

Abstract
Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) has become one major threat to human population health. The RNA-dependent RNA polymerase (RdRp) presents an ideal target of antivirals, whereas nucleoside analogs inhibitor is hindered by the proofreading activity of coronavirus. Herein, we report that corilagin (RAI-S-37) as a non-nucleoside inhibitor of SARS-CoV-2 RdRp, binds directly to RdRp, effectively inhibits the polymerase activity in both cell-free and cell-based assays, fully resists the proofreading activity and potently inhibits SARS-CoV-2 infection with a low 50% effective concentration (EC50) value of 0.13 μmol/L. Computation modeling predicts that RAI-S-37 lands at the palm domain of RdRp and prevents conformational changes required for nucleotide incorporation by RdRp. In addition, combination of RAI-S-37 with remdesivir exhibits additive activity against anti-SARS-CoV-2 RdRp. Together with the current data available on the safety and pharmacokinetics of corilagin as a medicinal herbal agent, these results demonstrate the potential of being developed into one of the much-needed SARS-CoV-2 therapeutics.
KEY WORDS: SARS-CoV-2, RdRp, Structure-based virtual screening, Viral replication, Non-nucleoside inhibitor, Drug combinations, Corilagin
Graphical abstract
We report that RAI-S-37 (corilagin) acts as a non-nucleoside inhibitor of SARS-CoV-2 RdRp, binds directly to RdRp, effectively inhibits the polymerase activity in both cell-free and cell-based assays, fully resists the proofreading activity and potently inhibits SARS-CoV-2 infection in vitro.

1. Introduction
The novel coronavirus disease 2019 (COVID-19) is caused by severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2)1,2. Since its outbreak in late 2019, SARS-CoV-2 has rapidly spread worldwide and has become one major public health threat3. As of November 30, 2020, approximately 62 million people have been diagnosed with SARS-CoV-2 infection, one million COVID-19 patients died, and the number of COVID-19 cases is steeply rising. Compared with SARS-CoV that emerged in 2002–2003, SARS-CoV-2 shows a much higher level of adaptation to humans and has far greater human to human transmission4, 5, 6, 7. Currently, there are no approved therapies specifically treating human coronaviruses. Although remdesivir was granted emergency use by the US Food and Drug Administration (FDA) in May 2020 to treat severely ill COVID-19 patients, further evaluations are ongoing to address its clinical benefit, safety, and optimal use8, 9, 10, 11. Thus, it is urgent to develop effective treatments to end this global pandemic.
SARS-CoV-2 is an enveloped, positive-sense, single-stranded RNA virus12. It belongs to the beta-coronavirus genus and is the seventh human coronavirus discovered to date1,4. Similar to other coronaviruses, the 30 kb SARS-CoV-2 RNA genome encodes unusually complex replication/transcription machinery that comprises several non-structural proteins (nsps)13,14. Among them, SARS-CoV-2 nsp12, also known as the RNA-dependent RNA polymerase (RdRp), plays a fundamental role in viral RNA synthesis. Moreover, RdRp has higher evolutionary stability and does not exist in mammalian cells, thus representing an extremely attractive drug target. Nucleos(t)ide inhibitors (NIs) such as remdesivir have been reported to suppress SARS-CoV-2 replication in vitro9,15,16. However, development of NIs that are incorporated into the product RNA chain is hampered by the proofreading activity of SARS-CoV-2 nsp14, which effectively decreases the incidence of mismatched nucleotides through its exoribonuclease (ExoN) activity, even that some of NIs exert antiviral activity through other mechanisms such as a chain terminator and exhibit less sensitive to the proofreading activity17, 18, 19. It is thus speculated that coronavirus may naturally have relatively high levels of resistance to nucleoside analogs. Unlike NIs that need to be incorporated into the growing viral RNA chain, non-nucleosides inhibitors (NNIs) usually exert antiviral activity by preventing RdRp conformational changes required for transcription20. It is thus proposed that NNIs may circumvent the resistance posed by the proofreading activity of coronavirus RNA transcription machinery. Unfortunately, no highly effective NNIs for SARS-CoV-2 currently exist.
Seeking for these NNIs has been facilitated by the rapid progress in the structural and enzymatic study of SARS-CoV-2 RdRp since the outbreak of COVID-19. Gao et al.21 described the first near atomic resolution structure of nsp12 catalytic subunit in complex with cofactors nsp7–nsp8. Sharing a configuration similar to other RNA polymerases, SARS-CoV-2 RdRp shows a typical right-hand conformation, consisting of the palm, the thumb, and the finger subdomains. The primer-template entry site, the nucleoside triphosphate (NTP) entry channel, and the nascent product exit path converge in a central cavity that is formed by the conserved motifs (A–G) within the fingers and palm domain. The divalent cation-binding residue D618 in motif A and the catalytic residues S759, D760, and D761 in motif C are highly conserved in most viral RdRps. Compared with the apo complex, these residues undergo structural rearrangements to position the template and primer for an in-line attack on the approaching nucleotide. Later on, Yin et al.22 reported the SARS-CoV-2 RdRp–RNA–remdesivir complex structure, in which the double-stranded RNA helix is inserted into the central channel of RdRp where the remdesivir monophosphate is covalently incorporated into the primer strand. Wang et al.23 further proposed a transition model from primase to polymerase complex, revealing that the SARS-CoV-2 nsp7, nsp8, and nsp12 undergo structural rearrangements to adapt RNA binding. Importantly, they also elucidated the delayed-chain-termination mechanism of remdesivir through structural and kinetic analyses23. Compounds that interfere with these RdRp conformational changes are expected to impair RdRp function and hereby inhibit SARS-CoV-2 infection.
In this study, we carried out computation modeling together with experimental validation in order to identify NNIs of SARS-CoV-2 RdRp. First, we performed structure-based virtual screening of a 15,220-compound library by targeting the central cavity of SARS-CoV-2 RdRp. Then, we selected the top 50 hits with strong binding energies and validated their binding affinities through the bio-layer interferometry (BLI) binding assay. Six out of 50 compounds showed direct binding to SARS-CoV-2 RdRp (KD = 0.54–220 μmol/L). Their inhibition of SARS-CoV-2 RdRp was measured with cell-free and cell-based polymerase activity assays. The most potent inhibitor, named RAI-S-37, binds to SARS-CoV-2 RdRp with a KD value of 0.54 μmol/L, and drastically inhibits the polymerase activity. Importantly, RAI-S-37 inhibits SARS-CoV-2 replication in cell culture with a 50% effective concentration (EC50) value of 0.13 μmol/L, thus warrants being further developed into effective SARS-CoV-2 therapeutics.
2. Materials and methods
2.1. Structure-based virtual screening
Starting from the cryo-electron microscopy structure of SARS-CoV-2 RdRp (PBD ID: 6M71), we employed the AutoDockTools (ADT; version 1.5.6) to prepare the protein and gird map parameters24. The PDB file of protein was converted to PDBQT format after being added polar hydrogens and Gasteiger charges. The central cavity of SARS-CoV-2 RdRp was designated as a box of 26 Å × 26 Å × 26 Å, centered near the active sites (x = 123.6 Å̊, y = 117.84 Å̊, z = 133.6 Å̊). For each compound, we utilized the Open Babel (version 2.3.2)25 to assign hydrogen appropriate for pH 7.4, minimize with MMFF94 force field26, and generate the PDBQT format input file. Then we performed structure-based virtual screening on Dell PowerEdge T630 Tower Server using Auto Dock Vina software package27. Total 15,220 compounds from DrugBank (version 5.1.5) and TargetMol Bioactive compounds Library (Catalog Nos. L4000 and L4150) were docked to SARS-CoV-2 RdRp central cavity defined above and ranked by their calculated binding free energies (ΔGADV). The protein–ligand interactions were visually inspected by using PyMOL (version 2.3.4) and Free Maestro (version 11.8.012). The top ranked hits with high structural diversity and favorable binding mode were chosen for further examination.
2.2. Molecular dynamic simulation
The generated SARS-CoV-2 RdRp/RAI-S-37 complex structure through molecular docking was further relaxed using molecule dynamic (MD) simulation28. Amber 11 software package was used to conduct all the simulations and analyze the results SARS-CoV-2 RdRp was described using the Amber ff99SB protein force field29. The missing hydrogen atoms of protein residues were added using LEAP module. The force field and internal coordinate preparation files of RAI-S-37 was generated utilizing the general Amber force field (GAFF) through the Antechamber module30,31. The restrained electrostatic potential charges of RAI-S-37 were fitted from quantum mechanism calculation at B3LYP/6-31G∗ level using Gaussian 09 program32. Total six Na+ counterions were added to SARS-CoV-2 RdRp/RAI-S-37 complex for charge neutralization. The complex was then solvated in a box filled with 28,791 TIP3P water molecules that extended 10 Å outside the protein33. Prior to MD simulation, the system was subjected to energy minimization as previously described34. Followed by this, the system was heated from 0 to 300 K for a period of 50 ps under constant volume periodic boundary conditions (NVT), with protein fixed with a force constant 10 kcal/(mol·Å2). Subsequently, the system was equilibrated for 500 ps at a constant pressure of 1 atm and temperature of 300 K (NPT). Finally, a production run of 10 ns simulations were performed with a time step of two fs under the same conditions. Throughout the simulation, the nonbonded interactions were evaluated with a cutoff of 10 Å̊, and the long-range electrostatic interactions were treated by Particle Mesh Ewald (PME) method. The SHAKE algorithm was applied to constrain the hydrogen bonds35.
