

Abstract

Anisotropic magnetic nanoparticles with a mesoporous silica shell have the combined merits of a magnetic core and a robust shell. Preparation of magnetically guidable core–shell nanostructures with a robust silica shell that contains well-defined, large, radially aligned silica pores is challenging, and hence this has rarely been described in detail. Herein, a dynamic soft-templating strategy is developed to controllably synthesize hierarchical, dual-mesoporous silica shells on diverse core nanoparticles, in terms of nanoparticle shape (i.e., spherical, chainlike, and disclike), magnetic properties (i.e., hard magnetic and superparamagnetic), and dimensions (i.e., from 3 nm to submicrometers). The developed interfacial coassembly method allows easy design of applicable silica shells containing tunable pore geometries with pore sizes ranging from below 5 nm to above 40 nm, with a specific surface area of 577 m2 g–1 and pore volume of 1.817 cm3 g–1. These are the highest values reported for magnetically guidable anisotropic nanoparticles. The versatility of the method is shown by transfer of the coating procedure to core particles as diverse as spherical superparamagnetic nanoparticles and their clusters as well as by ferromagnetic 3 nm thick hexaferrite nanoplatelets. This method can serve as a general approach for the fabrication of well-designed mesoporous silica coatings on a wide variety of core nanoparticles.
Keywords: interface assembly, iron oxide core nanoparticles, magnetic hexaferrites, mesoporous silica shell, radially aligned pores
Introduction
Highly organized and well-defined mesoporous materials are desired support for applications as diverse as those ranging from catalysis, energy harvesting, and water and soil remediation to medical applications such as targeted drug delivery.1−3 Frequently, mesoporous materials have been designed to carry large amounts (by weight) of functional molecules, ions, or catalysts on the basis of their inherent high surface area and large pore size and volume.4 Among the different porous materials available, such as zeolites, metal–organic frameworks, and nanocarbons, mesoporous silica-based nanoarchitectures represent the first choice for many applications. This is due to their well-accepted biocompatibility, chemical stability, easy surface functionalization, cost effectiveness, and straightforward synthesis scale-up.5 Mesoporous silica is usually synthesized by surfactant-supported soft-template methods that involve micelles and nanoscale droplets of organic solvents in an emulsion reaction system.6−9 Addition of the organic solvent into this reaction mixture greatly increases the pore size formed, as it promotes swelling of the soft templates and therefore represents a pore expander.10 In general, the addition of an organic solvent facilitates the formation of large pores (diameter > 10 nm), which can include trimethylbenzene, n-hexane and other long chain hydrocarbons, or block copolymers such as poly(alkylene oxide) triblock copolymers.11 In contrast, reaction systems without added pore expanders generate pores typically with a diameter of <5 nm.11 The formation of the mesoporous silica structure is governed by two major processes: self-assembly of supramolecular templates and hydrolysis of tetraethyl orthosilicate (TEOS) to silicic acid and its further condensation to silica.12 The reaction kinetics are controlled by the addition of the catalyst to promote TEOS hydrolysis and deposition of silica on the supramolecular template surfaces. As an alternative to soft templating methods, hard templating, combined soft and hard templating (i.e., multiple templating), and postsynthetic treatments, such as etching, can be used for preparation of mesoporous silica.2,13,14 However, the alternative strategies lack the simplicity and overall effectiveness of the soft templating methods. Indeed, hard templating is limited to preformed templates, and removal of the solid colloids is usually very challenging and time-consuming. Instead, the combined soft and hard templating methods allow the formation of advantageous architectures with hierarchical porosity, although they do not overcome the limitations related to hard templating. Postsynthetic treatments can be used to enlarge the pore diameters of materials that primarily form narrow pores. This, however, has few disadvantages because of the additional reaction steps needed and the low uniformity of the pores formed in the treated material. In general, centro-radial or stellate silica pores are highly advantageous, as they offer an accessible large pore compartment that can carry large therapeutic molecules, or they allow the rapid access of reactants into catalyst-decorated pores as well as the release of the products of the catalyzed reactions out of the pores to the reaction liquid.11,15
