

Abstract
Clinical response to chimeric antigen receptor (CAR) T cell therapy is correlated with CAR T cell persistence, especially for CAR T cells that target CD19+ hematologic malignancies. 4-1BB–costimulated CAR (BBζ) T cells exhibit longer persistence after adoptive transfer than do CD28-costimulated CAR (28ζ) T cells. 4-1BB signaling improves T cell persistence even in the context of 28ζ CAR activation, which indicates distinct prosurvival signals mediated by the 4-1BB cytoplasmic domain. To specifically study signal transduction by CARs, we developed a cell-free, ligand-based activation and ex vivo culture system for CD19-specific CAR T cells. We observed greater ex vivo survival and subsequent expansion of BBζ CAR T cells when compared to 28ζ CAR T cells. We showed that only BBζ CARs activated noncanonical nuclear factor κB (ncNF-κB) signaling in T cells basally and that the anti-CD19 BBζ CAR further enhanced ncNF-κB signaling after ligand engagement. Reducing ncNF-κB signaling reduced the expansion and survival of anti-CD19 BBζ T cells and was associated with a substantial increase in the abundance of the most pro-apoptotic isoforms of Bim. Although our findings do not exclude the importance of other signaling differences between BBζ and 28ζ CARs, they demonstrate the necessary and nonredundant role of ncNF-κB signaling in promoting the survival of BBζ CAR T cells, which likely underlies the engraftment persistence observed with this CAR design.
INTRODUCTION
Chimeric antigen receptor (CAR) T cell therapy targeting CD19 has induced complete tumor regression in a large percentage of patients suffering from hematologic malignancies, especially in children and young adults with acute lymphoblastic leukemia (ALL) (1). Event-free survival after CD19-specific CAR T cell therapy is highly correlated with the kinetics of T cell expansion (increase in cell number) and the area under the concentration-time curve (AUC) for the first 28 days after adoptive transfer (2). Although loss of antigen or epitope remains the most frequent mechanism of resistance to this therapy (3), relapse within the first 6 months is associated with the early loss of CAR T cell engraftment, illustrating the importance of persistent T cell engraftment to the success of adoptive cell therapy approaches (2, 4, 5).
The persistence of CAR T cells in vivo over time after adoptive transfer is influenced by CAR design. CARs are transmembrane proteins consisting of an antibody single-chain variable fragment (scFv), a hinge connecting to a transmembrane domain, and one or more intracellular signaling domains (6). The most commonly used designs combine the T cell receptor (TCR) subunit CD3ζ with the intracellular domain of either of the costimulatory molecules, CD28 or 4-1BB. Comparing CAR T cell persistence across human clinical trials using 28ζ or BBζ CD19-specific CARs, although fraught with pitfalls due to the other differences in the CARs used or in T cell manufacturing (for example, scFv, gene transfer vector, or culture conditions), suggests the greater persistence of 4-1BB–costimulated CAR T cell engraftment relative to CD28-costimulated CAR T cells (7, 8). This trend is more directly substantiated in murine adoptive transfer models, implying that costimulation directly affects CAR T cell persistence (9). When exchanged with CD28 costimulation, 4-1BB costimulation also rescues CAR T cell survival in the context of a basally signaling CAR (10), and basally signaling BBζ CAR T cells markedly expand ex vivo (11).
The signaling pathways that lead to the observed differences in CAR T cell persistence remain poorly understood. Salter et al. (12) demonstrated that 28ζ CARs drive both constitutive and CAR activation–induced proximal signaling through the kinases Lck and Zap-70 more potently than do the BBζ CARs. This potent proximal signaling promotes the terminal differentiation of 28ζ CAR T cells, which can be reversed by silencing two of the immunoreceptor tyrosine-based activation motif (ITAM) domains of the CD3ζ domain (13). This reversal suggests that CD28 signaling in the CAR is a dominant mechanism for the reduced persistence of these CAR T cells. However, 4-1BB costimulation enhances CAR T cell survival and persistence even in the context of the full 28ζ CAR signal (14, 15). This enhancement is consistent with the nonoverlapping roles that these costimulatory receptors play in normal immunity, especially for CD8+ T cell memory (16-20). Long-term CD8+ T cell survival after viral infections is supported by specific tumor necrosis factor receptor superfamily (TNFRSF) members, including 4-1BB (21, 22). Unlike CD28, a distinct property of these receptors, and one that was specifically observed in one report on murine 4-1BB, is the capacity to activate the non-canonical nuclear factor κB (ncNF-κB) pathway (23-26). Distinct from the classical NF-κB signaling pathway downstream of TCR and CD28 stimulation mediated by the CARMA1 complex and protein kinase Cθ (PKCθ) (23, 27-29), the ncNF-κB pathway promotes cell survival in numerous contexts, including normal lymphoid development under the control of TNFRSF members (30-32), Epstein-Barr virus infection (33), and malignancy (34-37).
ncNF-κB signaling is a complex pathway with slower activation kinetics compared with that of classical NF-κB signaling. It is initiated by the stabilization of NF-κB–inducing kinase (NIK), which is Lys48 (K48)–polyubiquitylated by the E3 ubiquitin ligases cellular inhibitors of apoptosis 1 (cIAP1) and 2 (cIAP2) and constitutively degraded by the proteasome (38). Upon ligand-dependent TNFRSF trimerization, differential TNF receptor–associated factor (TRAF) recruitment shifts cIAP1/2-mediated K48 polyubiquitylation from NIK to TRAF3, thereby alleviating the continuous proteasomal degradation of NIK. NIK accumulates and coordinates a protein complex including inhibitor of NF-κB kinase α (IKKα) and the Rel homology domain family members RelB and p100, the latter two of which form a heterodimer that is basally sequestered in the cytoplasm. Accumulating NIK phosphorylates IKKα, which, in turn, phosphorylates p100, generating a phosphodegron that leads to the partial degradation of p100 by the proteasome to form p52. The newly formed RelB-p52 heterodimer then translocates to the nucleus where it mediates transcriptional control of numerous genes involved in lymphoid development (38) and, in cancer cells, synergizes with extracellular signal–regulated kinase (ERK) to suppress genes expressing the proapoptotic proteins Bim and Bcl-2-modifying factor (BMF) (fig. S1) (35). Similar to 4-1BB signaling, ncNF-κB signaling is also essential for T cell–mediated immunity. Memory T cells require ncNF-κB signaling to survive both in mice (39, 40) and in humans; the latter requirement was revealed by a deficiency in long-lived memory T cells in two patients with biallelic, loss-of-function mutations in NIK (32). Similarly, mice deficient in NIK form few persistent memory T cells when challenged with acute lymphocytic choriomeningitis virus infection (40). Furthermore, T cell–specific NIK knockout mice have substantially fewer circulating memory T cells than their wild-type counterparts, suggesting that NIK supports T cell persistence through a cell-intrinsic mechanism (39).
On the basis of the importance of ncNF-κB signaling to T cell memory formation and the report of activation of this pathway by endogenous 4-1BB in murine T cells, we evaluated the activation and role of this pathway in human CAR T cells. We showed that ncNF-κB signaling played an essential role in supporting BBζ CAR T cell survival. In addition to providing greater insight into the costimulatory mechanisms of the CD28 and 4-1BB domains in CARs, the increased understanding of the role of this pathway in human T cells offers opportunities to manipulate it more broadly in T cell–based immunotherapies that rely upon T cell survival to mediate clinical benefit.
RESULTS
4-1BB costimulation enhances ex vivo CAR T cell expansion relative to CD28 costimulation and is associated with improved T cell survival
To study the ex vivo expansion of T cells after stimulation by CD19-specific CARs bearing different costimulatory domains, we used the approach of repeated in vitro stimulation (13-15, 41). We added irradiated Nalm6 leukemic cells bearing the CD19 target antigen to mCherry-expressing 28ζ or BBζ CAR T cell cultures (fig. S2) at 7-day intervals (Fig. 1A). Similar to previous studies comparing CD28- and 4-1BB–costimulated CARs, T cells bearing a 4-1BB–costimulated CAR (BBζ CAR T cells) accumulated over 14 days to yield about 1-log more T cells than CD28-costimulated CAR T cells (28ζ CAR T cells) (Fig. 1B). This increased expansion was associated with improved viability, which we observed by a 14% (±3.2%) absolute reduction in T cells that acquired 7-aminoactinomycin D (7AAD) fluorescence and lost mCherry on day 14 for BBζ CAR T cells relative to 28ζ CAR T cells (Fig. 1C). To determine whether there was also a difference in relative proliferation, we measured the abundance of proliferating cell nuclear antigen (PCNA) by Western blotting (Fig. 1D) and found no statistically significant difference in PCNA abundance between 28ζ and BBζ CAR T cells at day 0, 7, or 14 after CAR restimulation (Fig. 1E).
Fig. 1. 4-1BB–costimulated CAR activation drives greater ex vivo T cell expansion and survival than does CD28-costimulated CAR activation.

(A) Representative ex vivo expansion of the indicated CAR T cells stimulated by irradiated Nalm6 target cells at the indicated times at a T cell to target cell ratio of 2:1. (B) Quantification of T cell expansion after 14 days of ex vivo culture from the experiments represented in (A). (C) Quantification of cell death by measurement of the percentage of 7AAD+mCherry− cells in all events collected on day 14 of ex vivo culture from the experiments represented in (A). (D) Representative Western blotting analysis of PCNA abundance in the indicated CAR T cells at 0, 7, and 14 days of ex vivo culture with Nalm6 target cells. (E) Quantification of the relative amounts of PCNA in the indicated CAR T cells on day 14 of ex vivo culture with Nalm6 target cells. Data in (B), (C), and (E) are from three donors in two independent experiments and were analyzed by paired Student’s t test. (F) Representative ex vivo expansion of the indicated CAR T cells stimulated by anti-CD19 idiotype–coated target beads (at a T cell to bead ratio of 1:3) added at day 0 (green arrow) and removed at day 14 of culture (black arrow). (G) Quantification of T cell expansion after 14 days of ex vivo culture from the experiments represented in (F). (H) Quantification of cell death as assessed by measurement of the percentage of 7AAD+mCherry− cells in all events collected on day 14 of ex vivo culture from the experiments represented in (F). Data in (G) and (H) are from three donors in three independent experiments and were analyzed by paired Student’s t test. (I) Quantification of the relative abundance of PCNA as assessed by Western blotting analysis of the indicated CAR T cells on days 0, 7, and 14 of ex vivo culture with target beads. Data are from donors in three independent experiments and were analyzed by paired Student’s t test.
To identify signaling pathways that enhanced BBζ CAR T cell survival, we used a bead-based, artificial antigen-presenting cell (aAPC) approach for activating CD19-specific CAR T cells. We coated magnetic microbeads with anti-CD19 idiotype antibodies to cross-link the anti-CD19 CAR. This approach avoided the introduction of target cell proteins and RNA that confound analyses of signaling and did not add additional signals to the CAR T cells through other costimulatory interactions such as by integrin and CD28 activation (42). Furthermore, such beads have been used by our group and others to study CAR signaling and its effects on T cell expansion, metabolism, and differentiation (12, 43, 44). Similar to the studies using activation by Nalm6 leukemic cells, BBζ CAR T cells activated by these beads also expanded about a log more than did 28ζ CAR T cells (Fig. 1, F and G, and fig. S3). These expansions were less than those achieved with the irradiated target cells, likely due to a loss of additional costimulatory signals provided by those cells. Even so, we observed a 8.9% (±3.1%) absolute reduction in 7AAD+mCherry− cells on day 14 of BBζ CAR T cells relative to 28ζ CAR T cells (Fig. 1H) and did not detect a statistically significant difference in PCNA abundance (Fig. 1I). From these data, we conclude that our bead-based expansion system achieved similar outcomes as did the irradiated cell system.