The trajectories and presence of hydrogen bonds were analyzed using ptraj module in Amber. The representative structures of SARS-CoV-2 RdRp/RAI-S-37 complex were obtained through cluster analysis by using kclust module in MMTSB Tool Set36. The binding free energy decomposition were calculated with molecular mechanics/generalized born surface area (MM/GBSA) method as previously described37. All the simulation results were visualized using VMD (version 1.9.3) and PyMOL (version 2.3.4).
2.3. Cells, viruses, and antivirals
Human 293T (HEK293T, Cat# CRL-11268, ATCC, USA), Huh-7 (Cat# CCL-185, ATCC), and Vero (Cat# CCL-81, ATCC) cell lines were cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco, USA) supplemented with 10% fetal bovine serum (FBS; Gibco) at 37 °C in a humidified atmosphere of 5% CO2.
The SARS-CoV-2 used in this study was isolated from respiratory samples of confirmed COVID-19 patients and was as propagated in Vero cells as previously described38,39. All the experiments involved with SARS-CoV-2 were conducted in the biosafety level 3 (BSL-3) laboratory in the Institute of Pathogen Biology, Chinese Academy of Medical Sciences and Peking Union Medical College (Beijing, China). The HCoV-OC43 (Cat# VR-1558, ATCC) used in this study was kindly provided by Dr. Xuesen Zhao (Beijing Ditan hospital, Beijing, China).
Selected compounds from virtual screening procedure and remdesivir (Cat# T7766) were purchased from Target Molecule Corp (TargetMol, USA). All compounds were of a purity>95%.
2.4. BLI binding assay
The binding affinities between tested compounds and SARS-CoV-2 RdRp were measured using BLI-based Octet Platform (Octet Red 96, Fortebio). A temperature of 30 °C and a stirring speed of 1000 revolutions per minute (rpm) were used on Fortebio Octet Red 96 in black 96 well throughout the experiment. 1 × Phosphate-buffered saline (PBS; pH = 7.4) with 0.01% Tween-20, 0.1% bovine serum albumin (BSA), and 2% DMSO was used as assay buffer for all measurements. The purified His-tagged SARS-CoV-2 RdRp were captured via Ni-NTA (NTA) biosensors (Cat# 18-5101, ForteBio) at a concentration of 150 μg/mL, resulting in a saturation response of 5–6 nm after 300 s. Subsequently, the loaded biosensors were washed for 3 min in buffer to clear up loose nonspecifically bound SARS-CoV-2 RdRp and to establish a stable baseline. For binding kinetic measurements, the association of SARS-CoV-2 RdRp and tested compounds (3.125–100 μmol/L in assay buffer) was measured for 60–180 s and the dissociation of them was measured for 120 s in assay buffer. Reference wells that utilized buffer instead of tested compounds were also included to correct the baseline shift. A parallel set of Ni-NTA sensors that were incubated in buffer-only were prepared as the negative reference controls to correct the non-specific binding of the compounds to the biosensor surface. Raw kinetic data were analyzed using a double reference subtraction approach in which both the background and non-specific binding were subtracted. The binding affinity constant KD (KD = kdis/kon; kon is the association rate constant, kdis is the dissociation rate constant) values were calculated using 1:1 binding model through global fitting of multiple kinetic traces. Data Analysis 9.0 software was used to analyze the real-time monitoring data. All the measurements were performed in three independent experiments.
2.5. Protein expression and purification
The genes of SARS-CoV-2 nsp7 and nsp8 (GenBank: MN908947.3) were synthesized and cloned into pET-21a (+) vector (Genewiz Tec, Suzhou, China). The nsp7–6His–nsp8 gene, in which a 6 × histidine was introduced between the two subunits, was also prepared in the same manner. The SARS-CoV-2 nsp12 expression vector was kindly provided by Zihe Rao (Shanghai Tech University, Shanghai, China). Specifically, the genes of nsp12 (GenBank: MN908947) were cloned into a modified pET-22b vector with a 10 × His tag added at the C-terminus21,23.
The E. coli BL21 (DE3) cells transformed with these plasmids were cultured at 37 °C in lysogeny broth (LB) media containing 100 mg/L ampicillin until the optical density at 600 nm (A600) reached 0.6 to 0.8. The protein production was induced with 0.66 mmol/L isopropyl β-d-1-thiogalactopyranoside (IPTG) for more than 18 h at 16 °C. Subsequently, the cells were collected through centrifugation at 8422×g for 20 min, resuspended in buffer [20 mmol/L Tris-HCl (pH 8.0), 300 mmol/L NaCl, 4 mmol/L MgCl2, 2 mmol/L DTT], and lysed by sonication. Finally, the cell debris were removed through centrifugation at 10,397×g for 20 min.
For purification of nsp12, the supernatant was first subjected to the HisTrap column (GE Healthcare) to capture the target protein, and then purified bypassing through a HiTrap Q ion-exchange column (GE Healthcare). The eluate was subjected for further purification by loading onto a Superdex 200 10/300 Increase column (GE Healthcare) in buffer containing 20 mmol/L Tris-HCl (pH 8.0), 150 mmol/L NaCl, 4 mmol/L MgCl2, 2 mmol/L DTT. The final product of nsp12 was concentrated to a minimum of 5 mg/mL and stored at −80 °C for further use.
For purification of nsp7, nsp8, and nsp7–6His–nsp8, the target proteins were first purified through Ni-NTA affinity chromatography by loading onto the Histrap excel column (GE Healthcare, USA) and further purified by passing through a Hitrap Q ion-exchange column (GE Healthcare, USA). The final products were concentrated to more than 10 mg/mL and store at −80 °C.
2.6. In vitro polymerase activity assay
The in vitro measurement of SARS-CoV-2 polymerase activity was performed as previously described with slight modifications7. The activity assays were performed in the reaction buffer containing 20 mmol/L Tris-HCl (pH 8.0), 10 mmol/L KCl, 1 mmol/L DTT, and 4 mmol/L MgCl2. For assembling stable nsp7–nsp8 complex, purified nsp7 was incubated with nsp8 at a molar ratio of 1:1 for 5 h on ice. Primers labelled with FAM (5ʹ-FAM-GUCAUUCUCCUAAGAAGCUA-3ʹ) were annealed to the complementary template (5ʹ-CUAUCCCCAUGUGAUUUUAAUAGCUUCUUAGGAGAAUGAC-3ʹ) with a ratio of 1:1 by heating at 70 °C for 10 min and then cooling down to room temperature. To perform polymerase activity assay, nsp12 (1 μmol/L), nsp7 (2 μmol/L), and nsp8 (2 μmol/L) or nsp7–6His–nsp8 (2 μmol/L) were incubated with 0.25 μmol/L annealed template and 1 mmol/L NTP in reaction buffer for 30 min at 30 °C.
To examine the inhibitory effect of compounds against SARS-CoV-2 polymerase activity, the selected compounds were incubated with nsp12 for at least 1 h on ice prior to performing the primer extension assay. Afterward, 1 μmol/L nsp12 and 2 μmol/L nsp7–6His–nsp8 were incubated with 0.25 μmol/L annealed template and 1 mmol/L NTP in the presence of compounds (2.5, 10, and 40 μmol/L) in reaction buffer for 30 min at 30 °C. All samples were mixed with 2 × RNA loading buffer and denatured by boiling at 100 °C for 10 min. The products were separated in 20% polyacrylamide gel electrophoresis (PAGE) containing 8 mol/L urea. Images were collected with Bio-Rad ChemiDoc MP Imaging System and quantified by ImageJ software. In addition, the same samples were stained with Cybr Gold dye to mark the 20 nucleotides (nt) primer and the 40 nt template on PAGE.
2.7. Cell-based SARS-CoV-2 polymerase activity assay
We established a cell-based SARS-CoV-2 RdRp reporter assay system by modifying the previously developed system40,41. The plasmids nsp12, nsp7, nsp8, nsp10, and nsp14 were used to express codon-optimized Flag-nsp12, Flag-nsp7, Flag-nsp8, Flag-nsp10, and Flag-nsp14, respectively, all of which contain a Flag tag at the C-terminus. To generate plasmid pCoV-Gluc, which produces positive-strand of vRNA encoding Gaussia-luciferase (Gluc), 5ʹ untranslated region (UTR)–Gluc–3ʹ UTR was first synthesized (Sangon Biotech), then inserted into the BamH I and Not I sites of pRetroX-tight-Pur vector (kindly provided by Dr. Guo Fei). The primers are (5ʹ-GGCGGATCCATTAAAGGTTTATAC-3ʹ (forward) and 5ʹ-TTAGCGGCCGCGTCATTCTCCTAAGAA-3ʹ (reverse).
