HB, Branner S, Wiberg FC, & Wagtmann N (2003). Crystal structure of human dipeptidyl peptidase IV/CD26 in complex with a substrate analog. Nature Structural Biology 10, 19–25. [DOI] [PubMed] [Google Scholar]
- Reid JC, Tanasijevic B, Golubeva D, Boyd AL, Porras DP, Collins TJ, et al. (2018). CXCL12/CXCR4 signaling enhances human PSC-derived hematopoietic progenitor function and overcomes early in vivo transplantation failure. Stem Cell Reports 10, 1625–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rettenbacher M, & Reubi J (2001). Localization and characterization of neuropeptide receptors in human colon. Naunyn-Schmiedeberg’s Archives of Pharmacology 364, 291–304. [DOI] [PubMed] [Google Scholar]
- Ricard-Blum S, & Salza R (2014). Matricryptins and matrikines: biologically active fragments of the extracellular matrix. Experimental Dermatology 23, 457–463. [DOI] [PubMed] [Google Scholar]
- Richter R, Jochheim-Richter A, Ciuculescu F, Kollar K, Seifried E, Forssmann U, et al. (2014). Identification and characterization of circulating variants of CXCL12 from human plasma: Effects on chemotaxis and mobilization of hematopoietic stem and progenitor cells. Stem Cells and Development 23, 1959–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson E, Tate M, Lockhart S, McPeake C, O’Neill KM, Edgar KS, et al. (2016). Metabolically-inactive glucagon-like peptide-1(9–36)amide confers selective protective actions against post-myocardial infarction remodelling. Cardiovascular Diabetology 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy I, McAllister DM, Gorse E, Dixon K, Piper CT, Zimmerman NP, et al. (2015). Pancreatic cancer cell migration and metastasis is regulated by chemokine-biased agonism and bioenergetic signaling. Cancer Research 75, 3529–3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozenfeld R, & Devi LA (2010). Receptor heteromerization and drug discovery. Trends in Pharmacological Sciences 31, 124–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar N, Munoz D, Kallifatidis G, Singh RK, Jorda M, & Lokeshwar BL (2014). The chemokine receptor CXCR7 interacts with EGFR to promote breast cancer cell proliferation. Molecular Cancer 13, 198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Martin L, Sanchez-Mateos P, & Cabanas C (2012). CXCR7 impact on CXCL12Biology and disease. Trends in Molecular Medicine 19, 12–22. [DOI] [PubMed] [Google Scholar]
- Sauve M, Ban K, Momen MA, Zhou YQ, Henkelman RM, Husain M, et al. (2010). Genetic deletion or pharmacological inhibition of dipeptidyl peptidase-4 improves cardiovascular outcomes after myocardial infarction in mice. Diabetes 59, 1063–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J, Fukuhara A, Onodera T, Yokoyama C, Otsuki M, & Shimomura I (2017). Regulation of dipeptidyl peptidase-4, its substrate chemokines, and their receptors in adipose tissue of ob/ob mice. Hormone and Metabolic Research 49, 380–387. [DOI] [PubMed] [Google Scholar]
- Shin JH, Park YM, Kim DH, Moon GJ, Bang OY, Ohn T, et al. (2014). Ischemic brain extract increases SDF-1 expression in astrocytes through the CXCR2/miR-223/miR-27b pathway. Biochimica et Biophysica Acta 1839, 826–836. [DOI] [PubMed] [Google Scholar]
- Shioda T, Kato H, Ohnishi Y, Tashiro K, Ikegawa M, Nakayama EE, et al. (1998). Anti-HIV-1 and chemotactic activities of human stromal cell-derived factor 1alpha (SDF-1alpha) and SDF-1beta are abolished by CD26/dipeptidyl peptidase IV-mediated cleavage. Proceedings of the National Academy of Sciences of the United States of America 95, 6331–6336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirozu M, Nakano T, Inazawa J, Tashiro K, Tada H, Shinohara T, et al. (1995). Structure and chromosomal localization of the human stromal cell-derived factor 1 (SDF1) gene. Genomics 28, 495–500. [DOI] [PubMed] [Google Scholar]
- Singh P, Hoggatt J, Kamocka MM, Mohammad KS, Saunders MR, Li H, et al. (2017). Neuropeptide Y regulates a vascular gateway for hematopoietic stem and progenitor cells. The Journal of Clinical Investigation 127, 4527–4540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokos GG, Nikolaidis LA, Mankad S, Elahi D, & Shannon RP (2006). Glucagon-like peptide-1 infusion improves left ventricular ejection fraction and functional status in patients with chronic heart failure. Journal of Cardiac Failure 12, 694–699. [DOI] [PubMed] [Google Scholar]
- Sonne DP, Engstrøm T, & Treiman M (2008). Protective effects of GLP-1 analogues exendin-4 and GLP-1(9–36) amide against ischemia–reperfusion injury in rat heart. Regulatory Peptides 146, 243–249. [DOI] [PubMed] [Google Scholar]