CAR intracellular domains activate overlapping but kinetically and quantitatively distinct signaling pathways upon antigen stimulation
Both proximal and distal signaling pathways activated by the endogenous TCR and costimulatory molecules influence the proliferation and survival of T cells after CAR activation (45-47). To validate that our bead-based activation system stimulated similar T cell signaling pathways, we compared Zap-70, p65, and ERK1/2 phosphorylation in T cells expressing an anti-CD19 CAR containing no signaling domains (Δζ), the CD3 zeta chain alone (ζ), also known as a first-generation CAR, or one of two second-generation combinations, 28ζ or BBζ (fig. S4). Similar to recent work by Salter et al., we observed that 28ζ CAR–mediated Zap-70 phosphorylation was 4.5-fold greater than that mediated by the BBζ CAR 12 hours after bead addition and 3.7-fold greater than that mediated by the ζ CAR 30 min after bead addition (Fig. 2, A and B). The phosphorylation of p65 at Ser536, a measure of canonical NF-κB signaling, increased across all signaling CARs after bead addition, with a trend toward increased p65 phosphorylation early after activation of the 28ζ CAR and a small but statistically significant increase of 1.34-fold in the BBζ CAR T cells relative to that in the 28ζ CAR T cells at 12 hours (Fig. 2C). Also, like the work by Salter et al., ERK1 and ERK2 phosphorylation both increased with activation of all signaling CARs over time (Fig. 2, D and E), with ERK2 phosphorylation increasing 2.2- and 1.9-fold by 28ζ and BBζ CAR activation, respectively, relative to that by ζ-only CAR activation at 30 min (Fig. 2E). We conclude from these data that the bead-based aAPC system activated anti-CD19 CARs, leading to CAR signaling in a similar manner to that reported in the literature (12, 43), providing a robust system for evaluating signaling downstream of our CD19-specific CARs.
Fig. 2. Anti-CD19 idiotype target beads activate T cell signaling in anti-CD19 CAR T cells.

(A) Representative Western blotting of total and phosphorylated Zap-70, p65, and ERK1/2 proteins before and at 30 min and 12 hours after the addition of anti-CD19 beads to the indicated CAR T cells at a bead–to–T cell ratio of 5:1. The anti-CD19 CAR T cells used expressed CARs with no signaling domain (Δζ), the CD3ζ chain intracellular domain alone (ζ), the CD28 and CD3ζ chain intracellular domains (28ζ), or the 4-1BB and CD3ζ chain intracellular domains (BBζ). (B to E) Quantification of the relative abundances of phosphorylated Zap-70 (B), p65 (C), ERK1 (D), and ERK2 (E) normalized to their respective total proteins from the experiments represented in (A). P values were determined by two-way ANOVA with Tukey’s multiple comparisons test of data from three donors analyzed in three independent experiments. FC, fold change; TP, time point; NTD, non-transduced.
4-1BB, but not CD28, costimulation potentiates CAR-mediated ncNF-κB signaling
We hypothesized that the 4-1BB intracellular domain incorporated into a CAR specifically promoted CAR T cell survival and persistence through activation of ncNF-κB signaling. To interrogate this pathway and establish optimal time points for comparison across CAR constructs, we first determined the kinetics of activation after engagement of the BBζ CAR (fig. S5, A to D). We measured ncNF-κB signaling using the commonly reported metrics of p100 processing, p100 phosphorylation (Fig. 3, A and B), and p52/p100 ratio (Fig. 3, A and C) (48). We found that p100 phosphorylation was statistically significantly increased after bead addition, peaking at 6.1-fold over baseline at 12 hours (Fig. 3B). Similarly, the p52/p100 ratio was statistically significantly increased over the first 12 hours after BBζ CAR activation, with a peak of 1.87-fold over baseline at 12 hours (Fig. 3C). The abundance of the initiator kinase for this pathway, NIK, was statistically significantly increased after BBζ CAR activation with a peak of 8.7-fold over baseline also at 12 hours after CAR activation (Fig. 3, D and E). ncNF-κB signaling culminates in the nuclear translocation of RelB and p52. Therefore, we also measured RelB and p52 nuclear translocation in isolated nuclear fractions, as confirmed by loss of vinculin (Fig. 3F), at similar times after BBζ CAR activation using the same initial setup as for our assessment of NIK and p100 signal transduction. Cytoplasmic RelB increased and stayed above baseline after 8 hours of bead stimulation (Fig. 3F and fig. S5E), reflecting previously described increases in abundance after canonical NF-κB signaling (49). Furthermore, both RelB and p52 nuclear localizations were detectable before and increased after BBζ CAR activation (Fig. 3F and fig. S5E), indicating basal and ligand-induced activation of ncNF-κB signaling. Consistent with the delayed kinetics of this pathway relative to that of the canonical NF-κB pathway (24), we observed peak p100 processing and NIK stabilization 12 hours after BBζ CAR activation (Fig. 3, C and E).
Fig. 3. 4-1BB–costimulated anti-CD19 CAR activation stimulates CAR T cell ncNF-κB signaling.
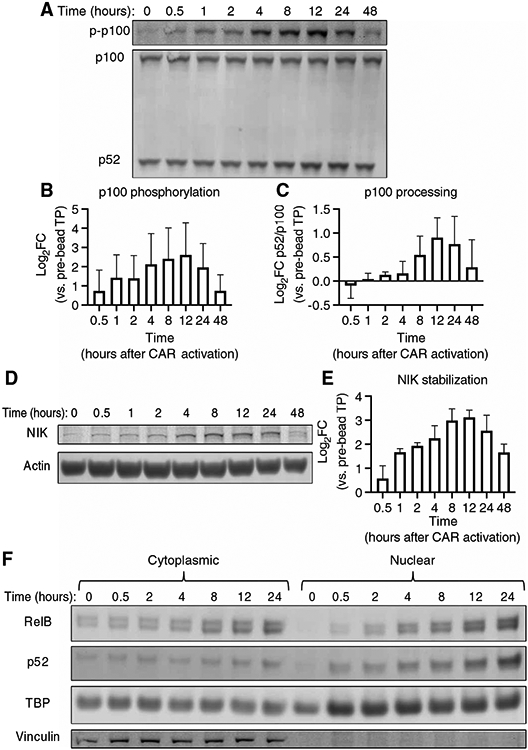
(A) Representative Western blotting analysis of p100 phosphorylation and the processing of p100 to p52 at the indicated times before and after the addition of target beads to anti-CD19 BBζ CAR T cells. (B and C) Quantitative analyses of the relative amounts of phosphorylated p100 (B) (P = 0.0397 as determined by analyzing the data from two independent experiments using CAR T cells derived from three healthy donors by one-way ANOVA with the factor being time, as is the case for all of the P values in this figure) and the relative amounts of p52 generated by p100 processing (C) (p52/p100 ratio, P = 0.0244) from the experiments represented in (A). (D) Representative Western blotting analysis of NIK protein at the indicated times before and after target bead addition to anti-CD19 BBζ CAR T cells. Actin was used as a loading control. (E) Quantitative analysis of the relative amounts of NIK protein at the indicated times after the addition of beads to the cells in the experiments represented in (D) (P = 0.011). (F) Representative Western blotting analysis of the cytoplasmic and nuclear fractions from anti-CD19 BBζ CAR T cells before and at the indicated times after the addition of target beads. The blot is representative of two separate experiments using CAR T cells generated from two healthy donors (see fig. S6 for the other experiment).
To test the hypothesis that ncNF-κB signaling occurred solely in 4-1BB–costimulated CAR T cells, we compared ncNF-κB signaling by anti-CD19–targeted Δζ, ζ, 28ζ, or BBζ CARs before activation and at 30 min and 12 hours after bead addition. We found that p100 phosphorylation was 3.2-fold greater 12 hours after bead addition in BBζ CAR T cells relative to that in 28ζ CAR T cells (Fig. 4, A and B). We observed a similar trend in p100 phosphorylation when comparing the BBζ and ζ-only CAR T cells. The p52/p100 ratio was statistically significantly increased in BBζ CAR T cells relative to that in 28ζ CAR T cells at all times, including before activation (1.8-fold greater) and 12 hours afterward (2.4-fold greater), whereas no difference in the p52/p100 ratio was observed between the 28ζ and ζ-only CAR T cells (Fig. 4C). Although NIK stabilization was enhanced after BBζ CAR activation relative to after activation of the other CARs (Fig. 4D), this trend was not statistically significant (Fig. 4E). The downstream basal ncNF-κB signaling in BBζ CAR T cells relative to that in 28ζ CAR T cells was also reflected in increased baseline RelB nuclear occupancy (Fig. 4F and fig. S6), which was 1.3-fold greater in BBζ CAR T cells relative to that in the 28ζ CAR T cells (Fig. 4G). RelB nuclear translocation increased further 12 hours after CAR activation and was 5.4-fold greater in BBζ CAR T cells relative to that in 28ζ CAR T cells (Fig. 4G). Although we observed increased p52 nuclear occupancy in BBζ CAR T cells at all times relative to that in the ζ and 28ζ CAR T cells (Fig. 4F), this increase reached statistical significance for BBζ versus 28ζ CAR T cells only at the 30-min time point and for BBζ versus ζ CAR T cells only at the 12-hour time point (Fig. 4H).
Fig. 4. 4-1BB, but not CD28, CAR costimulation drives basal and enhances CAR activation–induced ncNF-κB signaling.

(A) Representative Western blotting analysis of the phosphorylation of p100 and of its processing to p52 in the indicated CAR T cells at the indicated times before and after the addition of target beads at a bead–to–T cell ratio of 5:1. The anti-CD19 CARs used had no signaling domain (Δζ), the CD3ζ chain intracellular domain alone (ζ, green), the CD28 and CD3ζ chain intracellular domains (28ζ, red), or the 4-1BB and CD3ζ chain intracellular domains (BBζ, blue). (B and C) Quantitative analyses of the relative abundances of phosphorylated p100 (B) and of the ratio of p52 to p100 (C) from the experiments represented in (A). (D) Representative Western blotting analysis of NIK protein in the indicated CAR T cells at the indicated times before and after the addition of target beads as described for (A). Actin was used as a loading control. (E) Quantitative analysis of relative amounts of NIK protein in the indicated cells from the experiments represented in (D). (F) Representative Western blotting analysis of RelB, p52, and TBP (loading control) in nuclear fractions from the indicated anti-CD19 CAR T cells before and at the indicated times after the addition of target beads as described for (A). (G and H) Quantitative analyses of the relative amounts of nuclear RelB (G) and nuclear p52 (H) in the indicated cells from the experiments represented in (F). Data in (B), (C), (E), (G), and (H) are from three donors in three independent experiments. All log2(fold change relative to nonsignaling group) quantifications of band fluorescence intensities were normalized to those of the loading control. P values reported were determined by two-way ANOVA with Tukey’s multiple comparisons test.