HEK293T cells were transfected with CoV-Gluc, nsp12, nsp7, and nsp8 plasmids at the ratio of 1:10:30:30, or transfected with CoV-Gluc, nsp12, nsp7, nsp8, nsp10, and nsp14 plasmid DNA in a 1:20:60:60:50:50 ratio. At 12 h post transfection, cells were re-seeded in 96-well plates (104/well) and treated with serially diluted remdesivir and RAI-S-37 (from 0.39 to 50 μmol/L). After 24 h incubation, the cell supernatant was collected and measured for Gluc activity using Centro XS3 LB 960 microplate luminometer (Berthold Technologies, Bad Wildbad, Germany). The relative activity of SARS-CoV-2 RdRp is proportional to the intensity of the measured luciferase. EC50 values were obtained by non-linear regression analysis using GraphPad Prism 8.0 software.
2.8. Cell viability assay
Cell viability was examined with Cell Counting kit-8 (CCK-8, Beyotime), which is a water-soluble tetrazolium salt-8 (WST-8) reagent. HEK293T cells were seeded in 96-well plates with a density of 4 × 104 cells per well. one microliter of each tested compound was added to cells and incubated for 24 h. Then 10 μL of CCK-8 reagent was added into each well and incubated for 90 min at 37 °C. The absorbance at 450 nm was measured using the Enspire 2300 Multiable reader (PekinElmer).
2.9. SARS-CoV-2 infection assay
To evaluate the antiviral activity of the compounds, Vero cells were cultured overnight in a 48-well plate with 5 × 104 cells per well. Cells were pre-treated with different doses of the indicated compounds for 1 h, and then the virus [multiplicity of infection (MOI) of 0.05] was added to allow infection for 2 h. The virus and drug mixture were removed, and cells were further cultured with fresh drug-containing medium for 24 h. Viral RNA were quantified by TaqMan quantitative real-time RT-PCR (qRT-PCR) as reported with plotted standard curves using in vitro transcribed RNA. Briefly, the cell supernatants were collected and viral RNA in the cell supernatants were extracted by using Direct-zol RNA miniPrep kit (Zymo research, CA, USA) according to the manufacturer's instructions. The viral RNA was measured using primers and probe targeting the SARS-CoV-2 N by using the TaqMan Fast Virus 1-step Master Mix (Applied Biosystems, Foster City, CA) on CFX96TM Real-Time PCR system (Bio-Rad, Hercules, CA). The reference standard was ten-fold diluted from 1 × 109 copies to 1 × 104 copies. The primer sequence specific for the SARS-CoV-2 N was 5ʹ-GACCCCAAAATCAGCGAAAT-3ʹ (forward) and 5ʹ-TCTGGTTACTGCCAGTTGAATCTG-3ʹ (reverse). PCR amplification procedure was 50 °C, 15 min, 95 °C, 3 min, followed by 50 cycles of 95 °C for 15 s, 60 °C for 45 s. The copies of virus were calculated according to the standard curve. The dose–response curves were plotted for viral RNA copies versus the drug concentrations using GraphPad Prism 8.0 software.
2.10. Infection assay with HCoV-OC43
Huh-7 cells were seeded into 12-well plates at 1 × 105 per well for 24 h, before infection with HCoV-OC43 at a MOI of 0.1, in the presence of tested compounds with two-fold dilutions (from 0.625 to 80 μmol/L). After 4 h infection, the culture supernatants were removed, and replaced with fresh inhibitor-containing medium. At 48 h post-infection (hpi), cells were collected and lysed in lysis buffer. Total viral RNA was extracted by using the RNA extraction kit (Cat# 71202, Tiandz, China). The level of RNA in HCoV-OC43-infected Huh-7 cells was quantified by qRT-PCR using the one-step SYBR PrimeScript RT-PCR kit (Cat# RR066A, Takara). The primer pair (forward: 5ʹ-AGCAACCAGGCTGATGTCAATACC-3ʹ; reverse: 5ʹ-AGCAGACCTTCCTGAGCCTTCAAT-3ʹ) amplifies N gene of HCoV-OC43. Human glyceraldehyde-3-phosphate dehydrogenase (hGAPDH) was served as an internal control and amplified with primers 5ʹ-ATCATCCCTGCCTCTACTGG-3ʹ and 5ʹ-GTCAGGTCCACCACTGACAC-3ʹ.
2.11. HCoV-OC43 cytopathic effects (CPE) assay
2 × 104 per well of HCT-8 cells were seeded into 96-well plate for 24 h, and then infected with HCoV-OC43 at a MOI of 0.5 in the presence of two-fold serially diluted tested compound (from 0.097 to 200 μmol/L). After 6 days of infection, 20 μL of MTS solution was added into each well, and the plate was incubated for 3.5 h at 37 °C. Optical density at 490 nm wavelength was recorded using a SYNERGY H1 microplate reader (BioTek Instruments Inc., Winooski, VT, USA). HCT-8 cells without virus infection were used as positive control to indicate 100% CPE inhibition. HCT-8 cells with virus infection but without drugs were used as negative controls to indicate 0% CPE inhibition. The percentage CPE inhibition is shown in Eq. (1):
The percentage CPE inhibition=(A490 of samples − A490 of negative control)/(A490 of positive control − A490 of negative control) × 100(1).
2.12. Statistical analysis
Data are presented as mean ± standard deviation (SD) from at least three independent experiments unless otherwise indicated. Statistical differences are indicated as follows: *P < 0.05, **P < 0.01, ***P < 0.001. n.s. indicates non-significant.
3. Results
3.1. Structure-based virtual screening
To identify candidate inhibitors of SARS-CoV-2 RdRp, we first performed structure-based virtual screening using molecular docking calculation (Fig. 1A). The cryo-electron microscopy structure of full-length SARS-CoV-2 nsp12 (PDB ID 6M71) was used as the target. The polymerase central cavity, constituted by residues K551, R553, T556, V557, K621, C622, D623, S682, D760, and K798, was defined as the binding groove. Total 15,220 commercially available small molecules, including 8773 compounds from Drugbank database (version 5.1.5) and 6447 compounds from TargetMol bioactive compound library, were docked to this region, then ranked based on their ΔGADV. As shown in Figs. 1B and 3048 compounds have binding energy values between −8.0 to −10.4 kcal/mol. From these compounds, 176 were selected based on their structural diversity. Subsequently, we visually inspected the binding poses of these compounds and selected candidates that made favorable interactions with SARS-CoV-2 RdRp. Compounds that showed H-bond, π–π, or salt bridge interactions with the central cavity residues described above were retrieved. At the end, 50 compounds were selected for further experimental validation. The list of these compounds as well as their database ID, CAS number, calculated binding energies, and molecule weight are presented in Table S1 (Supporting Information).
Figure 1.

Structure-based virtual screening of the Drugbank database and TargetMol active compound library against SARS-CoV-2 RdRp. (A) Flowchart outlining the steps of the virtual screening and the selection of candidate molecules to be tested. (B) Results of virtual screening using AutoDock Vina (ADV) of the small molecule compound database against SARS-CoV-2 nsp12. Bars represent numbers of compounds with predicted free energies of binding in the indicated 1 kcal/mol bins.
3.2. Validation of virtual screening hits in the cell-free binding assay
To assess the ability of the selected 50 compounds to interact with SARS-CoV-2 RdRp, we developed a biosensor-based compound screening assay using BLI technology that can directly monitor the binding kinetics between compounds and the target protein in vitro. The NTA biosensor loaded with his-tagged SARS-CoV-2 RdRp were flowed through wells containing compounds. Meanwhile, the BLI response signals that reflect the binding interaction between tested compounds and SARS-CoV-2 RdRp were generated. We first performed a primary screen of the 50 compounds at a single concentration of 50 μmol/L. As shown in Fig. 2 and Table S1, most compounds were binned at 0 ± 0.02 nm of response. 34 out of 50 compounds that had BLI responses less than 0.02 nm were discarded because of relative weak binding. The remaining 16 compounds that have signals greater than 0.02 nm were further confirmed with a second screen on a range of concentrations to obtain the binding affinity constant, KD, for the SARS-CoV-2 RdRp. As a result, eight of the 16 compounds were identified to exhibit concentration response curves of binding with SARS-CoV-2 RdRp. The detected BLI KD, kon, and kdis values were shown in Table 1. KD is a ratio of kdis/kon. The association and dissociation curves and chemical structures were presented in Fig. 3 and Supporting Information Fig. S1. One of the hits, RAI-S-37, exhibited the strongest binding affinity to SARS-CoV-2 RdRp with fitted KD values of 0.54 μmol/L. Compounds RAI-S-11, RAI-S-36, and RAI-S-47 showed a moderate binding affinity, yielding KD values lower than 50 μmol/L. Compounds RAI-S-35 and RAI-S-45 bound weakly to SARS-CoV-2 RdRp with KD values around 200 μmol/L. The remaining two compounds RAI-S-19 and RAI-S-34 were also not pursued for their extremely weak binding affinity (KD > 1 mmol/L).