- Sparre-Ulrich AH, Gabe MN, Gasbjerg LS, Christiansen CB, Svendsen B, Hartmann B, et al. (2017). GIP(3–30)NH 2 is a potent competitive antagonist of the GIP receptor and effectively inhibits GIP-mediated insulin, glucagon, and somatostatin release. Biochemical Pharmacology 131, 78–88. [DOI] [PubMed] [Google Scholar]
- Stadlbauer U, Weber E, Langhans W, & Meyer U (2013). The Y2 receptor agonist PYY3–36 increases the behavioural response to novelty and acute dopaminergic drug challenge in mice. The International Journal of Neuropsychopharmacology 17, 407–419. [DOI] [PubMed] [Google Scholar]
- Stjernquist M, & Owman CH (1990). Further evidence for a prejunctional action of neuropeptide Y on cholinergic motor neurons in the rat uterine cervix. Acta Physiologica Scandinavica 138, 95–96. [DOI] [PubMed] [Google Scholar]
- Su J, Chen J, Lippold K, Monavarfeshani A, Carrillo GL, Jenkins R, et al. (2016). Collagen-derived matricryptins promote inhibitory nerve terminal formation in the developing neocortex. The Journal of Cell Biology 212, 721–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney EA, & Papayannopoulou T (2001). Increase in circulating SDF-1 after treatment with sulfated glycans. The role of SDF-1 in mobilization. Annals of the New York Academy of Sciences 938, 48–52 discussion 52–43. [DOI] [PubMed] [Google Scholar]
- Szpakowska M, Nevins AM, Meyrath M, Rhainds D, D’Huys T, Guite-Vinet F, et al. (2018). Different contributions of chemokine N-terminal features attest to a different ligand binding mode and a bias towards activation of ACKR3/CXCR7 compared with CXCR4 and CXCR3. British Journal of Pharmacology 175, 1419–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarantola E, Bertone V, Milanesi G, Capelli E, Ferrigno A, Neri D, et al. (2012). Dipeptidylpeptidase–IV, a key enzyme for the degradation of incretins and neuropeptides: activity and expression in the liver of lean and obese rats. European Journal of Histochemistry 56, e41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatemoto K (1982). Neuropeptide Y: complete amino acid sequence of the brain peptide.Proceedings of the National Academy of Sciences 79, 5485–5489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson NL, Hixson DC, Callanan H, Panzica M, Flanagan D, Faris RA, et al. (1991). A Fischer rat substrain deficient in dipeptidyl peptidase IV activity makes normal steady-state RNA levels and an altered protein. Use as a liver-cell transplantation model. The Biochemical Journal 273(Pt 3), 497–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi A, Vana PG, Chavan TS, Brueggemann LI, Byron KL, Tarasova NI, et al. (2015). Heteromerization of chemokine (C-X-C motif) receptor 4 with α1A/B-adrenergic receptors controls α1-adrenergic receptor function. Proceedings of the National Academy of Sciences 112, E1659–E1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troutt JS, Siegel RW, Chen J, Sloan JH, Deeg MA, Cao G, et al. (2011). Dual-monoclonal, sandwich immunoassay specific for glucose-dependent insulinotropic peptidebsubN1–42b/subN, the active form of the incretin hormone. Clinical Chemistry 57, 849–855. [DOI] [PubMed] [Google Scholar]
- Tschop M, Castaneda TR, Joost HG, Thone-Reineke C, Ortmann S, Klaus S, et al. (2004). Physiology: does gut hormone PYY3–36 decrease food intake in rodents? Nature 430 (1 p following 165; discussion 162 p following 165). [DOI] [PubMed] [Google Scholar]
- Tsuji E, Misumi Y, Fujiwara T, Takami N, Ogata S, & Ikehara Y (1992). An active-site mutation (Gly633 .fwdarw. Arg) of dipeptidyl peptidase IV causes its retention and rapid degradation in the endoplasmic reticulum. Biochemistry 31, 11921–11927. [DOI] [PubMed] [Google Scholar]
- Unniappan S, McIntosh CHS, Demuth HU, Heiser U, Wolf R, & Kieffer TJ (2006). Effects of dipeptidyl peptidase IV on the satiety actions of peptide YY. Diabetologia 49, 1915–1923. [DOI] [PubMed] [Google Scholar]
- Vahl TP, Paty BW, Fuller BD, Prigeon RL, & D’Alessio DA (2003). Effects of GLP-1-(7–36)NH2, GLP-1-(7–37), and GLP-1-(9–36)NH2on Intravenous Glucose Tolerance and Glucose-Induced Insulin Secretion in Healthy Humans. The Journal of Clinical Endocrinology & Metabolism 88, 1772–1779. [DOI] [PubMed] [Google Scholar]
- Vanheule V, Metzemaekers M, Janssens R, Struyf S, & Proost P (2018). How post-translational modifications influence the biological activity of chemokines. Cytokine 109, 29–51. [DOI] [PubMed] [Google Scholar]