To confirm the particular association between the BBζ CAR and ncNF-κB signaling in a CAR with an alternative scFv and antigen specificity (fig. S7), we compared the p52/p100 ratio in SS1-28ζ or SS1-BBζ CARs targeting mesothelin (fig. S7, A and C) before activation and at 30 min and 12 hours after the addition of mesothelin-coated beads (fig. S7, B and D to F). Similar to what we found with the CD19-specific CAR T cells, we observed a greater p52/p100 ratio in the SS1-BBζ CAR T cells compared to that in the SS1-28ζ CAR T cells from two separate donors before activation (fig. S7, E and F). Unlike in the anti-CD19 CAR T cells, the p52/p100 ratio decreased after the addition of mesothelin-coated beads to the SS1-BBζ CAR T cells, but it did not decrease in the SS1-28ζ CAR T cells under the same conditions. Together, these data suggest that the 4-1BB signaling domain within the CAR basally activates the ncNF-κB pathway regardless of the CAR target. Although the degree of antigen-induced enhancement of ncNF-κB signaling by BBζ CARs differs based on the scFv, nature of the ligand, or both, BBζ CAR T cells exhibited greater ncNF-κB signaling compared to that in 28ζ CAR T cells.
Expression of a dominant-negative NIK peptide reduces BBζ-mediated ncNF-κB signaling, ex vivo BBζ CAR T cell expansion, and survival
Targeting NIK has been a widely used strategy to block ncNF-κB signaling (50). Therefore, we used this approach to interrupt ncNF-κB signaling downstream of BBζ CAR activation by overexpressing the C terminus of NIK (dnNIK) in CAR T cells. dnNIK acts similarly to a dominant-negative mutant NIK protein by binding to p100 and thereby preventing downstream ncNF-κB signaling (51). Twenty-four hours after initially transducing primary human T cells with the BBζ CAR, we transduced the cells with either a control vector expressing mCherry alone or a bicistronic vector expressing dnNIK and mCherry. This strategy enabled us to compare ncNF-κB signaling in each group of cells by Western blotting analysis while maintaining equal CAR abundance across groups (fig. S8). We observed a 1.5-fold decrease in the p52/p100 ratio 12 hours after CAR activation in dnNIK-expressing T cells relative to that in control T cells (Fig. 5, A and B). Similarly, we observed 1.4- and 1.9-fold decreases in RelB nuclear occupancy 30 min and 12 hours, respectively, after BBζ CAR activation in dnNIK-expressing T cells relative to that in control T cells (Fig. 5, C and D, and fig. S9). We observed a similar trend in p52 nuclear occupancy, with statistically significant decreases of 1.9- and 2-fold at baseline and 12 hours after CAR activation, respectively, in dnNIK-expressing T cells relative to that in control T cells (Fig. 5, C and E, and fig. S9).
Fig. 5. Reducing 4-1BB costimulation–mediated ncNF-κB signaling diminishes BBζ CAR T cell ex vivo expansion and survival.

(A) Representative Western blotting analysis of p100 processing to p52 before, 30 min, and 12 hours after the addition of target beads to anti-CD19 BBζ CAR T cells expressing either mCherry (control) or mCherry and dnNIK. (B) Quantitative analysis of the ratio of p52 to p100 ratio from the experiments represented in (A). Data are from CAR T cells generated from three donors in three independent experiments. P value was determined by two-way ANOVA with Holm-Sidak’s multiple comparisons test. (C) Representative Western blotting analysis of RelB, p52, and TBP (loading control) in nuclear fractions of anti-CD19 BBζ CAR T cells treated as described for (A). (D and E) Quantitative analyses of the nuclear localization of RelB (D) and p52 (E) in cells from the experiments represented in (C) using TBP as the loading control. Data are from two independent experiments of CAR T cells generated from three donors. P values were determined by two-way ANOVA with Holm-Sidak’s multiple comparisons test. (F) Representative ex vivo expansion of control or dnNIK-expressing BBζ CAR T cells stimulated by anti-CD19 idiotype–coated target beads (at a T cell–to–bead ratio of 1:3) and added at day 0 (green arrow) and removed at day 14 of culture (black arrow). (G) Quantification of T cell expansion after 14 days of ex vivo culture from the experiments represented in (F). (H) Quantification of cell death by measurement of the percentage of 7AAD+mCherry− cells in all events collected on day 14 of the ex vivo culture represented in (F). (I) Representative ex vivo expansion of control or dnNIK-expressing 28ζ CAR T cells stimulated by anti-CD19 idiotype–coated target beads at a T cell–to–bead ratio of 1:3 and added at day 0 (green arrow) and removed at day 14 of culture (black arrow). (J) Quantification of T cell expansion after 14 days of ex vivo culture from the experiments represented in (I). (K) Quantification of cell death by measurement of the percentage of 7AAD+mCherry− cells in all events collected on day 14 of the ex vivo culture represented in (I). All P values were determined by paired Student’s t tests of data from four donors in four independent experiments.
Having observed a reduction in ncNF-κB signaling as a result of dnNIK expression, we used this approach in the bead-based CAR T cell expansion model to test the hypothesis that ncNF-κB signaling mediated anti-CD19 4-1BB–costimulated CAR T cell survival. We observed that dnNIK-expressing BBζ CAR T cells expanded 3.1-fold less than did control BBζ CAR T cells after 14 days of exposure to anti-CD19 idiotype beads (Fig. 5, F and G). This reduction in expansion was associated with a 6.2% absolute increase in cell death as assessed by measurement of 7AAD uptake by flow cytometry on day 14 (Fig. 5H). We did not detect a significant difference in PCNA expression between control and dnNIK-expressing BBζ CAR T cells before or 14 days after CAR T cell activation (fig. S10, A and B). To exclude the possibility that dnNIK nonspecifically caused CAR T cell death after CAR activation, we conducted these studies with anti-CD19 28ζ CAR T cells in parallel (Fig. 5, I to K). We observed no statistically significant differences in cell expansion (Fig. 5, I and J), 7AAD uptake (Fig. 5K), or PCNA abundance (fig. S10, C and D) between control and dnNIK-expressing 28ζ CAR T cells on day 14 of culture. These data indicate that 4-1BB–costimulated anti-CD19 CAR T cells specifically require ncNF-κB signaling to promote cell survival.
4-1BB costimulation–mediated ncNF-κB signaling represses the pro-apoptotic protein Bim
ncNF-κB signaling in multiple myeloma cell lines and 4-1BB costimulation in T cells both lead to enhanced cell survival by suppressing the production of the pro-apoptotic protein Bim (35, 52, 53). From our observation that ncNF-κB signaling supported BBζ CAR T cell survival, we hypothesized that the increased cell death observed in dnNIK-expressing BBζ CAR T cells was due, at least in part, to increased Bim abundance. To test this hypothesis, we assessed Bim abundance by Western blotting analysis at baseline and 14 days after the addition of anti-CD19 idiotype beads to CD19-specific BBζ (Fig. 6, A to E, and fig. S11, A to C) and 28ζ (Fig. 6, F to J, and fig. S11, D to F) CAR T cells transduced with a dnNIK-expressing vector or a control vector. No statistically significant difference was detected in the amounts of any of the Bim isoforms at baseline between dnNIK-expressing and control CAR T cells expressing either the BBζ (Fig. 6, B and C, and fig. S11B) or 28ζ (Fig. 6, G and H, and fig. S11E) CARs. The abundance of BimL was statistically significantly greater in dnNIK-expressing BBζ cells than in BBζ cells at day 14 (Fig. 6D), with a similar trend observed for BimS (Fig. 6E). No statistically significant differences in the abundances of any of the isoforms of Bim were apparent between dnNIK-expressing 28ζ CAR T cells and control vector–expressing 28ζ CAR T cells at day 14 (Fig. 6, I and J, and fig. S11F). Comparing Bim isoform abundances between 28ζ and BBζ CAR T cells at baseline (Fig. 6, K and L, and fig. S11, G and H) revealed a statistically significant decrease in BimS abundance in the BBζ cells with a similar trend observable for BimL (Fig. 6, K and L). Together, these observations of differential Bim isoform abundance before CAR activation between 28ζ and BBζ CAR T cells, especially with respect to the low amounts of BimS in BBζ cells and the increase in Bim isoform abundance in the context of dnNIK-expressing cells after CAR activation, are consistent with bcl2l11 (which encodes Bim) or its products as a target of ncNF-κB signaling in BBζ CAR T cells. The failure to detect a difference in Bim isoform abundance when ncNF-κB signaling was disrupted by the presence of dnNIK before CAR activation may be a result of the low overall amounts of BimS and BimL.
Fig. 6. 4-1BB costimulation–mediated ncNF-κB signaling opposes expression of the pro-apoptotic protein Bim.
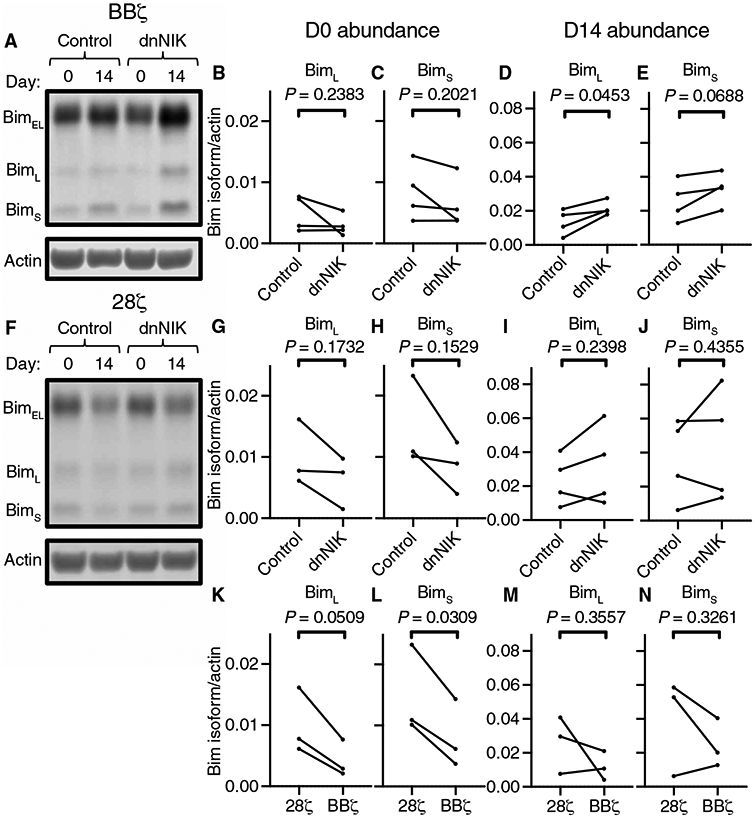
(A) Representative Western blotting analysis of Bim isoforms in control and dnNIK-expressing BBζ CAR T cells before and 14 days after ex vivo expansion in response to the addition of beads. (B to E) Quantitation of the relative basal amounts of BimL (B) and BimS (C), as well as of the amounts of BimL (D) and BimS (E) 14 days after the addition of beads from the experiments represented in (A). Data in (B) to (E) are from four donors in four independent experiments. All P values were derived from paired Student’s t tests. (F) Representative Western blotting analysis of Bim isoforms in control or dnNIK-expressing 28ζ CAR T cells before and 14 days after ex vivo expansion in response to the addition of beads. (G and H) Quantitation of the relative basal amounts of BimL (G) and BimS (H) from the experiments represented in (F). Data are from three independent experiments with three donors. P values were derived from paired Student’s t tests. (I and J) Quantitation of the relative amounts of BimL (I) and BimS (J) 14 days after bead addition from the experiments represented in (F). Data in (I) and (J) are from four donors in four independent experiments. No pre-bead sample was taken for fourth donor shown in (I) and (J). P values were derived from paired Student’s t tests. (K and L) Comparison of the basal amounts of BimL (K) and BimS (L) between 28ζ and BBζ CAR T cells. (M and N) Comparison of the relative amounts of BimL (M) and BimS (N) between 28ζ and BBζ CAR T cells 14 days after expansion in response to beads. Data in (K) to (N) are from three donors in three independent experiments. All P values were derived from paired Student’s t tests.
