

Abstract
Chimeric antigen receptor (CAR) T cell therapy has shown promise in hematologic malignancies, but its application to solid tumors has been challenging1–4. Given the unique effector functions of macrophages and their capacity to penetrate tumors5, we genetically engineered human macrophages with CARs to direct their phagocytic activity against tumors. We found that a chimeric adenoviral vector overcame the inherent resistance of primary human macrophages to genetic manipulation and imparted a sustained pro-inflammatory (M1) phenotype. CAR macrophages (CAR-Ms) demonstrated antigen-specific phagocytosis and tumor clearance in vitro. In two solid tumor xenograft mouse models, a single infusion of human CAR-Ms decreased tumor burden and prolonged overall survival. Characterization of CAR-M activity showed that CAR-Ms expressed pro-inflammatory cytokines and chemokines, converted bystander M2 macrophages to M1, upregulated antigen presentation machinery, recruited and presented antigen to T cells and resisted the effects of immunosuppressive cytokines. In humanized mouse models, CAR-Ms were further shown to induce a pro-inflammatory tumor microenvironment and boost anti-tumor T cell activity.
The solid tumor microenvironment (TME) actively recruits myeloid cells, leading to extensive infiltration with immunosuppressive macrophages6–8. Macrophages are abundant in the TME of most cancers, where they promote invasion and angiogenesis, facilitate metastasis and increase immunosuppression9. TME macrophages arise primarily from bone marrow-derived monocytes that are recruited by tumor or stroma-derived chemokines9,10. The importance of macrophages in the TME has generated interest in therapeutic approaches to deplete immunosuppressive tumor-associated macrophages (TAMs) or enhance macrophage phagocytosis11–13. Therapeutic approaches to deplete, repolarize or disinhibit the phagocytic activity of TAMs are in clinical development11,12,14,15. Current macrophage-based immunotherapeutic approaches are mechanistically dependent on TAMs, which express both activating and inhibitory Fc receptors, are polarized toward a pro-tumoral and immunosuppressive (M2) phenotype and lack tumor-associated antigen specificity16.
Outside the environment of established tumors, macrophages are central effectors and regulators of the innate immune system, capable of phagocytosis, cellular cytotoxicity, secretion of pro-inflammatory factors and antigen presentation to T cells17. Macrophages are critical effectors of targeted antibody-based cancer therapy14, and as professional antigen-presenting cells, activated macrophages can play a role in promoting an adaptive anti-tumor immune response18–20.
These considerations spurred previous attempts to adoptively transfer high numbers of autologous macrophages to patients with solid tumors21. These clinical trials demonstrated the feasibility and safety of infusing up to 3 × 109 autologous monocyte-derived macrophages but failed to demonstrate notable anti-tumor efficacy21–23. We hypothesized that the expression of CARs in human macrophages could redirect their phagocytic function and result in a targeted anti-tumor therapeutic effect with the potential to stimulate an adaptive immune response.
To model the potential for CAR-mediated redirection of macrophage phagocytosis, we transduced the human macrophage THP-1 cell line with a first-generation anti-CD19 CAR encoding the CD3ζ intracellular domain. CD3ζ has homology to the Fc common γ-chain, FcεRI-γ, a canonical signaling molecule for antibody-dependent cellular phagocytosis (ADCP) in macrophages. We measured the phagocytic potential of macrophages expressing an intact (CAR19ζ) or a truncated (CAR19Δζ) CAR lacking the CD3ζ intracellular domain (Supplementary Fig. 1a). CAR19ζ but neither CAR19Δζ nor control untransduced (UTD) macrophages phagocytosed antigen-bearing tumor cells in an antigen-specific manner (Fig. 1a,b). CAR-M phagocytosis was an active process requiring Syk, non-muscle myosin IIA and actin polymerization, similarly to Fc receptor-mediated ADCP (Supplementary Fig. 1b). CAR activity was equivalent regardless of the use of CD3ζ and Fcγ-based signaling domains (Supplementary Fig. 1c), and all subsequent experiments were performed using CD3ζ-based CARs. CAR-mediated macrophage phagocytosis was confirmed by video microscopy (Supplementary Videos 1 and 2 and Supplementary Fig. 1d) and by imaging flow cytometry (Supplementary Fig. 1e). CAR-Ms were capable of polyphagocytosis (representative images, Supplementary Fig. 1f). We next generated CAR-Ms targeting the solid tumor antigens mesothelin or HER2 and demonstrated phagocytosis of antigen-positive target cells (Fig. 1c,d). Together, these data demonstrated that CD3ζ-based CARs can direct anti-tumor phagocytic activity and provided support for subsequent efforts to translate this platform to primary human macrophages.
Fig. 1 |. Generation of human CAR-Ms and assessment of CAR-mediated tumor phagocytosis.
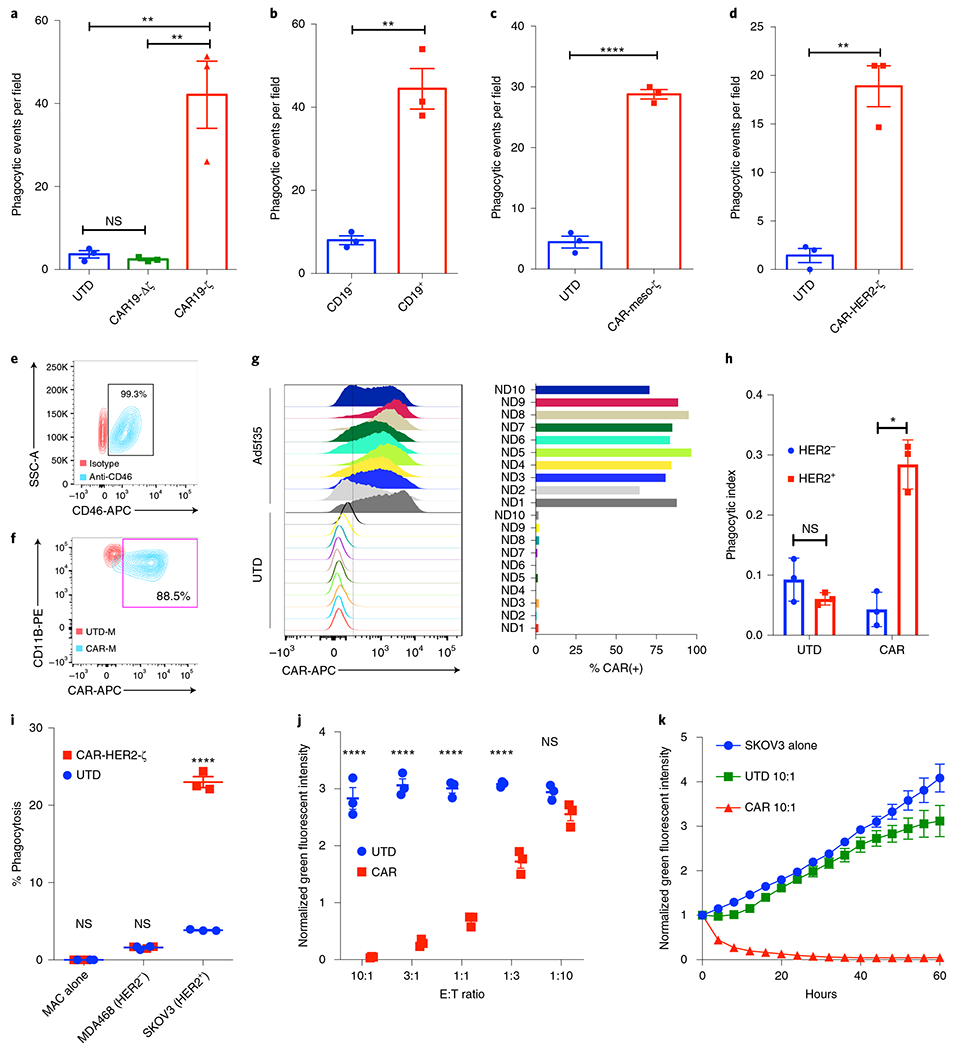