The scientific community is actively seeking versatile solutions for nanocomposite synthesis that combine advantageous properties such as high magnetic responsiveness of the core material and the cargo capabilities of the silica shell with preferentially, radially aligned mesopores.16−19 Magnetic nanocomposites can also be heated remotely by exploitation of magnetic hyperthermia to selectively heat only the attached catalyst that is close to the core nanoparticle hot spots, while the bulk of the reaction mixture remains unheated to avoid undesired reactions taking place in the reaction mixture during heterogeneous catalysis.20,21 Moreover, the shape anisotropy of core magnetic nanoparticles represents a novel means for controlled and remote cargo release.22,23 This feature can be exploited when nanoparticles are exposed to a low-frequency rotating magnetic field, as anisotropic magnetic nanoparticles (i.e., nanochains and nanoplatelets) can follow this rotational movement remotely and hence accelerate the transport of chemicals from the pores. A similar strategy was demonstrated by Wan et al., where an osteoclast inhibitor, zolendronate, was loaded into magnetic nanochains, followed by its exposure to a rotating magnetic field.24 The nanochains followed the rotational movement and provided a high degree of osteoclast inhibition compared to nonrotating zolendronate-loaded nanochains. This study also represents an elegant interfacial coassembly approach for the synthesis of radially aligned silica pores with a mean diameter of 7.3 nm on the surface of anisotropic core particles. The same group also demonstrated a coassembly method combined with a pore-expanding solvent where the pore size of the mesoporous silica shell expanded up to 18 nm.25
There is also a large literature on the synthesis of mesoporous silica nanoparticles, while reports on the synthesis of mesoporous silica shells with large radially aligned pores on core particles (diameter > 30 nm) are relatively scarce. To the best of our knowledge, the largest mean diameter of radially aligned pores achieved by simple soft templating is 30 nm, which was reported recently by Kwon et al.26 However, the largest pores achieved by using high-molecular-weight amphiphilic block copolymers as pore expanders were ∼36 nm in diameter, while the pore morphology was raspberry-like rather than stellate.27 In general, it is broadly accepted that cetyltrimethylammonium bromide (CTAB)-based systems allow the formation of pores that range in size from 4 to 7 nm, while addition of a pore expander increases this a little, to pores of 8 nm in diameter.7 Moreover, the current approaches for the synthesis of uniform layers of radially aligned large silica pores on the surfaces of core nanoparticles frequently form a mixed product that includes undesired extra silica nanoparticles.28 The formation of silica nanoparticles as a synthesis byproduct is related to the challenges faced by an incomplete understanding of the mechanisms of TEOS hydrolysis and silica nucleation and condensation in the presence of soft templates, which results in poor control of the relevant synthesis parameters. An in-depth understanding of the crucial synthesis parameters, and hence of the mechanisms involved to obtain a uniform layer of radially oriented large silica pores on the surface of core nanoparticles, will therefore be beneficial for the broader scientific community.
Here, a simple synthesis method is presented to obtain well-defined, dual-mesoporous, and radially aligned silica pores with diameters >40 nm. We believe that the silica shells of these magnetically guidable nanoparticles have the greatest diameter reported to date for such radially aligned silica pores (Figure 1). This study reveals the effects of each of the individual synthesis parameters on growth of these radially aligned silica pores on the surface of the magnetic nanochains, including for surfactant concentration, type and amount of catalyst and organic solvent, amount of silica precursor, reaction temperature and time, and stirring intensity. This synthesis protocol allows fine-tuning of the porosity of the silica layer, its thickness, and its specific surface area as well as the orientation of the pores, and it avoids the formation of a mixed product that contains undesired extra silica nanoparticles. Furthermore, the magnetic responsiveness of the nanochains is well preserved, which allows effective magnetic guidance that is suitable for a number of diverse applications in technology and medicine. Moreover, the versatility of the method is confirmed by its use with diverse core nanoparticles in terms of their magnetic properties (i.e., superparamagnetic and ferromagnetic), size (i.e., from a few nanometers to a submicrometer scale), and shape geometries (i.e., spherical, chainlike, and disclike). Finally, diverse core nanoparticles are successfully decorated with a shell with radially aligned silica pores, including 10 nm spherical nanoparticles, 92 nm superparamagnetic nanoparticles, and 3 nm thick ferromagnetic hexaferrite nanoplatelets (mean diameter 50 nm) (Figure 1).