Figure 2.
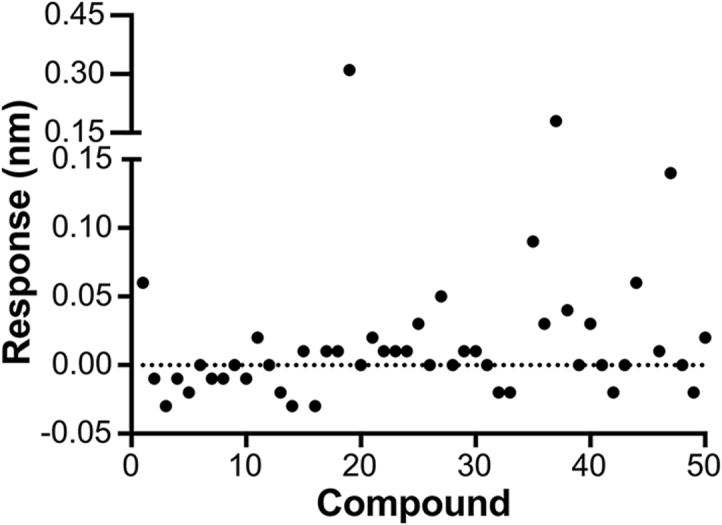
A plot of BLI responses with SARS-CoV-2 nsp12 for 50 compounds. The His-tagged SARS-CoV-2 RdRp captured onto NTA biosensors were applied to solutions containing compounds at a single concentration of 50 μmol/L. Double reference subtraction method was used to subtract the effect of baseline drift and non-specific binding. The data shown are a representative result of three independent experiments.
Table 1.
The molecule name (MolName), CAS, molecule weight (MW, g/mol), calculated binding energies (ΔGADV, kcal/mol), and BLI binding kinetics (KD, kon, kdis, and R2) of compounds selected from primary in vitro binding screen.
| No. | Mol. name | CAS | MW | ΔGADV | KD (μmol/L) | kon (L/(mol·s)) | kdis (1/s) | Full R2 |
|---|---|---|---|---|---|---|---|---|
| 1 | Pazopanib | 444731-52-6 | 437.5 | −9.2 | N.D.a | N.D. | N.D. | N.D. |
| 11 | Verbascoside | 61276-17-3 | 624.6 | −8.3 | 0.84 ± 0.85 | 108 | 9.05×10−5 | 98% |
| 19 | Zafirlukast | 107753-78-6 | 575.7 | −9.5 | >1000 | 8.43 | 4.99 | 87% |
| 21 | Paliperidone | 144598-75-4 | 426.5 | −8.6 | N.D. | N.D. | N.D. | N.D. |
| 25 | Pictilisib | 957054-30-7 | 513.6 | −8.7 | N.D. | N.D. | N.D. | N.D. |
| 27 | steviolbioside | 41093-60-1 | 642.7 | −9.4 | N.D. | N.D. | N.D. | N.D. |
| 34 | COH29 | 1190932-38-7 | 420.4 | −8.5 | >1000 | 8.10 | 0.189 | 87% |
| 35 | Oleanonic Acid | 17990-42-0 | 454.7 | −9.6 | 171.00 ± 35.90 | 98.4 | 1.68×10−2 | 84% |
| 36 | Forsythoside A | 79916-77-1 | 624.6 | −8.7 | 1.16 ± 0.44 | 137 | 1.59×10−4 | 99% |
| 37 | Corilagin | 23094-69-1 | 634.5 | −8.9 | 0.54 ± 0.01 | 496 | 2.68×10−4 | 97% |
| 38 | Ligustroflavone | 260413-62-5 | 724.7 | −10.1 | N.D. | N.D. | N.D. | N.D. |
| 40 | Ledipasviracetone | 1441674-54-9 | 947.1 | −9.5 | N.D. | N.D. | N.D. | N.D. |
| 44 | Narirutin | 14259-46-2 | 580.5 | −9.0 | N.D. | N.D. | N.D. | N.D. |
| 45 | MK-3903 | 1219737-12-8 | 454.9 | −8.9 | 220.00 ± 19.40 | 302 | 6.65×10−2 | 96% |
| 47 | T-5224 | 530141-72-1 | 517.5 | −8.6 | 38.70 ± 1.63 | 200 | 7.74×10−3 | 98% |
| 50 | AMG-900 | 945595-80-2 | 503.6 | −9.1 | N.D. | N.D. | N.D. | N.D. |
N.D. = not detected.
Figure 3.

Binding of RAI-S-37 to SARS-CoV-2 RdRp using cell-free BLI binding assay. (A) BLI binding kinetics of the interaction of RAI-S-37 with SARS-CoV-2 nsp12. The His-tagged SARS-CoV-2 RdRp captured onto NTA biosensors were applied to solutions containing RAI-S-37 from 3.125 to 100 μmol/L. Double reference subtraction method was used to subtract the effect of baseline drift and non-specific binding. KD was acquired from fitting into 1:1 binding model by global fitting of multiple kinetic traces and analyzed by Data Analysis 9.0 software. Date are expressed as mean ± SD. The data shown are a representative result of three independent experiments. (B) The chemical structure, molecule name, CAS number, and molecular weight of RAI-S-37.
3.3. RAI-S-37 inhibits SARS-CoV-2 RdRp in cell-free assay
Having found that compounds of RAI-S-11, RAI-S-35, RAI-S-36, RAI-S-37, RAI-S-45, and RAI-S-47 bind directly to SARS-CoV-2 nsp12 (Table 1), we established the primer-dependent RNA elongation reaction assay as previously described7, in order to assess the inhibitory effects of these compounds on RdRp and the contribution of compound–protein interaction to their anti-RdRp activity. Since cofactors nsp7 and nsp8 were reported to form hexadecameric primase complex in the early stage of reaction23, we purified recombinant nsp7, nsp8, and the fusion protein nsp7–6His–nsp8, in which a 6 × histidine linker was inserted between nsp7 and nsp8 sequences (Fig. 4A). In the cell-free polymerase activity assay, a 40 nt template RNA mimicking the 3ʹ UTR of SARS-CoV-2 RNA genome without the polyA tail and a 20 nt primer labeled with FAM were used to monitor the polymerase activity of RdRp, which generated the 40 nt FAM-labeled product.
Figure 4.

RAI-S-37 inhibits SARS-CoV-2 RdRp in the cell-free viral RNA synthesis assay. (A) SDS-PAGE analysis of purified SARS-CoV-2 RdRp subunits nsp12, nsp8, nsp7, and nsp7–6His–nsp8. Standard protein markers are shown in the last lane. (B) and (C) Cell-free polymerase activity of nsp7, nsp8, nsp7–6His–nsp8, nsp12 in various combinations. A 40 nt template RNA mimics the 3ʹ UTR of SARS-CoV-2 RNA genome without the polyA tail, was used together with a 20 nt primer labeled with FAM to monitor the polymerase activity, which was determined by measuring the 40 nt FAM RNA product. In addition to the 40 nt fully elongated RNA product, some prematurely terminated products (shorter than 40 nt) were also observed. (D)–(I) Inhibitory effects of RAI-S-11, RAI-S-35, RAI-S-36, RAI-S-37, RAI-S-45, and RAI-S-47 on SARS-CoV-2 RdRp at the concentration of 40, 10, and 2.5 μmol/L, respectively. Polymerase activity was determined by quantifying the intensity of the fully elongated 40 nt RNA product bands. Data are expressed as mean ± SD (n = 3). ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001. n.s. indicates non-significant.
As shown in Fig. 4B and C, nsp12 itself was capable of RNA synthesis albeit with low efficiency. Including the nsp7 and nsp8 proteins enhanced the polymerase activity of nsp12 by ∼1.9-fold. Stronger RdRp activity of nsp12 was observed with the addition of nsp7–6His–nsp8 fusion protein, which increased the polymerase activity by ∼1.3-fold compared to addition of nsp7 and nsp8.
To measure the direct inhibition of the polymerase activity of SARS-CoV-2 RdRp by the selected compounds, nsp12 was pre-incubated with different concentrations (2.5, 10, and 40 μmol/L) of these compounds prior to polymerization reaction. Results in Fig. 4D and E showed that, at 40 μmol/L, compounds RAI-S-37, RAI-S-45, and RAI-S-47 inhibited the polymerase activity by 83%, 70%, 60%, respectively. At 10 μmol/L, RAI-S-45 and RAI-S-47 lost their inhibition, whereas RAI-S-37 still exhibited 73% inhibition of the polymerization reaction (Fig. 4F and G). Even at 2.5 μmol/L, RAI-S-37 reduced the activity of SARS-CoV-2 RdRp by 24% (Fig. 4H and I). Given the strong binding affinity of RAI-S-37 to SARS-CoV-2 RdRp, these functional data suggest that RAI-S-37 is a potentially effective SARS-CoV-2 RdRp inhibitor.
