

Abstract
Tumor-associated M2-macrophages are one of the most abundant immunosuppressive cell types in the PDAC TME. However, the molecular mechanisms responsible for the generation of M2-macrophages are unclear. Here we demonstrated that overexpression of DCLK1-isoform2 in AsPC1 and MIA PaCa2 cells resulted in the polarization of M1-macrophages towards an M2-phenotype via secreted chemokines/cytokines. These M2-macrophages enhanced parental PDAC cell migration, invasion, and self-renewal and this was associated with increased expression of Snail and Slug. We observed distinct expression of Dclk-isoform2, marked infiltration of M2-macrophages and a marginal increase of CD8+ T-cells in 20-week-old KPCY mice pancreas compared to 5-week-old. Utilizing an autochthonous mouse model of pancreatic adenocarcinoma, we observed distinct immunoreactive Dclk1 and arginase1 in tissues where CD8+ T-cell infiltration was low and observed a paucity of DCLK1 and arginase1 staining where CD8+ T-cell infiltration was high. Finally, we found that DCLK1-isoform2 tumor educated M2-macrophages inhibits CD8+ T-cells proliferation and Granzyme-B activation. Inhibition of DCLK1 in an organoid co-culture system enhanced CD8+ T-cell activation and associated organoid death. We conclude that DCLK1-isoform2 is a novel initiator of alternate macrophage activation that contributes to the immunosuppression observed in the PDAC TME. These data suggest that tumor DCLK1-isoform2 may be an attractive target for PDAC therapy, either alone or in conjunction with immunotherapeutic strategies.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is associated with an immunosuppressive tumor microenvironment (TME), which potentially explains the poor response to immunotherapeutic agents including checkpoint inhibitors (1). PDAC TME attracts several immunosuppressive cell types that coordinately act to inhibit the effectiveness of cytotoxic CD8+ T-cells, thus, attenuating their anti-tumor function (1,2). These immune cell types include myeloid-derived suppressor cells (MDSCs), regulatory T-cells (Treg), and tumor-associated (M2) macrophages (1,3). M2-macrophages are an abundant immunosuppressive cell type in the PDAC TME (4). In this report, we investigated the mechanism by which PDAC cells regulate M2-macrophage generation (a process called polarization) and its tumor immunosuppression activity.
Macrophages regularly sense their microenvironment and, depending on the inflammatory signal, adopt either an M1 or M2 phenotype (5). M1-macrophages are pro-inflammatory, whereas M2-macrophages are anti-inflammatory, pro-tumorigenic and immunosuppressive (5). The density of M2-macrophages in the PDAC TME is highly correlated with a poor prognosis (6). Although the origin of M2-macrophages is not fully understood, experimental evidence suggests that tumor-dependent mechanisms are responsible for macrophage polarization (7). M2-macrophages facilitate tumor invasion, metastasis, and enhance chemoresistance (5,7). Recent reports suggest that M2-macrophages can suppress the anti-tumor activity of cytotoxic CD8+ T-cells within tumors (8). Reversing the immunosuppressive effects of M2-macrophages offers an attractive therapeutic strategy to enhance host anti-tumor immunity.
Doublecortin-like kinase 1 (DCLK1) is a marker of tumor stem cells, which identifies the progenitors and cell of origin in inflammation-associated mouse pancreatic cancer (9–11). DCLK1 is overexpressed in the tumors and pancreatic intraepithelial (PanIN) lesions of various pancreatic cancer models (9,10,12). Pancreatic cancer patients with DCLK1 expressing tumors exhibit poor survival compared to patients without DCLK1-expressing tumors (13). DCLK1 inhibition in pancreatic cancer cells blocks tumor cell migration/invasion and results in tumor xenograft growth arrest (14). Recent studies have identified the important function of DCLK1 expressing intestinal tuft cells in regulating the immune response after infection and or injury (15). Furthermore, it is reported that DCLK1 expressing tuft cells are the major local source of IL17 and COX1/COX2 mediated PGE2 which play crucial roles in regulating pro-inflammatory molecules and immune cells (16,17). It is demonstrated that KrasG12D activation in the pancreas is associated with the abnormal generation of pancreatic tuft cells (16) and these cells are identified as progenitor cells and tumor-initiating cells after injury (10). Although earlier studies have suggested the critical role for Dclk1+ cells in pancreatic cancer development after severe injury, their role in the regulation of immune cells for the development of immunosuppressive tumor microenvironment in PDAC has not been established.
Human DCLK1 consists of four primary isoforms, each with a shared kinase domain, driven from two promoter regions termed α and β (Supplementary Figure 1) (18). The α-promoter drives the expression of isoforms termed α-long (isoform2) and α-short (isoform1) and the β-promoter drives the expression of isoforms termed β-long (isoform4) and β-short (isoform3). However, the potential tumorigenic function and tumor immune regulation of these isoforms are not clear.
In this report, we analyzed the role of DCLK1-isoform2 expressing PDAC cells in macrophage polarization and examined its immunosuppression for tumor progression. Our findings suggest that DCLK1-isoform2 expression in PDAC cells induce polarization of macrophages that contribute to the aggressiveness of PDAC cells and inhibition of cytotoxic T-cell function.
Materials and Methods
Experimental animals
The KRasLSLG12Dp53LSLR172HPdx1CreRosa26YFP (KPCY) and control mice were described previously (19). Mice were housed under controlled conditions and were monitored regularly. All animal experiments were performed with the approval and authorization from the Institutional Animal Care and Use Committee (IACUC) of the University of Oklahoma Health Sciences Center.
Mouse samples
Fixed CD8+ T-cell high (“T-cell high”) and CD8+ T-cell low (“T-cell low”) pancreatic adenocarcinoma tissues from autochthonous mouse model (20) were generously provided by Ben Z. Stanger, MD, Ph.D., of the Perelman School of Medicine, University of Pennsylvania.
Determination of DCLK1-correlated immunosuppressive markers and Macrophage polarization markers in TCGA PAAD patient data
The PAAD RNA-seq datasets in the TCGA dataset was downloaded through the UCSC cancer genome browser, as previously described (21). The corrplot function (R package corrplot) was used to confirm the correlation between the expression levels of DCLK1 and other genes.
Analysis of DCLK1 isoforms expression
TSVdb is a web-based tool (22), which enables to readily access, analyze, and interpret alternative splicing based on TCGA PAAD samples for DCLK1. We interpret DCLK1 isoforms expression variations between solid tumor tissues and normal adjacent tissues. TSVdb is available at http://www.tsvdb.com.
Cell culture
THP-1 cells were purchased on 2018 from ATCC, USA. THP1 cells were treated with 100 ng/ml of Phorbol-12-myristate-13-acetate (Sigma-Aldrich, Santa Clara, CA) for 48 hours. After then M1-macrophages were generated by the addition of IFN-γ (100 ng/ml). Macrophage polarization was confirmed by M1 and M2 markers expression. Human PDAC cell lines, AsPC-1 and MIA PaCa were obtained from ATCC on 2017. PDAC cells were grown in DMEM, supplemented with 10% FBS (Sigma) at 37°C and 5% CO2. Human CD8+ T-cells (TLL-104) were obtained on 2018 from ATCC and grown in T-cell specific Iscove’s modified Dulbecco’s medium. Lentivirus containing human DCLK1isoform2 cDNA sequence was constructed as described previously (23). All cell lines were used within 25 passages. All the cells were confirmed to be negative for mycoplasma using the MycoAlert Mycoplasma Detection Kit (Lonza). AsPC-1 and MIA PaCa2 were infected with lentivirus to overexpress DCLK1-isoform2-RFP fusion protein (AsPC1-DCLK1-isoform2 / MIA PaCa2-DCLK1isoform2) or RFP (AsPC1-RFP / MIA PaCa2-RFP) as control, and selected with puromycin to establish stable cell lines. Co-culture experiments, those that physically interact (juxtacrine): M1-macrophages or M2-macrophages (1×105 cells per 6 well plate) were plated in a 6 well plate and after 24h, AsPC1 or MIA PaCa2 or T-cells at a concentration of 1×105 cells were seeded for co-culture. Dual-culture experiments, those that physically separate from one another using transwell plates that allow communication only via soluble factors (paracrine): M1-macrophages or M2-macrophages (1×105 cells per 6 well plate) were plated in the 6 well plates and AsPC1 or MIA PaCa2 or T-cells at a concentration of 1×105 cells were seeded in the transwell inserts for dual-culture. Co-culture experiments, those that physically interact (juxtacrine): M1-macrophages or M2-macrophages (1×105 cells per 6 well plate) were plated in a 6 well plate and after 24h, AsPC1 and T-cells at a concentration of 1×105 cells were seeded for co-culture.
siRNA-mediated knockdown of DCLK1
PDAC cells were seeded into 6 cm Petri-dishes and allowed to attach overnight. Following attachment, 1nM of commercially validated siRNA targeting human DCLK1 or 1nM human scrambled sequence (siSCR) not targeting any known genes (Santa Cruz, Biotech., USA) were complexed with Lipofectamine 3000 (Invitrogen) and added to the dishes in fresh cell culture MEM medium. After 48h cells were used for further analysis.