Figure 1.

(A–C) Schematic representations of the differently shaped core magnetic nanoparticles used in this study (A, C) and a single radially aligned pore (B) obtained by using nanoscale droplets of the organic solvent spatially confined as a dense cover on the surface of the core nanoparticles—an interface coassembly. (D) Corresponding representative transmission electron micrographs of the nanoparticles coated with silica with radially aligned pores. Scale bars: 50 nm.
Experimental Section
Materials
CTAB, citric acid, and aqueous ammonia solution (25%) were from VWR Int. GmbH (Vienna, Austria); cyclohexane, n-hexane, and toluene were from Merck KGaA (Darmstadt, Germany); triethylamine (TEA), triethanolamine (TEOA), 2-amino-2-(hydroxymethyl)propane-1,3-diol (TRIS), iron(III) sulfate, and TEOS were from Sigma-Aldrich (St. Louis, MO); sodium hydroxide (NaOH) was from Labochem Int. (Heidelberg, Germany); dibenzyl ether, iron(II) sulfate, barium nitrate, iron(III) nitrate nonahydrate, scandium(III) nitrate, and sodium dodecyl sulfate (SDS) were from Alfa Aesar (Kandel, Germany); and ethyl acetate, nitric acid (65%), and ethanol (absolute) were from Carlo Erba Reagents (Milan, Italy). All of the reactants and reagents were used without additional purification. Purified water was obtained from an Adrona E30 system (Adrona Ltd., Riga, Latvia). Superparamagnetic nanochains (iNANOvative|chains) and superparamagnetic nanoparticle clusters (iNANOvative|silica cr) were provided by Nanos SCI (Nanos Scientificae Ltd., Ljubljana, Slovenia).
Core Nanoparticles
To show the versatility of this method, different core nanomaterials were used. Superparamagnetic nanochains (estimated particle density, 4030 kg m–3) and superparamagnetic nanoparticle clusters (estimated particle density, 4260 kg m–3) were designed and synthesized by the company Nanos SCI. The synthesis protocols have been described in detail previously.29,30
The hexaferrite nanoplatelets were synthesized by using a hydrothermal method, as reported previously.31,32 Briefly, barium nitrate, iron(III) nitrate, and scandium(III) nitrate were dissolved in water (200 mL) to 125 mmol L–1 Ba2+, 562.5 mmol L–1 Fe3+, and 62.5 mmol L–1 Sc3+ and coprecipitated with NaOH (1.13 mol) dissolved in water (200 mL). The reaction mixture was then transferred to an autoclave (1 L; Inconel; Parr Instruments, Moline, IL), and the autoclave was sealed and heated to 240 °C at a heating rate of 3 °C min–1. The autoclave was allowed to cool to room temperature; the nanoplatelets formed were washed several times with water and 0.1 mol L–1 nitric acid and then dispersed in water (288 mL). The nanoplatelets were further stabilized with citric acid. Here, 0.5 g mL–1 aqueous citric acid (12 mL) was added to the water suspension of nanoplatelets. The pH was increased to 5.1 by using aqueous ammonia solution (∼25%). The reaction mixture was heated to 80 °C and vigorously stirred (∼750 rpm) for 90 min. The reaction mixture was then allowed to cool to room temperature, and the pH was increased further to 10.1 by using aqueous ammonia solution (∼25%), followed by centrifugation at 3000g for 5 min, to remove the excess citric acid. The nanoplatelets collected were redispersed with dilute aqueous ammonia solution (30 mL) at pH 10.1. The citric acid-stabilized barium hexaferrite nanoplatelets were coated with a 2 nm thick layer of nonporous silica by using a modified Stöber reaction. The citric acid-stabilized nanoplatelets were dispersed in a mixture of absolute ethanol (960 mL), water (210 mL), and TEOS (1.8 mL). The coating reaction was initiated by addition of aqueous ammonia solution (∼25%, 9 mL), and the reaction mixture was stirred at room temperature overnight. Finally, the silica-coated nanoplatelets were washed several times with water.