3.4. RAI-S-37 inhibits SARS-CoV-2 RdRp and resists the proofreading activity in cell-based assay
To further measure the efficiency of RAI-S-37 against SARS-CoV-2 RdRp, we developed a cell-based reporter assay with Gluc as the reporter to evaluate the anti-SARS-CoV-2 RdRp activity. The luciferase expression reporter plasmid pCoV-Gluc was generated by inserting the coding sequence of Gluc between the 5ʹ UTR and 3ʹ UTR of SARS-CoV-2, such that pCoV-Gluc expresses Gluc under the control of SARS-CoV-2 UTRs. Upon expression of SARS-CoV-2 RdRp, the Gluc RNA is further amplified by viral RdRp, resulting in the increase of Gluc mRNA and Gluc protein. Therefore, the increased Gluc activity represents the activity of SARS-CoV-2 RdRp. Using this system, we measured SARS-CoV-2 RdRp activity in cells that were exposed to different doses of the selected compounds. The RdRp inhibitor remdesivir was used as a positive control. As shown in Fig. 5A, remdesivir markedly suppressed the polymerization reaction with an EC50 value of 1.81 μmol/L. RAI-S-37 also inhibited SARS-CoV-2 RdRp activity with an EC50 value of 3.33 μmol/L, which corroborated with the result of the cell-free RdRp activity assay (Fig. 4H). Neither remdesivir nor RAI-S-37 showed cytotoxicity on HEK293T cells below 50 μmol/L (Supporting Information Fig. S2). Together, these data demonstrate that RAI-S-37 is able to inhibit SARS-CoV-2 RdRp activity both in cell-free and cell-based assays.
Figure 5.

Inhibitory effect of remdesivir provided in combination with RAIS-S-37 on SARS-CoV-2 RdRp activity. (A) HEK293T cells were transfected with CoV-Gluc, nsp12, nsp7, and nsp8 plasmid DNA at the ratio of 1:10:30:30. (B) HEK293T cells were transfected with CoV-Gluc, nsp12, nsp7, nsp8, nsp10, and nsp14 plasmid DNA at the ratio of 1:20:60:60:50:50. At 12 h post transfection, cells were re-seeded in 96-well plates (104/well) and treated with serially diluted remdesivir and RAI-S-37. After 24 h incubation, Gluc activity in supernatants was determined. EC50 values were obtained by non-linear regression analysis using GraphPad Prism 8.0. The Data are expressed as mean ± SD (n = 3).
SARS-CoV-2 nsp10–nsp14 exoribonuclease complex plays a pivotal role in decreasing the incidence of mismatched nucleotides. Unlike nucleoside analogs that are sensitive to this exoribonuclease activity, non-nucleoside inhibitors may circumvent the proofreading activity of coronavirus. To address this hypothesis, we introduced nsp10–nsp14 into cell-based RdRp assay, and then tested the sensitivity of SARS-CoV-2 RdRp to RAI-S-37 with expression of nsp10–nsp14. As shown in Fig. 5B, compared with the results without nsp10–nsp14 (Fig. 5A), expression of nsp10–nsp14 leads to a 2.1-fold increase in EC50 value (from 1.81 to 3.57 μmol/L) of remdesivir. In contrast, the same EC50 values of RAI-S-37 were observed in the absence and presence of the nsp10–nsp14, corroborating that the non-nucleoside inhibitor RAI-S-37 is resistant to the proofreading activity.
Because remdesivir and RAI-S-37 inhibit SARS-CoV-2 RdRp activity through different mechanism, we also tested whether the combination treatment using both could increase the inhibitory effect. As shown in Fig. 5A, the combination of RAI-S-37 with remdesivir can enhance the inhibitory effects (EC50 = 1.25 μmol/L). The additive interaction was also observed in the presence of nsp10–nsp14 (Fig. 5B). The dose–response curve for drugs remdesivir and RAI-S-37 administered together is very close to the sum of the two-individual dose–response curves.
3.5. RAI-S-37 inhibits SARS-CoV-2 infection
To determine whether RAI-S-37 inhibits the replication of SARS-CoV-2, we infected Vero cells with SARS-CoV-2 at a MOI of 0.05 in the presence of increasing concentrations of RAI-S-37. Levels of SARS-CoV-2 production was determined by quantification of viral RNA copy numbers in the cell supernatant via qRT-PCR. As shown in Fig. 6A, remdesivir inhibited SARS-CoV-2 infection in Vero cells with an EC50 value of 0.06 μmol/L. Under the same condition, RAI-S-37 suppressed SARS-CoV-2 production in dose-dependent manner with an EC50 value of 0.13 μmol/L.
Figure 6.

The antiviral activities of RAI-S-37 against SARS-CoV-2 and HCoV-OC43. (A) Vero E6 cells were infected with SARS-CoV-2 at the MOI of 0.05 with the treatment of different doses of the indicated drugs for 48 h. Levels of SARS-CoV-2 in the cell supernatant was determined by qRT-PCR. Data of the graphs represent mean % inhibition of virus yield. The experiments were performed in triplicates. (B) Huh-7 cells were infected with HCoV-OC43 at the MOI of 0.1 in the presence of increasing concentrations of the tested compounds. DMSO was used as vehicle control. The Data are expressed as mean ± SD (n = 3).
RdRp sequences are highly conserved across different CoVs. To determine whether RAI-S-37 inhibits other human CoVs, we assessed the activity of RAI-S-37 against HCoV-OC43, one beta CoV, in Huh-7 cells by qRT-PCR. As shown in Fig. 6B, both remdesivir and RAI-S-37 inhibited HCoV-OC43 infection in a dose-dependent manner with EC50 values of 2.49 and 4.96 μmol/L, respectively. The anti-HCoV-OC43 activity of both compounds was also shown by the results of the CPE assay (Supporting Information Fig. S3). Remdesivir and RAI-S-37 protected cells from CPE induced by HCoV-OC43 infection at 0.39 and 0.78 μmol/L, respectively. Together, these data demonstrate that RAI-S-37 effectively inhibits both SARS-CoV-2 and HCoV-OC43, with the potential as a broad CoV inhibitor.
3.6. Predicted binding model of RAI-S-37 to RdRps of SARS-CoV-2 and HCoV-OC43
To better understand the interaction of RAI-S-37 with SARS-CoV-2 RdRp, we computed the structure of SARS-CoV-2 RdRp in complex with RAI-S-37 using the published structure of SARS-CoV-2 RdRp (PDB ID 6M71) and the AutoDock Vina program, through docking RAI-S-37 to the ligand binding region of polymer. The structure was then relaxed using molecule dynamic simulation for 10 ns. Afterwards, MM/GBSA computational method was used to calculate the binding free energy of RAI-S-37 and decompose it at the amino acid level. The root-mean-square deviation (RMSD) value of the backbone atoms against initial protein-ligand complex is 2.1 Å, indicating great stability of RAI-S-37 in SARS-CoV-2 RdRp binding sites (Supporting Information Fig. S4).
As shown in Fig. 7A, the binding pocket of RAI-S-37 is located in the SARS-CoV-2 RdRp palm subdomain. Superimposition of the structures of SARS-CoV-2 RdRp/RAI-S-37 and SARS-CoV-2 RdRp/template-RTP (PDB ID 7BV2) revealed that RAI-S-37 is at the gate of the nucleotide entry channel to the active site and may block the entry of nucleotide triphosphate to the active site (Fig. 7B). Specifically, RAI-S-37 is adjacent to the highly conserved SDD sequence (residues 759 to 761) in motif C. Cavity analysis of the binding site (Table 2) indicated that the key residues that make main contribution to RAI-S-37 binding are G616 (−1.47 kcal/mol), D761 (−1.34 kcal/mol), K798 (−1.27 kcal/mol), W617 (−1.24 kcal/mol), W800 (−1.18 kcal/mol), D618 (−0.92 kcal/mol), S814 (−0.81 kcal/mol), E811 (−0.75 kcal/mol), S549 (−0.72 kcal/mol), C799 (−0.71 kcal/mol), and A550 (−0.55 kcal/mol). Consistent with the cavity analysis, the main interaction involves a very strong hydrogen bond between the hydroxyl hydrogen atom of RAI-S-37 and the hydroxyl group of K794 residue (Fig. 7C and D). The percentage occupation of this H-bond is 71% during 10 ns MD and the average bond length is 2.8 Å. In addition, G616, E811, together with the conserved residues D761 in motif C, are well positioned to form H-bonds with RAI-S-37.
Figure 7.