- Varin EM, Mulvihill EE, Beaudry JL, Pujadas G, Fuchs S, Tanti JF, et al. (2018). Circulating Levels of Soluble Dipeptidyl Peptidase-4 Are Dissociated from Inflammation and Induced by Enzymatic DPP4 Inhibition. Cell Metabolism 29(2), 320–334. [DOI] [PubMed] [Google Scholar]
- Vergote D, Butler GS, Ooms M, Cox JH, Silva C, Hollenberg MD, et al. (2006). Proteolytic processing of SDF-1alpha reveals a change in receptor specificity mediating HIV-associated neurodegeneration. Proceedings of the National Academy of Sciences of the United States of America 103, 19182–19187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlestedt C, Pich E, Koob G, Yee F, & Heilig M (1993). Modulation of anxiety and neuropeptide Y-Y1 receptors by antisense oligodeoxynucleotides. Science 259, 528–531. [DOI] [PubMed] [Google Scholar]
- Wang W, Choi K,BK, Li W, Lao Z, Lee AYH, Souza SC, et al. (2014). Quantification of intact and truncated stromal cell-derived factor-1α in circulation by immunoaffinity enrichment and tandem mass spectrometry. Journal of the American Society for Mass Spectrometry 25, 614–625. [DOI] [PubMed] [Google Scholar]
- Xu G, Kaneto H, Laybutt DR, Duvivier-Kali VF, Trivedi N, Suzuma K, et al. (2007). Downregulation of GLP-1 and GIP receptor expression by hyperglycemia: Possible contribution to impaired incretin effects in diabetes. Diabetes 56, 1551–1558. [DOI] [PubMed] [Google Scholar]
- Xue L, Wang J, Wang W, Yang Z, Hu Z, Hu M, et al. (2014). The effect of stromal cell-derived factor 1 in the migration of neural stem cells. Cell Biochemistry and Biophysics 70, 1609–1616. [DOI] [PubMed] [Google Scholar]
- Yang F, Xue F, Guan J, Zhang Z, Yin J, & Kang Q (2018). Stromal-cell-derived factor (SDF) 1-alpha overexpression promotes bone regeneration by osteogenesis and angiogenesis in osteonecrosis of the femoral head. Cellular Physiology and Biochemistry 46, 2561–2575. [DOI] [PubMed] [Google Scholar]
- Yang H (2002). Central and peripheral regulation of gastric acid secretion by peptide YY.Peptides 23, 349–358. [DOI] [PubMed] [Google Scholar]
- Yu L, Cecil J, Peng SB, Schrementi J, Kovacevic S, Paul D, et al. (2006). Identification and expression of novel isoforms of human stromal cell-derived factor 1. Gene 374, 174–179. [DOI] [PubMed] [Google Scholar]
- Yu Y, Wu R-X, Gao L-N, Xia Y, Tang H-N, & Chen F-M (2016). Stromal cell-derived factor-1-directed bone marrow mesenchymal stem cell migration in response to inflammatory and/or hypoxic stimuli. Cell Adhesion & Migration 10, 342–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabel BA, Wang Y, Lewen S, Berahovich RD, Penfold ME, Zhang P, et al. (2009). Elucidation of CXCR7-mediated signaling events and inhibition of CXCR4-mediated tumor cell transendothelial migration by CXCR7 ligands. Journal of Immunology 183, 3204–3211. [DOI] [PubMed] [Google Scholar]
- Zhang K, McQuibban GA, Silva C, Butler GS, Johnston JB, Holden J, et al. (2003). HIV-induced metalloproteinase processing of the chemokine stromal cell derived factor-1 causes neurodegeneration. Nature Neuroscience 6, 1064–1071. [DOI] [PubMed] [Google Scholar]
- Zhang X, Tu H, Yang Y, Fang L, Wu Q, & Li J (2017). Mesenchymal stem cell-derived extracellular vesicles: Roles in tumor growth, progression, and drug resistance. Stem Cells International 2017, 1758139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziarek JJ, Kleist AB, London N, Raveh B, Montpas N, Bonneterre J, et al. (2017). Structural basis for chemokine recognition by a G protein-coupled receptor and implications for receptor activation. Science Signaling, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziarek JJ, Veldkamp CT, Zhang F, Murray NJ, Kartz GA, Liang X, et al. (2013). Heparin oligosaccharides inhibit chemokine (CXC motif) ligand 12 (CXCL12) cardioprotection by binding orthogonal to the dimerization interface, promoting oligomerization, and competing with the chemokine (CXC motif) receptor 4 (CXCR4) N terminus. The Journal of Biological Chemistry 288, 737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlotnik A, & Yoshie O (2000). Chemokines: a new classification system and their role in immunity. Immunity 12, 121–127. [DOI] [PubMed] [Google Scholar]
- Zukowska-Grojec Z, Karwatowska-Prokopczuk E, Rose W, Rone J, Movafagh S, Ji H, et al. (1998). Neuropeptide Y: A Novel Angiogenic Factor From the Sympathetic Nerves and Endothelium. Circulation Research 83, 187–195. [DOI] [PubMed] [Google Scholar]