Cell proliferation assays
Post-treatment, 10 μl of TACS MTT Reagent (RND Systems) was added to each well and the cells were incubated at 37°C until a dark crystalline precipitate became visible in the cells. Then 100 μl of 266 mM NH4OH in DMSO was added to the wells and placed on a plate shaker at low speed for 5 minutes. After shaking, the plate was allowed to incubate for 10 minutes protected from light and the OD550 for each well was read using a microplate reader. The results were averaged and calculated as percentage cell proliferation.
Clonogenic assay
Reduced growth‐factor matrigel was mixed with cell suspensions containing RPMI medium (volume 1:1). A hundred microliters of the mixture were pipetted into 48‐well plates at 500 cells/well. After matrigel solidification, a 500 μL RPMI medium with serum supplement was added to each well. The plates were then incubated at 37°C under 5% CO2. The media was refreshed weekly and wells monitored for spheroid formation, as described previously (17,21,24–26).
Organoid co-culture
Organoids were generated as described in the clonogenic assay. Tumor organoid and immune cell co-culture were carried to create a tumor immunosuppressive microenvironment and to understand the role of DCLK1-isoform2 in such an environment. Organoids generated were counted and wells were selected with an equal number of organoids. Selected wells with an equal number of organoids were seeded with M2-macrophages and T-cells at a concentration of 1×105 cells per well of 48 well plates. After 24h, 1nM of commercially validated siRNA targeting human DCLK1 or 1nM human scrambled sequence (siSCR) were complexed with Lipofectamine 3000 and added to the wells. After 72h organoids-co-culture was used for cell viability and granzyme B activity.
Cell viability assay
Viability of cells and spheroids using CellTiter-Glo® 3D Cell Viability assay quantifying intra-tissue ATP content and this assay combines the enhanced penetration and lytic activity required for efficient lysis of 3D cell culture with the generation of the stable ATP-dependent luminescent signal. Add a volume of CellTiter-Glo® 3D Reagent equal to the volume of cell culture medium present in each well (1:1). Plates were vigorously mixed for 5 min using plate shaker and incubated for 25 min at room temperature. Record luminescence and results represented in percentage viability.
Human cytokine array
The human cytokine array (ThermoFisher Scientific, USA) for 36 cytokine/chemokines was carried out according to the manufacturer’s instructions. Briefly, the membrane was blocked using blocking buffer for an hour, then the samples mixed with array antibodies were added to the membrane and incubated overnight at 4°C. Then the membranes were washed with wash buffer and developed using ECL and images were captured and quantified.
Granzyme-B activity assay
The human Granzyme-B enzyme hydrolyzes the specific substrate (Ac-IEPD-AFC) to release the quench of the fluorescent group, which can be detected fluorometrically at Ex/Em = 380/500 nm. The Granzyme-B activity assay was carried out according to the manufacturer instructions (Abcam, USA). Briefly, the reaction mixture was added to each well-containing cell lysates or positive controls or blank. Plates were incubated for 60 min at 37°C and fluorescence was measured at Ex/Em=380/500 nm).
Migration and invasion assay
For the invasion assay, matrigel-coated Transwells (BD Biosciences) were prepared by retrieving in serum-free media for 2 h at 37°C. For the migration assay, Transwells (BD Biosciences) were used. Cell culture medium containing 10% FBS was added to the bottom of each well as a chemoattractant, and the cells were incubated in the transwell inserts for 24 h at 37°C under 5% CO2. Afterward, a cotton swab was used to scrape non-invasive/migratory cells off the top of Transwells; the remaining cells were fixed and stained with 0.1% crystal violet, and allowed to dry. After drying, all invading/migrating cells were counted from each Transwell. Results are reported as the percentage of cells invaded and/or migrated.
Quantitative real-time RT-PCR
Total RNA was isolated from a tumor or immune cells using Tri Reagent (MRC) per the manufacturer’s instructions. First-strand cDNA synthesis was carried out using SuperScript II Reverse Transcriptase and random hexanucleotide primers (Invitrogen). The complementary DNA was subsequently used to perform RT-PCR on an iCycler IQ5 Thermal Cycler (BioRad) using SYBR Green (Molecular Probes) with gene-specific primers and JumpStart™ Taq DNA polymerase (Sigma). The crossing threshold value assessed was normalized to β-actin and quantitative changes in mRNA were expressed as fold-change relative to control ± SD value.
Immunoblot Analysis
Twenty-five micrograms of the total protein were size-separated in a 4%–12% SDS polyacrylamide gel and transferred electrophoretically onto a PVDF membrane with a wet-blot transfer apparatus (Bio-Rad, Hercules, CA). The membrane was blocked and incubated overnight with a primary antibody and was subsequently incubated with horseradish peroxidase-conjugated secondary antibody. The proteins were detected using ECL Western blotting detection reagents (Amersham-Pharmacia, Piscataway, NJ). Protein density quantification was performed using GelQuant software. Actin (42-kD) was used as a loading control.
Immunohistochemistry/immunofluorescence
Standard immunohistochemistry protocols were used with specific antibodies, as described previously (17,27,28). Immunohistochemistry: Heat-induced epitope retrieval was performed on 4-μm formalin-fixed, paraffin-embedded sections utilizing a pressurized Decloaking Chamber (Biocare Medical LLC, Concord, CA) in citrate buffer (pH 6.0) at 99 °C for 18 minutes. Brightfield: Slides were incubated in 3% hydrogen peroxide at room temperature for 10 min. After incubation with the primary antibody overnight at 4 °C, slides were incubated in a Promark peroxidase-conjugated polymer detection system (Biocare Medical LLC) for 30 min at room temperature. After washing, slides were devolved with diaminobenzidine (Sigma–Aldrich). Fluorescence: Slides were incubated in normal serum and BSA blocking step at room temperature for 20 min. After incubation with primary antibody overnight at 4 °C, slides were labeled with Alexa Fluor® dye-conjugated secondary antibody and mounted with ProLong Gold (Invitrogen). Image Acquisition: Slides were examined on the Nikon Eclipse Ti motorized microscope paired with image app operated by the NIS-Elements Microscope Imaging Software platform (Nikon Instruments, Melville, NY).
Statistical analyses
All statistical analyses were performed in GraphPad Prism 6.0, SPSS Statistics 22, and Microsoft Excel. One-way ANOVA and the Student’s T-test were used to determine statistical significance. Pearson product-moment correlation was used for analysis and correlation of gene expressions between two groups. P values of <0.05 = *, <0.01 = **, and 0.001 = *** were considered statistically significant.