a, Assessment of indicated THP-1 macrophage phagocytosis against CD19+ K562 target cells using fluorescent microscopy. Data are represented as the mean ± s.e.m. of n = 3 technical triplicates, with three random fields of view assessed per replicate. Statistical significance was calculated with one-way ANOVA with multiple comparisons. b, Phagocytosis of CD19+ or control CD19− K562 target cells by CAR19ζ-expressing THP-1 macrophages. Data are represented as the mean ± s.e.m. of n = 3 technical triplicates, with three random fields of view assessed per replicate. Statistical significance was calculated using a two-sided t-test. c,d, In vitro phagocytosis by UTD or CAR-meso-ζ THP-1 macrophages of mesothelin+ve K562 cells (c) or by CAR-HER2-ζ macrophages of HER2+ K562 cells (d). Data are represented as mean ± s.e.m. of n = 3 technical replicates, with three random fields of view assessed per replicate. Statistical significance was calculated using a two-sided t-test. e, Representative FACS plot of CD46 expression on primary human macrophages (blue) or isotype control (red). This graph is representative of n = 10 human donors. f, Representative FACS plot of CAR-HER2 expression on Ad5f35-transduced primary human macrophages (blue, CAR-M; red, UTD-M). This graph is representative of n = 10 human donors. g, CAR expression on macrophages derived from n = 10 human donors 48 h after Ad5f35-CAR transduction. h, Incucyte-based phagocytosis of HER2 functionalized or control pH-Rodo-labeled beads by UTD or CAR-M. Data are represented as mean ± s.e.m. of n = 3 technical replicates and are representative of three experiments. Statistical analysis was calculated using a two-sided t-test. i, FACS-based phagocytosis of HER2− (MDA-468) or HER2+ (SKOV3) target tumor cells by UTD or CAR-M. Statistical significance was calculated with one-way ANOVA with multiple comparisons, and data represent n = 3 technical replicates (representative of at least three individual experiments). j, Incucyte-based killing assay of GFP+ SKOV3 by UTD or CAR-M after 48 h of co-culture at different E:T ratios. Statistical significance was calculated with one-way ANOVA with multiple comparisons, and data represent the mean ± s.e.m. of n = 3 technical replicates (representative of at least three individual experiments). k, Incucyte-based killing kinetics of UTD or CAR-M against SKOV3 at a 10:1 E:T ratio. Data are represented as the mean ± s.e.m. of n = 3 technical replicates (representative of at least three individual experiments). For all panels, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Gene transfer into primary human macrophages has been a longstanding challenge. Given that macrophages express CD46 (Fig. 1e), the docking protein for group B adenoviruses such as Ad35 (ref. 24), we evaluated a replication-incompetent chimeric adenoviral vector (Ad5f35)25 as a method of CAR delivery to macrophages. Primary human macrophages were generated from peripheral blood CD14+ monocytes with GM-CSF, as GM-CSF is associated with an M1-prone differentiation status26. We engineered an anti-HER2 CAR (Supplementary Fig. 1g) into an Ad5f35 vector and demonstrated that Ad5f35-transduced human macrophages expressed CAR with high efficiency (Fig. 1f) and reproducibility between donors (Fig. 1g). The resultant primary human anti-HER2 CAR-Ms demonstrated antigen-specific phagocytosis of HER2+ beads and tumor cells (Fig. 1h,i). CAR-M eradicated SKOV3, a HER2+ ovarian cancer cell line, in a dose- and time-dependent manner (Fig. 1j,k). The level of tumor phagocytosis and killing directly correlated with the level of CAR expression (Supplementary Fig. 1h) and with the level of target antigen expression (Supplementary Fig. 1i,j). Notably, macrophages transduced by empty Ad5f35 (containing no transgene) at an equivalent multiplicity of infection (MOI) lacked anti-tumor activity (Supplementary Fig. 1k). To evaluate the potential for on-target, off-tumor toxicity, we evaluated the HER2 expression and anti-HER2 CAR-M phagocytosis of a panel of 20 representative normal human tissues and found that normal cells were not phagocytosed (Supplementary Fig. 2a,b).
We next tested the anti-tumor activity of CAR-Ms in vivo using two distinct models in NOD-scid IL2Rgnull-3/GM/SF (NSGS) mice27. In the first approach, SKOV3 cells were injected intravenously (IV) leading to lung metastases. Mice then received a single IV injection of saline, UTD macrophages (UTD), macrophages transduced with empty Ad5f35 vector (Empty) or anti-HER2 CAR-Ms (Fig. 2a). CAR-treated mice demonstrated a marked reduction in tumor burden (Fig. 2b). Although all mice eventually progressed, a single infusion of CAR-Ms led to a prolongation of overall survival (Fig. 2c). Given that empty transduced macrophages did not demonstrate anti-tumor activity in vitro or in vivo, in all future experiments we compared UTD with CAR. Co-localization and anti-tumor activity of CAR-M were evaluated by immunohistochemical analysis of explanted lungs, revealing the presence of multiple metastatic tumor nests in this model (Fig. 2d), with a significant reduction in metastatic tumor burden after CAR-M treatment (Fig. 2e).
Fig. 2 |. Evaluation of anti-tumor activity, persistence and trafficking of primary human anti-HER2 CAR-Ms in xenograft models.
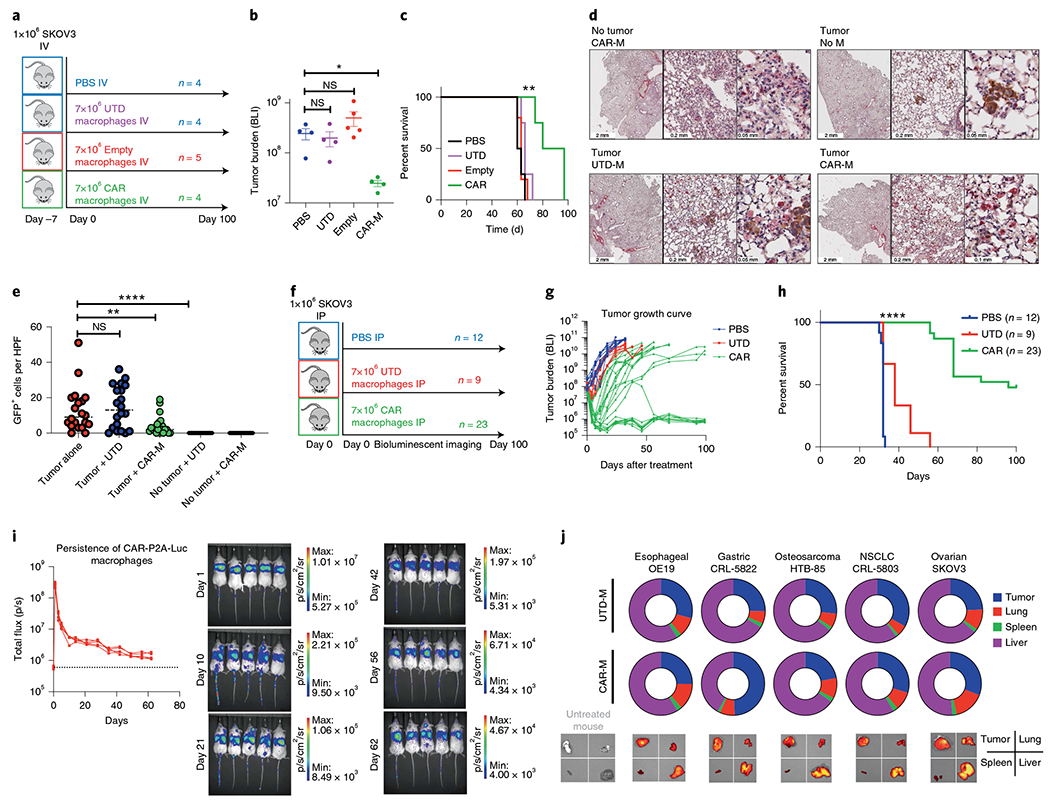
a, NSGS mice were IV injected with luciferase(+) SKOV3 and treated with IV PBS, UTD, empty vector transduced (empty) or CAR-HER2 macrophages 7 d later with n as shown. b, Tumor burden (total flux) by bioluminescent imaging 31 d after treatment. Data are represented as mean ± s.e.m., and statistical significance was calculated with ANOVA with multiple comparisons. c, Kaplan-Meier survival curve (median survival, 88.5 d (CAR) versus 63 d (Empty), P = 0.0047). Statistical significance was calculated using the log-rank Mantel-Cox test with df = 2. d, Lungs from mice with metastatic disease were assessed with dual immunohistochemical analysis (tumor: anti-GFP, brown; macrophage detection: anti-human CD68, red). Each panel shows low (left), medium (middle) and high (right) power view. The experiment was performed once. e, Quantification of GFP+ tumor cells in n = 5 random ×20 high-power fields per slice, with two slices per mouse and two mice per group. Analysis was done by an observer who was blinded to treatment group allocation. Data are presented as the mean, with statistical significance calculated using multiple two-sided t-tests. f, NSGS mice were injected with SKOV3 IP 2-4 h before receiving injections of PBS, control (UTD) or CAR-HER2 human macrophages IP as shown. g, Tumor burden measured by bioluminescence (total flux) over 100 d. h, Kaplan-Meier