

Abstract
Background:
Selpercatinib (LOXO-292) and pralsetinib (BLU-667) are highly potent RET-selective protein tyrosine kinase inhibitors (TKIs) for treating advanced RET-altered thyroid cancers and non-small-cell lung cancer (NSCLC). It is critical to analyze RET mutants resistant to these drugs and unravel the molecular basis to improve patient outcomes.
Patients and methods:
Cell-free DNAs (cfDNAs) were analyzed in a RET-mutant medullary thyroid cancer (MTC) patient and a CCDC6-RET fusion NSCLC patient who had dramatic response to selpercatinib and later developed resistance. Selpercatinib-resistant RET mutants were identified and cross-profiled with pralsetinib in cell cultures. Crystal structures of RET-selpercatinib and RET-pralsetinib complexes were determined based on high-resolution diffraction data collected with synchrotron radiation.
Results:
RETG810C/S mutations at the solvent front and RETY806C/N mutation at the hinge region were found in cfDNAs of an MTC patient with RETM918T/V804M/L, who initially responded to selpercatinib and developed resistance. RETG810C mutant was detected in cfDNAs of a CCDC6-RET-fusion NSCLC patient who developed acquired resistance to selpercatinib. Five RET kinase domain mutations at three non-gatekeeper residues were identified from 39 selpercatinib-resistant cell lines. All five selpercatinib-resistant RET mutants were cross-resistant to pralsetinib. X-ray crystal structures of the RET-selpercatinib and RET-pralsetinib complexes reveal that, unlike other TKIs, these two RET TKIs anchor one end in the front cleft and wrap around the gate wall to access the back cleft.
Conclusions:
RET mutations at the solvent front and the hinge are resistant to both drugs. Selpercatinib and pralsetinib use an unconventional mode to bind RET that avoids the interference from gatekeeper mutations but is vulnerable to non-gatekeeper mutations.
Keywords: RET-selective inhibitor, selpercatinib, pralsetinib, acquired resistance, mutation, crystal structure
INTRODUCTION
Genetic alterations of the rearranged during transfection (RET) gene occur in diverse cancers.1–4 Several multi-targeted tyrosine kinase inhibitors (TKIs) with RET inhibitor activity, such as vandetanib, cabozantinib, lenvatinib, and RXDX-105, have been tested in the clinic with modest efficacy.1,5,6 In addition to dose-limiting off-target effects, these TKIs are subject to resistance by gatekeeper mutations.1,7 Structurally, this is partly because these TKIs are known or predicted to occupy both the front and back drug-binding clefts of the RET kinase domain by going through the gate that separates these two clefts.8–12
Selpercatinib13–15 and pralsetinib16 are two highly selective and potent RET TKIs. Recently, selpercatinib received the United States Food and Drug Administration (FDA) approval for treating metastatic RET fusion-positive non-small-cell lung cancer (NSCLC), advanced/metastatic RET-altered medullary thyroid cancer (MTC), and papillary thyroid carcinoma, while pralsetinib was approved by the FDA for treating RET fusion-positive NSCLC. The US FDA approved for pralsetinib was updated on December 1, 2020 to include advanced/metastatic RET-altered MTC and PTC.
Protein tyrosine kinase-targeted cancer therapies are subject to acquired resistance. Besides activation of alternative mechanisms bypassing the targeted kinase,17 a mechanism is secondary on-target mutations that interfere with drug binding.18,19 Identifying and characterizing resistance mechanisms, discovering new drugs, and translating the drugs to the clinic to overcome the resistance are critical for improving patient outcomes.
Herein, we report acquired RET mutations in a RET-mutant MTC patient and a RET fusion-positive NSCLC following treatment with selpercatinib. In preclinical experiments, we identified five selpercatinib-resistant mutants in the RET kinase domain and cross-profiling drug sensitivities of these mutants with pralsetinib. Four of the five selpercatinib-resistant RET mutations found in preclinical experiments were found as acquired RET mutations in the two patients.We also determined crystal structures of the RET-selpercatinib and RET-pralsetinib complexes with high-resolution X-ray diffraction data. These two crystal structures detail a novel kinase inhibitor binding mode not previously seen in other TKIs, and reveal the vulnerability of selpercatinib and pralsetinib to non-gatekeeper RET mutations.
PATIENTS AND METHODS
Patients
Patient 1 was a 49-year-old male who developed neck swelling and was diagnosed with sporadic MTC, harboring RET M918T mutation described previously.13 With lymph node and liver metastases, the patient was sequentially treated with six TKIs: sorafenib [best response of progressive disease (PD)], vandetanib [stable disease (SD)], cabozantinib (SD), MGCD-516 (PD), RXDX-105 [partial response (PR)], and vandetanib plus everolimus (PD). Molecular analysis of cell-free DNA (cfDNA) (Guardant360VR® [Guardant Health, Redwood City, CA]) isolated from blood taken before vandetanib plus everolimus treatment identified the founder RET M918T mutation and an acquired RET V804M gatekeeper mutation. A RET V804L gatekeeper mutant was detected subsequently. As the patient’s performance status was rapidly declining, he was initiated on selpercatinib on a single-patient protocol with rapid dose escalation to 160 mg twice daily (Figure 1A).13 The patient tolerated therapy well, had a dramatic improvement in clinical status, and confirmed PR for 24 months (Figure 1B–D). Around 25 months (30 cycles), his liver metastases increased, but he was clinically stable (Figure 1B–D). A biopsy sample was non-diagnostic and inconclusive. At 30 months (34 cycles), the patient developed hyperbilirubinemia and transaminitis, and his clinical status rapidly declined (supplementary Figure S1A, available at https://doi.org/10.1016/j.annonc.2020.10.599). The patient’s characteristics are summarized in supplementary Table S1, available at https://doi.org/10.1016/j.annonc.2020.10.599.
Figure 1. Acquired resistance to selpercatinib in an MTC patient.