Results
Higher expression of DCLK1 in human pancreatic cancer predicts immunosuppressive phenotype
We previously reported that DLCK1 is highly expressed in human and mouse models of PDAC (13,19). To address whether DCLK1-isoform2 an alternative splice variant of DCLK1 is highly expressed in patients with PDAC, we used TSVdb (22), an interactive web-portal, to perform a comparative analysis on DCLK1 splicing variants from TCGA pancreatic cancer (PAAD) RNA-Seq datasets. We found that DCLK1-isoform2 is highly expressed in pancreatic cancer, whereas DCLK1-isoform1 is marginally increased compared to solid tissue normal (Figure 1A; Supplementary Figure 2). We observed increased immunostaining for DCLK1-isoform2 in epithelial and, stromal cells in human PDAC tissues (total 103 cases) compared to scant staining in normal adjacent tissues (Figure 1B, 1C) using Abcam-31704 Ab which detects DCLK1-isoform2/4 (23). We analyzed the correlation between DCLK1 mRNA expression and immuno-suppressive gene expressions using the TCGA-PAAD RNA-seq database. The dataset revealed that increased DCLK1 was associated with increased expression of immunosuppressive markers (ARG1, ARG2, CCL17, CCL22, IL10, IL13, IL25, IL16, CCL1, CCL2, MIP, CXCL12, CD4, CD6, CTLA4, ITGAM) (Figure 1D). Furthermore, a strong correlation was noted between DCLK1 mRNA and the M2-macrophages markers (CD163, MRC1) compared with M1-macrophage markers (IL23A, IL1A) (Figure 1E). Collectively, our data for the first time indicate a potential link between DCLK1 and macrophage polarization towards an immunosuppressive M2 phenotype.
Fig. 1: DCLK1-isoform2 expression increased in PDAC and associates with immunosuppressive phenotype.
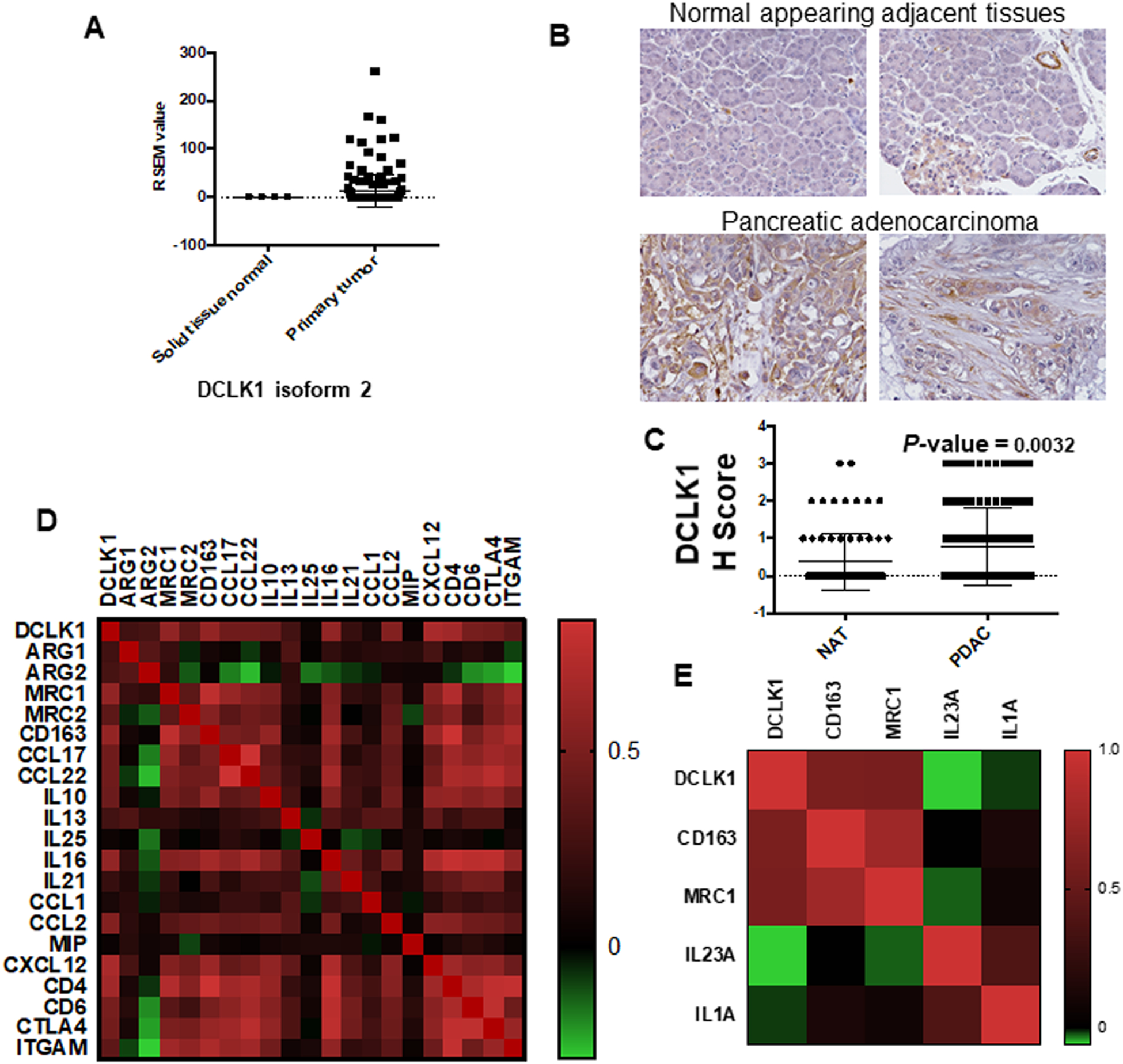
A) DCLK1-isoform2 mRNA is highly expressed in pancreatic adenocarcinoma compared to adjacent solid pancreatic normal tissue in the TCGA-PAAD dataset analyzed from TSVdb webtool. B) Human tissue microarray (TMA) of pancreatic adenocarcinoma and adjacent normal tissues were stained for DCLK1-isoform2 and the intensity was scored. Immunohistochemistry reveals that DCLK1-isoform2 is highly expressed in pancreatic adenocarcinoma compared to normal adjacent pancreas issues. C) Representative graph of DCLK1 expression in normal and pancreatic adenocarcinoma TMA. D) DCLK1 mRNA and mRNA of immunosuppressive markers (ARG1, ARG2, CCL17, CCL22, IL10, IL13, IL25, IL16, CCL1, CCL2, MIP, CXCL12, CD4, CD6, CTLA4, ITGAM) and (E) macrophage markers (CD163, MRC1, IL23A, IL1A) were downloaded from PAAD dataset of TCGA database. DCLK1 expression is positively correlated with genes of immunosuppressive markers and polarized macrophage M2 markers; Color indicates a correlation of DCLK1 and other genes, negative (green), and positive (red).
Generation and viability of M1-macrophages
M1-macrophages were generated from THP1 cells and confirmed by both morphology and the expression of M1 markers (CD86 and HLA-DR) (Figure 2A and 2B). The viability of M1-macrophages was assessed by MTT assay at 0, 6, 12, 24, 48, 72, 96 and 120h. We observed a 5–8% reduction in the viability of M1-macrophages at 120h (Figure 2C). Our data suggest that M1-macrophages derived from THP1 cells can survive up to 5 days without a significant loss in viability.
Fig. 2: Co-culture of DCLK1-isoform2 overexpressing AsPC1 cells or MIA PaCA2 cells polarize M1-macrophages into an M2 phenotype.

A) Morphological difference between THP1 cells and THP-1 derived macrophages (M1). B) Protein expression of M1-macrophage markers HLA-DR and CD86 between THP1 and M1-macrophages by immunoblot analysis. C) The line graph represents % macrophage viability up to 120h. D) Morphological difference between AsPC1 vector control and AsPC1-DCLK1-isoform2 overexpressing cells. E) Protein expression of DCLK1-isoform2 in the AsPC1 vector control cells and AsPC1-DCLK1-isoform2 overexpressing cells by immunoblot analysis. F) mRNA expression of DCLK1-isoform2 in the AsPC1 vector control cells and AsPC1-DCLK1-isoform2 overexpressing cells by RT-PCR analysis. G) Protein expression of CD163 and CD206 (M2 markers) and CD86 and HLA-DR (M1 markers) in the total protein lysates of AsPC1 vector control cells+M1-macrophages treated with siScramble, AsPC1 vector control cells+M1-macrophages treated with siDCLK1, AsPC1-DCLK1-isoform2 cells+M1-macrophages treated with siScramble and AsPC1-DCLK1-isoform2 cells+M1-macrophages treated with siDCLK1. H) Protein expression of CD163 and CD206 (M2 markers) and CD86 and HLA-DR (M1 markers) in the total protein lysates of MIA PaCa2 vector control cells+M1-macrophages treated with siScramble, MIA PaCa2 vector control cells+M1-macrophages treated with siDCLK1, MIA PaCa2-DCLK1-isoform2 cells+M1-macrophages treated with siScramble and MIA PaCa2-DCLK1-isoform2 cells+M1-macrophages treated with siDCLK1. All quantitative data are expressed as means ± SD of a minimum of three independent experiments. P values of <0.05 were considered statistically significant.