Predicted binding sites for RAI-S-37 in SARS-CoV-2 RdRp. (A) Predicted structure of SARS-CoV-2 RdRp in complex with RAI-S-37. (B) Superimposition of the structures of SARS-CoV-2 RdRp/RAI-S-37 and SARS-CoV-2 RdRp/template-RTP. RAI-S-37 and RTP are shown as cyan dots and magenta stick, respectively. (C) The polymerases are shown in cartoon representation with the backbone atoms depicted in purple. RAI-S-37 and the amino acid residues involved the polymerase–ligand interactions are shown as sticks. (D) The 2D ligand–polymerase interaction diagrams. Residues within 4 Å of RAI-S-37 are displayed. Hydrogen bond is represented by the arrows with the distance between the donor and the acceptor. Dark blue indicates positive charge, red indicates negative charge, light blue for polar, green for hydrophobic, and white for glycine. (E) Superposition of the SARS-CoV-2-nsp12/RAI-S-37 and HCoV-OC43-nsp9/RAI-S-37 complexes. SARS-CoV-2-nsp12 and HCoV-OC43-nsp9 are shown as slate and yellow cartoons, and the bound RAI-S-37 is shown as cyan and salmon sticks, respectively. (F) Comparisons of protein–ligand interactions of complexes SARS-CoV-2-nsp12/RAI-S-37 and HCoV-OC43-nsp9/RAI-S-37.
Table 2.
Cavity analysis for the predicted complex of RAI-S-37-bound SARS-CoV-2 RdRp. The total energy (kcal/mol) is the sum of van der Waals, electrostatic, polar solvation, and non-polar solvation. The residues are ranked by total energy. Only residues with binding energy greater than 0.5 kcal/mol are shown.
| Residue | van der Waals | Electrostatic | Polar solvation | Non-polar solvation | Total |
|---|---|---|---|---|---|
| G616 | −0.31 | −1.20 | 0.06 | −0.01 | −1.47 |
| D761 | −0.70 | −12.41 | 12.01 | −0.24 | −1.34 |
| K798 | −0.43 | −3.28 | 2.57 | −0.13 | −1.27 |
| W617 | −1.45 | −1.31 | 1.60 | −0.09 | −1.24 |
| W800 | −1.09 | −0.09 | 0.08 | −0.07 | −1.18 |
| D618 | −2.73 | 0.74 | 1.42 | −0.36 | −0.92 |
| S814 | −1.47 | 0.15 | 0.82 | −0.31 | −0.81 |
| E811 | −2.52 | −8.50 | 10.69 | −0.42 | −0.75 |
| S549 | −0.58 | −0.71 | 0.70 | −0.13 | −0.72 |
| C799 | −0.55 | 0.04 | −0.19 | −0.01 | −0.71 |
| A550 | −0.47 | −0.43 | 0.44 | −0.08 | −0.55 |
Sequence alignment of SARS-CoV-2 and HCoV-OC43 RdRps showed 72% homology (Supporting Information Fig. S5), suggesting that RAI-S-37 may inhibit HCoV-OC43 RdRp through interactions similar to those with SARS-CoV-2 RdRp, which is supported by the dose-dependent inhibition of HCoV-OC43 by RAI-S-37 (Fig. 6B). To shed more light on these interactions, we computed the 3D structure of HCoV-OC43-RdRp starting from SARS-CoV-2 nsp12 and then docked RAI-S-37 to the central cavity of the predicted structure. The ligand–polymerase interaction diagram is depicted in Supporting Information Fig. S6. The calculated binding free energy (ΔGADV = −8.6 kcal/mol) was equivalent to that of SARS-CoV-2 RdRp (ΔGADV = −8.8 kcal/mol). Superposition of these two complexes showed similar structural features at the RAI-S-37 binding sites (Fig. 7E). Notably, RAI-S-37 forms hydrogen bonds with D756, W796, and E807, which are equivalent to D760, W800, and E811 in SARS-CoV-2 RdRp. Furthermore, the top 10 amino acids identified for SARS-CoV-2 RdRp/RAI-S-37 binding are conserved in HCoV-OC43 RdRp (Fig. 7F and Supporting Information Fig. S5). This further supports a similar mechanism behind the binding RAI-S-37 to RdRps of SARS-CoV-2 and HCoV-OC43, which leads to the inhibition of both viral RdRps.
4. Discussion
The novel coronavirus SARS-CoV-2 poses a severe threat to public health. There is an urgent need for the development of COVID-19 therapeutics. Virtual screening has become an important tool in drug development for its knowledge-driven, cost-effective, and time-saving advantages. Using this technique, we successfully identified a strong RdRp inhibitor of SARS-CoV-2. Since the three-dimensional structure of target protein is available, we applied structure-based virtual screening of commercial chemical databases (total 15,220 compounds) against the cavity center of SARS-CoV-2 RdRp. A small set of 50 compounds were selected and further examined using in vitro BLI binding assays. At the end, six out of these 50 compounds were identified for direct binding to SARS-CoV-2 RdRp with micromolar range affinities. In particular, RAI-S-37, which showed the strongest binding affinity to RdRp (KD = 0.54 μmol/L), effectively inhibited SARS-CoV-2 RdRp in Vero cells with an EC50 value of 0.13 μmol/L. It has been reported that compounds of high docking scores may fail to bind the target protein in the in vitro binding assay42. For example, we did not detect the binding of RAI-S-38, a compound that was predicted to bind to SARS-CoV-2 RdRp with the most favorable binding energy (ΔGADV = −10.1 kcal/mol). It is known that the driving forces that dictate protein–ligand binding are the sum of diverse interactions and energy exchanges among the protein, ligand, water, and buffer ions43,44. The ADV docking procedure did not consider the energy of solvated sate, thus provides an estimate, not an exact value of binding affinity. The most precise method to compute relative binding energy is the use of free-energy perturbation techniques45, 46, 47. However, due to the high computational cost, we did not include the solvent factors, such as liquid water and buffer ions in the primary virtual screening procedure.
RAI-S-37 differs from the existing SARS-CoV-2 RdRp inhibitors, such as ribavirin and remdesivir, because it is a NNI, thus can circumvent the proofreading activity of coronavirus. The nsp14 of SARS-CoV-2 plays a pivotal role in decreasing the incidence of mismatched nucleotides through its ExoN activity17,18. It is thus speculated that coronavirus may generally have a relatively high level of resistance to nucleoside analogs. For example, the NI ribavirin poorly inhibits SARS-CoV-2 in vitro with a high EC50 value of 109.5 μmol/L9. Although remdesivir has been shown to be more effective than other NIs, it is also sensitive to the proofreading activity of ExoN and thus requires rigorous clinical trial evaluation before approved for regular clinical use48. Data in our study confirm that expression of nsp10–nsp14 leads to a 2.1-fold increase in EC50 value of remdesivir in cell-based RdRp activity assay (Fig. 5). In contrast, the EC50 values of RAI-S-37 remain the same in the absence and presence of the nsp10–nsp14, suggesting that the NNI RAI-S-37 is resistant to the coronavirus proofreading activity. Given that RAI-S-37 and remdesivir use different mechanisms to inhibit the viral activity, it was hypothesized that using both of them may reduce the chances of drug resistance. In this regard, we examined the inhibitory effect of remdesivir provided in combination with RAIS-S-37 on SARS-CoV-2 RdRp activity. The results showed that when remdesivir is combined with RAI-S-37, it has additive inhibitory effect against SARS-CoV-2 RdRp (Fig. 5). We also evaluated the combinational effect of these two drugs against HCoV-OC43 infection in cells. Interestingly, RAI-S-37 showed a moderate synergistic effect with remdesivir on inhibiting HCoV-OC43 infection, with maximal ZIP-scores of 11.7 (Supporting Information Fig. S7). Further studies are warranted to evaluate the anti-SARS-CoV-2 efficacy of RAI-S-37 alone, and/or in combination with remdesivir. Combined, these results indicate that the non-nucleoside inhibitor RAI-S-37 provides a complementary treatment strategy for COVID-19.
Drug repurposing is a promising strategy to develop antiviral agents as new therapeutics49. In this study, we identified that corilagin (RAI-S-37), a promising herbal medicine, strongly inhibits SARS-CoV-2 replication in Vero cells. Oral administration of corilagin in BALB/c mice demonstrated that corilagin exhibits excellent tolerability and systemic bioavailability50. Specifically, corilagin did not show any toxicity in acute toxicity studies. An scute does of corilagin up to 3500 mg/kg body weight (b.wt.) showed maximum tolerance (LD50 = 3500–5000 mg/kg b. wt.)50,51. Besides, orally administered corilagin can be systemically transported to mice and rat blood plasma. Reddy et al.50 reported that the maximum plasma concentration (Cmax) in mice and rats are 57.0 and 52.37 μg/mL, respectively, with the time to Cmax (Tmax) averaging 2 h. The half lifetime of the compound (t1/2) was found to be about 6 h. The calculated EC50 and EC90 values of RAI-S-37 against SARS-CoV-2 are 0.13 μmol/L (0.08 μg/mL) and 1 μmol/L (0.63 μg/mL), respectively. These values are well below the concentrations of corilagin in BALB/c mice trials, suggesting that RAI-S-37 holds great promise of becoming an effective SARS- CoV-2 therapeutics.