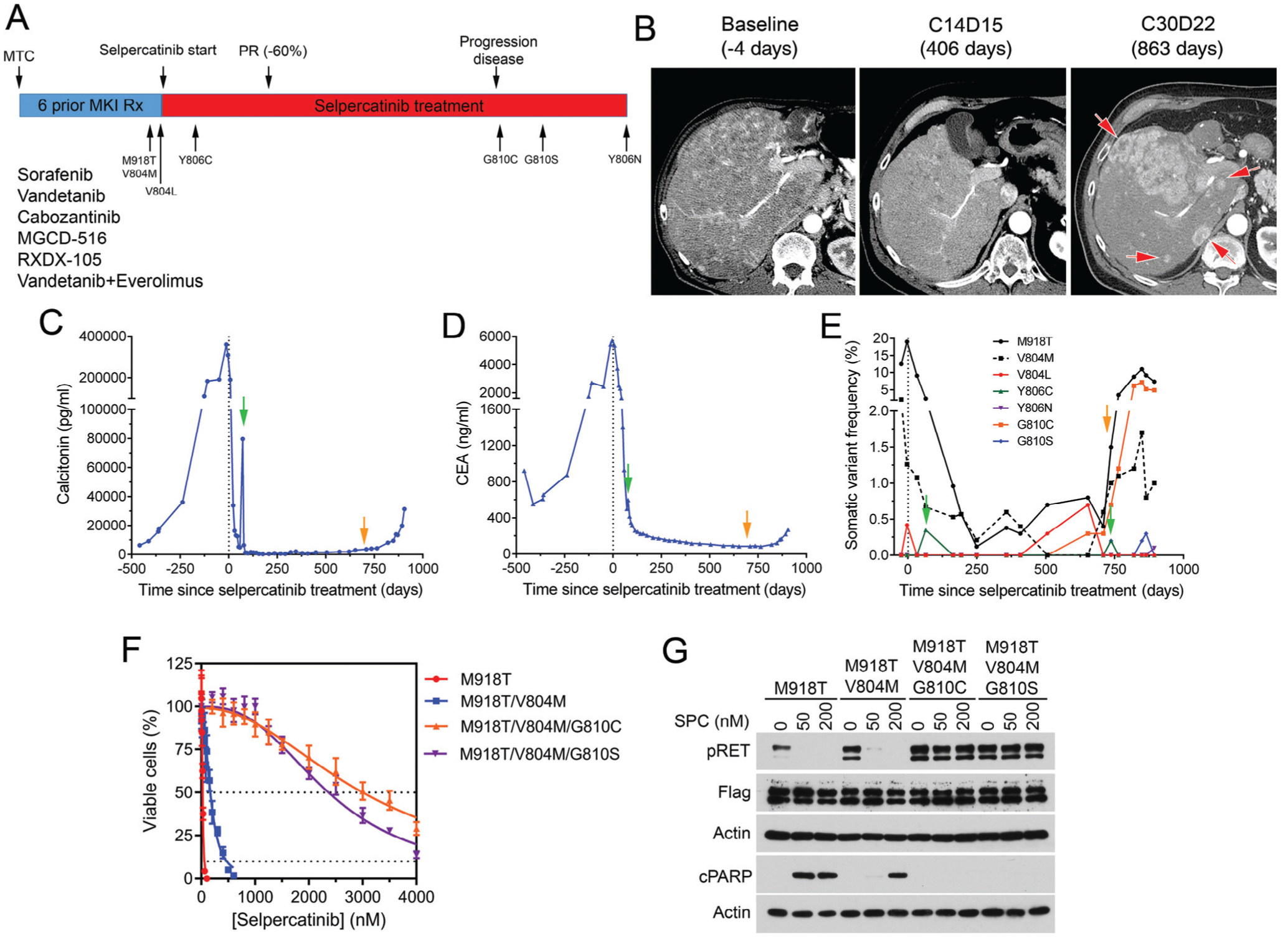
(A) Treatment history of an MTC patient treated with selpercatinib. (B) Axial arterial phase computed tomography images show innumerable enhancing hepatic metastases at baseline (left) before selpercatinib treatment that improved on cycle 14, day 15 (middle). A follow-up scan on cycle 30, day 22 demonstrated several new avidly enhancing lesions (right, red arrows). (C, D, E) Monitoring of calcitonin (C), carcinoembryonic antigen (CEA, D), and RET mutations in plasma cell-free DNA (cfDNA)(E). Green arrows indicate time points that the RETY806C mutation was detected. Orange arrows indicate the time point of tumor progression. (F) Comparison of IC50s of selpercatinib in BaF3 cells expressing the indicated RET mutants. (G) Immunoblotting analysis of phospho-RET (pY905), cleaved PARP, and b-actin after treatment of BaF3 cells expressing the indicated RET mutants with selpercatinib.
CEA, carcinoembryonic antigen; cPARP, cleaved poly(ADP-ribose) polymerase; MKI, multikinase inhibitor; MTC, medullary thyroid cancer; PR, partial response; pRET, phospho-RET (pY905); Rx, treatment; SPC, selpercatinib.
Patient 2 was a 66-year-old male never smoker who presented with back and chest pain. A complete workup and biopsy revealed Stage IV metastatic thyroid transcription factor 1-positive lung adenocarcinoma. Immunostain for programmed cell death ligand 1 was negative. Molecular testing was negative for epidermal growth factor receptor (EGFR) or BRAF mutation, and ALK or ROS1 gene rearrangement. He was treated with eight cycles of carboplatin/pemetrexed/bevacizumab and achieved PR (Figure 2A). He was then switched to maintenance with pemetrexed and bevacizumab for four more cycles until the tumors progressed, clinical status declining with increasing pain, cough with worsening bone, and lung and liver metastases. Next-generation sequencing of plasma cfDNA revealed CCDC6-RET fusion,TP53 G244S/R181G, CDKN2A H83R, MET, and EGFR amplification (supplementary Table S1, available at https://doi.org/10.1016/j.annonc.2020.10.599). The patient was enrolled in the selpercatinib trial. In a few weeks, the patient’s performance status rapidly improved, pain and cough resolved, his tumors had a deep response of 64% reduction and confirmed PR per RECIST V1.1 (Figure 2A–C). After 18 cycles of selpercatinib, his pain started to come back, his PS declined (Figure 2D, supplementary Figure S1B, available at https://doi.org/10.1016/j.annonc.2020.10.599), and his scans showed new bilobar liver metastases (Figure 2C).
Figure 2. Acquired resistance to selpercatinib in a CCDC6-RET-positive NSCLC patient.