DCLK1-isoform2 overexpressing AsPC1 cells polarize M1-macrophage into M2-phenotype
Our recent study reported increased expression of DCLK1-isoform2 and -isoform4 in renal cancer (23). The present data (Figure 1A) indicates that only DCLK1-isoform2 is highly expressed in PDAC. To investigate its role in the polarization of M1-macrophages, DCLK1-isoform2 was overexpressed in AsPC1 cells (AsPC1-DCLK1-isoform2) and MIA PaCa2 cells (MIA PaCa2-DCLK1-isoform2). Measurement of protein and mRNA expression of DCLK1-isoform2 confirmed overexpression (Figure 2E, F). AsPC1-DCLK1-isoform2 cells appear morphologically similar by 80–90% compared to the vector control cells, however, 10–20% displayed a spindle-like morphology (Figure 2D). The co-culture of AsPC1-DCLK1-isoform2 cells or MIA PaCa2-DCLK1-isoform2 cells with M1-macrophages resulted in a marked increase in expression of the M2-macrophage markers CD163 and CD206, and the reduction in the M1 markers, CD86, and HLA-DR (Figure 2G, H). This increase in M2 marker expression was blocked when AsPC1-DCLK1-isoform2 cells or MIA PaCa2-DCLK1-isoform2 cells were treated with siRNA against DCLK1 before co-culture (Figure 2G, H). Taken together, these data strongly suggest that overexpression of DCLK1-isoform2 in the tumor cells promotes macrophage polarization into an M2-phenotype in vitro.
DCLK1-isoform2 promotes macrophage polarization via secreted cytokines/chemokines
To determine whether AsPC1-DCLK1-isoform2 cells require physical interaction or paracrine interaction to polarize macrophages, we performed dual-culture experiments where M1-macrophages and AsPC1-DCLK1-isoform2 cells were grown in trans-wells (Figure 3A). We observed a distinct upregulation of M2 markers in the macrophages when dual-cultured with AsPC1-DCLK1-isoform2 cells, but not with vector control AsPC1 cells similar to that observed in co-culture experiments (Figure 3B), suggesting that secretory factors generated by AsPC1-DCLK1-isoform2 cells were responsible for the polarization. To identify the secreted cytokines/chemokines involved in this process we performed cytokine protein profiling from the spent media of AsPC1-DCLK1-isoform2 cells and observed nine key chemokines/cytokines (CCL1, CCL2, MIP1a/b, CXL12, G-CSF, IL13. IL25, IL16, and IL21) that were greater than 2-fold compared to vector control cells (Figure 3C, D). These key cytokines/chemokines have been reported previously to play an important role in macrophage polarization towards the M2 phenotype (29). Further studies are underway to specifically characterize the role of each soluble factor in DCLK1 dependent macrophage polarization.
Fig. 3: DCLK1-isoform2 overexpressing AsPC1 cells secrete cytokines/chemokines to polarize macrophages in a dual culture system.
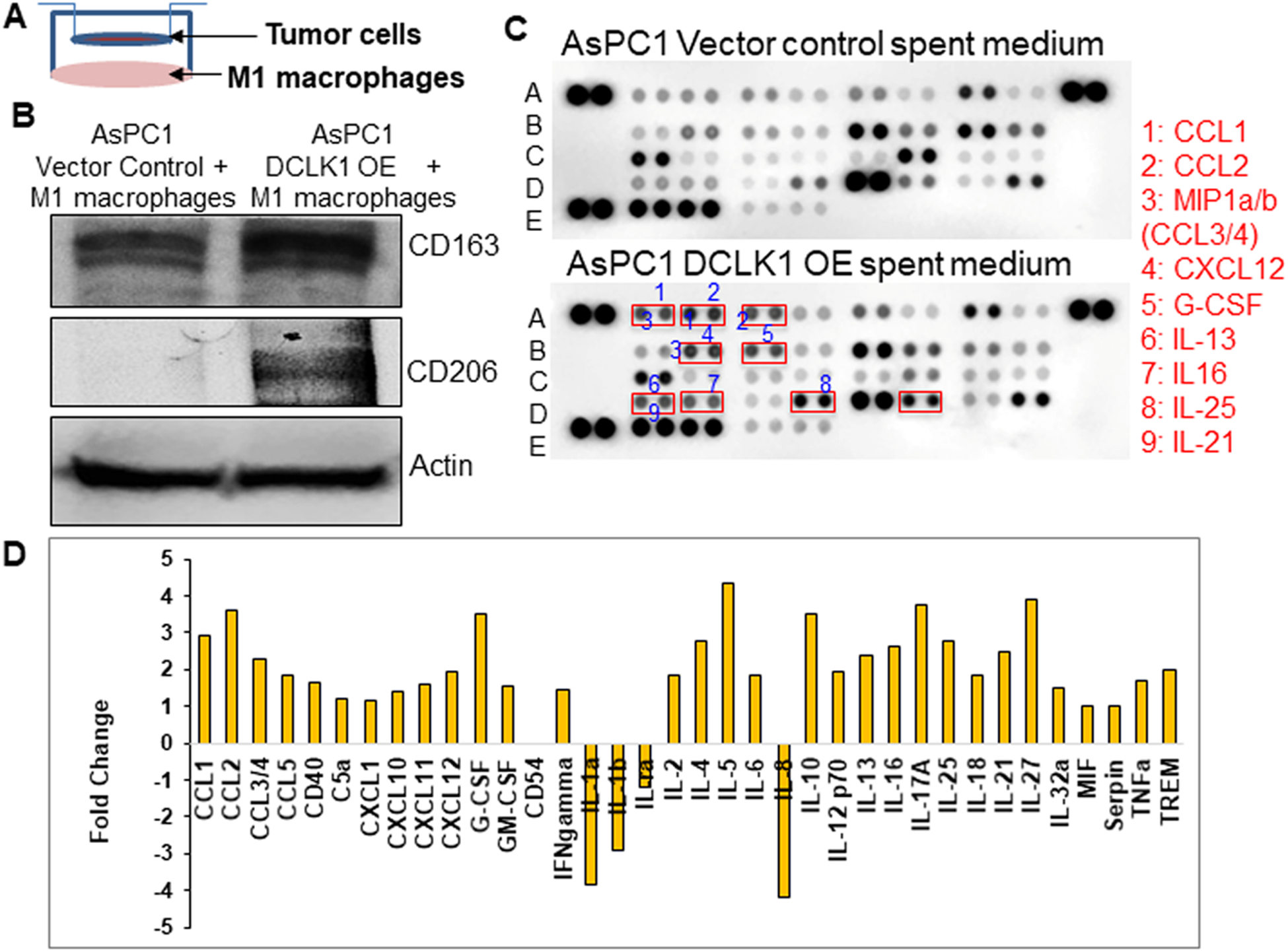
A) Dual cell culture model. B) Protein expression of CD163 and CD206 (M2 markers) in the total protein lysates of M1-macrophages dual cultured with AsPC1 vector control cells or M1-macrophages dual cultured with AsPC1-DCLK1-isoform2 cells. C) Human cytokine array by dot blot analysis, conditioned media collected from AsPC1 vector control cell culture or AsPC1-DCLK1-isoform2 overexpressing cell culture utilized for the array. Cytokines/chemokines with 2-fold differences in their increase were numbered in the blot and named. D) Arrays were quantified using the densitometric analysis to represent a fold change in a bar graph.