Although previous work with multiple myeloma cell lines demonstrated cooperative suppression of Bim production by ERK and ncNF-κB signaling (35), previous studies of T cells have only addressed ERK-mediated Bim suppression (53-55). Therefore, we assessed ERK phosphorylation in the dnNIK-expressing BBζ CAR T cells to determine whether dnNIK expression affected BBζ CAR–mediated ERK activation. Thirty minutes after bead addition, we observed a 1.2-fold reduction in ERK1 phosphorylation in dnNIK-expressing CAR T cells relative to that in control cells but saw no statistically significant difference in ERK1 phosphorylation before or 12 hours after bead addition (fig. S12, A and B). We also observed no statistically significant difference in ERK2 phosphorylation at any time (fig. S12C). Thus, although the reduction in ERK1 phosphorylation upon activation in dnNIK-expressing CAR T cells was small and transient, we cannot completely rule out that the observed reductions in Bim abundance are not, in part, due to small reductions in ERK signaling.
Another transcription factor known to drive Bim abundance in T cells is Forkhead box O3a (FOXO3a) (56). FOXO3a is regulated by numerous signaling pathways, including by γ-chain cytokines through phosphorylation of Thr32 and subsequent proteasomal degradation (56, 57). To determine whether γ-chain cytokine signaling was affected by dnNIK and affected Bim abundance through FOXO3a regulation, we assessed FOXO3a phosphorylation at Thr32 and total FOXO3a abundance in control and dnNIK-expressing BBζ CAR T cells at days 0 and 14 of expansion (fig. S13, A and B). We observed no statistically significant differences in FOXO3a abundance between the control and dnNIK-expressing cells at either time (fig. S13B), instead observing a reduction in the amount of FOXO3a from days 0 to 14. We were also unable to detect any phosphorylation of FOXO3a at Thr32 in any of the samples (fig. S13A). Instead, we detected the phosphorylation of FOXO1 at Thr34, which can be distinguished by its smaller size relative to that of FOXO3a in Western blotting analysis (fig. S13A). From these data, we conclude that FOXO3a was unaffected by dnNIK and is unlikely to contribute to changes in Bim abundance.
DISCUSSION
Here, we showed that the 4-1BB cytoplasmic domain incorporated into a CAR potently activates ncNF-κB signaling in human T cells. This activation appeared to be constitutive across two 4-1BB–costimulated CARs with independent antigenic specificity, and ncNF-κB signaling was also increased in CD19-specific BBζ CAR T cells upon CAR engagement with ligand when compared with that in T cells expressing a first-generation CAR or a CD28-costimulated CAR. Moreover, disruption of this pathway substantially impaired anti-CD19 BBζ CAR T cell survival after CAR-mediated activation, leading to reduced T cell expansion ex vivo. This reduced expansion and survival was associated with the increased abundance of the pro-apoptotic protein Bim, the suppression of which mediates the prosurvival effects of ncNF-κB signaling in other systems (35, 52). Thus, we have demonstrated that the 4-1BB costimulatory domain within a CAR directly promotes T cell survival through the ncNF-κB pathway, consistent with previous reports demonstrating that murine endogenous 4-1BB costimulation activates the ncNF-κB signaling (24), whereas endogenous CD28 costimulation does not (23, 58). Our findings also add to our understanding of CAR costimulation because previously published reports have implicated faster and more potent proximal signaling by CD28-costimulated CARs to explain the differences between 4-1BB– and CD28-costimulated CAR T cell survival (12), whereas our results provide an additional mechanism directed by the 4-1BB domain itself.
A frequently observed feature of 4-1BB–costimulated CARs is basal signaling in the absence of antigen, so-called tonic signaling (11, 41, 59). Our data are consistent with these previous reports, and we also observed CD28-costimulated CAR basal signaling through the Zap-70 pathway (Fig. 2, A and B) (12). 4-1BB–costimulated CAR basal signaling appeared to occur through both the canonical NF-κB (Fig. 2) (60) and ncNF-κB (Figs. 3 and 4 and fig. S7) pathways, with the latter assessed by the detection of increased baseline p100 processing (Fig. 4C and fig. S7, E and F) and RelB nuclear localization (Fig. 4G). This basal ncNF-κB signaling is consistent across anti-CD19 and anti-mesothelin CARs. Note that not all pathways downstream of CAR activation were basally active. ERK phosphorylation was undetectable before CAR activation (Fig. 2, A, D, and E), and ERK2 phosphorylation, specifically at 30 min after CAR activation, was statistically significantly increased in both 4-1BB– and CD28-costimulated CAR T cells compared to that in first-generation CAR T cells (Fig. 2E). These distinct signaling modalities of CARs may arise from the constitutive dimerization of the CAR caused by disulfide bridges between the CD8 hinge domains or associations between neighboring scFv domains (61, 62). Although native 4-1BB can form dimers linked by disulfide bonds (63), trimerization by its ligand 4-1BBL initiates downstream signaling (63-67). Whereas CD28-costimulated CAR basal signaling is detrimental to CAR T cell function and persistence (10, 68), basal ncNF-κB signaling enhances the survival of multiple cell types, including T cells (37, 52). These observations are consistent with the BBζ CAR–associated enhanced survival of both anti-CD19 (2, 41) and anti-mesothelin (69) CAR T cells. ncNF-κB signaling integrates with ERK and other signaling pathways to achieve these effects (35, 70-73), emphasizing the importance of further understanding how CARs activate some pathways in the absence of antigen and others only upon crosslinking, and how these signals combine to affect T cell function.
Whereas the basal activation of ncNF-κB signaling was consistent between two CARs with different scFv domains and antigen specificity, the observed difference in ligand-induced enhancement of the pathway between the CD19-specific and mesothelin-specific CARs was unexpected. We previously reported on the greater tonic signaling of the mesothelin-specific BBζ CAR incorporating the SS1 scFv as compared to that of CARs incorporating the anti-CD19 FMC63 scFv (11). Although speculative, this increased tonic signaling may lead to increased basal ncNF-κB signaling that is closer to the maximal response, thereby blunting our ability to see a difference upon activation. In this case, the small reduction that we observed in the p52/p100 ratio after bead-based activation could be the result of increased p100 abundance due to increased canonical NF-κB signaling (60, 74). Alternatively, differences in the way that the CAR engages antigen or in the way that the antigen is presented for these two scFv-ligand systems might also influence the ligand-dependent activation of the ncNF-κB pathway downstream of a BBζ CAR. In particular, CARs internalize after antigen engagement, similarly to the TCR-CD3 complex, and the degree of internalization has been associated with ligand density and receptor affinity, which were not controlled in our activation system (75, 76). This internalization can negatively affect CAR function (76) and could lead to diminished ncNF-κB signaling.
It is likely that the signaling downstream of 4-1BB costimulation that we observed will extend to other TNFRSFs. Transient expression of OX40L on both activated CD4 and CD8 T cells (77, 78) can activate OX40 and downstream ncNF-κB signaling (79). This OX40-OX40L interaction may explain the small increase in ncNF-κB signaling that we observed in the ζ-chain and 28ζ CARs (Fig. 4) because both OX40 and OX40L are expressed at the cell surface upon T cell activation (77). Other researchers have incorporated OX40 costimulation directly into the CAR, thereby achieving enhancements in CAR T cell function and persistence (80), likely, at least in part, by activation of ncNF-κB signaling.
Approaches to pharmacologically activate the ncNF-κB pathway may also have utility, especially in T cell–based immunotherapies that do not use TNFRSF-based costimulation. One class of molecules, SMAC mimetics, has already been leveraged in combination with CAR T cells to enhance target-negative tumor cell killing (81). SMAC mimetics activate ncNF-κB signaling (82) and enhance T cell expansion and function (83). On the basis of our observations that 4-1BB costimulation–mediated ncNF-κB signaling is a driver of anti-CD19 CAR T cell survival, it may be possible to use SMAC mimetics to similarly enhance non-TNFRSF–costimulated T cells. This combination may be especially beneficial in transgenic TCR–directed T cell therapies, which have no synthetic costimulation (84). The tumor responses induced by these therapies correlate with cell persistence (85), which, based on our findings and previous reports of the effects of SMAC mimetic on T cells (83, 86), could be increased by SMAC mimetic–mediated ncNF-κB signaling.
Last, our study has provided some insight into the mechanism by which ncNF-κB signaling by the 4-1BB domain of the CAR promotes T cell survival. The pro-apoptotic protein Bim plays a central role in T cell survival, is increased in abundance with T cell activation, and is repressed by ERK signaling (87-92). ERK-mediated Bim suppression was previously identified as a key mechanism by which endogenous murine 4-1BB enhances T cell survival (53); however, ncNF-κB signaling is implicated with ERK signaling in Bim regulation within multiple myeloma, suggesting that these two pathways are not mutually exclusive (35). In particular, ERK influences the cellular abundance of Bim in at least two ways: cooperation with RelB-p52 dimers to inhibit bcl2l11 transcription (35) and through the phosphorylation and subsequent degradation of the BimEL isoform (54, 93). This latter function of ERK may explain why we did not observe differences in BimEL abundance between anti-CD19 28ζ and BBζ CAR T cells or between dnNIK-expressing BBζ CAR T cells and their controls (fig. S11), given that ERK was active in both settings (Fig. 2, D and E, and fig. S12) (12). The observed increases in BimL and BimS abundances in dnNIK-expressing BBζ CAR T cells relative to those in control cells after CAR activation (Fig. 6, D and E) are consistent with a transcriptional effect. In addition, the BimL and BimS isoforms are even more potent sensitizers of cells to apoptosis than is BimEL (94) and are capable of diminishing T cell survival in the absence of BimEL (95). We were unable to detect a difference in the amounts of BimL and BimS between anti-CD19 28ζ and BBζ CAR T cells after CAR-mediated expansion, despite showing baseline differences, which may be due to selection events occurring in the 28ζ CAR T cell culture as was observed with the early loss of cells upon bead stimulation (Fig. 5I) and consistent with previous work describing rapid 28ζ CAR T cell effector differentiation and cell loss (12, 43). Bim suppression is an important downstream effect of 4-1BB–induced ncNF-κB signaling and likely contributes to BBζ CAR–mediated T cell survival, and further studies are needed to identify additional ncNF-κB–responsive genes that may also contribute to this survival.
Our system enabled us to identify ncNF-κB signaling as a specific and critical mediator of 4-1BB–costimulated CAR T cell survival, but we must also acknowledge some of its limitations. In this report, we focused on anti-CD19 CARs, which our group (41, 96) and Salter et al. (12, 44) have used to study CAR signaling. Although it is possible that our findings regarding CAR T cell survival are limited to the anti-CD19 CAR, 4-1BB–costimulated CAR T cells targeting multiple other antigens have been described as having similar enhanced survival (69, 97-99). We overexpressed the C terminus of NIK to inhibit ncNF-κB signaling for our studies due to the well-described effects of this approach in disrupting ncNF-κB (51) and the lack of commercially available pharmacological inhibitors (100). Although protein overexpression and dominant-negative mutants can have unintended, off-target effects, the absence of an appreciable effect of dnNIK expression on both polyclonal T cell expansion in response to anti-CD3/anti-CD28 beads or 28ζ CAR–mediated expansion supports the generally nontoxic and targeted nature of our approach (Fig. 5, I and J).