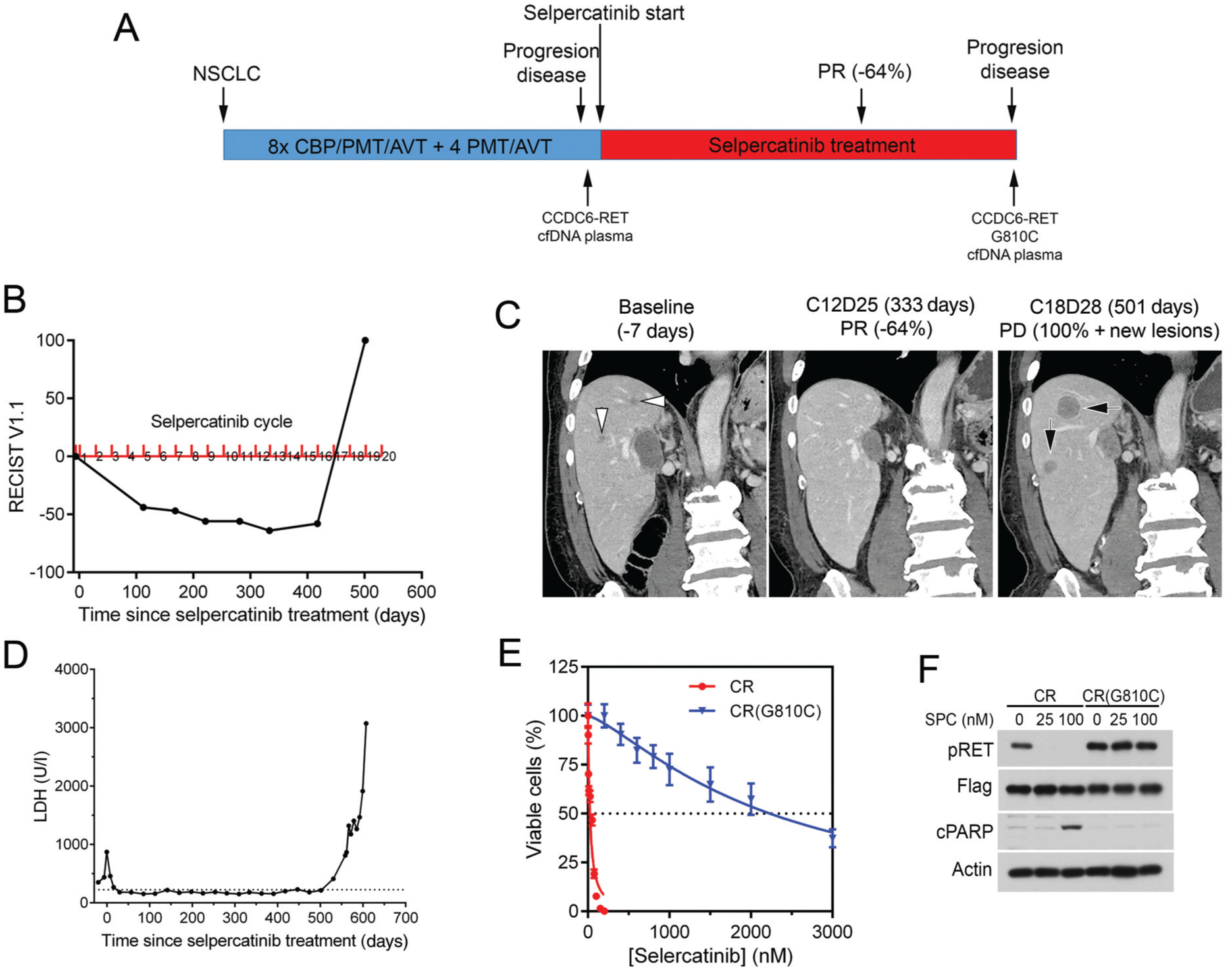
(A) Treatment history and RET alterations. (B) RECIST v 1.1 measurements. (C) Coronal reformations from contrast-enhanced abdominal computed tomography (CT) images show hepatic metastases at baseline before selpercatinib treatment (left, white arrowheads) that decreased in size and disappeared by cycle 12, day 25 (middle). Follow-up imaging on cycle 18, day 28 shows new lesions (right, black arrows). (D) Test results of lactate dehydrogenase (LDH). LDH was used as a non-specific marker of tumor burden. (E, F) Cell viability and immunoblotting analyses of BaF3/CCDC6-RET (CR) and BaF3/CCDC6-RETG810C [CR(G810C)] cells.
AVT, avastin; CBP, carboplatin; cfDNA, cell-free DNA; cPARP, cleaved poly(ADP-ribose) polymerase; NSCLC, non-small cell lung cancer; PD, progressive disease; PMT, pemetrexed; PR, partial response; pRET, phospho-RET (pY905); SPC, selpercatinib.
Clinical methods
Patients provided written informed consent to participate in the clinical trial. Molecular tests were carried out in accordance with protocols approved by the institutional review board at UT MD Anderson Cancer Center (supplementary materials, available at https://doi.org/10.1016/j.annonc.2020.10.599).
Isolation and characterization of drug-resistant RET mutations in cell cultures
Selpercatinib-resistant RET mutations were identified by sequencing of genomic DNA from BaF3/KIF5B-RET cell lines that could grow at >10× IC50s concentrations of selpercatinib (supplementary materials, available at https://doi.org/10.1016/j.annonc.2020.10.599). The CCDC6-RETG810C, full-length RETM918T/V804M, RETM918T/V804M/G810C, and RETM918T/V804M/G810S mutants cDNA were made by site-specific mutagenesis and expressed in BaF3 for analysis. In IC50s and immunoblotting assays were as described.7,10,20
Crystal structure determination
The purified RET kinase protein contained amino acid residues 705–1013 of RET (GenPept ID: NP_066124). The atomic coordinates and structure factors of selpercatinib-bound RET and pralsetinib-bound RET have been deposited in the Protein Data Bank21 under accession numbers 7JU6 (RET-selpercatinib) (https://www.rcsb.org/structure/7JU6) and 7JU5 (RET-pralsetinib) (https://www.rcsb.org/structure/7JU5). The detailed procedures of structural studies are provided in the supplementary materials, available at https://doi.org/10.1016/j.annonc.2020.10.599.
RESULTS
Acquired RET mutations detected in cfDNA
Patient 1: plasma cfDNA analyses detected RETY806C mutation at 2 months, which corresponded to a spike of calcitonin and carcinoembryonic antigen (Figure 1C–E). The RETY806C mutation reappeared at 24.5 months. RETG810C/S mutations emerged at 22 and 24 months, respectively. The RETY806N mutation was detected in cfDNA at 29 months (Figure 1E, supplementary Table S2, available at https://doi.org/10.1016/j.annonc.2020.10.599).