survival curve over 100 d. Statistical significance was calculated using the log-rank Mantel-Cox test (CAR versus UTD, P < 0.0001 with df = 2). i, Persistence and biodistribution of CAR-P2A-luciferase-expressing human macrophages after IV administration in n = 5 tumor-free NSGS mice. Luciferase signal was tracked over the course of 62 d (dotted line represents background luminescence). Macrophages initially transited through the lung and accumulated in the liver of tumor-free mice. The experiment was performed twice with similar results. j, Tumor trafficking and biodistribution of VivoTrack680-labeled UTD or CAR-M in five tumor xenograft models. NSGS mice were killed and organs/tumors were explanted for ex vivo imaging 5 d after a single injection of 5 × 106 UTD or CAR-M, n = 3–5 mice per treatment group per tumor type; the experiment was performed once. For all panels, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
In the second model, intraperitoneal (IP) carcinomatosis was modeled in NSGS mice via IP injection with SKOV3. Mice were treated 2–4 h later with a single IP injection of saline, UTD macrophages or anti-HER2 CAR-Ms (Fig. 2f). CAR-Ms, but not UTD macrophages, led to marked tumor regression in most treated mice as demonstrated by serial bioluminescent imaging over 100 d (Fig. 2g). The treatment was not associated with significant toxicity as demonstrated by body weights (Supplementary Fig. 2c) and led to significantly improved overall survival in the CAR treatment group (Fig. 2h).
We next sought to evaluate the persistence, biodistribution and trafficking of CAR-M after systemic administration. Using luciferase/CAR co-expressing human macrophages, CAR-M persisted for at least 62 d in vivo in tumor-free NSGS mice (Fig. 2i). To evaluate the ability of human UTD or CAR-M to traffic to established tumor xenografts of multiple types, fluorescently labeled macrophages were administered and the relative signal in the tumor, liver, spleen and lung was evaluated in explanted whole-organ tissue on day 5. Macrophages trafficked to all five solid tumor types that were evaluated (Fig. 2j). Adenoviral transduction did not affect the pattern of macrophage chemokine receptor expression (Supplementary Fig. 2d) and did not affect the trafficking capability of macrophages. In all mice, the liver was the primary normal tissue where macrophages accumulated after IV administration.
Macrophage phenotype is plastic and can change in response to cytokines, pathogen-associated molecular patterns, metabolic cues, cell–cell interactions and tissue-specific signals28. Given that adenoviral infection activates the inflammasome, we hypothesized that exposure to Ad5f35, regardless of the encoded transgene, might induce a pro-inflammatory (M1) macrophage phenotype29,30. Nonbiased hierarchical clustering of macrophage transcriptomes from four human donors showed that transduced macrophages clustered distinctly from UTD macrophages (Supplementary Fig. 3a). Furthermore, when UTD, Ad5f35-CAR transduced, empty-vector Ad5f35 transduced (Empty), classically activated (M1) and alternatively activated (M2) macrophage transcriptomes from multiple human donors were subjected to nonbiased principal component analysis, adenovirally transduced macrophages clustered toward the classically activated and away from the alternatively activated macrophages, regardless of CAR expression (Fig. 3a). Transduction led to the induction of many interferon-associated genes (Fig. 3b), consistent with a classically activated M1 phenotype, and pathway analysis confirmed the induction of M1-associated pathways, such as interferon signaling, pattern recognition receptor signaling, Th1 pathway and iNOS signaling (Supplementary Fig. 3b). Furthermore, key components of the antigen-presentation machinery such as co-stimulatory ligands, antigen processing and presentation and MHC-Class I/II genes were induced (Supplementary Fig. 3c). We validated the induction of a pro-inflammatory M1 phenotype by quantitative PCR and flow cytometry and demonstrated a dose-dependent response (Supplementary Fig. 3d,e). Induction and repression of these markers were equivalent between CAR and empty vector Ad5f35, demonstrating that the phenotype is induced by the viral vector rather than CAR expression per se in macrophages (Supplementary Fig. 3f). These findings were validated in macrophages from ten human donors (Supplementary Fig. 3g), and the M1 phenotype was maintained for at least 40 d after transduction (Supplementary Fig. 3h).
Fig. 3 |. Adenovirally transduced CAR-Ms are M1 polarized, modify the TME and cross-present tumor antigens.
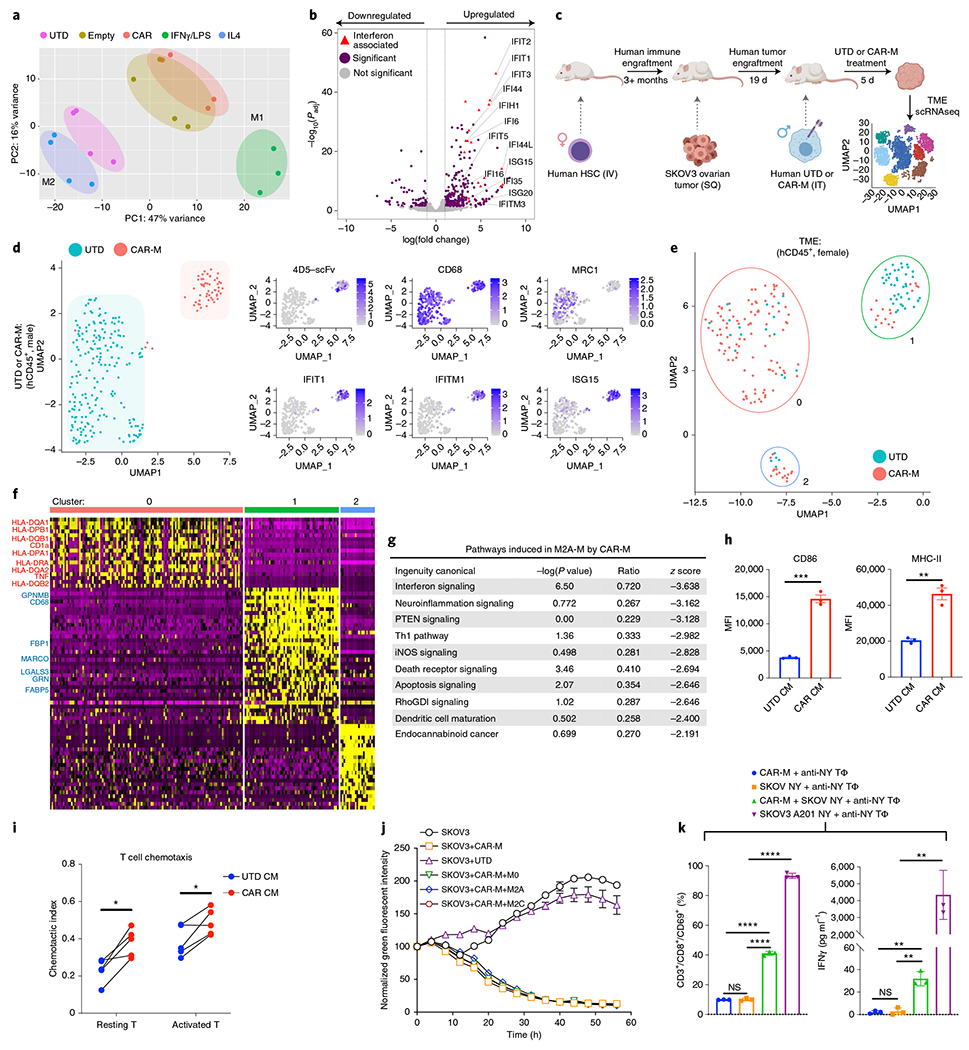
a, Gene expression principal component analysis clustering from UTD, Ad5f35-empty-vector transduced, Ad5f35-CAR-HER2-ζ transduced, classically activated M1 or alternatively activated M2 human macrophages. n = 3–4 human donors. b, Volcano plot of differentially expressed genes in UTD versus CAR-M. Purple indicates Padj < 0.05 and log2 fold change >1 or <−1 Red triangles indicate significantly upregulated interferon-associated genes (n = 4 human donors). Statistical significance was calculated using the Wald test for DESeq2 data. c, Schema of HIS TME model generation. Mice were engrafted with human female CD34+ cells, and after human myeloid engraftment was confirmed, SKOV3 tumors were established in the subcutaneous flank. Nineteen days after tumor administration, 1 × 107 UTD or CAR macrophages from a male human donor were injected intratumorally. Tumors were harvested 5 d after treatment, and scRNAseq was performed on the tumor samples. Adoptively transferred macrophages were distinguished from macrophages in the TME by Y-chromosome gene mapping. d, Cluster plots demonstrating the phenotypic distinction of UTD from CAR macrophages within the TME (left). On the right, the relative