The superparamagnetic iron oxide nanoparticles (SPIONs) were prepared by coprecipitation of Fe2+ and Fe3+ ions from aqueous solution, as reported previously.33 Briefly, iron(II) and iron(III) sulfates were dissolved to final concentrations of 27 mmol L–1 Fe2+ and 14 mmol L–1 Fe3+. Precipitation was initiated by using aqueous ammonia (∼25%) in two steps.33 First, the pH was adjusted to 3 and kept constant for 30 min. Then, the pH was increased to 11.6. After an additional 30 min, the nanoparticles formed were collected by using a NdFeB magnet (Supermagnete, Webcraft GmbH, Gottmadingen, Germany), washed three times with diluted aqueous ammonia solution at pH 10.5, and then finally dispersed in water (120 mL). The washed nanoparticles were further stabilized with citric acid. Here, 0.5 g mL–1 citric acid aqueous solution (5 mL) was added to the suspension of nanoparticles in water (120 mL), and the pH was adjusted to 5.2 with aqueous ammonia solution (∼25%). The reaction mixture was stirred in an oil bath at 80 °C for 90 min. The pH was then raised to 10.2 with aqueous ammonia solution (∼25%). Next, the citric acid-stabilized SPIONs were coated with 2 nm thick nonporous silica.33 The pH of the SPION suspension was adjusted to 11 with aqueous ammonia solution (∼25%), followed by addition of TEOS (2.5 mL) dissolved in absolute ethanol (25 mL). The reaction mixture was stirred at room temperature overnight. Finally, the nanoparticles were washed several times with water and then dispersed in water.
General Procedure: Synthesis of Mesoporous Silica on Core Nanoparticles
A general procedure was developed for synthesis of the mesoporous silica shell on all three types of core nanoparticles used in this study. The supportive reactions were conducted to understand how each individual reaction parameter influences the final properties and morphology of the silica shells formed. These were based on changes to the individual reaction parameters of the general procedure, such as surfactant, catalyst, organic solvent, amount of silica precursor, reaction temperature, reaction time, and stirring intensity. These were examined separately, one at a time, while the other reaction settings were kept constant according to the general procedure.
In the general procedure, the CTAB surfactant (14.1 mmol) was dissolved in water (45 mL) in a 250 mL flat-bottomed flask. When studying the effects of surfactant on the silica shell properties, the CTAB was increased by 2-fold (28.2 mmol) and 3-fold (42.3 mmol) and reduced by a factor of 20 (0.71 mmol). An equimolar amount (14.1 mmol) of SDS surfactant was also studied to determine the effects of oppositely charged surfactants. Complete surfactant dissolution was achieved in all cases except when CTAB was used at 3-fold. Then, the core nanoparticles (60 mg) were dispersed by the surfactant solution. After 30 s of sonication (Sonis 4; Iskra Pio, Slovenia) the flask was placed in an oil bath at the set temperature of 65 °C, as used for the general procedure. The other studied temperatures were room temperature, 40 °C, 75 °C, and 90 °C. Then, the base was added as the reaction catalyst. In the general procedure, TEOA (0.9 mmol) was used as the catalyst. To determine the effects of the amount of catalyst, the TEOA added was increased by 3-fold (2.7 mmol) and 6-fold (5.4 mmol) and reduced by a factor of 3 (0.3 mmol) compared to TEOA in the general procedure. Moreover, a series of different catalysts were studied, as TRIS, TEA, aqueous ammonia solution (∼25%), and NaOH, which were used in equimolar amounts to the TEOA used in the general procedure (TEOA, 0.9 