We had identified G810C/S and Y806C/N as selpercatinib-resistant RET mutants in preclinical experiments using the KIF5B-RET oncogene as a model (see later). To verify that the results are applicable in the context of the RETM918T/V804M oncogene, we tested the RETG810C/S mutants in the context of RETM918T/V804M. BaF3 cell lines expressing the full-length RETM918T, RETM918T/V804M, RETM918T/V804M/G810C, and RETM918T/V804M/G810S were established, and the IC50s of selpercatinib in these cells were measured. Selpercatinib inhibited BaF3/RETM918T cells with IC50s of 23 ± 1 nM. The selpercatinib IC50s were increased 8-, 131-, and 102-fold in BaF3/RETM918T/V804M, BaF3/RETM918T/V804M/G810C, and BaF3/RETM918T/V804M/G810S cells, respectively (Figure 1F). In immunoblotting assays, while autophosphorylation of RETM918T and RETM918T/V804M mutants were inhibited by selpercatinib at the test concentrations (50–200 nM), the triple mutants were not (Figure 1G). Consistently, the BaF3/RETM918T/V804M/G810C and BaF3/RETM918T/V804M/G810S cells were resistant to selpercatinib-induced apoptosis (Figure 1G).
Patient 2: when the disease progressed during the 18th cycle of selpercatinib treatment, plasma cfDNA had CCD6-RET fusion and a novel RET NM_020975.4(RET): c.2428G>Tp.G810C Exon 14 SNV Missense RETG810C solvent-front mutation; CDKN2A NM_000077.4(CDKN2A):c.248A>Gp.H83R; TP53 NM_000546.5(TP53):c.730G>A p.G244S Exon 7 (Figure 2A, supplementary Table S1, available at https://doi.org/10.1016/j.annonc.2020.10.599).
To assess whether the CCDC6-RETG810C mutant found in patient 2 was resistant to selpercatinib, BaF3/CCDC6-RET and BaF3/CCDC6-RETG810C cells were generated and analyzed. The results showed that cells expressing CCDC6-RETG810C had a 93-fold higher IC50 than cells expressing CCDC6-RET (Figure 2E). The CCDC6-RETG810C kinase was resistant to selpercatinib, and the BaF3/CCDC6-RETG810C cells were resistant to apoptosis induced by selpercatinib (Figure 2F).
Identification and characterization of selpercatinib- and pralsetinib-resistant RET mutants in cell cultures and in tumor xenografts
By screening a random mutation library of BaF3/KIF5B-RET cells, we identified four selpercatinib-resistant mutations in the RET kinase domain from 36 cell lines (Table 1). In an alternative approach, we cultured BaF3/KIF5B-RET cells with stepwise increasing concentrations of selpercatinib and established three selpercatinib-resistant cell lines that identified another RET mutation (Table 1). Thus, a total of five different selpercatinib-resistant RET mutations were identified. Cross-profiling of IC50s with pralsetinib showed that these five selpercatinib-resistant mutants were also resistant to pralsetinib (Table 1).
Table 1.
Identification of selpercatinib-resistant RET mutants and cross-profiling with pralsetinib
| Method | Mutant | Selecting selpercatinib concentration (nM) | Mutant clone count | IC50 (nM) (fold: mutant/wt) | ||
|---|---|---|---|---|---|---|
| 150 | 300 | Selpercatinib | Pralsetinib | |||
| Isolation from mutation library | V738A | 2 | 0 | 2 | 238.8 ± 7.2 (29) | 177.5 ± 6.7 (19) |
| Y806C | 1 | 0 | 1 | 174.4 ± 5.4 (21) | 295.8 ± 10.7 (32) | |
| Y806N | 2 | 0 | 2 | 149.8 ± 6.3 (18) | 292.5 ± 5.9 (32) | |
| G810S | 20 | 11 | 31 | 880.2 ± 25.6 (107) | 390.6 ± 10.8 (42) | |
| Cell culture with selpercatinib | G810S | 120 | 2 | 880.2 ± 25.6 (107) | 390.6 ± 10.8 (42) | |
| G810C | 120 | 1 | 1227 ± 44.1 (150) | 641.7 ± 19.1 (70) | ||
| N/A | G810R | N/A | N/A | 2744 ± 160.6 (334) | 2650 ± 287.3 (288) | |
| V804L | N/A | N/A | 17.2 ± 0.5 (2) | 1.8 ± 0.6 (2) | ||
| V804M | N/A | N/A | 55.9 ± 1.6 (7) | 16.8 ± 0.8 (2) | ||
| wt | N/A | N/A | 8.2 ± 0.4 (1) | 9.2 ± 0.4 (1) | ||
IC50, concentration that causes 50% inhibition of growth; N/A, not applicable; wt, wildtype.
The five selpercatinib- and pralsetinib-resistant mutations were located at the β2 strand (RETV738A), hinge (RETY806C/N), and solvent front (RETG810C/S) sites in the RET kinase domain (see later). No gatekeeper mutation was found in resistant cell lines, consistent with the RET gatekeeper mutants being sensitive to selpercatinib. A BaF3/KIF5B-RETG810R mutation cell line was also tested. Cell lines containing these mutations had 18- to 334-fold higher IC50s for selpercatinib or pralsetinib (Table 1,Figure 3A and C). Immunoblotting assays of RET kinase activity and apoptosis showed that these mutated KIF5B-RET fusion kinases were less sensitive to inhibition by selpercatinib and pralsetinib and that the cells expressing these mutants were resistant to apoptosis induced by these drugs (Figure 3B and D).
Figure 3. Sensitivities of RET mutants to selpercatinib and pralsetinib.