expression of the indicated genes was mapped onto CAR-M (top cluster) or UTD (bottom cluster). The data represent three mice and two mice for the CAR-M and UTD arms, respectively. e, Cluster plots demonstrating the phenotypic distinction of human TME after UTD versus CAR-M treatment. Cluster 0 was enriched in the CAR-M-treated arm (86% events from the CAR cohort), whereas cluster 1 was enriched in events from the UTD-treated arm (71% events from the UTD cohort). Cluster 2 was mixed and had few events. f, Gene expression heat map from each TME scRNAseq cluster from e. Pro-inflammatory genes were found in cluster 0 (annotated in red). Potential anti-inflammatory or M2-associated genes in cluster 1 are annotated in blue. For panels c-f, n = 3 mice per group; the experiment was performed once. g, Ingenuity pathway analysis of the transcriptome of in vitro-polarized M2A macrophages after exposure to conditioned media from UTD or CAR-M for 48 h; n = 3 technical replicates. Statistical significance was calculated using Fisher’s exact test. h, Human immature dendritic cells were treated with UTD or CAR-M conditioned media for 48 h, and activation markers CD86 and MHC-II (HLA-DR) were assessed by FACS. Data are represented as the mean ± s.e.m. from n = 3 technical replicates. Statistical significance was calculated using a two-tailed t-test. i, Chemotaxis of resting or activated CD3+ T cells by UTD or CAR-M conditioned media. Data represent T cells from n = 4 human donors, and statistical significance was calculated using a paired t-test. j, Incucyte-based killing assay of SKOV3 by CAR-M alone or in the presence of M0, M2a or M2c macrophages at an E:T:M2 ratio of 3:1:1. Data are represented as the mean ± s.e.m. of n = 3 technical replicates. k, CAR-M cross-presentation assay showing CD69 induction (left) and IFNγ secretion (right) by anti-NYESO TCR+ T cells 24 h after co-culture with HLA-A201(+) CAR-M (CAR-M, blue), HLA-A201(−)NYESO(+) SKOV3 (SKOV NY, orange) or HLA-A201(+) CAR-M co-cultured with SKOV NY for 48 h (green). Anti-NYESO T cells exposed to HLA-A201(+)NYESO(+) SKOV3 alone were used as positive control (purple). Data are represented as the mean ± s.e.m. of n = 3 technical replicates. Statistical analysis was performed using ANOVA with multiple comparisons. For all panels, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. CM, conditioned media.
Given that CAR-Ms become pro-inflammatory, we sought to evaluate the bidirectional interaction of CAR-Ms with the human TME in a humanized immune system (HIS) mouse model. HIS mice were generated by transplanting female human CD34+ hematopoietic stem cells (HSCs) into NSG mice. After confirmation of human myeloid cells in the peripheral blood, SKOV3 ovarian tumors were engrafted in the subcutaneous flank and male human UTD or CAR macrophages were injected intratumorally after 19 d. Five days later, tumors were harvested and subjected to single-cell RNA sequencing. The use of female HSCs and male UTD or CAR macrophages allowed the distinction of HSC-derived TME macrophages from adoptively transferred macrophages (Fig. 3c). UTD and CAR macrophages (confirmed by scFv mapping) from within the TME were phenotypically distinct. Although both UTD and CAR macrophages expressed the macrophage identity marker CD68, only UTD expressed the M2 marker MRC1 (CD206), and only CAR-Ms expressed M1-associated interferon response genes (IFIT1, ISG15 and IFITM1) (Fig. 3d). These data demonstrate that CAR-Ms maintained a pro-inflammatory phenotype within the human TME.
We validated phenotypic plasticity by challenging control or Ad5f35-transduced human macrophages with M2-inducing factors in vitro. Upon stimulation with IL4, IL13 or SKOV3 conditioned media, only UTD macrophages upregulated the canonical M2 marker CD206 (Supplementary Fig. 4a). Furthermore, upon stimulation with IL4, only UTD macrophages increased their basal oxygen consumption rate, an expected characteristic of IL4-induced M2 macrophages (Supplementary Fig. 4b)31. Transcriptome analysis revealed that significantly fewer genes were induced by IL4 or IL13 in CAR versus UTD macrophages (Supplementary Fig. 4c,d), including the CD206-encoding M2-associated gene MRC1 (Supplementary Fig. 4e,f).
Using the humanized TME mouse model, the effect of CAR-Ms on the surrounding TME was interrogated at single-cell resolution. Adoptively transferred macrophages were removed from analysis by excluding all human male cells. The human TME grouped into two large clusters (cluster 0 and cluster 1) and one small cluster of cells that was excluded from analysis (cluster 2). Cluster 0 was enriched in the CAR-M-treated arm (86% CAR, 14% UTD), whereas cluster 1 was enriched in the UTD-treated arm (71% UTD, 29% CAR) (Fig. 3e). Gene expression analysis of cluster 0 revealed an enrichment of pro-inflammatory genes such as MHC-II and TNF, demonstrating that CAR-Ms remodeled the TME toward a pro-inflammatory state (Fig. 3f).
To validate these in vivo findings, we evaluated the effect of CAR-Ms on M2 macrophages in vitro and found that CAR-M conditioned media induced a phenotypic shift in M2A, M2C and M2D macrophages (Supplementary Fig. 5a). CAR-Ms induced pro-inflammatory pathways such as interferon signaling, TH1 pathway and iNOS signaling in M2 macrophages (Fig. 3g). Pro-inflammatory marker induction in all M2 macrophage subtypes that were exposed to CAR-Ms was confirmed by FACS (Supplementary Fig. 5b). Additionally, CAR-Ms induced activation and maturation markers in immature human dendritic cells (Fig. 3h) and directly induced the recruitment of both resting and activated human T cells in an in vitro chemotaxis assay (Fig. 3i). Finally, having shown that CAR-Ms can exert a dominant effect on surrounding immune cells, we demonstrated that CAR-Ms maintained their anti-tumor activity in the presence of human M2 macrophages (Fig. 3j).