M2-macrophages enhance PDAC cell aggressiveness
To determine whether DCLK1-isoform2 polarized M2-macrophages contribute to tumor cell aggressiveness, we dual-cultured M2-macrophages or M1-macrophages with parental AsPC1 cells and evaluated the self-renewal and invasion/ migration of tumor cells. We noted a marked increase in tumor cell invasion and migration (2–3fold) when dual-cultured with DCLK1-isoform2 polarized M2-macrophages (Figure 4A, B). We also observed an increased expression of EMT factors SNAIL and SLUG in the PDAC cells (Figure 4D). Self-renewal of parental AsPC1 cells increased 3fold when dual-cultured with DCLK1-isoform2 induced M2-macrophages (Figure 4F, G). Interestingly, DCLK1 silencing in parental AsPC1 cells reversed the M2-macrophage-mediated increase in SNAIL and SLUG, invasion, migration and self-renewal (Figure 4B, C, E, F, G). To investigate the complex cytokine network between cancer cells and macrophages, we performed cytokine protein profiling from the spent media of M2-macrophages and observed key chemokines/cytokines (Fold reduced: MIP1a/b (CCL3/4), CCL5, CXCL1, CXCL10, CXCL12, G-CSF, CD54, IL-1b, IL-8, IL-13, IL-18, IL-21, IL-32a, Serpin E1; Fold increased: IL-1ra, IL-4, IL-16) that were greater than 2-fold compared to M1-macrophages (Figure 4H, I). These key cytokines/chemokines have been reported to play an important role in tumor aggression (30–32).
Fig. 4: DCLK1-isoform2 mediated M2-macrophages enhance PDAC aggression via soluble secretory factors.
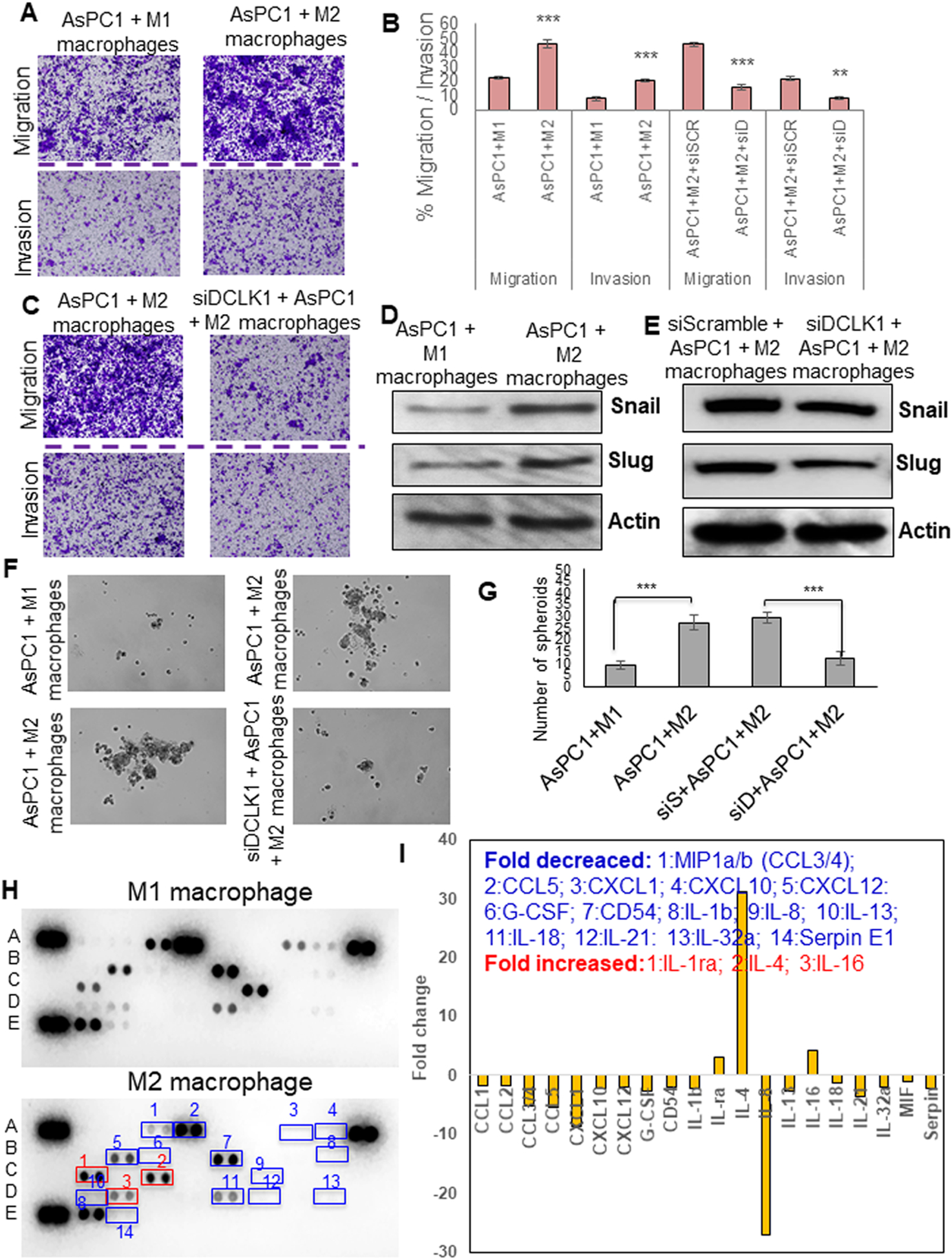
A) Migration and invasion of AsPC1 cells from M1-macrophage or M2-macrophage dual-culture. B) Bar graph represents % tumor cell migrated or invaded. C) Migration and invasion of AsPC1 cells either treated with siScramble or siDCLK1 from M2-macrophage dual-culture. D) Protein expression of SNAIL and SLUG in the AsPC1 cells dual cultured with M1 or M2-macrophages. E) Protein expression of SNAIL and SLUG in the AsPC1 cells treated with siScramble or siDCLK1 from a dual culture experiment with M2-macrophages. F) Self-renewal of AsPC1 cells from M1-macrophage or M2-macrophage dual-culture; self-renewal of AsPC1 cells treated with siScramble or siDCLK1 from M2-macrophage dual-culture.G) Bar graph represents the number of spheroids formed. H) Human cytokine array by dot blot analysis, conditioned media collected from M1 or M2 macrophages utilized for the array. Cytokines/chemokines with 2-fold differences in their increase or decrease were numbered in the blot and named. I) Arrays were quantified using the densitometric analysis to represent a fold change in a bar graph. All quantitative data are expressed as means ± SD of a minimum of three independent experiments. P values of <0.05 = *, <0.01 = **, and 0.001 = *** were considered statistically significant.
Dclk1 and tumor immunosuppressive PDAC phenotype in vivo
We investigated the expression of Dclk1 and the relative abundance of the immunosuppressive cells of the pancreatic TME in KPCY (KRasLSLG12Dp53LSLR172HPdx1CreRosa26YFP) mice at 5 weeks (pre-cancerous) and 20 weeks (PDAC) of age. We observed increased immunoreactive Dclk1-isoform2 in epithelial and stromal tissues of PDAC from 20-week-old KPCY mice compared to 5-week-old (Figure 5A; Supplementary Figure 3). We observed a marked increase of the pan-myeloid marker (CD11B), which identifies myeloid, macrophage and dendritic cells in 20-week-old KPCY tumors compared to 5-week-old (Figure 5A; Supplementary Figure 3). We observed a distinct immunoreactive CD8 in 20-week-old KPCY mice tumor compared to 5-week-Old (Figure 5B; Supplementary Figure 3). However, within PDAC tumors there is marked heterogeneity of T-cells infiltration. We obtained fixed CD8+ T-cell high (“T-cell high”) and CD8+ T-cell low (“T-cell low”) tumor tissues of autochthonous mouse model of pancreatic adenocarcinoma (20) as a generous gift from Ben Z. Stanger, MD, Ph.D., at the University of Pennsylvania. We performed IHC in these tissues and observed that the expression of DCLK1-isoform2 was inversely correlated with the infiltration of CD8+ T cells. DCLK1 staining was higher in CD8+ T-cell low tumor tissues and lower in CD8+ T-cell high tumor tissues (Figure 5C; Supplementary Figure 4). Following this observation, we observed a significant increase in the expression of arginase1 (M2 marker) in CD8+ T-cell low tumor tissues in contrast to CD8+ T-cell high tumor tissues (Figure 5D; Supplementary Figure 5). This suggests that the expression of DCLK1-isoform2 prevents the infiltration of CD8+ T-cells through M2-macrophages.