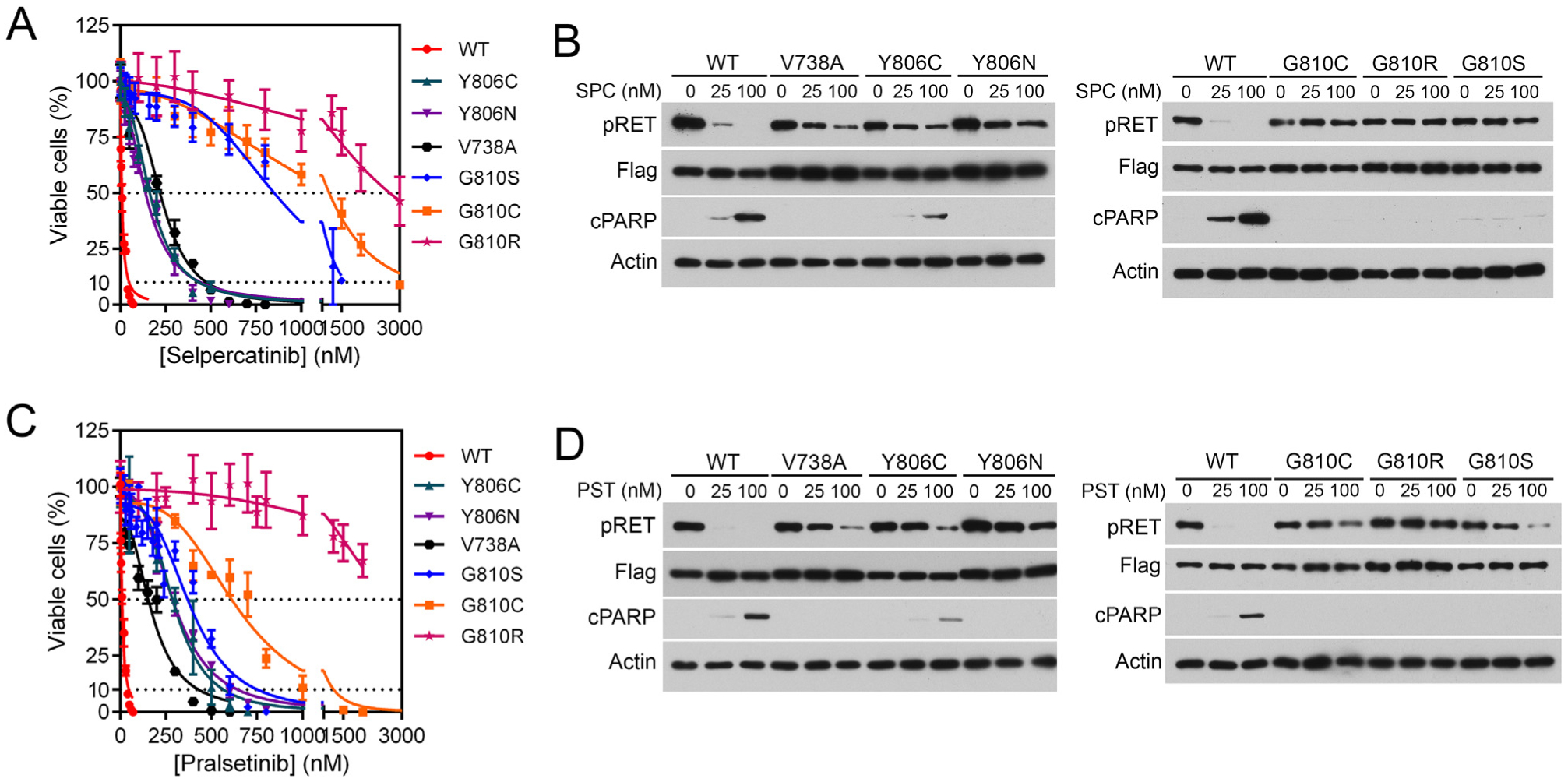
(A, C) BaF3 cells expressing KIF5B-RET (WT) or KIF5B-RET; the indicated mutations were treated with various concentrations of selpercatinib (A) or pralsetinib (C) for 3 days, and viable cells were measured. (B, D) Immunoblotting analysis of RET tyrosine phosphorylation and apoptosis after treatment of cells with selpercatinib (SPC) (B) or pralsetinib (PST) (D). Cells were treated with the drug for 4 h for analyzing RET phosphorylation or 24 h for analyzing cPARP.
cPARP, cleaved poly(ADP-ribose) polymerase; pRET, phospho-RET (pY905); WT, wildtype.
Structural basis of resistance to selpercatinib and pralsetinib
We determined the crystal structures of the RET kinase-selpercatinib and the RET kinase-pralsetinib complexes based on the high-resolution diffraction data collected with synchrotron radiation (supplementary Table S3, available at https://doi.org/10.1016/j.annonc.2020.10.599, PDB codes 7JU6 and 7JU5, respectively). The best crystal of the RET kinase-selpercatinib complex gave diffraction data to 2.06 Å, while the best crystal of the RET kinase-pralsetinib complex gave diffraction data to 1.9 Å. Selpercatinib and pralsetinib bound the RET kinase similarly in a novel binding mode that occupied both front and back pockets in the active site clefts without passing through the gate between V804 and K758 into BP-I (Figure 4A, B, F).8 Instead, these compounds accessed the back pocket by wrapping around the gate wall K758 residue. This avoids steric clash with gatekeeper V804L/M mutations. Compared with pralsetinib, the two central rings of selpercatinib were buried deeper in the ligand-binding cleft. This placed them further from the solvent edge of the binding cleft (Figure 4G).
Figure 4. Crystal structures of the RET-selpercatinib and RET-pralsetinib complexes.
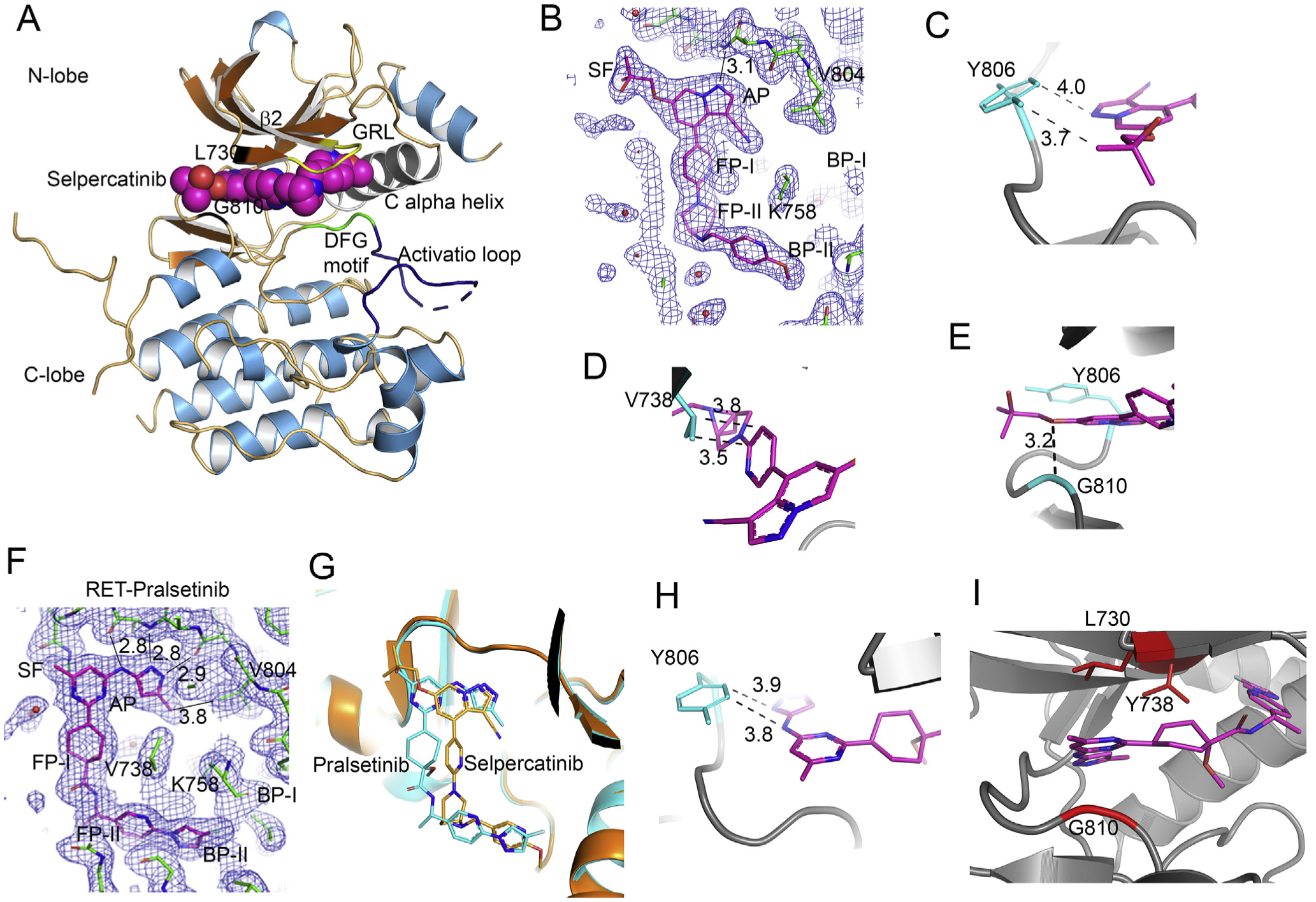
(A) Ribbon diagram of RET kinase-selpercatinib complex (7JU6:chain B). (B) View of the 2mFo-DFc electron density map contoured at the 1 sigma level showing the fit of selpercatinib in the 7JU6:chain B. One H-bond occurred between the drug and the protein in the adenosine pocket (AP) site. The gate is between K758 and V804. (C, D,E) Close-up views of areas of Y806, V738, and G810. Dash lines are van der Waals interactions. Distances are in angstroms (Å). (F) View of the 2mFo-DFc electron density map contoured at the 1 sigma level showing the fit of pralsetinib in 7JU5:chain A. The three left-most distances are H-bonds. The distance on the right is a van der Waals interaction. (G) Superposition of the RET kinase-pralsetinib complex (carbon atoms colored cyan) and the RET kinase-selpercatinib complex (carbon atoms colored orange). The pralsetinib and selpercatinib were not used in the least-squares fit. (H) Close-up of pralsetinib (colored magenta) in the vicinity of Y806 (colored cyan). The distances are van der Waals interactions. (I) Close-up of pralsetinib in the vicinity of V738 and G810.