Given the upregulation of co-stimulatory ligands and antigen processing and presentation genes and the established role of macrophages as professional antigen-presenting cells (APCs), we sought to test the capacity for CAR-Ms to co-stimulate and present antigens to T cells. To evaluate whether Ad5f35 activation affects the ability of human macrophages to activate T cells, macrophages transduced with the tumor-associated antigen NY-ESO1 and with HLA-A2*01 (Supplementary Fig. 5c,d) were then transduced with Ad5f35, or not (UTD), and co-cultured with autologous antigen-specific T cells (made by transducing CD8+ T cells with clone 1G4 anti-NYESO1 TCR construct32). Ad5f35-transduced NY-ESO1-expressing macrophages induced significantly more proliferation of 1G4 CD8+ T cells than NY-ESO1-expressing control macrophages or Ad5f35-transduced macrophages lacking NY-ESO1 (Supplementary Fig. 5e).
To test CAR-M cross-presentation of intracellular tumor antigens ingested during phagocytosis of whole tumor cells, we generated SKOV3 expressing NY-ESO-1 only (SKOV-NY), SKOV3 expressing HLA-A201 and NY-ESO-1 (SKOV-A201-NY) and CAR-Ms from an HLA-A201+ donor. TRAC knockout anti-NYESO1 TCR T cells were incubated with CAR-Ms (macrophage control), SKOV-NY (tumor control), SKOV-A2-NY (positive control) or CAR-Ms that were fed SKOV-NY for 48 h. CD8+ anti-NYESO1 T cells were activated (as determined by CD69 induction and production of IFNγ) by CAR-Ms that ingested SKOV-NY (Fig. 3k). Anti-NYESO1 T cells were not activated by SKOV-NY or CAR-Ms alone, demonstrating that macrophages were able to cross-present tumor-derived antigens after phagocytosis, suggesting that CAR-Ms might lead to epitope spreading.
To test the potential of CAR-Ms to stimulate T cells in vivo, NSGS mice were engrafted with metastatic SKOV3 and treated with CAR-Ms, CAR-Ms plus donor-derived polyclonal T cells or T cells alone. Mice that received CAR-Ms and donor-derived T cells had deeper anti-tumor responses than those that received either CAR-Ms or T cells alone at a late timepoint (Supplementary Fig. 5f). Given that the T cells were not engineered to specifically recognize SKOV3 antigens in this in vivo model, the synergistic effect between CAR-Ms and T cells was likely due to CaR-M-mediated potentiation of nonspecific (that is, allogeneic) T cell anti-tumor activity. The interaction of CAR-Ms with endogenous T cells will be evaluated in syngeneic immunocompetent murine tumor models in future studies.
There is increasing interest in depleting or re-polarizing TAMs to generate an anti-tumor pro-inflammatory effect. However, it is likely that this approach would be successful only if removal of an inhibitory cell type (TAM) were complemented by the addition of a positive stimulus. We hypothesized that human peripheral blood monocytes could be differentiated to macrophages, endowed with tumor antigen-specific domains and allowed to traffic into tumors. To test this hypothesis, we developed an efficient and robust gene transfer method that had the added benefit of conferring on the engineered macrophages a robust pro-inflammatory signature with the potential to further polarize the surrounding microenvironment. In addition to direct anti-tumor activity by CAR-Ms, we found evidence for cross-presentation and for T cell co-stimulation, thereby raising the possibility that CAR-Ms could directly diminish tumor burden, sculpt the TME and generate a vaccinal effect. Having established a CAR-M platform, in future work we will study the potential mechanisms of tumor progression and evaluate rational combinations with agents that affect cell death pathways, phagocytosis or T cell-based immunity.
Methods
Cell lines.
The THP-1, SKOV3, K562, MDA-468, CRL-2351, HTB-20, HTB-85, CRL-5803, CRL-5822, CRL-1555, HTB-131, HTB-20 and CRL-1902 cell lines were purchased from the American Type Culture Collection (ATCC). Cells were cultured in RPMI media with 10% FBS, penicillin, streptomycin, 1× GlutaMAX and 1× HEPES unless otherwise recommended by the ATCC. All cell lines were transduced with a lentiviral vector co-encoding click beetle green (CBG) luciferase and green florescent protein (GFP) under an EF1α promoter, separated by a P2A sequence. Transduced target cell lines were FACS sorted for 100% GFP positivity before use as targets in vitro and in vivo. THP-1 cells were lentivirally transduced, FACS sorted and maintained in liquid culture. CAR expression and purity were routinely validated.
Plasmid construction and virus.
For lentivirus production, CAR constructs were cloned into the third-generation pTRPE lentiviral backbone using standard molecular biology techniques. All CAR constructs used a CD8 leader sequence, (GGGGS)3 linker, CD8 hinge and CD8 transmembrane domain and were expressed under the control of an EF1α promoter. Lentivirus was packaged in HEK293 cells and purified and concentrated as described previously33. In indicated experiments, Vpx was incorporated into lentivirus at the packaging stage as previously described34. Cell lines were transduced with lentivirus MOI 3–5 unless otherwise noted. For replication-deficient adenovirus production, anti-HER2 CAR was cloned into the pShuttle transfer plasmid using Xba-I and Sal-I and subsequently cloned into pAd5f35 using I-Ceu I and PI-Sce I. All cloning steps were validated by restriction enzyme digest and sequencing. pAd5f35 is first-generation E1/E3 deleted adenoviral backbone. Ad5f35-CAR-HER2-ζ was generated, expanded, concentrated and purified using standard techniques in 293 cells by the Baylor Vector Development Laboratory. All adenoviral batches were verified negative for replication-competent adenovirus and passed sterility and endotoxin analysis. Adenoviral titer was determined using Adeno-X Rapid Titer Kit (Clontech) and validated by functional transgene expression in human macrophages. An MOI of 1,000 plaque-forming units (PFU) per cell was used unless otherwise stated.
Animal studies.
All mouse studies were conducted in accordance with national guidelines for the humane treatment of animals and were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Pennsylvania. Schemas of the used xenograft models are shown in detail in the first panel of each relevant figure. NOD/SCID Il2rg−/− hIL3-hGMCSF-hSF (NSG-SM3 or NSGS) mice originally obtained from Jackson Laboratories were purchased and bred by the Stem Cell and Xenograft Core at the University of Pennsylvania. Cells (SKOV3 tumor cells, human macrophages or human T cells) were injected in 200–300 ul PBS for both IP and IV tail vein injections. IV injections of human macrophages were split into consecutive injections to attain the target dose. Bioluminescent imaging was performed at least weekly using an IVIS Spectrum (PerkinElmer) and analysis was performed using LivingImage v4.3.1 (Caliper Life Sciences). Mice were weighed weekly and were subject to routine veterinary assessment for signs of overt illness. Animals were killed at experimental termination or when predetermined IACUC rodent health endpoints were reached.
Flow cytometry.
Primary human macrophages were tested for CAR-HER2 expression using a two-step staining protocol: human HER2/ERBB2 Protein-His tag (10004-H08H-100, Sino Biological) primary stain followed by Human TruStain FcX (422302, BioLegend) and Anti-His Tag APC (IC050A, R&D Systems) secondary stain. TruStain FcX (422302, BioLegend) was always used for FACS staining of monocytes, macrophages or monocytic cell lines expressing Fc receptors. Macrophage purity was tested using the following panel: Anti-CD11b PE (301306, BioLegend), Anti-CD14 BV711 (301838, BioLegend), Anti-CD3 FITC (11-0038-42, eBioscience), Anti-CD19 PE-CY7 (25-0198-42, eBioscience), Anti-CD66b PerCP-CY5.5 (305108, BioLegend), Anti-CD56 BV605 (318334, BioLegend) and Live/Dead Fixable Aqua (L/D Aqua) Dead Cell Stain Kit (L34957, Thermo Fisher). The same panel was used for testing the monocyte purity after CD14 MACS selection, before seeding for differentiation. M1 and M2 markers on primary human macrophages were detected with the following panel: Anti-CD11B PE (301306, BioLegend), Anti-CD80 BV605 (305225, BioLegend), Anti-CD86 BV711 (305440, BioLegend), Anti-CD206 BV421 (321126, BioLegend), Anti CD163 APC-CY7 (333622, BioLegend), Anti-HLA-DR BV785 (307642, BioLegend), Anti-HLA ABC PE/CY7 (311430, BioLegend) and Live/Dead Fixable Aqua Dead Cell Stain Kit. CD46 expression was detected with Anti-CD46 APC (352405, BioLegend) and CXADR was detected with Anti-CAR PE (FCMAB418PE-I, EMD Millipore). Appropriate fluorescence-matched isotype controls were acquired from BioLegend. Surface HER2 was detected using Anti-Human CD340/HER2 APC (324408, BioLegend). Flow cytometry data were acquired on a BD Fortessa with HTS (BD Biosciences) and analyzed with FlowJo X10 (FlowJo).