Fig. 5: DCLK1-isoform2 expression positively and negatively correlates with M2-macrophages and CD8+ T-cells respectively to induce immunosuppression in PDAC.
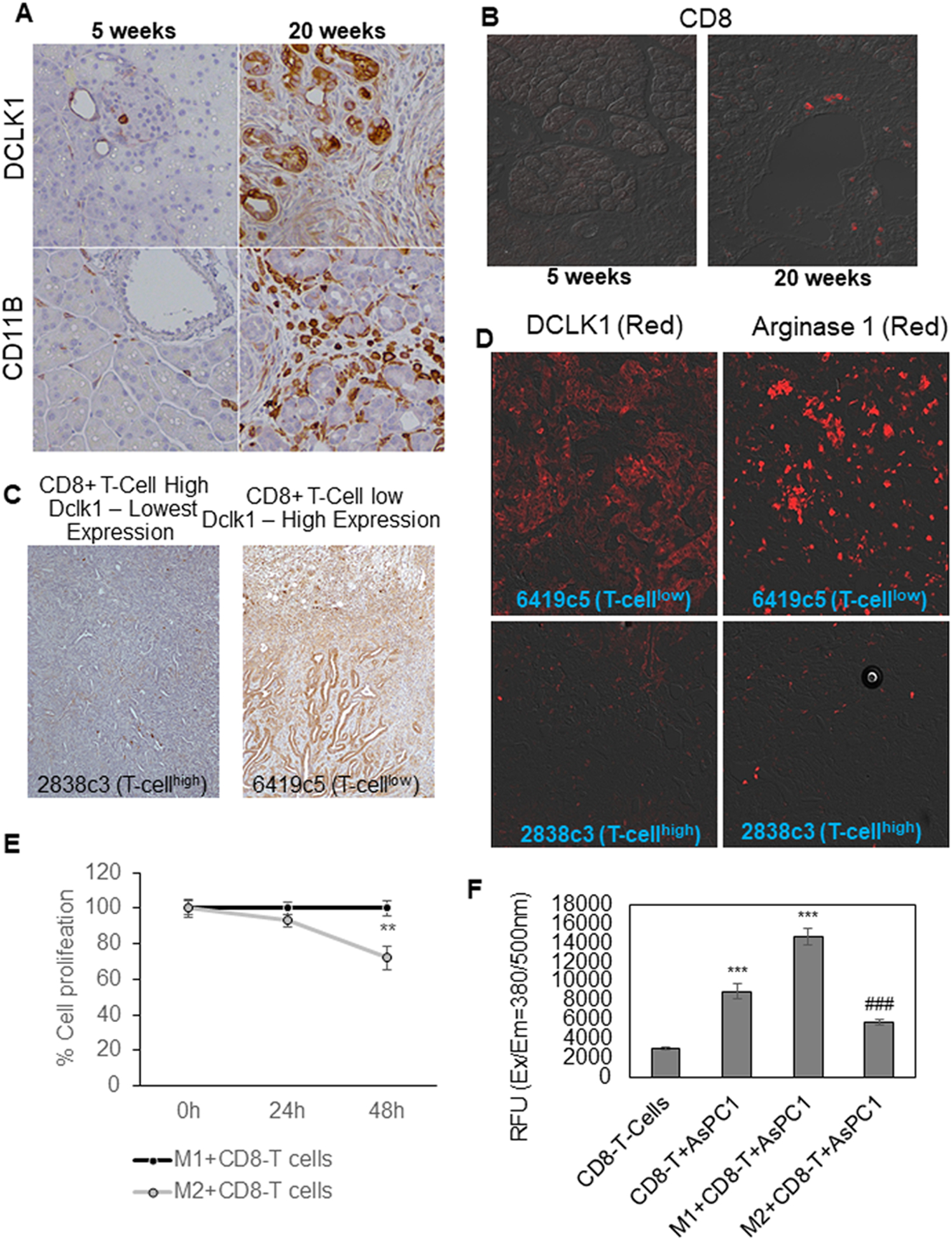
A) IHC was performed to stain Dclk1 and CD11B in the pancreatic tissues collected from 5 weeks (precancerous) and 20 weeks (PDAC) old KPCY mice (n=5). B) CD8 staining was performed in the pancreatic tissues collected from 5 weeks and 20 weeks old KPCY mice. C) Fixed tumor tissues with CD8+ T-cell high (“T-cell high”) and CD8+ T-cell low (“T-cell low”) from an autochthonous mouse model (n=5) of pancreatic adenocarcinoma were stained for Dclk1. D) Fixed tumor tissues with CD8+ T-cell high and CD8+ T-cell low from an autochthonous mouse model of pancreatic adenocarcinoma were stained for Dclk1 and Arginase1. E) Line graph represents the percentage proliferation of CD8+ T-cells collected from M1-macrophage dual-culture and M2-macrophage dual-culture. F) Bar graph of relative fluorescence units (RFU) represents the GranzymeB activity of CD8+ T-cells co-cultured with AsPC1 or AsPC1+M1-macrophages or AsPC1+M2-macrophages. *** = compared to CD8+ T-cells; ### = compared to M1+T-cells+AsPC1 culture. All quantitative data are expressed as means ± SD and P values of <0.05 = *, <0.01 = **, and 0.001 = *** were considered statistically significant.
M2-macrophages inhibit CD8+ T cell activity
We investigate whether DCLK1-isoform2 polarized M2-macrophages can inhibit CD8+ T-cells proliferation and the cytotoxic activity in PDAC. CD8+ T-cells when dual-cultured with tumor-educated M2-macrophages exhibited a 25% reduction in its proliferation compared to M1-macrophage dual-culture (Figure 5F). The activity of Granzyme-B was significantly reduced (2.5fold) in CD8+ T-cells when co-cultured with AsPC1 cells+M2-macrophages compared to CD8+ T-cells co-cultured with AsPC1 cells+M1-macrophages (Figure 5G). Taken together, DCLK1-isoform2 expressing tumor cells induced M2-macrophages inhibits proliferation and cytotoxic function of CD8+ T-cells.
Tumor organoid co-culture model as immunosuppressive TME and role of DCLK1
To further understand the relevant immune-modulatory role of DCLK1-isoform2 in PDAC TME, we used a 3D co-culture model that contains tumor organoids, CD8+ T cells, and M2 macrophages. Three-dimensional organoid models are relevant models to test for therapeutic strategies in a co-cultures system (33,34). Immune cells including M2 macrophages and CD8+ T cells were co-cultured with tumor organoids in a 3D environment which recapitulates the tumor micro-environment and its complex immune and cellular interactions (Figure 6A). After 24h of the addition of immune cells into the organoids, 3D cultures were treated with siScramble or siDCLK1 and the cultures were monitored for 72h. We observed a significant reduction in the number of organoids in siDCLK1 treated 3D cultures compared to siScramble treatment (Figure 6B, C). Compared to 0h, siScramble treatment enhanced the organoid numbers at 72h which suggests that M2 macrophages are immunosuppressive and enhance tumor aggressiveness. On the other hand, inhibition of DCLK1 enhanced the cytotoxic activity of CD8+ T cells by increasing the GranzymeB activity (Figure 6D) and reduced the viability of 3D cultures (Figure 6E). Our 3D TME data, which enables a better understanding of the immune-modulatory profile of DCLK1-isoform2 and can be used to test DCLK1 related drug testing or as adjuvant therapy with other immune-modulatory therapies.