In summary, we have identified an essential and nonredundant mechanism by which 4-1BB costimulation within a CAR supports T cell survival, namely, by mediating both basal and ligand-dependent activation of ncNF-κB signaling, which is associated with the reduced abundance of the most pro-apoptotic isoforms of Bim. ncNF-κB signaling by 4-1BB–costimulated CARs also likely influences gene expression in CAR T cells beyond those genes that affect survival and persistence because this pathway regulates the expression of many genes, such as those encoding chemokines and cytokines that are important for T cell trafficking and function (38). Although not evaluated in our study, ncNF-κB signaling has been reported to be required for the production of granulocyte-macrophage colony-stimulating factor (GM-CSF) by T helper 17 (TH17) cells (101), suggesting that the 4-1BB–induced activation of this pathway could play a role in cytokine release syndrome that depends on this cytokine (102, 103). Our work therefore represents only a beginning for the study of this unique and complex signaling pathway in CAR T cells that clearly deserves further exploration.
MATERIALS AND METHODS
Primary cells and Nalm6 cells
Primary human T cells were procured and cultured as described previously (41). Briefly, peripheral blood mononuclear cells were collected from anonymous healthy donors by aphaeresis. T cells were isolated from these aphaeresis products by the University of Pennsylvania Human Immunology Core using Lymphoprep and RosetteSep Human T Cell Enrichment Cocktail kits (STEMCELL Technologies) according to the manufacturer’s instructions. Primary human T cells were cultured at 37°C in RPMI 1640 medium (Gibco) supplemented with 10% fetal bovine serum, 10 mM Hepes, penicillin (100 U/ml), and streptomycin (100 μg/ml). Nalm6 cells (American Type Culture Collection) were cultured at 37°C in the same medium that was used for T cells and refed every other day to a concentration of 0.25 × 106 cells/ml. Immediately before being used as stimulator cells, Nalm6 cells were irradiated with 100 Gy using an x-ray irradiator.
Generation of high-titer lentiviral vectors and T cell transduction
All CAR constructs were constructed as previously described (fig. S14) (41, 96). All lentiviral vectors were produced and used to transduce primary human T cells for all single transduced T cell assays as previously described (41). Dual-transduced CAR T cells were generated as follows. We transduced T cells with anti-CD19 CARs containing either CD28 or 4-1BB costimulatory domains from healthy donor bulk T cells by lentiviral transduction, as described previously (41). These cells were subsequently transduced a second time with either a control lentiviral vector expressing mCherry or a bicistronic vector expressing dnNIK and mCherry, which were separated in the vector by a T2A site. These dual-transduced cells were cultured for 5 to 8 days, sorted to purity based on the abundance of mCherry, and further expanded with interleukin-7 (IL-7) and IL-15 (final concentration of 10 ng/ml; premium grade, Miltenyi Biotec) added with R10 medium every other day to maintain a starting concentration of 0.75 × 106 cells/ml. This expansion regimen was maintained until the average cell volume plateaued, as measured on a Coulter Counter (Beckman Coulter) for two consecutive measurements. These cells were either immediately used in assays (described as “fresh”) or cryopreserved as described previously (41). Cells were enumerated by combining counts collected on a Coulter Counter (Beckman Coulter) with viability as assessed by flow cytometry using 7AAD (Cell Viability Solution, BD Via-Probe) exclusion (fig. S15A). CD4 (BUV395, RPA-T4, BD Biosciences), CD8 (BV785, RPA-T8, BioLegend), and CD19 (APC, HIB19, STEMCELL Technologies) cell surface expression were also assessed by flow cytometry. Elimination of irradiated Nalm6 stimulator cells by day 3 after addition was confirmed by the absence of CD19+ cells (fig. S15B).
Preparation and use of stimulation beads
Stimulator beads were prepared as described previously (43). Briefly, Dynabeads M-450 Tosylactivated (Invitrogen) were washed and resuspended in a borate buffer (pH 9.5) at 4 × 108 beads/ml and then incubated overnight with 150 μg of anti-CD19 idiotype antibody (Novartis) or mesothelin-Fc at 37°C with constant mixing. The beads were then washed three times in wash buffer [3% human AB serum, 0.1% sodium azide, and 2 mM EDTA in phosphate-buffered saline (PBS)] at 4°C for 10 min each with constant mixing, followed by an overnight wash at 4°C in the same buffer with constant mixing. Before use, the beads were washed three times in PBS and then resuspended in RPMI 1640–based medium as described earlier. Before use in experiments, the beads were subjected to quality control by being mixed with BBζ CAR T cells at a 3:1 bead-to-cell ratio. Bead activity was assessed by measuring T cell size with a Coulter Counter after 3 days and comparing it to the size of the same BBζ CAR T cells mixed with previous batches of beads at the same ratio used in parallel as a positive control, as well as to the size of BBζ CAR T cells cultured alone as a negative control. Beads were then used at a 3:1 bead-to-cell ratio for all bead-based expansion assays, whereas they were used at a 5:1 bead-to-cell ratio for all signaling assays. T cells for expansion assays were mixed with beads and cultured as described earlier. T cells for stimulation assays were mixed with beads and spun down to synchronize bead-cell contact. They were then incubated at 37°C for the times indicated in the figure legends.
Whole-cell lysis
Radioimmunoprecipitation assay (RIPA) buffer stock was made at 1× consisting of 150 mM NaCl, 1% NP-40, 0.5% deoxycholate, 0.1% SDS, and 50 mM tris-HCl (pH 8.0). The stock was stored at 4°C. The night before use, one tablet each of phosphatase inhibitor (PhosSTOP, Roche) and proteasome inhibitor (cOmplete, Roche) was added to a working volume of RIPA buffer. On the day of lysate collection, T cells were harvested from the culture, washed with ice-cold PBS (Corning), and resuspended in the working stock of ice-cold RIPA buffer. Lysates were incubated on ice for 30 min while being vortexed at maximum speed for 10 s at 10-min intervals. The cells were then centrifuged at 16,000g for 10 min in a refrigerated centrifuge. The supernatants were collected, aliquoted, and immediately frozen at −80°C.
Nuclear and cytoplasmic fractionation
Our nuclear-cytoplasmic fractionation technique was adapted from a previously described method (104). Cytoplasmic buffer stock (1×) was as follows: 10 mM Hepes, 10 mM KCl, and 0.1 mM EDTA (pH 7.9). Nuclear buffer stock (1×) was as follows: 20 mM Hepes, 0.42 M NaCl, and 0.1 mM EDTA (pH 7.9). Both buffers were stored at 4°C. The night before use, one tablet each of phosphatase inhibitor (PhosSTOP, Roche) and proteasome inhibitor (cOmplete, Roche) was added to a working volume of each buffer. On the day of lysate collection, T cells were harvested from the culture, washed with ice-cold PBS (Corning), and resuspended in the working stock of ice-cold cytoplasmic buffer. Samples were incubated on ice for 10 min. A final concentration of 0.1% NP-40 (Thermo Fisher Scientific) was added to each sample. Each sample was then incubated at room temperature for 5 min, followed by a 10-s maximum speed vortex and immediate centrifugation at 4000g for 90 s at 4°C. Supernatants were collected on ice, aliquoted, and frozen at −80°C for later use as the cytoplasmic fraction. The remaining nuclear pellets were washed twice with ice-cold cytoplasmic buffer and transferred to a fresh tube. They were then resuspended in ice-cold nuclear buffer and shaken at 4°C for 30 min at 1400 rpm. Samples were then centrifuged at 16,000g for 15 min at 4°C. Supernatants were collected, aliquoted on ice, and stored at −80°C to be used as nuclear fractions.
Western blotting analysis
Cell lysates were thawed on ice, and protein concentration was assessed by DC Protein Assay (Bio-Rad) according to the manufacturer’s instructions. Equal quantities of protein were added to Laemmli buffer (Bio-Rad) containing 2-mercaptoethanol (Bio-Rad) and boiled at 95°C for 5 min. Protein was separated in NuPAGE gels using the Cell SureLock Mini-Cell (Invitrogen) and transferred to a methanol-activated PVDF-FL membrane (Millipore Sigma) in the Mini Trans-Blot Electrophoretic Transfer Cell (Bio-Rad) in 10% methanol NuPAGE transfer buffer (Thermo Fisher Scientific). After transfer, the membranes were incubated in methanol for 1 min, followed by a deionized water wash, and then incubated in tris-buffered saline (TBS) for 2 min before being blocked in TBS Odyssey blocking buffer (LI-COR) for 1 hour. Proteins were visualized with the primary antibodies against the following targets and incubated overnight at 4°C in TBS Odyssey blocking buffer (LI-COR) with 0.5% Tween 20 (Bio-Rad): PCNA (1:2000; PC10, Cell Signaling Technology), actin (1:1000; 8H10D10, Cell Signaling Technology), pZap-70 (1:1000; Tyr319, 65E4, Cell Signaling Technology), Zap-70 (1:1000; L1E5, Cell Signaling Technology), pp65 (1:1000; Ser536, 93H1, Cell Signaling Technology), p65 (1:1000; L8F6, Cell Signaling Technology), pp44/42 mitogen-activated protein kinase (MAPK) (ERK1/2) (1:2000; Tyr202/Tyr204, D13.14.4E, Cell Signaling Technology), p44/42 MAPK (ERK1/2) (1:2000; L34F12, Cell Signaling Technology), NIK (1:750; Cell Signaling Technology), pNF-κB2 p100 (1:1000; Ser866/Ser870, Cell Signaling Technology), NF-κB p52 (1:1000; Millipore Sigma), RelB (1:1000; D7D7W, Cell Signaling Technology), and Bim (1:1000; C34C5, Cell Signaling Technology). Each membrane was only incubated with one primary antibody from a single origin species (rabbit or mouse) at one time. The membranes were washed with TBS containing 0.1% Tween 20 and incubated with donkey anti-rabbit (1:15,000; IRDye 800CW, LI-COR), donkey anti-mouse (1:15,000; IRDye 680RD, LI-COR), or both secondary antibodies in TBS Odyssey blocking buffer (LI-COR) with 0.5% Tween 20 and 0.1% SDS (Thermo Fisher Scientific) for 40 min at room temperature. The membranes were then washed again with TBS containing 0.1% Tween 20, rinsed once in TBS, and then kept in TBS at 4°C until imaging. Membranes were imaged with an Odyssey CLx (LI-COR), and fluorescence was evaluated with Image Studio (LI-COR). When membranes were reused to visualize additional proteins, they were first reblocked in TBS Odyssey blocking buffer (LI-COR) for 1 hour at room temperature and then processed as described earlier.
Statistical analysis
Results are expressed as means ± SD. All LogFC values are derived as follows: Log2 of ((time of interest of experimental group: raw fluorescence of the protein of interest/raw fluorescence of relevant loading control)/(control time point of control group: raw fluorescence of protein of interest/raw fluorescence of relevant loading control)). Analysis of relative protein abundance in Fig. 5 (D and E) was determined by calculating the ratio of the fluorescence intensity of the stated protein sample to the fluorescence intensity of the same protein with the lowest abundance sample in the experiment. Statistical comparisons were made as noted in the figure legends, with the factors of two-way analysis of variance (ANOVA) analyses being CAR and time using Prism (GraphPad).
Supplementary Material
Fig. S1. The ncNF-κB pathway.
Fig. S2. CAR expression on T cells from three donors expanded by irradiated Nalm6 cells.
Fig. S3. CAR expression on T cells from three donors expanded by anti-CD19 beads.
Fig. S4. CAR expression on T cells from three donors activated by anti-CD19 beads for signaling assays.
Fig. S5. Anti-CD19 BBζ CAR activation induces T cell signaling.
Fig. S6. Additional representative Western blotting analysis of nuclear and cytoplasmic fractions to compare anti-CD19 CAR signaling.
Fig. S7. Anti-mesothelin BBζ CAR also drives ncNF-κB signaling.
Fig. S8. Dual-transduced T cell production scheme, day 0 CAR, and mCherry expression.