FACS-based phagocytosis assay.
Next, 1 × 105 UTD or CAR-HER2-ζ human monocyte-derived macrophages (48 h after transduction) were co-cultured with media (MAC alone), 1 × 105 GFP+ MDA-468 cells (HER2−) or 1 × 105 GFP+ SKOV3 (HER2+) target cells for 3–4 h at 37 °C in triplicate. After co-culture, cells were harvested with Accutase (Innovative Cell Technologies), stained with Anti-CD11b APC-CY7 (301342, BioLegend) and analyzed with FACS using a BD Fortessa (Becton Dickinson). The percent of GFP+ events within the CD11b+ population was plotted as percentage phagocytosis. Data are represented as mean ± standard error of triplicate wells. Statistical significance between CAR-HER2-ζ and UTD was calculated using ANOVA with multiple comparisons; ****P < 0.0001; NS, nonsignificant.
Bead-based phagocytosis assay.
Strepavidin-coated polystyrene microparticles (5.0–5.9-μm diameter, Spherotech) were sterilized for 20 min in 70% isopropanol. Beads were spun down and resuspended in 0.1 M sodium bicarbonate buffer (pH 8.5) and labeled with 10 μM pHrodo SE (Thermo Fisher) for 30 min in the dark. Beads were spun down to remove free dye and resuspended in 0.1 PB (pH 6.5). Biotinylated proteins from ACROBiosystems were added to the beads at a concentration sufficient to occupy one quarter of the binding sites. Beads were incubated with protein for 1 h, washed and resuspended in PBS for use in experiments.
UTD or CAR macrophages were thawed and plated at a density of 2 × 104 viable cells per well in a 96-well plate and allowed to adhere overnight. The next day, the media was aspirated and previously functionalized or blank beads in TexMACS and 10% FBS were added to obtain a 5:1 bead-to-cell ratio. Changes in fluorescence were monitored with IncuCyte Live-Cell Analysis System (Essen BioScience). The phagocytic index was calculated by subtracting the red image mean of wells containing only beads from that of wells with cells and beads and then dividing by the red image mean of the bead-only wells.
Primary cells.
Normal donor apheresis was either performed at the hematology unit at the Hospital of the University of Pennsylvania under an institutional review board-approved protocol through the Human Immunology Core of the University of Pennsylvania or acquired and shipped fresh from HemaCare. Apheresis-derived leukopacs were subjected to elutriation using an Elutra Cell Separation System (Terumo BCT) to reduce erythrocytes, platelets, lymphocytes and granulocytes. Monocyte-enriched fractions were pooled and subjected to MACS CD14-positive selection (Miltenyi Biotec) according to the manufacturer’s instructions. The preselection and post-selection (positive and negative fraction) purity was tested using flow cytometry. Selected CD14 monocytes were seeded in Cell Differentiation Bags (Miltenyi Biotec) in RPMI with 10% FBS, penicillin, streptomycin, 1× GlutaMAX, 1× HEPES and 10 ng ml−1 recombinant human GM-CSF (300-03, PeproTech) for 7 d. Differentiation was monitored by light microscopy. Adenovirus was added on day 5 at an MOI of 1 × 103 based on PFU titer. Differentiated macrophages were harvested at day 7 and tested for CAR expression, differentiation and macrophage purity by FACS. For smaller-scale experiments, macrophages were plated directly in tissue-culture-treated well plates or flasks and transduced at an MOI of 1,000 PFU directly in well plates or flasks. CD3-selected T cells were provided by the University of Pennsylvania Human Immunology Core and were expanded and transduced as previously described33. Immature dendritic cells were generated from CD14+ monocytes using GM-CSF (100 ng ml−1) and IL-4 (20 ng ml−1) for 9 d.
Microscopy.
Microscopy-based phagocytosis assay.
Control or CAR-expressing monomeric red fluorescent protein-positive (mRFP+) THP1 cells were plated at 7.5 × 104 per well in 48-well plates and differentiated with 1 ng ml−1 phorbol 12-myristate 13-acetate (PMA) in RPMI with 10% FBS for 48 h. After differentiation, PMA was washed out with media and 7.5 × 104 control or target GFP+ K562 tumor cells were added and co-cultured for 4 h at 37 °C. After 4 h, tumor cells (nonadherent) were washed out and the plate was imaged for mRFP and GFP fluorescence. The average number of phagocytic events in three random fields of view per well were averaged, in triplicate wells, on a ×10 field of view. Cells were imaged using an EVOS FL Auto 2 Imaging System (AMAFD2000, Thermo Fisher). Data represent the mean ± standard error of triplicate wells. Statistical significance was calculated using a t-test.
Live video imaging microscopy.
Next, 3.0 × 105 CAR or control mRFP+ THP-1 cells were differentiated as above in six-well plates and co-cultured with 3.0 × 105 control or target GFP+ K562 cells for 16–24 h in an incubated 37 °C live imaging chamber and imaged every 30–120 s for mRFP and GFP using the EVOS FL Auto 2 Live Imaging System (Thermo Fisher) using the ×10 lens.
In vitro cytotoxicity assay.
CBG/GFP double-positive SKOV3, HTB-20 and CRL-2351 tumor cells were used as targets in luciferase-based killing assays by control (UTD) or CAR-HER2-ζ (CAR) macrophages. The effector-to-target (E:T) ratio was serially titrated from 10:1 down to 1:30 for both effector groups. Bioluminescence was measured using an IVIS Spectrum (PerkinElmer). Percent specific lysis was calculated on the basis of luciferase signal (total flux) relative to tumor alone, using the following formula.
Data are shown as mean ± s.e.m., with each condition in triplicate. Negative specific lysis values indicate more signal than in the tumor-alone wells. Statistical significance was calculated using ANOVA with multiple comparisons; ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05; NS, nonsignificant.
Image cytometry.
Control or CAR mRFP+ THP-1s were differentiated and co-cultured with CD19+GFP+ K562 target cells as described above. After 4 h of co-culture, cells were washed and harvested with trypsin-EDTA and stained with L/D Aqua for viability. Imaging cytometry was performed on Amnis ImageStreamX (EMD Millipore). Cells were gated for mRFP+GFP+ double-positive events and the phagocytosis-identification algorithm (Amnis ImageStreamX) was applied, which identifies GFP signal within an mRFP+ event.
Macrophage polarization.
For M1 or classically activated macrophage polarization, human monocyte-derived macrophages were exposed to 20 ng ml−1 recombinant interferon-γ (300–02, PeproTech) and 100 ng ml−1 lipopolysaccharide (tlrl-eklps, InvivoGen) in RPMI with 10% FBS for 24 h. Where described, human monocyte-derived macrophages were exposed to 20 ng ml−1 recombinant human IL4 (200-04, PeproTech) or IL13 (200-13, PeproTech). In some experiments, 48-h conditioned media from SKOV3 was used (50% diluted in RPMI with 10% FBS) to polarize macrophages toward M2 for 24 h. In experiments where control or CAR macrophages were challenged with M2-inducing cytokines, cells were treated with cytokine for 24 h, 48 h after viral transduction. To generate different subtypes of M2 macrophages, the below protocols were used. All human cytokines were purchased from PeproTech.