The nine-membered pyrazolo ring of selpercatinib occupied the adenosine pocket (AP) that is bordered by Y806 on one side (Figure 4B). The side chain of Y806 had van der Waals interactions with the pyrazolo[1,5-a]pyridine ring and the hydroxymethyl group (Figure 4C), which would be disrupted by substitution of Y806 with cysteine or asparagine that have non-hydrophobic, shorter side chains. The pyridine ring and the 6-[6-(6-methoxypyridin-3-yl)methyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl group occupied FP-I and FP-II, respectively. The pyridine ring made two van der Waals interactions with the side chain of V738 located on the β2 strand (Figure 4A and D). Replacement of V738 with alanine, which has a shorter side chain, will weaken the van der Waals interactions. Selpercatinib has a hydroxymethylpropoxyl group attached to the pyrazolo[1,5-a]pyridine ring. The hydroxymethyl group projected through the solvent front at the mouth of the AP (Figure 4A, B, E). G810 is the C-lobe residue at the solvent front site. Substitution of the single hydrogen atom side chain of glycine with bulky side chains like those of cysteine, serine, and arginine would cause a steric clash with the hydroxymethylpropoxyl group.
The aminopyrimidinyl and methylaminopyrazol rings of pralsetinib occupied the AP (Figure 4F). The hydrophobic side of the side chain of Y806 made hydrophobic interactions with a face of the methylaminopyrazol ring in the AP site (Figure 4H). Mutation of Tyr-806 to Cys or Asn that contain non-hydrophobic, shorter side chains would disrupt these hydrophobic interactions. One face of the methylamino-pyrimidine ring had pi-H interactions with C-alpha carbon atoms of G810 on the C-lobe side of the solvent front, and the other face of the ring had hydrophobic interactions with both terminal methyl groups of the side chain of L730 of the β1 strand in the N-lobe side of the solvent front (Figure 4I). The methyl group of the methylpyrimidine extended into the first hydration shell at the solvent front. Substitution of G810 with an amino acid that has a larger side chain, such G810C/S/R, would introduce severe steric clashes with the methylpyrimidine ring, whereas substitution of L730 with isoleucine or valine would weaken the hydrophobic interactions and the introduced CG1 and CG2 carbon atoms would cause new steric clashes. The cyclohexane ring of pralsetinib occupied FP-I and the pyridine ring occupied FP-II (Figure 4I). The cyclohexane ring packed against the back-bone of L730 and G731 at the solvent front on the N-lobe of the active site cleft. The CG2 carbon atom of the V738 side chain formed a hydrophobic interaction with the edge of the pyridine ring (Figure 4I). These hydrophobic interactions would be lost when the V738 residue is replaced by an alanine.
DISCUSSION
While avoiding the interference of the gatekeeper mutations, selpercatinib and pralsetinib were subject to resistance caused by non-gatekeeper mutations. Four of five selpercatinib-resistant RET kinase domain mutants that we identified in the laboratory were also detected in the MTC patient and in the NSCLC patient. The fifth mutation identified in the laboratory, RETV738A, is located outside the RET coverage region of clinical cfDNA assays and thus would evade detection in these patients. Recently, the RETG810C/S solvent front mutants have also been found in the cfDNA of two patients with CCDC6-RET-positive NSCLC that acquired pralsetinib-resistance.22
The RET mutations at the C-lobe solvent front (RETG810C/S/R), hinge (RETY806C/N), and β2 strand (RETV738A) shared resistance to both selpercatinib and pralsetinib. The IC50 fold changes of RETG810C/S/R mutants were consistently higher for selpercatinib than for pralsetinib, suggesting that mutations at the C-lobe solvent front site have a higher impact on selpercatinib than on pralsetinib.