Fig. 6: Inhibition of DCLK1-isoform2 in the PDAC organoid co-culture system enhanced the cytotoxic function of CD8+ T-cells and reduced viability of tumor organoid.

A) Organoids generated from MIA PaCa2 cells and organoids were co-cultured with M2 macrophages and CD8+ T cells. B) Organoid co-cultures were treated with siScramble or siDCLK1 and monitored up to 72h. C) siDCLK1 reduced the number of organoids compared to siScramble treatment to the organoid co-culture system, the bar graph represents the number of organoids. D) Bar graph of relative fluorescence units (RFU) represents the GranzymeB activity of CD8+ T-cells co-cultured with organoids+M2-macrophages treated with siScramble or siDCLK1. E) Bar graph of percentage cell viability between organoid co-cultures treated with siScramble or siDCLK1 at 0h and 72h. All quantitative data are expressed as means ± SD and P values of <0.05 = *, <0.01 = **, and 0.001 = *** were considered statistically significant.
Discussion
There is a large body of evidence that the TME has a tremendous role in regulating tumor mediated immunosuppression. This complex interaction between the tumor and several immune cell types creates an enormous challenge for the development of novel therapeutic agents to treat PDAC. In this study, we describe a new role for DCLK1-isoform2 in promoting macrophage polarization towards the immunosuppressive phenotype via a paracrine dependent mechanism. These M2-macrophages advance cancer cell invasion, migration, and self-renewal and reduce cytotoxic T-cell function. Our studies illuminate the mechanistic interaction of M2-macrophages and the tumor epithelium. Interestingly, we demonstrate that these interactions are mediated via DCLK1-isoform2 dependent mechanism.
Macrophages are an incredibly diverse set of immune cells that have been hijacked by tumor cells to favor tumor progression and induce immunosuppression (29). However, the mechanism by which tumor cells polarize macrophages towards an M2-phenotype has not yet been fully explored. Indeed, in PDAC, TAMs are the most abundant immune cell population in the TME (4,35). We used co-culture and dual-culture system to evaluate the role of DCLK1-isoform2 in macrophage polarization. We reveal that overexpression of DCLK1-isoform2 in PDAC cells plays a major role in macrophage polarization by enhancing the secretion of key cytokines/chemokines that have been associated with the macrophage polarization process (29). Silencing DCLK1 inhibits this process and influences macrophages to retain the M1 phenotype. Interestingly our studies reveal the findings that these DCLK1-isoform2 polarized M2-macrophages possess the ability to increase the aggressiveness (invasion/migration) and self-renewal capacity of the parental pancreatic cancer cell line. DCLK1 silencing in the parental pancreatic cancer cell line albeit low at baseline completely abrogated M2-macrophage ability to enhance the aggressiveness and self-renewal of PDAC cells. These data suggest that M2-macrophages are a major component of pancreatic cancers and can promote tumorigenesis by facilitating invasion, migration and self-renewal via key secretory cytokines (31,36). Our data also suggest that interaction between DCLK1-isoform2 expressing epithelial cells and M1-macrophages creates a feed-back loop that involves signaling from the tumor cells to M1-macrophages and communication between M2-macrophages and epithelial cells that require DCLK1-isoform2. The studies presented here demonstrate that in mouse pancreatic tumor tissues, elevated Dclk1-isoform2 expression is associated with high levels of M2-macrophages and low levels of CD8+ T-cell infiltration. In fact, there is an inverse correlation observed between Dclk1-isoform2 or M2-macrophages and T-cell infiltration. One main function of the M2-macrophage is to induce immunosuppression by either inhibits the cytotoxic activity of CD8+ T-cells or keep T-cells non-response and senescence (5,8,37). We report here the immunosuppressive role of DCLK1-isoform2 overexpressing PDAC cell educated M2-macrophages which reduced the proliferation and cytotoxic activity of CD8+ T-cells. However, further molecular studies are required to identify the key factors from M2-macrophages involved in the de-activation of CD8+ T-cells. Furthermore, to understand the immune-modulatory role of DCLK1-isoform2 in the PDAC TME we developed 3D co-cultures of tumor organoids and immune cells and inhibited DCLK1 by siRNA treatment. We demonstrated that our 3D co-culture model is relevant in vitro tools for testing human immune therapies including DCLK1 therapy. We demonstrated that inhibition of DCLK1-isoform2 in the 3D co-culture enhanced the cytotoxic function of CD8+ T cells and thus reduced the number of organoids.
Conclusion
Our studies link DCLK1-isoform2 expression in tumor cells to the regulation of a major component of the innate immune system, the M2-macrophage, which is a predominant immune cell type in PDAC. Our findings thus signify the potential role of DCLK1-isoform2 in tumor cell-mediated macrophage polarization and associated immunosuppression for tumor aggressiveness. Although future studies are required to definitively link DCLK1-isoform2 and pro-tumorigenic features, targeting DCLK1-isoform2 in PDAC may represent a novel therapeutic approach to reactivating host anti-tumor immunity.
Supplementary Material
Acknowledgment:
This work was supported by DOD grant # W81XWH-18-1-0457 (PI: Chandrakesan) and NIH grant # 1R01CA182869-01A1 (PI: C Houchen). We thank Dr. Stanley Lightfoot, OUHSC, Oklahoma, USA, for the histology and pathology evaluation. We thank Landon Moore and Naushad Ali, OUHSC, Oklahoma, USA, for language edit.
Footnotes
Conflict of interests: Courtney Houchen is the co-founder of COARE Holdings, Inc. USA. The other authors declare no conflict of interest.
References
- 1.Karamitopoulou E Tumour microenvironment of pancreatic cancer: immune landscape is dictated by molecular and histopathological features. Br J Cancer 2019;121(1):5–14 doi 10.1038/s41416-019-0479-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fukumura D, Kloepper J, Amoozgar Z, Duda DG, Jain RK. Enhancing cancer immunotherapy using antiangiogenics: opportunities and challenges. Nat Rev Clin Oncol 2018;15(5):325–40 doi 10.1038/nrclinonc.2018.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med 2013;19(11):1423–37 doi 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Farajzadeh Valilou S, Keshavarz-Fathi M, Silvestris N, Argentiero A, Rezaei N. The role of inflammatory cytokines and tumor associated macrophages (TAMs) in microenvironment of pancreatic cancer. Cytokine Growth Factor Rev 2018;39:46–61 doi 10.1016/j.cytogfr.2018.01.007. [DOI] [PubMed] [Google Scholar]
- 5.Pathria P, Louis TL, Varner JA. Targeting Tumor-Associated Macrophages in Cancer. Trends Immunol 2019;40(4):310–27 doi 10.1016/j.it.2019.02.003. [DOI] [PubMed] [Google Scholar]
- 6.Hu H, Hang JJ, Han T, Zhuo M, Jiao F, Wang LW. The M2 phenotype of tumor-associated macrophages in the stroma confers a poor prognosis in pancreatic cancer. Tumour Biol 2016;37(7):8657–64 doi 10.1007/s13277-015-4741-z. [DOI] [PubMed] [Google Scholar]
- 7.Goswami KK, Ghosh T, Ghosh S, Sarkar M, Bose A, Baral R. Tumor promoting role of anti-tumor macrophages in tumor microenvironment. Cell Immunol 2017;316:1–10 doi 10.1016/j.cellimm.2017.04.005. [DOI] [PubMed] [Google Scholar]