Fig. S9. Western blotting analysis of nuclear and cytoplasmic fractions to compare control and dnNIK-expressing anti-CD19 BBζ T cells.
Fig. S10. dnNIK does not affect PCNA abundance in CAR T cells.
Fig. S11. BimEL abundance in anti-CD19 CAR T cells.
Fig. S12. ERK1/2 phosphorylation in control and dnNIK-expressing BBζ CAR T cells.
Fig. S13. FOXO3a abundance and phosphorylation in control and dnNIK-expressing BBζ CAR T cells.
Fig. S14. CAR constructs.
Fig. S15. Representative gating strategy to quantify cell death by flow cytometry and assess the elimination of CD19+ Nalm6 cells.
Acknowledgments:
We thank A. Wang and M. Hresko for technical assistance and V. Bhoj, S. Richman, S. Ghassemi, S. Nunez-Cruz, S. Gill, T. Kambayashi, and C. van Dang for helpful discussions. We also thank S.-C. Sun for sharing the original construct from which we cloned the complementary DNA encoding dnNIK.
Funding: This research was supported by grants from the University of Pennsylvania-Novartis Alliance (to C.H.J. and M.C.M.) and NIH grants (R01CA226983 to R.S.O. and C.H.J.; P01CA214278 to M.C.M. and C.H.J.; and R01CA172921 to S.M.A.).
Footnotes
Competing interests: C.H.J. reports receiving commercial research funding from Tmunity Therapeutics and has an ownership interest and intellectual property (IP) licensed to Novartis. M.C.M. reports receiving commercial research funding from Novartis and has an ownership interest and IP licensed to Cabaletta Bio. The other authors declare that they have no competing interests.
Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. The anti-CD19 idiotype antibody from Novartis requires a material transfer agreement.
SUPPLEMENTARY MATERIALS
REFERENCES AND NOTES
- 1.Feins S, Kong W, Williams EF, Milone MC, Fraietta JA, An introduction to chimeric antigen receptor (CAR) T-cell immunotherapy for human cancer. Am. J. Hematol 94, S3–S9 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Mueller KT, Waldron E, Grupp SA, Levine JE, Laetsch TW, Pulsipher MA, Boyer MW, August KJ, Hamilton J, Awasthi R, Stein AM, Sickert D, Chakraborty A, Levine BL, June CH, Tomassian L, Shah SS, Leung M, Taran T, Wood PA, Maude SL, Clinical pharmacology of tisagenlecleucel in B-cell acute lymphoblastic leukemia. Clin. Cancer Res 24, 6175–6184 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Orlando EJ, Han X, Tribouley C, Wood PA, Leary RJ, Riester M, Levine JE, Qayed M, Grupp SA, Boyer M, De Moerloose B, Nemecek ER, Bittencourt H, Hiramatsu H, Buechner J, Davies SM, Verneris MR, Nguyen K, Brogdon JL, Bitter H, Morrissey M, Pierog P, Pantano S, Engelman JA, Winckler W, Genetic mechanisms of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nat. Med 24, 1504–1506 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Mueller KT, Maude SL, Porter DL, Frey N, Wood P, Han X, Waldron E, Chakraborty A, Awasthi R, Levine BL, Melenhorst JJ, Grupp SA, June CH, Lacey SF, Cellular kinetics of CTL019 in relapsed/refractory B-cell acute lymphoblastic leukemia and chronic lymphocytic leukemia. Blood 130, 2317–2325 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Turtle CJ, Hanafi L-A, Berger C, Gooley TA, Cherian S, Hudecek M, Sommermeyer D, Melville K, Pender B, Budiarto TM, Robinson E, Steevens NN, Chaney C, Soma L, Chen X, Yeung C, Wood B, Li D, Cao J, Heimfeld S, Jensen MC, Riddell SR, Maloney DG, CD19 CAR-T cells of defined CD4+: CD8+composition in adult B cell ALL patients. J. Clin. Invest 126, 2123–2138 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van der Stegen SJC, Hamieh M, Sadelain M, The pharmacology of second-generation chimeric antigen receptors. Nat. Rev. Drug Discov 14, 499–509 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, Braunschweig I, Oluwole OO, Siddiqi T, Lin Y, Timmerman JM, Stiff PJ, Friedberg JW, Flinn IW, Goy A, Hill BT, Smith MR, Deol A, Farooq U, McSweeney P, Munoz J, Avivi I, Castro JE, Westin JR, Chavez JC, Ghobadi A, Komanduri KV, Levy R, Jacobsen ED, Witzig TE, Reagan P, Bot A, Rossi J, Navale L, Jiang Y, Aycock J, Elias M, Chang D, Wiezorek J, Go WY, Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N. Engl. J. Med 377, 2531–2544 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, Bader P, Verneris MR, Stefanski HE, Myers GD, Qayed M, De Moerloose B, Hiramatsu H, Schlis K, Davis KL, Martin PL, Nemecek ER, Yanik GA, Peters C, Baruchel A, Boissel N, Mechinaud F, Balduzzi A, Krueger J, June CH, Levine BL, Wood P, Taran T, Leung M, Mueller KT, Zhang Y, Sen K, Lebwohl D, Pulsipher MA, Grupp SA, Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N. Engl. J. Med 378, 439–448 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weinkove R, George P, Dasyam N, McLellan AD, Selecting costimulatory domains for chimeric antigen receptors: Functional and clinical considerations. Clin. Transl. Immunol 8, e1049 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, Smith JP, Walker AJ, Kohler ME, Venkateshwara VR, Kaplan RN, Patterson GH, Fry TJ, Orentas RJ, Mackall CL, 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med 21, 581–590 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frigault MJ, Lee J, Basil MC, Carpenito C, Motohashi S, Scholler J, Kawalekar OU, Guedan S, McGettigan SE, Posey AD, Ang S, Cooper LJN, Platt JM, Johnson FB, Paulos CM, Zhao Y, Kalos M, Milone MC, June CH, Identification of chimeric antigen receptors that mediate constitutive or inducible proliferation of T cells. Cancer Immunol. Res 3, 356–367 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Salter AI, Ivey RG, Kennedy JJ, Voillet V, Rajan A, Alderman EJ, Voytovich UJ, Lin C, Sommermeyer D, Liu L, Whiteaker JR, Gottardo R, Paulovich AG, Riddell SR, Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. Sci. Signal 11, eaat6753 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feucht J, Sun J, Eyquem J, Ho Y-J, Zhao Z, Leibold J, Dobrin A, Cabriolu A, Hamieh M, Sadelain M, Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency. Nat. Med 25, 82–88 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao Z, Condomines M, van der Stegen SJC, Perna F, Kloss CC, Gunset G, Plotkin J, Sadelain M, Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T cells. Cancer Cell 28, 415–428 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Drent E, Poels R, Ruiter R, van de Donk NWCJ, Zweegman S, Yuan H, de Bruijn J, Sadelain M, Lokhorst HM, Groen R, Mutis T, Themeli M, Combined CD28 and 4-1BB costimulation potentiates affinity-tuned chimeric antigen receptor-engineered T cells. Clin. Cancer Res 25, 4014–4025 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee H-W, Park S-J, Choi BK, Kim HH, Nam K-O, Kwon BS, 4-1BB promotes the survival of CD8+ T lymphocytes by increasing expression of Bcl-xL and Bfl-1. J. Immunol 169, 4882–4888 (2002). [DOI] [PubMed] [Google Scholar]
- 17.Shuford WW, Klussman K, Tritchler DD, Loo DT, Chalupny J, Siadak AW, Brown TJ, Emswiler J, Raecho H, Larsen CP, Pearson TC, Ledbetter JA, Aruffo A, Mittler RS, 4-1BB costimulatory signals preferentially induce CD8+ T cell proliferation and lead to the amplification in vivo of cytotoxic T cell responses. J. Exp. Med 186, 47–55 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Giardino Torchia ML, Munitic I, Castro E, Herz J, McGavern DB, Ashwell JD, c-IAP ubiquitin protein ligase activity is required for 4-1BB signaling and CD8+ memory T-cell survival. Eur. J. Immunol 45, 2672–2682 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bertram EM, Lau P, Watts TH, Temporal segregation of 4-1BB versus CD28-mediated costimulation: 4-1BB ligand influences T cell numbers late in the primary response and regulates the size of the T cell memory response following influenza infection. J. Immunol 168, 3777–3785 (2002). [DOI] [PubMed] [Google Scholar]
- 20.Hurtado JC, Kim YJ, Kwon BS, Signals through 4-1BB are costimulatory to previously activated splenic T cells and inhibit activation-induced cell death. J. Immunol 158, 2600–2609 (1997). [PubMed] [Google Scholar]
- 21.Hendriks J, Xiao Y, Rossen JWA, van der Sluijs KF, Sugamura K, Ishii N, Borst J, During viral infection of the respiratory tract, CD27, 4-1BB, and OX40 collectively determine formation of CD8+ memory T cells and their capacity for secondary expansion. J. Immunol 175, 1665–1676 (2005). [DOI] [PubMed] [Google Scholar]
- 22.Snell LM, Lin GHY, McPherson AJ, Moraes TJ, Watts TH, T-cell intrinsic effects of GITR and 4-1BB during viral infection and cancer immunotherapy. Immunol. Rev 244, 197–217 (2011). [DOI] [PubMed] [Google Scholar]
- 23.Li Y, Sedwick CE, Hu J, Altman A, Role for protein kinase Cθ (PKCθ) in TCR/CD28-mediated signaling through the canonical but not the non-canonical pathway for NF-κB activation. J. Biol. Chem 280, 1217–1223 (2005). [DOI] [PubMed] [Google Scholar]
- 24.McPherson AJ, Snell LM, Mak TW, Watts TH, Opposing roles for TRAF1 in the alternative versus classical NF-κB pathway in T cells. J. Biol. Chem 287, 23010–23019 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiao X, Shi X, Fan Y, Wu C, Zhang X, Minze L, Liu W, Ghobrial RM, Lan P, Li XC, The costimulatory receptor OX40 inhibits interleukin-17 expression through activation of repressive chromatin remodeling pathways. Immunity 44, 1271–1283 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ramakrishnan P, Wang W, Wallach D, Receptor-specific signaling for both the alternative and the canonical NF-kappaB activation pathways by NF-κB-inducing kinase. Immunity 21, 477–489 (2004). [DOI] [PubMed] [Google Scholar]
- 27.Kane LP, Lin J, Weiss A, It’s all Rel-ative: NF-κB and CD28 costimulation of T-cell activation. Trends Immunol. 23, 413–420 (2002). [DOI] [PubMed] [Google Scholar]
- 28.Sun Z, Arendt CW, Ellmeier W, Schaeffer EM, Sunshine MJ, Gandhi L, Annes J, Petrzilka D, Kupfer A, Schwartzberg PL, Littman DR, PKC-theta is required for TCR-induced NF-kappaB activation in mature but not immature T lymphocytes. Nature 404, 402–407 (2000). [DOI] [PubMed] [Google Scholar]
- 29.Schimmack G, Eitelhuber AC, Vincendeau M, Demski K, Shinohara H, Kurosaki T, Krappmann D, AIP augments CARMA1-BCL10-MALT1 complex formation to facilitate NF-κB signaling upon T cell activation. Cell Commun. Signal 12, 49 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shinkura R, Kitada K, Matsuda F, Tashiro K, Ikuta K, Suzuki M, Kogishi K, Serikawa T, Honjo T, Alymphoplasia is caused by a point mutation in the mouse gene encoding Nf-κ b-inducing kinase. Nat. Genet 22, 74–77 (1999). [DOI] [PubMed] [Google Scholar]