Selpercatinib and pralsetinib use a binding mode that is very different from other TKIs. Previously determined structures of complexes with TKIs show that the TKIs occupy both the front and back clefts of the drug-binding pockets by passing through the gate that separates the front and back clefts (e.g. vandetanib, PDB code 2IVU; nintedanib, PDB code 6NEC)9,10 or bind only the front cleft, such as alectinib (PDB code 3AOX),23 certinib (PDB code 4MKC),24 osimertinib (PDB code 4ZAU),25 and entrectinib (PDB code 5KVT). In contrast, selpercatinib and pralsetinib dock one end in the front cleft without inserting through the gate, and wrap around the area outside the gate wall formed by the side chain of K758 and bury the other end in the BP-II pocket of the back cleft.8 This novel binding mode allows high-affinity binding while avoiding disruption of gatekeeper mutations. Nevertheless, this novel kinase inhibitor binding mode remains liable to resistance from mutations at several non-gatekeeper residues identified in this study.
Information on acquired resistance mechanisms to RET, both on target and off target, are emerging. Previous reports have shown RETV804M and RETS904F as resistance mechanisms to vandetanib and acquired RETG810R/C/S/V mutations in RET fusion-positive NSCLC patients whose tumors developed resistance to selpercatinib.26–28 More recently, acquired RET kinase domain mutations have been found in pralsetinib-treated NSCLC patients.22 Another recent study analyzing eighteen RET fusion-positive patients who received selpercatinib or pralsetinib revealed acquired RET G810 solvent front mutations in two cases (10%), three resistant cases (15%) with acquired MET amplification, and one specimen had acquired KRAS amplification.29 Another four cases of acquired selpercatinib resistance with MET amplification were reported and, it was demonstrated that this could be overcome by combining selpercatinib with crizotinib.30 Given that >1000 patients with RET alterations were enrolled on selective RET inhibitor trials globally, these trials are still in progress, and the resistance mechanisms have been reported in just a few patients so far, the frequencies of various resistance mechanisms remain to be determined.
While the most resistant mutant G810C is the principal event in the two clinical cases here, the MTC patient who ultimately progressed harbored RET M918T/V804L/V804M/Y806C/Y806N/G810C/G810S mutations. It is envisioned that the less strong selpercatinib-resistant G810S and Y804C/N could play a significant role in patients treated with lower doses of selpercatinib. Our findings point to the need to develop next-generation RET TKIs covering both gatekeeper and non-gatekeeper mutations for on-target resistance, in addition to deciphering patterns of off-target resistance by alternative mechanisms for combination therapies.
Supplementary Material
FUNDING
This work was supported by a Presbyterian Health Foundation Team Science grant (to BHMM and JW), National Institutes of Health (NIH) (grant number R01CA242845) to (BHMM, VS, and JW), the Oklahoma Tobacco Settlement Endowment Trust (to the Stephenson Cancer Center), the Cancer Prevention and Research Institute of Texas (grant number RP1100584), the Sheikh Khalifa Bin Zayed Al Nahyan Institute for Personalized Cancer Therapy (grant number 1U01 CA180964), the National Center for Advancing Translational Sciences (grant number UL1 TR000371), and the MD Anderson Cancer Center (support grant number P30 CA016672). Use of the Laboratory of the Biomolecular Structure and Function at the University of Oklahoma Health Sciences Center was supported by a grant from the National Institute of General Medical Sciences (grant number P20GM103640). Use of the Stanford Synchrotron Radiation Lightsource (SSRL), SLAC National Accelerator Laboratory, was supported by the U.S. Department of Energy (DOE), Office of Basic Energy Sciences under contract number DE-AC02-76SF00515. The SSRL Structural Molecular Biology (SMB) Program was supported by the DOE Office of Biological and Environmental Research, and the National Institute of General Medical Sciences (including grant number P41GM103393). We thank Drs Silvia Russi and Doukov Tzanko of the SMB program for their assistance with data collection. The shared resources at the University of Oklahoma Health Sciences Center were supported by NIH/National Institute of General Medical Sciences (grant number P20GM103639) and the NIH/National Cancer Institute (grant number P30CA225520).
DISCLOSURE
VS: research funding/grant support for clinical trials: Roche/Genentech, Novartis, Bayer, GlaxoSmithKline, Nanocarrier, Vegenics, Celgene, Northwest Biotherapeutics, Berghealth, Incyte, Fujifilm, Pharmamar, D3, Pfizer, Multivir, Amgen, Abbvie, Alfa-sigma, Agensys, Boston Biomedical, Idera Pharma, Inhibrx, Exelixis, Blueprint Medicines, Loxo Oncology, Medimmune, Altum, Dragonfly Therapeutics, Takeda and, National Comprehensive Cancer Network, NCI-CTEP and UT MD Anderson Cancer Center, Turning Point Therapeutics, Boston Pharmaceuticals; travel: Novartis, Pharmamar, ASCO, ESMO, Helsinn, Incyte; consultancy/advisory board: Helsinn, LOXO Oncology/Eli Lilly, R-Pharma US, INCYTE, QED Pharma, Medimmune, Novartis. Other: Medscape. MH has participated in advisory boards for Blueprint Medicines Corporation, Eli Lilly and Company, and Loxo Oncology, and has served as a consultant for Veracyte. MC, receiving grant support, paid to her institution, from Eisai, Exelixis, Genentech USA, Kura Oncology, and Merck, and advisory board fees from Ignyta and Loxo Oncology. FM-B reports consulting: Aduro BioTech Inc., DebioPharm, eFFECTOR Therapeutics, F. Hoffman-La Roche Ltd., Genentech Inc., IBM Watson, Jackson Laboratory, Kolon Life Science, OrigiMed, PACT Pharma, Parexel International, Pfizer Inc., Samsung Bioepis, Seattle Genetics Inc., Tyra Biosciences, Xencor, Zymeworks. Advisory committee: Immunomedics, Inflection Biosciences, Mersana Therapeutics, Puma Biotechnology Inc., Seattle Genetics, Silverback Therapeutics, Spectrum Pharmaceuticals. Sponsored research: Aileron Therapeutics, Inc., AstraZeneca, Bayer Healthcare Pharmaceutical, Calithera Biosciences Inc., Curis Inc., CytomX Therapeutics Inc., Daiichi Sankyo Co. Ltd., Debiopharm International, eFFECTOR Therapeutics, Genentech Inc., Guardant Health Inc., Millennium Pharmaceuticals Inc., Novartis, Puma Biotechnology Inc., Taiho Pharmaceutical Co. Honoraria: Chugai Biopharmaceuticals, Mayo Clinic, Rutgers Cancer Institute of New Jersey. All remaining authors have declared no conflicts of interest.