M2A: 100 ng ml−1 M-CSF (7 d); 20 ng ml−1 IL-13 and 20 ng ml−1 IL-4 (2 d)
M2C: 100 ng ml−1 M-CSF (7 d); 10 ng ml−1 IL-10 and 10 ng ml−1 TGF-β1 (2 d)
M2D: 100 ng ml−1 M-CSF (7 d); 50 ng ml−1 IL-6; 20 ng ml−1 LIF (2 d)
RNA sequencing of human macrophages.
RNA was isolated from human macrophages from matched donors, treated as described in each figure and polarized and challenged as above using Ambion RiboPure RNA Purification Kit (AM1924, Thermo Fisher). RNA-seq libraries were generated using TruSeq RNA Library Prep Kit (RS-122-2001/2, Illumina) and validated with BioA by the University of Pennsylvania Next Generation Sequencing Core facility before sequencing. The libraries were sequenced on 75-bp single-end reads using a NextSeq sequencer (Illumina). Low-quality reads were trimmed using Trimmomatic (v0.36) and mapped to human genome (hg38) using STAR (v2.6.0c) with default parameters. Gene count was calculated using featureCounts (v1.6.1)35. Nonexpressed genes with read counts <1 in all samples were removed before differential expression analysis. DESeq2 with log fold change of 1 and adjusted P value of 0.05 was used to identify differentially expressed genes.
For genome browser tracks, bam files were first converted into bed files using bedtools (v2.27.1). Normalized bedgraph tracks were generated using makeUCSCfile with 10,000,000 normalization factor (Homer v2) and converted into bigwig format for integrative genomics viewer (IGV; Broad Institute) usage. Reads were mapped to the human genome (hg38) using RUM before using DegSeq and EdgeR for differential analysis. Group auto-scale was applied to all conditions for y-axis equalization in IGV. Ingenuity Pathway Analysis (Qiagen) was used to map differentially expressed genes to canonical pathways.
Real-time PCR.
RNA was isolated using Ambion RiboPure RNA Purification Kit (AM1924, Thermo Fisher) and reverse transcribed using iScript RT Supermix for RT-qPCR (1708841, Bio-Rad). For quantitative PCR, template cDNA, primers, Taqman Gene Expression primer/probe and Taqman Gene Expression Master Mix (4369016, Applied Biosystems) were used according to the manufacturer’s instructions. The following human primer/probes from Applied Biosystems were used: TNF (Hs00174128_m1), IL12A (Hs01073447_m1), GAPDH (Hs02786624_G1), TAP1 (Hs00388675_m1), CD206 (Hs00267207_m1), CD80 (Hs01045161_m1) and IFNβ (Hs01077958_s1).
T cell stimulation assays.
NY-ESO-1 antigen processing and presentation assay.
Primary human macrophages were transduced with HLA-A201-P2A-NY-ESO1 Vpx lentivirus or not (Ag and No Ag, respectively). 1G4 NY-ESO-1 TCR T cells were generated as previously described and stained with CellTrace Violet (CTV) Cell Proliferation Kit (C34557, Thermo Fisher) according to the manufacturers instructions36. Forty-eight hours after lentiviral transduction, macrophages were transduced with Ad5f35-CAR-HER2-ζ for polarization, or not, for an additional 48 h before the addition of CTV-labeled 1G4 anti-NY-ESO1 TCR autologous T cells for 5 d. Proliferation of anti-NYESO1 TCR+CD8+ T cells was determined by FACS by measuring dilution of CTV.
NY-ESO-1 cross-presentation assay.
CAR-Ms were generated from an HLA-A0201+ normal donor leukopack. SKOV3 was transduced with lentiviral vectors to express NY-ESO-1 only or HLA-A0201 and NY-ESO-1. NY-ESO-1-specific T cells were generated by transducing human T cells with a lentivirus encoding the 1G4 anti-NYESO-1 specific TCR and knocking out TRAC using CRISPRγCas9 to reduce background activation, as previously described37. Next, 5 × 104 CAR-Ms were cultured with 1 × 104 SKOV-NY or SKOV-A0201-NY for 48 h in 96-well plates. As controls, macrophages and SKOV variants were also cultured separately. Then, 1 × 105 T cells were added to each condition. Twenty-four and 96 h later, T cells were analyzed for activation markers using FACS. The following antibodies were used: human CD4 PerCP/Cy5.5 (344608, BioLegend), human CD8 BV421 (344748, BioLegend), human CD3 BV711 (317328, BioLegend), human CD11b FITC (101206, BioLegend), human CD69 PE (3110906, BioLegend) and L/D Aqua (Thermo Fisher). Additionally, T cell activation was evaluated by measurement of IFN in the supernatant using MSD U-Plex Kits (Meso Scale Diagnostics).
Mitochondrial respiratory analysis in human macrophages.
Mitochondrial function was assessed using an extracellular flux analyzer (Agilent Seahorse Bioscience). Primary human control or CAR macrophages (48 h after transduction), with or without 24-h exposure to 20 ng ml−1 recombinant human IL-4 (200-04, PeproTech) were seeded at 1 × 105 cells per well onto XF96 cell culture microplates. To assay mitochondrial function, the medium was replaced with XF assay base medium supplemented with 5.5 mM glucose, 2 mM L-glutamine and 1 mM sodium pyruvate. Before use, XF96 assay cartridges were calibrated in accordance with the manufacturers instructions. During instrument calibration (60 min) the cells were switched to a CO2-free, 37 °C incubator. Cellular oxygen consumption rates and extracellular acidification levels were measured under basal conditions and after treatment with 1.5 μM oligomycin, 1.5 μM fluoro-carbonyl cyanide phenylhydrazone and 40 nM rotenone, with 1 μM antimycin A.
Single-cell RNA-seq.
Raw sequencing reads were mapped to the customized human reference genome (GRCH38) using CellRanger V3 (10× Genomics). CellRanger-filtered cells and their gene expression profiles were further processed using Seurat v3.0.1 (https://doi.org/10.1101/460147). For additional quality control, any cells with fewer than 500 expressed genes, with an unusually high number of unique molecular identifier counts (above 3 median absolute deviations) or with high mitochondrial RNA expression (above 3 median absolute deviations) were removed. To compare T cells between UTD and CAR macrophage samples, libraries from UTD and CAR macrophage samples were first merged and corrected for batch effect using the fastMNN package (https://doi.org/10.1038/nbt.4091) from Seurat Wrappers. Next, all cells were clustered with 0.4 resolution and visualized using UMAP reduction with reduction = mnn and dims = 1:30 parameters. Then, T cells that were GFP negative, RPS4Y1 negative and positive for CD3D and PTPRC were isolated and reclustered. Finally, differentially expressed genes among clusters were identified using FindMarkers function with threshold values of min.pct = 0.25 and log fold change = 0.5.
T cell chemotaxis.
Human CD3+ cells were separated using CD3 microbeads (Miltenyi Biotec) in a MACS cell separation column (Miltenyi Biotec) according to the manufacturer’s instructions. Purity of the retained fraction was more than 95% CD3+, as assessed by flow cytometry. Resting T cells were labeled using CFSE (Thermo Fisher) according to the manufacturer’s instructions and used immediately after being resuspended in RPMI and 0.5% BSA. In some cases, T cells were activated before CFSE labeling using CD3/CD28 Dynabeads (Thermo Fisher) along with 30 U ml−1 IL-2 (PeproTech) for 72 h.
Next, 96-well transwell systems (Corning) were functionalized with 10 μg ml−1 of fibronectin (R&D Systems) in PBS for 3 h at 37 °C. The PBS solution was carefully aspirated, and labeled T cells were added to the top of the inserts. Conditioned media was then added to the bottom of the wells. After 4 h, the insert was removed and the media in the bottom reservoir was read for CFSE using a plate reader. The chemotactic index was defined as the fractional difference in fluorescence between treatment conditions and RPMI control, divided by the fluorescence of the control.
Statistics.