Unlike some other viral RdRps, such as poliovirus 3Dpol, that can synthesize RNA by themselves, SARS-CoV-2 nsp12 requires viral cofactors including nsp7 and nsp8 to prompt its polymerase activity. Hillen et al.52 reported that the helical extensions of nsp8 protrude along the exiting RNA and can be regarded as a positively charged "sliding poles”, which is required for replicating the long SARS-CoV-2 RNA genome. Wang et al.23 further showed that the N-terminal helical extension of nsp8 undergoes structural arrangement and forms a stable platform that may enable progressive replication of viral RNA. In agreement with these biochemical studies, our study further demonstrates the essential roles of the nsp7 and nsp8 in stimulating the polymerase activity of nsp12. As shown in Fig. 4B and C, SARS-CoV-2 nsp12 itself is capable of conducting RNA polymerization with low efficiency, whereas inclusion of nsp7 and nsp8 boosted the polymerase activity of nsp12 by ∼1.9-fold. Notably, nsp7–6His–nsp8 further enhanced the efficiency of RNA synthesis by ∼1.3-fold, suggesting that the nsp7–nsp8 complex enhances processivity of nsp12-mediated RNA synthesis. Inserting a linker of six histidine between nsp7 and nsp8 was used to better assimilate the association of nsp7 and nsp8, which is believed to help achieve optimal RNA polymerase activity. These data are consistent with the reported transition model, in which the nsp7–nsp8 hexadecameric complex operates as a primase before transition to the nsp12–nsp7–nsp8 catalytic complex23.
The predicted binding sites of RAI-S-37 in SARS-CoV-2 RdRp are located at the palm subdomain. Specifically, the pocket consists of G616 to Y619 in motif A, D761 to V763 in motif C, K798 in motif D, E811 to S814 in motif E, and I548 to K551 in motif F. RAI-S-37 forms a strong hydrogen bond with D761, a key catalytic residue in the SDD motif (residues 759–761). In addition, D618, the classic divalent cation-binding residue, is also in close contact with RAI-S-37. It has been reported that D618, D760, and D761 are involved in coordinating two magnesium ions at the catalytic center21. Given the close interactions between RAI-S-37 and these residues, it is posited that the binding of RAI-S-37 may block the conformational change that is needed to coordinate the divalent cations during the catalytic process. Multiple interactions of RAI-S-37 with motif F residues may block the entry of NTP to the active side, thus contributing to the inhibition of viral RNA synthesis. Of note, corilagin is reported to be a potential inhibitor of SARS-CoV-2 main protease (Mpro) via in silico molecular docking studies. For this reason, we performed SARS-CoV-2 Mpro enzyme activity inhibition assay to assess the efficacy of corilagin. However, even at a high concentration of 10 μmol/L, corilagin did not show any inhibitory effect on SARS-CoV-2 Mpro (Supporting Information Fig. S8). Together, these results suggest that RAI-S-37 exerts its antiviral activity by preventing conformational changes of RdRp which are obligatory for nucleotide incorporation into the growing viral RNA chain, while the detailed mechanism awaits further investigation.
5. Conclusions
We have identified a novel NI of SARS-CoV-2 RdRp, called corilagin (RAI-S-37), through structure-based virtual screening and experimental validation. We were able to show that RAI-S-37 binds directly to SARS-CoV-2 RdRp, effectively inhibits the polymerase activity in both cell-free and cell-based polymerase activity assays, and that RAI-S-37 exhibits does-dependent inhibition of SARS-CoV-2 infection with a very low EC50 value of 0.13 μmol/L. We further predicted the potential binding modes of RAI-S-37 to viral RdRp and found that RAI-S-37 may function by preventing the conformational change of RdRp which is essential for synthesizing the long viral RNA genome. In addition, combination of RAI-S-37 with remdesivir exhibits additive activity against anti-SARS-CoV-2 RdRp. Combined, these results indicate that RAI-S-37 holds great promise of becoming a new and effective anti-SARS-CoV-2 drug to treat COVID-19 patients.
Acknowledgments
We thank National Infrastructure of Microbial Resources (NIMR-2014-3, China) and CAMS Collection Center of Pathogenic Micro-organisms (CAMS-CCPM-A, China) for providing valuable reagents. This work was supported by the National Mega-Project for Infectious Disease (2018ZX10301408, China), the National Mega-Project for Significant New Drug Discovery (2018ZX09711003-002-002, China), the National Natural Science Foundation of China (81802019 and 81902075), the Beijing Natural Science Foundation (7184228, China), CAMS Innovation Fund for Medical Sciences (2018-I2M-3-004 and 2020-I2M-2-010, China) and the Peking Union Medical College Youth Fund (3332016063 and 3332018096, China) .
Author contributions
Quanjie Li performed the structure-based virtual screening, designed the BLI binding assay, evaluated the binding kinetics of compounds, predicted the binding mode of compounds, and wrote the original draft; Dongrong Yi designed the expression constructs, purified nsp12, nsp7, nsp8, and nsp7–nsp8, and performed the in vitro polymerase activity assay; Jianyuan Zhao initiated the cell-based polymerase activity assay; Xiangling Cui participated in expression and purification of proteins; Yongxin Zhang evaluated the cytotoxicity and antiviral activities of tested compounds; Xiaobo Lei, Xia Xiao, Tao Jiao, and Xiaojing Dong conducted the experiments on SARS-CoV-2 in P3 laboratory. Xuesen Zhao, Hui Zeng, Lili Ren, Chen Liang, Fei Guo, and Xiaoyu Li participated in data analysis; Shan Cen and Jianwei Wang conceived and supervised the project and wrote the inputs from all authors.
Conflicts of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting data to this article can be found online at https://doi.org/10.1016/j.apsb.2021.02.011.
Contributor Information
Jianwei Wang, Email: wangjw28@163.com.
Shan Cen, Email: shancen@imb.pumc.edu.cn.
Appendix ASupplementary data
The following is the Supplementary data to this article:
References
- 1.Zhou P., Yang X.L., Wang X.G., Hu B., Zhang L., Zhang W. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Csg of the International CSG The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol. 2020;5:536–544. doi: 10.1038/s41564-020-0695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Holshue M.L., DeBolt C., Lindquist S., Lofy K.H., Wiesman J., Bruce H. First case of 2019 novel coronavirus in the United States. N Engl J Med. 2020;382:929–936. doi: 10.1056/NEJMoa2001191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhong N.S., Zheng B.J., Li Y.M., Poon L.L.M., Xie Z.H., Chan K.H. Epidemiology and cause of severe acute respiratory syndrome (SARS) in Guangdong, Peopleʹs Republic of China, in February, 2003. Lancet. 2003;362:1353–1358. doi: 10.1016/S0140-6736(03)14630-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perlman S., Netland J. Coronaviruses post-SARS: Update on replication and pathogenesis. Nat Rev Microbiol. 2009;7:439–450. doi: 10.1038/nrmicro2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Van Doremalen N., Bushmaker T., Morris D.H., Holbrook M.G., Gamble A., Williamson B.N. Aerosol and surface stability of SARS-CoV-2 as compared with SARS-CoV-1. N Engl J Med. 2020;382:1564–1567. doi: 10.1056/NEJMc2004973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peng Q., Peng R., Yuan B., Zhao J., Wang M., Wang X. Structural and biochemical characterization of the nsp12–nsp7–nsp8 core colymerase complex from SARS-CoV-2. Cell Rep. 2020;31:107774. doi: 10.1016/j.celrep.2020.107774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Y., Zhang D., Du G., Du R., Zhao J., Jin Y. Remdesivir in adults with severe COVID-19: A randomised, double-blind, placebo-controlled, multicentre trial. Lancet. 2020;395:1569–1578. doi: 10.1016/S0140-6736(20)31022-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang M.L., Cao R.Y., Zhang L.K., Yang X.L., Liu J., Xu M.Y. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020;30:269–271. doi: 10.1038/s41422-020-0282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McCreary E.K., Angus D.C. Efficacy of remdesivir in COVID-19. J Am Med Assoc. 2020;324:1041–1042. doi: 10.1001/jama.2020.16337. [DOI] [PubMed] [Google Scholar]