REFERENCES
- 1.Drilon A, Hu ZI, Lai GGY, Tan DSW. Targeting RET-driven cancers: lessons from evolving preclinical and clinical landscapes. Nat Rev Clin Oncol. 2018;15:151–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Subbiah V, Cote GJ. Advances in targeting RET-dependent cancers. Cancer Discov. 2020;10:498–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kohno T, Tabata J, Nakaoku T. REToma: a cancer subtype with a shared driver oncogene. Carcinogenesis. 2020;41:123–129. [DOI] [PubMed] [Google Scholar]
- 4.Liu X, Hu X, Shen T, et al. RET kinase alterations in targeted cancer therapy. Cancer Drug Resist. 2020;3:472–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gautschi O, Milia J, Filleron T, et al. Targeting RET in patients with RET-rearranged lung cancers: results from the global, multicenter RET registry. J Clin Oncol. 2017;35:1403–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Drilon A, Fu S, Patel MR, et al. A phase I/Ib trial of the VEGFR-sparing multikinase RET inhibitor RXDX-105. Cancer Discov. 2019;9:384–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu X, Shen T, Mooers BHM, et al. Drug resistance profiles of mutations in the RET kinase domain. Br J Pharmacol. 2018;175:3504–3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Linden OP, Kooistra AJ, Leurs R, et al. KLIFS: a knowledge-based structural database to navigate kinase-ligand interaction space. J Med Chem. 2014;57:249–277. [DOI] [PubMed] [Google Scholar]
- 9.Knowles PP, Murray-Rust J, Kjaer S, et al. Structure and chemical inhibition of the RET tyrosine kinase domain. J Biol Chem. 2006;281: 33577–33587. [DOI] [PubMed] [Google Scholar]
- 10.Terzyan SS, Shen T, Liu X, et al. Structural basis of resistance of mutant RET protein-tyrosine kinase to its inhibitors nintedanib and vandetanib. J Biol Chem. 2019;294:10428–10437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roskoski R Jr, Sadeghi-Nejad A. Role of RET protein-tyrosine kinase inhibitors in the treatment RET-driven thyroid and lung cancers. Pharmacol Res. 2018;128:1–17. [DOI] [PubMed] [Google Scholar]
- 12.Li GG, Somwar R, Joseph J, et al. Antitumor activity of RXDX-105 in multiple cancer types with RET rearrangements or mutations. Clin Cancer Res. 2017;23:2981–2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Subbiah V, Velcheti V, Tuch BB, et al. Selective RET kinase inhibition for patients with RET-altered cancers. Ann Oncol. 2018;29:1869–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wirth LJ, Sherman E, Robinson B, et al. Efficacy of selpercatinib in RET-altered thyroid cancers. N Engl J Med. 2020;383:825–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Drilon A, Oxnard GR, Tan DSW, et al. Efficacy of selpercatinib in RET fusion-positive non-small-cell lung cancer. N Engl J Med. 2020;383: 813–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Subbiah V, Gainor JF, Rahal R, et al. Precision targeted therapy with BLU-667 for RET-driven cancers. Cancer Discov. 2018;8:836–849. [DOI] [PubMed] [Google Scholar]
- 17.Nelson-Taylor SK, Le AT, Yoo M, et al. Resistance to RET-inhibition in RET-rearranged NSCLC is mediated by reactivation of RAS/MAPK signaling. Mol Cancer Ther. 2017;16:1623–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Katayama R, Lovly CM, Shaw AT. Therapeutic targeting of anaplastic lymphoma kinase in lung cancer: a paradigm for precision cancer medicine. Clin Cancer Res. 2015;21:2227–2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Awad MM, Katayama R, McTigue M, et al. Acquired resistance to crizotinib from a mutation in CD74-ROS1. N Engl J Med. 2013;368: 2395–2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang Q, Schneeberger VE, Luetteke N, et al. Preclinical modeling of KIF5B-RET fusion lung adenocarcinoma. Mol Cancer Ther. 2016;15: 2521–2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.ww PDBc. Protein Data Bank: the single global archive for 3D macro-molecular structure data. Nucleic Acids Res. 2019;47:D520–D528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gainor J, Curigliano G, Doebele RC, et al. Analysis of resistance mechanisms to pralsetinib in patients with RET fusion-positive non-small cell lung cancer (NSCLC) from the ARROW study. IASLC 2020 North American Conferenceon LungCancer (October 16–17, 2020) 2020; Abstract OA05.02. [Google Scholar]
- 23.Sakamoto H, Tsukaguchi T, Hiroshima S, et al. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell. 2011;19:679–690. [DOI] [PubMed] [Google Scholar]
- 24.Friboulet L, Li N, Katayama R, et al. The ALK inhibitor ceritinib overcomes crizotinib resistance in non-small cell lung cancer. Cancer Discov. 2014;4:662–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yosaatmadja Y, Silva S, Dickson JM, et al. Binding mode of the break-through inhibitor AZD9291 to epidermal growth factor receptor revealed. J Struct Biol. 2015;192:539–544. [DOI] [PubMed] [Google Scholar]
- 26.Dagogo-Jack I, Stevens SE, Lin JJ, et al. Emergence of a RET V804M gatekeeper mutation during treatment with vandetanib in RET-rearranged NSCLC. J Thorac Oncol. 2018;13:e226–e227. [DOI] [PubMed] [Google Scholar]
- 27.Nakaoku T, Kohno T, Araki M, et al. A secondary RET mutation in the activation loop conferring resistance to vandetanib. Nat Commun. 2018;9:625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Solomon BJ, Tan L, Lin JJ, et al. RET solvent front mutations mediate acquired resistance to selective RET inhibition in RET-driven malignancies. J Thorac Oncol. 2020;15:541–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin JJ, Liu SV, McCoach CE, et al. Mechanisms of resistance to selective RET tyrosine kinase inhibitors in RET fusion-positive non-small cell lung cancer. Ann Oncol. 2020;31:1725–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rosen EY, Johnson ML, Clifford SE, et al. Overcoming MET-dependent resistance to selective RET inhibition in patients with RET fusion-positive lung cancer by combining selpercatinib with crizotinib. Clin Cancer Res. 2020. 10.1158/1078-0432.CCR-20-2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.