mmol). Also, the silica precursor (i.e., TEOS) was dissolved in an organic solvent and added to the reaction mixture in two equal additions over 90 min. In the general procedure, TEOS (1.5 mmol) was dissolved in cyclohexane (12 mL), and the reaction was initiated once the TEOS dissolved in the organic solvent was added into the reaction mixture. Where the volume of organic solvent was studied, the amount of TEOS was unchanged (1.5 mmol), while the volume of cyclohexane was increased by 3-fold (36 mL) and 6-fold (72 mL) and reduced by a factor of 6 (2 mL) compared to the volume of cyclohexane in the general procedure. Additionally, cyclohexane was replaced by different types of organic solvents, as n-hexane, toluene, dibenzyl ether, and ethyl acetate. n-Hexane, toluene, and dibenzyl ether were studied at the same volume as cyclohexane in the general procedure (12 mL). Ethyl acetate was also studied with the volume reduced by a factor of 6 (2 mL) compared to the volume of cyclohexane in the general procedure. Where the amount of silica precursor was studied, the volume of cyclohexane used in the general procedure was unchanged (12 mL), while the TEOS was increased by 3-fold (4.5 mmol) and 9-fold (13.5 mmol) and reduced by a factor of 3 (0.5 mmol) compared to the TEOS in the general procedure. For all of the experiments, the reaction mixtures were stirred at 600 rpm overnight, with the exception of stirring at 300 and 900 rpm when the effects of the mixing rate were examined. Once the reactions were complete, the core–shell nanoparticles were collected from the suspension by using a NdFeB magnet (Supermagnete, Webcraft GmbH, Gottmadingen, Germany) and washed with ethanol, acetone, and distilled water.
Characterization of Nanoparticle Suspensions
Sample micrographs were obtained by using transmission electron microscopy (TEM; JEM 2100; JEOL, Akishima, Japan) at 200 kV. The nanoparticle suspensions were deposited on copper TEM grids coated with carbon (mesh, 200; SPI Supplies, West Chester, PA) and air-dried. The mesoporous silica coating thickness was determined by analysis of the TEM micrographs using the DigitalMicrograph software (Gatan, Plaesanton, CA) and an image analyzer (Fiji ImageJ).34 Gas adsorption–desorption analysis was performed with nitrogen gas using a surface area and pore size analyzer (NOVA 2000e; Quantachrome Instruments, Boynton Beach, FL) to determine the pore size and size distribution, the total pore volume (measured at P/P0 = 0.99), and the specific surface area (determined by the Brunauer–Emmett–Teller method). The mean pore sizes were calculated from the Barrett–Joyner–Halenda theoretical model (addition of ca. 0.7 nm for the Broekhoff and de Boer or the density functional theory equivalents). Of note, the Barrett–Joyner–Halenda method is good for large pore quantification but can underestimate the sizes of small pores, and so this needs to be taken into account because these mesoporous core–shell nanomaterials have a dual-mesoporous structure. The nanomaterials were washed with acetone and ethanol to remove the residual surfactants from the silica pores and then freeze-dried under vacuum before the gas adsorption–desorption analysis. The hydrodynamic particle size was determined by dynamic light scattering (DLS) (Analysette DynaSizer; Fritsch GmbH, Weimar, Germany). Zeta-potential measurements were performed by using a particle size analyzer (Litesizer 500; Anton Paar, Graz, Austria). Magnetic measurements were performed at room temperature on a vibrating-sample magnetometer (7307 VSM; Lake Shore Cryotronics, Westerville, OH), with a maximum field of 10 kOe.