- 8.Peranzoni E, Lemoine J, Vimeux L, Feuillet V, Barrin S, Kantari-Mimoun C, et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proc Natl Acad Sci U S A 2018;115(17):E4041–E50 doi 10.1073/pnas.1720948115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Westphalen CB, Takemoto Y, Tanaka T, Macchini M, Jiang Z, Renz BW, et al. Dclk1 Defines Quiescent Pancreatic Progenitors that Promote Injury-Induced Regeneration and Tumorigenesis. Cell Stem Cell 2016;18(4):441–55 doi 10.1016/j.stem.2016.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bailey JM, Alsina J, Rasheed ZA, McAllister FM, Fu YY, Plentz R, et al. DCLK1 marks a morphologically distinct subpopulation of cells with stem cell properties in preinvasive pancreatic cancer. Gastroenterology 2014;146(1):245–56 doi 10.1053/j.gastro.2013.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chandrakesan P, Panneerselvam J, Qu D, Weygant N, May R, Bronze MS, et al. Regulatory Roles of Dclk1 in Epithelial Mesenchymal Transition and Cancer Stem Cells. J Carcinog Mutagen 2016;7(2) doi 10.4172/2157-2518.1000257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sureban SM, May R, Lightfoot SA, Hoskins AB, Lerner M, Brackett DJ, et al. DCAMKL-1 regulates epithelial-mesenchymal transition in human pancreatic cells through a miR-200a-dependent mechanism. Cancer Res 2011;71(6):2328–38 doi 10.1158/0008-5472.CAN-10-2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weygant N, Ge Y, Qu D, Kaddis JS, Berry WL, May R, et al. Survival of Patients with Gastrointestinal Cancers Can Be Predicted by a Surrogate microRNA Signature for Cancer Stem-like Cells Marked by DCLK1 Kinase. Cancer Res 2016;76(14):4090–9 doi 10.1158/0008-5472.CAN-16-0029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sureban SM, May R, Qu D, Weygant N, Chandrakesan P, Ali N, et al. DCLK1 regulates pluripotency and angiogenic factors via microRNA-dependent mechanisms in pancreatic cancer. PLoS One 2013;8(9):e73940 doi 10.1371/journal.pone.0073940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schneider C, O’Leary CE, Locksley RM. Regulation of immune responses by tuft cells. Nat Rev Immunol 2019. doi 10.1038/s41577-019-0176-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Y, Zoltan M, Riquelme E, Xu H, Sahin I, Castro-Pando S, et al. Immune Cell Production of Interleukin 17 Induces Stem Cell Features of Pancreatic Intraepithelial Neoplasia Cells. Gastroenterology 2018;155(1):210–23 e3 doi 10.1053/j.gastro.2018.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chandrakesan P, May R, Weygant N, Qu D, Berry WL, Sureban SM, et al. Intestinal tuft cells regulate the ATM mediated DNA Damage response via Dclk1 dependent mechanism for crypt restitution following radiation injury. Sci Rep 2016;6:37667 doi 10.1038/srep37667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Omori Y, Suzuki M, Ozaki K, Harada Y, Nakamura Y, Takahashi E, et al. Expression and chromosomal localization of KIAA0369, a putative kinase structurally related to Doublecortin. J Hum Genet 1998;43(3):169–77 doi 10.1007/s100380050063. [DOI] [PubMed] [Google Scholar]
- 19.Qu D, Johnson J, Chandrakesan P, Weygant N, May R, Aiello N, et al. Doublecortin-like kinase 1 is elevated serologically in pancreatic ductal adenocarcinoma and widely expressed on circulating tumor cells. PLoS One 2015;10(2):e0118933 doi 10.1371/journal.pone.0118933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li J, Byrne KT, Yan F, Yamazoe T, Chen Z, Baslan T, et al. Tumor Cell-Intrinsic Factors Underlie Heterogeneity of Immune Cell Infiltration and Response to Immunotherapy. Immunity 2018;49(1):178–93 e7 doi 10.1016/j.immuni.2018.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chandrakesan P, Yao J, Qu D, May R, Weygant N, Ge Y, et al. Dclk1, a tumor stem cell marker, regulates pro-survival signaling and self-renewal of intestinal tumor cells. Mol Cancer 2017;16(1):30 doi 10.1186/s12943-017-0594-y. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 22.Sun W, Duan T, Ye P, Chen K, Zhang G, Lai M, et al. TSVdb: a web-tool for TCGA splicing variants analysis. BMC Genomics 2018;19(1):405 doi 10.1186/s12864-018-4775-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ge Y, Weygant N, Qu D, May R, Berry WL, Yao J, et al. Alternative splice variants of DCLK1 mark cancer stem cells, promote self-renewal and drug-resistance, and can be targeted to inhibit tumorigenesis in kidney cancer. Int J Cancer 2018;143(5):1162–75 doi 10.1002/ijc.31400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chandrakesan P, May R, Qu D, Weygant N, Taylor VE, Li JD, et al. Dclk1+ small intestinal epithelial tuft cells display the hallmarks of quiescence and self-renewal. Oncotarget 2015;6(31):30876–86 doi 10.18632/oncotarget.5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chandrakesan P, Roy B, Jakkula LU, Ahmed I, Ramamoorthy P, Tawfik O, et al. Utility of a bacterial infection model to study epithelial-mesenchymal transition, mesenchymal-epithelial transition or tumorigenesis. Oncogene 2014;33(20):2639–54 doi 10.1038/onc.2013.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chandrakesan P, Weygant N, May R, Qu D, Chinthalapally HR, Sureban SM, et al. DCLK1 facilitates intestinal tumor growth via enhancing pluripotency and epithelial mesenchymal transition. Oncotarget 2014;5(19):9269–80 doi 10.18632/oncotarget.2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chandrakesan P, Ahmed I, Anwar T, Wang Y, Sarkar S, Singh P, et al. Novel changes in NF-{kappa}B activity during progression and regression phases of hyperplasia: role of MEK, ERK, and p38. J Biol Chem 2010;285(43):33485–98 doi 10.1074/jbc.M110.129353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chandrakesan P, Ahmed I, Chinthalapally A, Singh P, Awasthi S, Anant S, et al. Distinct compartmentalization of NF-kappaB activity in crypt and crypt-denuded lamina propria precedes and accompanies hyperplasia and/or colitis following bacterial infection. Infect Immun 2012;80(2):753–67 doi 10.1128/IAI.06101-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murray PJ. Macrophage Polarization. Annu Rev Physiol 2017;79:541–66 doi 10.1146/annurev-physiol-022516-034339. [DOI] [PubMed] [Google Scholar]
- 30.Babic A, Schnure N, Neupane NP, Zaman MM, Rifai N, Welch MW, et al. Plasma inflammatory cytokines and survival of pancreatic cancer patients. Clin Transl Gastroenterol 2018;9(4):145 doi 10.1038/s41424-018-0008-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bazzichetto C, Conciatori F, Falcone I, Cognetti F, Milella M, Ciuffreda L. Advances in Tumor-Stroma Interactions: Emerging Role of Cytokine Network in Colorectal and Pancreatic Cancer. J Oncol 2019;2019:5373580 doi 10.1155/2019/5373580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qu X, Tang Y, Hua S. Immunological Approaches Towards Cancer and Inflammation: A Cross Talk. Front Immunol 2018;9:563 doi 10.3389/fimmu.2018.00563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neal JT, Li X, Zhu J, Giangarra V, Grzeskowiak CL, Ju J, et al. Organoid Modeling of the Tumor Immune Microenvironment. Cell 2018;175(7):1972–88 e16 doi 10.1016/j.cell.2018.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Courau T, Bonnereau J, Chicoteau J, Bottois H, Remark R, Assante Miranda L, et al. Cocultures of human colorectal tumor spheroids with immune cells reveal the therapeutic potential of MICA/B and NKG2A targeting for cancer treatment. J Immunother Cancer 2019;7(1):74 doi 10.1186/s40425-019-0553-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Foucher ED, Ghigo C, Chouaib S, Galon J, Iovanna J, Olive D. Pancreatic Ductal Adenocarcinoma: A Strong Imbalance of Good and Bad Immunological Cops in the Tumor Microenvironment. Front Immunol 2018;9:1044 doi 10.3389/fimmu.2018.01044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kawakami K, Kawakami M, Husain SR, Puri RK. Targeting interleukin-4 receptors for effective pancreatic cancer therapy. Cancer Res 2002;62(13):3575–80. [PubMed] [Google Scholar]
- 37.Li X, Yao W, Yuan Y, Chen P, Li B, Li J, et al. Targeting of tumour-infiltrating macrophages via CCL2/CCR2 signalling as a therapeutic strategy against hepatocellular carcinoma. Gut 2017;66(1):157–67 doi 10.1136/gutjnl-2015-310514. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.