- 31.Claudio E, Brown K, Park S, Wang H, Siebenlist U, BAFF-induced NEMO-independent processing of NF-κ B2 in maturing B cells. Nat. Immunol 3, 958–965 (2002). [DOI] [PubMed] [Google Scholar]
- 32.Willmann KL, Klaver S, Doğu F, Santos-Valente E, Garncarz W, Bilic I, Mace E, Salzer E, Conde CD, Sic H, Májek P, Banerjee PP, Vladimer GI, Haskoloğlu S, Bolkent MG, Küpesiz A, Condino-Neto A, Colinge J, Superti-Furga G, Pickl WF, van Zelm MC, Eibel H, Orange JS, Ikincioğulları A, Boztug K, Biallelic loss-of-function mutation in NIK causes a primary immunodeficiency with multifaceted aberrant lymphoid immunity. Nat. Commun 5, 5360 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eliopoulos AG, Caamano JH, Flavell J, Reynolds GM, Murray PG, Poyet J-L, Young LS, Epstein-Barr virus-encoded latent infection membrane protein 1 regulates the processing of p100 NF-κB2 to p52 via an IKKγ/NEMO-independent signalling pathway. Oncogene 22, 7557–7569 (2003). [DOI] [PubMed] [Google Scholar]
- 34.Cormier F, Monjanel H, Fabre C, Billot K, Sapharikas E, Chereau F, Bordereaux D, Molina TJ, Avet-Loiseau H, Baud V, Bendall L, Frequent engagement of RelB activation is critical for cell survival in multiple myeloma. PLOS ONE 8, e59127 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vallabhapurapu SD, Noothi SK, Pullum DA, Lawrie CH, Pallapati R, Potluri V, Kuntzen C, Khan S, Plas DR, Orlowski RZ, Chesi M, Kuehl WM, Bergsagel PL, Karin M, Vallabhapurapu S, Transcriptional repression by the HDAC4-RelB-p52 complex regulates multiple myeloma survival and growth. Nat. Commun 6, 8428 (2015). [DOI] [PubMed] [Google Scholar]
- 36.Ranuncolo SM, Pittaluga S, Evbuomwan MO, Jaffe ES, Lewis BA, Hodgkin lymphoma requires stabilized NIK and constitutive RelB expression for survival. Blood 120, 3756–3763 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wharry CE, Haines KM, Carroll RG, May MJ, Constitutive non-canonical NFκB signaling in pancreatic cancer cells. Cancer Biol. Ther 8, 1567–1576 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun S-C, The non-canonical NF-κB pathway in immunity and inflammation. Nat. Rev. Immunol 17, 545–558 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li Y, Wang H, Zhou X, Xie X, Chen X, Jie Z, Zou Q, Hu H, Zhu L, Cheng X, Brightbill HD, Wu LC, Wang L, Sun S-C, Cell intrinsic role of NF-κB-inducing kinase in regulating T cell-mediated immune and autoimmune responses. Sci. Rep 6, 22115 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rowe AM, Murray SE, Raué H-P, Koguchi Y, Slifka MK, Parker DC, A cell-intrinsic requirement for NF-κB-inducing kinase in CD4 and CD8 T cell memory. J. Immunol 191, 3663–3672 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, Samanta M, Lakhal M, Gloss B, Danet-Desnoyers G, Campana D, Riley JL, Grupp SA, June CH, Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol. Ther 17, 1453–1464 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li W, Fan D, Yang M, Yan Y, Shi R, Cheng J, Li Z, Zhang M, Wang J, Xiong D, Cytosine arabinoside promotes cytotoxic effect of T cells on leukemia cells mediated by bispecific antibody. Hum. Gene Ther 24, 751–760 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kawalekar OU, O’Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD, Patel PR, Guedan S, Scholler J, Keith B, Snyder N, Blair I, Milone MC, June CH, Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity 44, 380–390 (2016). [DOI] [PubMed] [Google Scholar]
- 44.Ramello MC, Benzaïd I, Kuenzi BM, Lienlaf-Moreno M, Kandell WM, Santiago DN, Pabón-Saldaña M, Darville L, Fang B, Rix U, Yoder S, Berglund A, Koomen JM, Haura EB, Abate-Daga D, An immunoproteomic approach to characterize the CAR interactome and signalosome. Sci. Signal 12, eaap9777 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Salojin KV, Zhang J, Delovitch TL, TCR and CD28 are coupled via ZAP-70 to the activation of the Vav/Rac-1-/PAK-1/p38 MAPK signaling pathway. J. Immunol 163, 844–853 (1999). [PubMed] [Google Scholar]
- 46.D’Souza WN, Chang C-F, Fischer AM, Li M, Hedrick SM, The Erk2 MAPK regulates CD8 T cell proliferation and survival. J. Immunol 181, 7617–7629 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu X, Berry CT, Ruthel G, Madara JJ, MacGillivray K, Gray CM, Madge LA, McCorkell KA, Beiting DP, Hershberg U, May MJ, Freedman BD, T cell receptor-induced nuclear factor κB (NF-κB) signaling and transcriptional activation are regulated by STIM1- and Orai1-mediated calcium entry. J. Biol. Chem 291, 8440–8452 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Remouchamps C, Dejardin E, Methods to assess the activation of the alternative (noncanonical) NF-κB pathway by non-death TNF receptors. Methods Mol. Biol 1280, 103–119 (2015). [DOI] [PubMed] [Google Scholar]
- 49.Sanchez-Paulete AR, Labiano S, Rodriguez-Ruiz ME, Azpilikueta A, Etxeberria I, Bolaños E, Lang V, Rodriguez M, Aznar MA, Jure-Kunkel M, Melero I, Deciphering CD137 (4-1BB) signaling in T-cell costimulation for translation into successful cancer immunotherapy. Eur. J. Immunol 46, 513–522 (2016). [DOI] [PubMed] [Google Scholar]
- 50.Sun S-C, The noncanonical NF-κB pathway. Immunol. Rev 246, 125–140 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xiao G, Harhaj EW, Sun SC, NF-κB-inducing kinase regulates the processing of NF-kappaB2 p100. Mol. Cell 7, 401–409 (2001). [DOI] [PubMed] [Google Scholar]
- 52.Wang Z, Zhang B, Yang L, Ding J, Ding H-F, Constitutive production of NF-κB2 p52 is not tumorigenic but predisposes mice to inflammatory autoimmune disease by repressing Bim expression. J. Biol. Chem 283, 10698–10706 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sabbagh L, Pulle G, Liu Y, Tsitsikov EN, Watts TH, ERK-dependent Bim modulation downstream of the 4-1BB-TRAF1 signaling axis is a critical mediator of CD8 T cell survival in vivo. J. Immunol 180, 8093–8101 (2008). [DOI] [PubMed] [Google Scholar]
- 54.Luciano F, Jacquel A, Colosetti P, Herrant M, Cagnol S, Pages G, Auberger P, Phosphorylation of Bim-EL by Erk1/2 on serine 69 promotes its degradation via the proteasome pathway and regulates its proapoptotic function. Oncogene 22, 6785–6793 (2003). [DOI] [PubMed] [Google Scholar]
- 55.O’Reilly LA, Kruse EA, Puthalakath H, Kelly PN, Kaufmann T, Huang DCS, Strasser A, MEK/ERK-mediated phosphorylation of Bim is required to ensure survival of T and B lymphocytes during mitogenic stimulation. J. Immunol 183, 261–269 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Riou C, Yassine-Diab B, Van Grevenynghe J, Somogyi R, Greller LD, Gagnon D, Gimmig S, Wilkinson P, Shi Y, Cameron MJ, Campos-Gonzalez R, Balderas RS, Kelvin D, Sekaly R-P, Haddad EK, Convergence of TCR and cytokine signaling leads to FOXO3a phosphorylation and drives the survival of CD4+ central memory T cells. J. Exp. Med 204, 79–91 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sunters A, Fernández de Mattos S, Stahl M, Brosens JJ, Zoumpoulidou G, Saunders CA, Coffer PJ, Medema RH, Coombes RC, Lam EW-F, FoxO3a transcriptional regulation of Bim controls apoptosis in paclitaxel-treated breast cancer cell lines. J. Biol. Chem 278, 49795–49805 (2003). [DOI] [PubMed] [Google Scholar]
- 58.Marinari B, Costanzo A, Marzano V, Piccolella E, Tuosto L, CD28 delivers a unique signal leading to the selective recruitment of RelA and p52 NF-κB subunits on IL-8 and Bcl-xL gene promoters. Proc. Natl. Acad. Sci. U.S.A 101, 6098–6103 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ajina A, Maher J, Strategies to address chimeric antigen receptor tonic signaling. Mol. Cancer Ther 17, 1795–1815 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li G, Boucher JC, Kotani H, Park K, Zhang Y, Shrestha B, Wang X, Guan L, Beatty N, Abate-Daga D, Davila ML, 4-1BB enhancement of CAR T function requires NF-κB and TRAFs. JCI Insight 3, 1999 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Snow PM, Terhorst C, The T8 antigen is a multimeric complex of two distinct subunits on human thymocytes but consists of homomultimeric forms on peripheral blood T lymphocytes. J. Biol. Chem 258, 14675–14681 (1983). [PubMed] [Google Scholar]
- 62.Harris DT, Kranz DM, Adoptive T cell therapies: A comparison of T cell receptors and chimeric antigen receptors. Trends Pharmacol. Sci 37, 220–230 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bitra A, Doukov T, Croft M, Zajonc DM, Crystal structures of the human 4-1BB receptor bound to its ligand 4-1BBL reveal covalent receptor dimerization as a potential signaling amplifier. J. Biol. Chem 293, 1317–1329 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Won E-Y, Cha K, Byun J-S, Kim D-U, Shin S, Ahn B, Kim YH, Rice AJ, Walz T, Kwon BS, Cho H-S, The structure of the trimer of human 4-1BB ligand is unique among members of the tumor necrosis factor superfamily. J. Biol. Chem 285, 9202–9210 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gilbreth RN, Oganesyan VY, Amdouni H, Novarra S, Grinberg L, Barnes A, Baca M, Crystal structure of the human 4-1BB/4-1BBL complex. J. Biol. Chem 293, 9880–9891 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zheng C, Kabaleeswaran V, Wang Y, Cheng G, Wu H, Crystal structures of the TRAF2: cIAP2 and the TRAF1: TRAF2: cIAP2 complexes: Affinity, specificity, and regulation. Mol. Cell 38, 101–113 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zapata JM, Perez-Chacon G, Carr-Baena P, Martinez-Forero I, Azpilikueta A, Otano I, Melero I, CD137 (4-1BB) signalosome: Complexity is a matter of TRAFs. Front. Immunol 9, 2618 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zheng W, O’Hear CE, Alli R, Basham JH, Abdelsamed HA, Palmer LE, Jones LL, Youngblood B, Geiger TL, PI3K orchestration of the in vivo persistence of chimeric antigen receptor-modified T cells. Leukemia 32, 1157–1167 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang E, Wang L-C, Tsai C-Y, Bhoj V, Gershenson Z, Moon E, Newick K, Sun J, Lo A, Baradet T, Feldman MD, Barrett D, Purá E, Albelda S, Milone MC, Generation of potent T-cell immunotherapy for cancer using DAP12-based, multichain, chimeric immunoreceptors. Cancer Immunol. Res 3, 815–826 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li Y, Zhou Q-L, Sun W, Chandrasekharan P, Cheng HS, Ying Z, Lakshmanan M, Raju A, Tenen DG, Cheng S-Y, Chuang K-H, Li J, Prabhakar S, Li M, Tergaonkar V, Non-canonical NF-κB signalling and ETS1/2 cooperatively drive C250T mutant TERT promoter activation. Nat. Cell Biol 17, 1327–1338 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.House CD, Grajales V, Ozaki M, Jordan E, Wubneh H, Kimble DC, James JM, Kim MK, Annunziata CM, IKKε cooperates with either MEK or non-canonical NF-kB driving growth of triple-negative breast cancer cells in different contexts. BMC Cancer 18, 595 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shinzawa M, Konno H, Qin J, Akiyama N, Miyauchi M, Ohashi H, Miyamoto-Sato E, Yanagawa H, Akiyama T, Inoue J.-i., Catalytic subunits of the phosphatase calcineurin interact with NF-κB-inducing kinase (NIK) and attenuate NIK-dependent gene expression. Sci. Rep 5, 10758 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liu Z, Mar KB, Hanners NW, Perelman SS, Kanchwala M, Xing C, Schoggins JW, Alto NM, A NIK-SIX signalling axis controls inflammation by targeted silencing of non-canonical NF-κB. Nature 22, 249–253 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mukherjee T, Chatterjee B, Dhar A, Bais SS, Chawla M, Roy P, George A, Bal V, Rath S, Basak S, A TNF-p100 pathway subverts noncanonical NF-κB signaling in inflamed secondary lymphoid organs. EMBO J., 3501–3516 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liu H, Rhodes M, Wiest DL, Vignali DA, On the dynamics of TCR:CD3 complex cell surface expression and downmodulation. Immunity 13, 665–675 (2000). [DOI] [PubMed] [Google Scholar]