Results and Discussion
The present study was designed and developed by using nanochains as the core particles, which are commercially available, magnetically guidable, and anisotropic by shape and have superparamagnetic properties. The nanochains were synthesized by magnetic assembly where a few spherical nanocrystal clusters (i.e., 6 ± 3) with mean diameter of 92 ± 16 nm were aligned in a magnetic field and fixed with a 5 nm thick silica shell to form nanochains of submicrometer length (Figure 2A,B).35 The individual nanocrystal clusters were first self-assembled with ∼60 superparamagnetic iron oxide nanocrystals (diameter 10.4 ± 1.6 nm) that were the primary building blocks for the assembled nanochains. The nanochains must be superparamagnetic because their processing demands good colloidal stability of their aqueous suspensions. The assembly of a large number of magnetic nanocrystals into single entities increases the particle magnetic volume, which results in effective translational movement of the nanochains in a magnetic field gradient (Supporting Information, Figure S1). The nanochains had a high saturation magnetization MS of ∼48 emu g–1 and were therefore ideal candidates for in-depth studies of the relevant synthesis parameters used to obtain dual-mesoporous silica shells with radially aligned pores. This is because of their high magnetic responsiveness, which allows rapid magnetic separation and therefore easy purification after synthesis. These nanochains are anisotropic, and so once they are exposed to a low-frequency rotating magnetic field, they behave as tiny “nanomixers” because they can follow the rotational field movement remotely. This magnetic responsiveness is a prerequisite for the development of advanced drug delivery and catalytic systems where substantial amounts of active compounds, such as drugs or catalysts, can be incorporated inside well-organized and radially aligned silica pores. Large radially aligned pores (diameter > 15 nm) on magnetic nanochains are especially attractive for these applications.
Figure 2.

(A–D) Representative transmission electron microscopy micrographs of magnetic nanochains before the coating with mesoporous silica (A, B) and when coated with silica (using the general procedure) (C, D), showing the radially aligned pores. Scale bars: 100 nm. (E, F) Representative pore-size distribution (E) and the nitrogen adsorption–desorption isotherms of the corresponding sample (F).
The general procedure yielded silica shells with radially aligned (stellate) silica pores on the surface of the nanochains, with an ∼95 nm thick shell and dual mean silica pore diameters of 3.4 and 17 nm, with no extra silica nanoparticles formed (Figure 2C,D). The central sample produced by following the optimized procedure is indicated as prepared by the general procedure that is described in detail in the Experimental Section. The further investigation of the relevant synthesis parameters was based on the general procedure developed as the starting point for evaluation of the effects of each individual parameter on the silica shell morphology.
The approach for the formation of the silica shells with defined pores on the surface of the core particles was based on the well-accepted soft-template method.11 An interfacial coassembly of soft-template components, such as surfactant-based micelles and small nanoscale droplets of the organic solvent, has a crucial role in the heterogeneous deposition and condensation of the silica precursors on the surface of the core particles (Figure 3).