- 11.Grein J., Ohmagari N., Shin D., Diaz G., Asperges E., Castagna A. Compassionate use of remdesivir for patients with severe Covid-19. N Engl J Med. 2020;382:2327–2336. doi: 10.1056/NEJMoa2007016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vicenzi E., Canducci F., Pinna D., Mancini N., Carletti S., Lazzarin A. Coronaviridae and SARS-associated coronavirus strain HSR1. Emerg Infect Dis. 2004;10:413–418. doi: 10.3201/eid1003.030683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ziebuhr J. The coronavirus replicase. Curr Top Microbiol Immunol. 2005;287:57–94. doi: 10.1007/3-540-26765-4_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Snijder E.J., Decroly E., Ziebuhr J. The nonstructural proteins directing coronavirus RNA synthesis and processing. Adv Virus Res. 2016;96:59–126. doi: 10.1016/bs.aivir.2016.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sheahan T.P., Sims A.C., Zhou S., Graham R.L., Pruijssers A.J., Agostini M.L. An orally bioavailable broad-spectrum antiviral inhibits SARS-CoV-2 in human airway epithelial cell cultures and multiple coronaviruses in mice. Sci Transl Med. 2020;12 doi: 10.1126/scitranslmed.abb5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Choy K.T., Wong A.Y.L., Kaewpreedee P., Sia S.F., Chen D., Hui K.P.Y. Remdesivir, lopinavir, emetine, and homoharringtonine inhibit SARS-CoV-2 replication in vitro. Antivir Res. 2020;178:104786. doi: 10.1016/j.antiviral.2020.104786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ma Y., Wu L., Shaw N., Gao Y., Wang J., Sun Y. Structural basis and functional analysis of the SARS coronavirus nsp14–nsp10 complex. Proc Natl Acad Sci U S A. 2015;112:9436–9441. doi: 10.1073/pnas.1508686112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eckerle L.D., Becker M.M., Halpin R.A., Li K., Venter E., Lu X. Infidelity of SARS-CoV nsp14-exonuclease mutant virus replication is revealed by complete genome sequencing. PLoS Pathog. 2010;6 doi: 10.1371/journal.ppat.1000896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Subissi L., Posthuma C.C., Collet A., Zevenhoven-Dobbe J.C., Gorbalenya A.E., Decroly E. One severe acute respiratory syndrome coronavirus protein complex integrates processive RNA polymerase and exonuclease activities. Proc Natl Acad Sci U S A. 2014;111:E3900–E3909. doi: 10.1073/pnas.1323705111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sofia M.J., Chang W., Furman P.A., Mosley R.T., Ross B.S. Nucleoside, nucleotide, and non-nucleoside inhibitors of hepatitis C virus NS5B RNA-dependent RNA-polymerase. J Med Chem. 2012;55:2481–2531. doi: 10.1021/jm201384j. [DOI] [PubMed] [Google Scholar]
- 21.Gao Y., Yan L., Huang Y., Liu F., Zhao Y., Cao L. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science. 2020;368:779–782. doi: 10.1126/science.abb7498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yin W., Mao C., Luan X., Shen D.D., Shen Q., Su H. Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science. 2020;368:1499–1504. doi: 10.1126/science.abc1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Q., Wu J., Wang H., Gao Y., Liu Q., Mu A. Structural basis for RNA replication by the SARS-CoV-2 polymerase. Cell. 2020;182:417–428. doi: 10.1016/j.cell.2020.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huey R., Morris G.M. Using AutoDock 4 with AutoDocktools: A tutorial. Scripps Res Inst USA. 2008;1:54–56. [Google Scholar]
- 25.O'Boyle N.M., Banck M., James C.A., Morley C., Vandermeersch T., Hutchison G.R., Open Babel An open chemical toolbox. J Cheminf. 2011;3:33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Halgren T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J Comput Chem. 1996;17:490–519. [Google Scholar]
- 27.Trott O., Olson A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alonso H., Bliznyuk A.A., Gready J.E. Combining docking and molecular dynamic simulations in drug design. Med Res Rev. 2006;26:531–568. doi: 10.1002/med.20067. [DOI] [PubMed] [Google Scholar]
- 29.Lindorff-Larsen K., Piana S., Palmo K., Maragakis P., Klepeis J.L., Dror R.O. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins. 2010;78:1950–1958. doi: 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang J., Wang W., Kollman P.A., Case D.A. Antechamber: An accessory software package for molecular mechanical calculations. J Am Chem Soc. 2001;222:U403. [Google Scholar]
- 31.Wang J., Wolf R.M., Caldwell J.W., Kollman P.A., Case D.A. Development and testing of a general amber force field. J Comput Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 32.Frisch M., Trucks G., Schlegel H.B., Scuseria G.E., Robb M.A., Cheeseman J.R. Gaussian. Inc.; Wallingford CT: 2009. Gaussian 09, Revision d. 01.https://gaussian.com/g09citation/ [Google Scholar]
- 33.Mark P., Nilsson L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J Phys Chem. 2001;105:9954–9960. [Google Scholar]
- 34.Yi D., Li Q., Pang L., Wang Y., Zhang Y., Duan Z. Identification of a broad-spectrum viral inhibitor targeting a novel allosteric site in the RNA-dependent RNA polymerases of dengue virus and norovirus. Front Microbiol. 2020;11:1140. doi: 10.3389/fmicb.2020.01440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miyamoto S., Kollman P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J Comput Chem. 1992;13:952–962. [Google Scholar]
- 36.Feig M., Karanicolas J., Brooks C.L., III MMTSB Tool Set: Enhanced sampling and multiscale modeling methods for applications in structural biology. J Mol Graph Model. 2004;22:377–395. doi: 10.1016/j.jmgm.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 37.Genheden S., Ryde U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expet Opin Drug Discov. 2015;10:449–461. doi: 10.1517/17460441.2015.1032936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ren L.L., Wang Y.M., Wu Z.Q., Xiang Z.C., Guo L., Xu T. Identification of a novel coronavirus causing severe pneumonia in human: A descriptive study. Chin Med J (Engl) 2020;133:1015–1024. doi: 10.1097/CM9.0000000000000722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lei X., Dong X., Ma R., Wang W., Xiao X., Tian Z. Activation and evasion of type I interferon responses by SARS-CoV-2. Nat Commun. 2020;11:3810. doi: 10.1038/s41467-020-17665-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gao Q., Wang Z., Liu Z., Li X., Zhang Y., Zhang Z. A cell-based high-throughput approach to identify inhibitors of influenza A virus. Acta Pharm Sin B. 2014;4:301–306. doi: 10.1016/j.apsb.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Min J.S., Kim G.W., Kwon S., Jin Y.H. A cell-based reporter assay for screening inhibitors of MERS coronavirus RNA-dependent RNA polymerase activity. J Clin Med. 2020;9:2399. doi: 10.3390/jcm9082399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hou T., Xu X. Recent development and application of virtual screening in drug discovery: An overview. Curr Pharmaceut Des. 2004;10:1011–1033. doi: 10.2174/1381612043452721. [DOI] [PubMed] [Google Scholar]
- 43.Gilson M.K., Zhou H.X. Calculation of protein–ligand binding affinities. Annu Rev Biophys Biomol Struct. 2007;36:21–42. doi: 10.1146/annurev.biophys.36.040306.132550. [DOI] [PubMed] [Google Scholar]
- 44.Du X., Li Y., Xia Y.L., Ai S.M., Liang J., Sang P. Insights into protein–ligand interactions: Mechanisms, models, and methods. Int J Mol Sci. 2016;17:144. doi: 10.3390/ijms17020144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ewing T.J., Lybrand T.P. A comparison of perturbation methods and Poisson-Boltzmann electrostatics calculations for estimation of relative solvation free energies. J Phys Chem. 1994;98:1748–1752. [Google Scholar]
- 46.Reddy M.R., Singh U., Erion M.D. Ab initio quantum mechanics-based free energy perturbation method for calculating relative solvation free energies. J Comput Chem. 2007;28:491–494. doi: 10.1002/jcc.20510. [DOI] [PubMed] [Google Scholar]
- 47.Wang L., Wu Y., Deng Y., Kim B., Pierce L., Krilov G. Accurate and reliable prediction of relative ligand binding potency in prospective drug discovery by way of a modern free-energy calculation protocol and force field. J Am Chem Soc. 2015;137:2695–2703. doi: 10.1021/ja512751q. [DOI] [PubMed] [Google Scholar]
- 48.Shannon A., Le N.T., Selisko B., Eydoux C., Alvarez K., Guillemot J.C. Remdesivir and SARS-CoV-2: Structural requirements at both nsp12 RdRp and nsp14 exonuclease active-sites. Antivir Res. 2020;178:104793. doi: 10.1016/j.antiviral.2020.104793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pushpakom S., Iorio F., Eyers P.A., Escott K.J., Hopper S., Wells A. Drug repurposing: Progress, challenges and recommendations. Nat Rev Drug Discov. 2019;18:41–58. doi: 10.1038/nrd.2018.168. [DOI] [PubMed] [Google Scholar]
- 50.Reddy B.U., Mullick R., Kumar A., Sharma G., Bag P., Roy C.L. A natural small molecule inhibitor corilagin blocks HCV replication and modulates oxidative stress to reduce liver damage. Antivir Res. 2018;150:47–59. doi: 10.1016/j.antiviral.2017.12.004. [DOI] [PubMed] [Google Scholar]
- 51.Gupta A., Singh A.K., Kumar R., Ganguly R., Rana H.K., Pandey P.K. Corilagin in cancer: A critical evaluation of anticancer activities and molecular mechanisms. Molecules. 2019;24:3399. doi: 10.3390/molecules24183399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hillen H.S., Kokic G., Farnung L., Dienemann C., Tegunov D., Cramer P. Structure of replicating SARS-CoV-2 polymerase. Nature. 2020;584:154–156. doi: 10.1038/s41586-020-2368-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.