Statistical analysis was performed in GraphPad Prism 6.0 (GraphPad). Each figure legend denotes the statistical test used. All central tendencies indicate the mean, and all error bars indicate s.e.m. unless otherwise indicated. ANOVA multiple-comparison P values were generated using Tukey’s multiple-comparisons test. All t-tests were two-sided unless otherwise indicated. For all figures, * indicates P < 0.05, ** indicates P < 0.01, *** indicates P < 0.001 and **** indicates P < 0.0001.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
RNA sequencing data that support the findings of this study will be deposited and made publicly available in the NCBI Gene Expression Omnibus repository under accession numbers GSE120084 and GSE120086.
Supplementary Material
Acknowledgements
We acknowledge technical support from the University of Pennsylvania (UPenn) Cell and Vaccine Production Facility, UPenn Human Immunology Core, UPenn Stem Cell and Xenograft Core, UPenn Microscopy Core, UPenn Small Animal Imaging Facility, UPenn Flow Cytometry Core, Wistar Institute Flow Cytometry Core and Baylor Vector Development Laboratory. The Vpx plasmid was a kind gift of N. Landau (Department of Microbiology, New York University). Funding was provided by Abramson Cancer Center and Carisma Therapeutics, Inc. M.K. was partially supported by NIH T32 #T32GM008076. Drawings were created with BioRender (www.biorender.com). We thank S. Kelly, B. Peacock, D. Mitchell, N. Minutolo, A. Smole and L. Sepgyr for review of the manuscript.
Footnotes
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41587-020-0462-y.
Competing interests
M.K. and S.G. are scientific co-founders and Scientific Advisory Board members and hold equity in Carisma Therapeutics. C.J. is a member of the Scientific Advisory Board of Carisma Therapeutics. M.K., S.G. and C.J. are inventors on intellectual property related to this work. S.G. has received research funding from Carisma Therapeutics. A.B., M.Z., M.S., K.G., N.A., Y.O. and M.K. are employees of Carisma Therapeutics. Carisma Therapeutics is a company pursuing the commercial development of this technology.
Supplementary information is available for this paper at https://doi.org/10.1038/s41587-020-0462-y.
References
- 1.Maude SL et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N. Engl. J. Med 378, 439–448 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schuster SJ et al. Chimeric antigen receptor T cells in refractory B-cell lymphomas. N. Engl. J. Med 377, 2545–2554 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beatty GL & O’Hara M Chimeric antigen receptor-modified T cells for the treatment of solid tumors: defining the challenges and next steps. Pharmacol. Ther 166, 30–39 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kingwell K CAR T therapies drive into new terrain. Nat. Rev. Drug Discov 16, 301–304 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Ritchie D et al. In vivo tracking of macrophage activated killer cells to sites of metastatic ovarian carcinoma. Cancer Immunol. Immunother 56, 155–163 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Biswas SK, Allavena P & Mantovani A Tumor-associated macrophages: functional diversity, clinical significance, and open questions. Semin. Immunopathol 35, 585–600 (2013). [DOI] [PubMed] [Google Scholar]
- 7.Zhang Q et al. Prognostic significance of tumor-associated macrophages in solid tumor: a meta-analysis of the literature. PLoS One 7, e50946 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Medrek C, Pontén F, Jirström K & Leandersson K The presence of tumor associated macrophages in tumor stroma as a prognostic marker for breast cancer patients. BMC Cancer 12, 306 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Noy R & Pollard JW Tumor-associated macrophages: from mechanisms to therapy. Immunity 41, 49–61 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee H-W, Choi H-J, Ha S-J, Lee K-T & Kwon Y-G Recruitment of monocytes/macrophages in different tumor microenvironments. Biochim. Biophys. Acta 1835, 170–179 (2013). [DOI] [PubMed] [Google Scholar]
- 11.Mantovani A, Marchesi F, Malesci A, Laghi L & Allavena P Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol 14, 399–416 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morrison C Immuno-oncologists eye up macrophage targets. Nat. Rev. Drug Discov 15, 373–374 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Weiskopf K Cancer immunotherapy targeting the CD47/SIRPα axis. Eur. J. Cancer 76, 100–109 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Weiskopf K & Weissman IL Macrophages are critical effectors of antibodies therapies for cancer. MAbs 7, 303–310 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weiskopf K et al. Engineered SIRPα variants as immunotherapeutic adjuvants to anticancer antibodies. Science 341, 88–91 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roghanian A, Stopforth RJ, Dahal LN & Cragg MS New revelations from an old receptor: immunoregulatory functions of the inhibitory Fc gamma receptor, FcγRIIB (CD32B). J. Leukoc. Biol 103, 1077–1088 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Franken L, Schiwon M & Kurts C Macrophages: sentinels and regulators of the immune system. Cell. Microbiol 18, 475–487 (2016). [DOI] [PubMed] [Google Scholar]
- 18.Barrio MM et al. Human macrophages and dendritic cells can equally present MART-1 Antigen to CD8+ T cells after phagocytosis of gamma-irradiated melanoma cells. PLoS One 7, e40311 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tang-Huau T-L et al. Human in vivo-generated monocyte-derived dendritic cells and macrophages cross-present antigens through a vacuolar pathway. Nat. Commun 9, 2570 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hume DA Macrophages as APC and the dendritic cell myth. J. Immunol 181, 5829–5835 (2008). [DOI] [PubMed] [Google Scholar]
- 21.Andreesen R, Hennemann B & Krause SW Adoptive immunotherapy of cancer using monocyte-derived macrophages: rationale, current status, and perspectives. J. Leukoc. Biol 64, 419–426 (1998). [DOI] [PubMed] [Google Scholar]
- 22.Thiounn N et al. Adoptive immunotherapy for superficial bladder cancer with autologous macrophage activated killer cells. J. Urol 168, 2373–2376 (2002). [DOI] [PubMed] [Google Scholar]
- 23.Burger M et al. The application of adjuvant autologous antravesical macrophage cell therapy vs. BCG in non-muscle invasive bladder cancer: a multicenter, randomized trial. J. Transl. Med 8, 54 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gaggar A, Shayakhmetov DM & Lieber A CD46 is a cellular receptor for group B adenoviruses. Nat. Med 9, 1408–1412 (2003). [DOI] [PubMed] [Google Scholar]
- 25.Nilsson M et al. Development of an adenoviral vector system with adenovirus serotype 35 tropism; efficient transient gene transfer into primary malignant hematopoietic cells. J. Gene Med 6, 631–641 (2004). [DOI] [PubMed] [Google Scholar]
- 26.Fleetwood AJ, Lawrence T, Hamilton JA & Cook AD Granulocyte-macrophage colony-stimulating factor (CSF) and macrophage CSF-dependent macrophage phenotypes display differences in cytokine profiles and transcription factor activities: implications for CSF blockade in inflammation. J. Immunol 178, 5245–5252 (2007). [DOI] [PubMed] [Google Scholar]
- 27.Wunderlich M et al. AML xenograft efficiency is significantly improved in NOD/SCID-IL2RG mice constitutively expressing human SCF, GM-CSF and IL-3. Leukemia 24, 1785–1788 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mosser DM & Edwards JP Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol 8, 958–969 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lam E, Stein S & Falck-Pedersen E Adenovirus detection by the cGAS/STING/TBK1 DNA sensing cascade. J. Virol 88, 974–981 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Muruve DA et al. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature 452, 103–107 (2008). [DOI] [PubMed] [Google Scholar]
- 31.Kelly B & O’Neill LA Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 25, 771–784 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Robbins PF et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: long-term follow-up and correlates with response. Clin. Cancer Res 21, 1019–1027 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gill S et al. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor-modified T cells. Blood 123, 2343–2354 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bobadilla S, Sunseri N & Landau NR Efficient transduction of myeloid cells by an HIV-1-derived lentiviral vector that packages the Vpx accessory protein. Gene Ther. 20, 514–520 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liao Y, Smyth GK & Shi W featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 36.Rapoport AP et al. NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat. Med 21, 914–921 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ren J et al. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin. Cancer Res 23, 2255–2266 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA sequencing data that support the findings of this study will be deposited and made publicly available in the NCBI Gene Expression Omnibus repository under accession numbers GSE120084 and GSE120086.