- 76.Walker AJ, Majzner RG, Zhang L, Wanhainen K, Long AH, Nguyen SM, Lopomo P, Vigny M, Fry TJ, Orentas RJ, Mackall CL, Tumor antigen and receptor densities regulate efficacy of a chimeric antigen receptor targeting anaplastic lymphoma kinase. Mol. Ther 25, 2189–2201 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Soroosh P, Ine S, Sugamura K, Ishii N, OX40-OX40 ligand interaction through T cell-T cell contact contributes to CD4 T cell longevity. J. Immunol 176, 5975–5987 (2006). [DOI] [PubMed] [Google Scholar]
- 78.Kondo K, Okuma K, Tanaka R, Zhang LF, Kodama A, Takahashi Y, Yamamoto N, Ansari AA, Tanaka Y, Requirements for the functional expression of OX40 ligand on human activated CD4+ and CD8+ T cells. Hum. Immunol 68, 563–571 (2007). [DOI] [PubMed] [Google Scholar]
- 79.Murray SE, Polesso F, Rowe AM, Basak S, Koguchi Y, Toren KG, Hoffmann A, Parker DC, NF-κB–inducing kinase plays an essential T cell–intrinsic role in graft-versus-host disease and lethal autoimmunity in mice. J. Clin. Invest 121, 4775–4786 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hombach AA, Abken H, Costimulation by chimeric antigen receptors revisited the T cell antitumor response benefits from combined CD28-OX40 signalling. Int. J. Cancer 129, 2935–2944 (2011). [DOI] [PubMed] [Google Scholar]
- 81.Michie J, Beavis PA, Freeman AJ, Vervoort SJ, Ramsbottom KM, Narasimhan V, Lelliott EJ, Lalaoui N, Ramsay RG, Johnstone RW, Silke J, Darcy PK, Voskoboinik I, Kearney CJ, Oliaro J, Antagonism of IAPs enhances CAR T-cell efficacy. Cancer Immunol. Res 7, 183–192 (2019). [DOI] [PubMed] [Google Scholar]
- 82.Tchoghandjian A, Jennewein C, Eckhardt I, Rajalingam K, Fulda S, Identification of non-canonical NF-κB signaling as a critical mediator of Smac mimetic-stimulated migration and invasion of glioblastoma cells. Cell Death Dis. 4, e564 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dougan M, Dougan S, Slisz J, Firestone B, Vanneman M, Draganov D, Goyal G, Li W, Neuberg D, Blumberg R, Hacohen N, Porter D, Zawel L, Dranoff G, IAP inhibitors enhance co-stimulation to promote tumor immunity. J. Exp. Med 207, 2195–2206 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Krummel MF, Heath WR, Allison J, Differential coupling of second signals for cytotoxicity and proliferation in CD8+ T cell effectors: Amplification of the lytic potential by B7. J. Immunol 163, 2999–3006 (1999). [PubMed] [Google Scholar]
- 85.Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, Kammula US, Royal RE, Sherry RM, Wunderlich JR, Lee C-CR, Restifo NP, Schwarz SL, Cogdill AP, Bishop RJ, Kim H, Brewer CC, Rudy SF, VanWaes C, Davis JL, Mathur A, Ripley RT, Nathan DA, Laurencot CM, Rosenberg SA, Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 114, 535–546 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Beug ST, Beauregard CE, Healy C, Sanda T, St-Jean M, Chabot J, Walker DE, Mohan A, Earl N, Lun X, Senger DL, Robbins SM, Staeheli P, Forsyth PA, Alain T, LaCasse EC, Korneluk RG, Smac mimetics synergize with immune checkpoint inhibitors to promote tumour immunity against glioblastoma. Nat. Commun 8, 14278 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wojciechowski S, Jordan MB, Zhu Y, White J, Zajac AJ, Hildeman DA, Bim mediates apoptosis of CD127lo effector T cells and limits T cell memory. Eur. J. Immunol 36, 1694–1706 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hildeman DA, Zhu Y, Mitchell TC, Bouillet P, Strasser A, Kappler J, Marrack P, Activated T cell death in vivo mediated by proapoptotic bcl-2 family member bim. Immunity 16, 759–767 (2002). [DOI] [PubMed] [Google Scholar]
- 89.Bouillet P, Purton JF, Godfrey DI, Zhang L-C, Coultas L, Puthalakath H, Pellegrini M, Cory S, Adams JM, Strasser A, BH3-only Bcl-2 family member Bim is required for apoptosis of autoreactive thymocytes. Nature 415, 922–926 (2002). [DOI] [PubMed] [Google Scholar]
- 90.Masson F, Kupresanin F, Mount A, Strasser A, Belz GT, Bid and Bim collaborate during induction of T cell death in persistent infection. J. Immunol 186, 4059–4066 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sandalova E, Wei C-H, Masucci MG, Levitsky V, Regulation of expression of Bcl-2 protein family member Bim by T cell receptor triggering. Proc. Natl. Acad. Sci. U.S.A 101, 3011–3016 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Snow AL, Oliveira JB, Zheng L, Dale JK, Fleisher TA, Lenardo MJ, Critical role for BIM in T cell receptor restimulation-induced death. Biol. Direct 3, 34 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ley R, Hadfield K, Howes E, Cook SJ, Identification of a DEF-type docking domain for extracellular signal-regulated kinases 1/2 that directs phosphorylation and turnover of the BH3-only protein BimEL. J. Biol. Chem 280, 17657–17663 (2005). [DOI] [PubMed] [Google Scholar]
- 94.O’Connor L, Strasser A, O’Reilly LA, Hausmann G, Adams JM, Cory S, Huang DC, Bim: A novel member of the Bcl-2 family that promotes apoptosis. EMBO J. 17, 384–395 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Clybouw C, Merino D, Nebl T, Masson F, Robati M, O’Reilly L, Hübner A, Davis RJ, Strasser A, Bouillet P, Alternative splicing of Bim and Erk-mediated Bim(EL) phosphorylation are dispensable for hematopoietic homeostasis in vivo. Cell Death Differ. 19, 1060–1068 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, Varela-Rohena A, Haines KM, Heitjan DF, Albelda SM, Carroll RG, Riley JL, Pastan I, June CH, Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc. Natl. Acad. Sci. U.S.A 106, 3360–3365 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Guedan S, Posey AD, Shaw C, Wing A, Da T, Patel PR, McGettigan SE, Casado-Medrano V, Kawalekar OU, Uribe-Herranz M, Song D, Melenhorst JJ, Lacey SF, Scholler J, Keith B, Young RM, June CH, Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight 3, e96976 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Song D-G, Ye Q, Carpenito C, Poussin M, Wang L-P, Ji C, Figini M, June CH, Coukos G, Powell DJ, In vivo persistence, tumor localization, and antitumor activity of CAR-engineered T cells is enhanced by costimulatory signaling through CD137 (4-1BB). Cancer Res. 71, 4617–4627 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Priceman SJ, Gerdts EA, Tilakawardane D, Kennewick KT, Murad JP, Park AK, Jeang B, Yamaguchi Y, Yang X, Urak R, Weng L, Chang W-C, Wright S, Pal S, Reiter RE, Wu AM, Brown CE, Forman SJ, Co-stimulatory signaling determines tumor antigen sensitivity and persistence of CAR T cells targeting PSCA+ metastatic prostate cancer. Oncoimmunology 7, e1380764 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Paul A, Edwards J, Pepper C, Mackay S, Inhibitory-κB Kinase (IKK) α and nuclear factor-κB (NFκB)-inducing kinase (NIK) as anti-cancer drug targets. Cell 7, 176 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yu J, Zhou X, Nakaya M, Jin W, Cheng X, Sun S-C, T cell-intrinsic function of the noncanonical NF-κB pathway in the regulation of GM-CSF expression and experimental autoimmune encephalomyelitis pathogenesis. J. Immunol 193, 422–430 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sterner RM, Sakemura R, Cox MJ, Yang N, Khadka RH, Forsman CL, Hansen MJ, Jin F, Ayasoufi K, Hefazi M, Schick KJ, Walters DK, Ahmed O, Chappell D, Sahmoud T, Durrant C, Nevala WK, Patnaik MM, Pease LR, Hedin KE, Kay NE, Johnson AJ, Kenderian SS, GM-CSF inhibition reduces cytokine release syndrome and neuroinflammation but enhances CAR-T cell function in xenografts. Blood 133, 697–709 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sachdeva M, Duchateau P, Depil S, Poirot L, Valton J, Granulocyte-macrophage colony-stimulating factor inactivation in CAR T-cells prevents monocyte-dependent release of key cytokine release syndrome mediators. J. Biol. Chem 294, 5430–5437 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Madge LA, May MJ, Inflammation and Cancer (Humana Press, 2009), pp. 209–232. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. The ncNF-κB pathway.
Fig. S2. CAR expression on T cells from three donors expanded by irradiated Nalm6 cells.
Fig. S3. CAR expression on T cells from three donors expanded by anti-CD19 beads.
Fig. S4. CAR expression on T cells from three donors activated by anti-CD19 beads for signaling assays.
Fig. S5. Anti-CD19 BBζ CAR activation induces T cell signaling.
Fig. S6. Additional representative Western blotting analysis of nuclear and cytoplasmic fractions to compare anti-CD19 CAR signaling.
Fig. S7. Anti-mesothelin BBζ CAR also drives ncNF-κB signaling.
Fig. S8. Dual-transduced T cell production scheme, day 0 CAR, and mCherry expression.
Fig. S9. Western blotting analysis of nuclear and cytoplasmic fractions to compare control and dnNIK-expressing anti-CD19 BBζ T cells.
Fig. S10. dnNIK does not affect PCNA abundance in CAR T cells.
Fig. S11. BimEL abundance in anti-CD19 CAR T cells.
Fig. S12. ERK1/2 phosphorylation in control and dnNIK-expressing BBζ CAR T cells.
Fig. S13. FOXO3a abundance and phosphorylation in control and dnNIK-expressing BBζ CAR T cells.
Fig. S14. CAR constructs.
Fig. S15. Representative gating strategy to quantify cell death by flow cytometry and assess the elimination of CD19+ Nalm6 cells.