Figure 3.
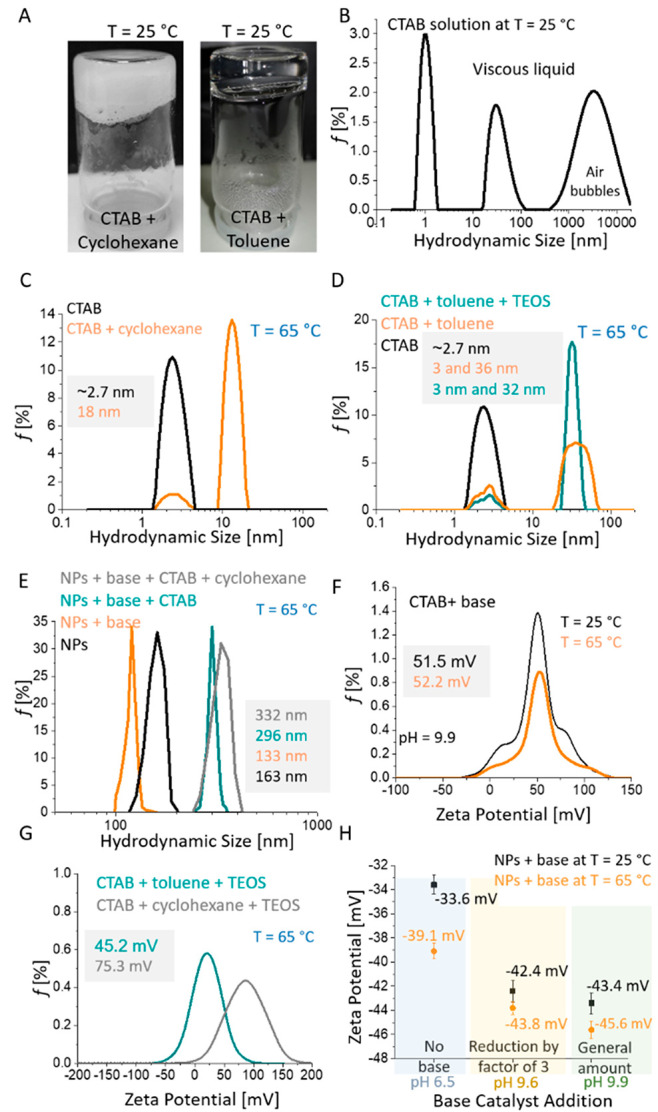
(A) Photographs of CTAB plus cyclohexane and CTAB plus toluene gel-like formations performed spontaneously at 25 °C. (B–E) Hydrodynamic size distributions of the combinations of the different components of the reaction mixture at the concentrations of the general procedure. (B) Micellar structures in aqueous CTAB solution at 25 °C. (C, D) Aqueous CTAB solution ± cyclohexane (C) and ± toluene and ± tetraethyl orthosilicate (TEOS) (D) at 65 °C. (E) Aqueous nanoparticle cluster suspensions ± base catalyst ± CTAB and ± cyclohexane at 65 °C. (F, G) Zeta-potential distributions of the different reaction mixture components. (F) Effects of temperature (25 and 65 °C) on aqueous CTAB solution plus reaction catalyst at the concentration of the general procedure. (G) Effects of organic solvent additions (cyclohexane and toluene) on aqueous CTAB solution at 65 °C. (H) Effects of specific base catalyst additions on zeta-potential of 92 nm nanoparticle clusters (NPs) at 25 and 65 °C.
Dynamic light scattering revealed the presence of particularly small template components (∼3 nm) in reaction mixtures containing either an aqueous solution of CTAB surfactant or an emulsion of CTAB solution and organic solvent at 65 °C (reaction temperature of the general procedure) (Figure 3C,D). The nanoscale colloid size of ∼3 nm corresponds to CTAB micelles that are the smallest soft-template components and the crucial element for formation of the silica shell with dual porosity. Interestingly, addition of the organic solvents into the aqueous CTAB solution formed gel-like structures at 25 °C, which made DLS analysis impossible and silica-coating reactions particularly difficult (Figure 3A and Figure S8A). Furthermore, the DLS suggested the need to use the optimal reaction temperature, not only because of gelling at 25 °C but also because colloids with hydrodynamic sizes from ∼20 to ∼100 nm, and also >1 μm, were detected in the aqueous CTAB solution at 25 °C (Figure 3B). However, only a single monodispersed population of micelles with mean hydrodynamic sizes of ∼3 nm was detected at 65 °C (Figure 3C,D). As the CTAB solution was relatively viscous at 25 °C, we assumed that these large objects that were detected were air droplets formed by the intensive stirring (at the same speed as for the reaction mixture) before the DLS measurements were performed (Figure 3B). High surfactant concentrations that exceed the critical micellar concentration by over 300-fold result in the formation of numerous micelles and also in the emulsifying of the organic solvent into relatively uniform nanoscale droplets at 65 °C (Figure 3C,D).36 The hydrodynamic sizes of the nanoscale droplets (i.e., reaction mixture without nanoparticles) were from 10 to 25 nm when cyclohexane was used as organic solvent, while toluene resulted in the formation of larger nanodroplets, with sizes from 15 to 60 nm at the set temperature of 65 °C. Addition of TEOS to the cyclohexane solvent resulted in very monodispersed nanodroplets of the organic phase, with a smaller mean hydrodynamic size of 32 nm compared to the emulsion system with cyclohexane only, where the mean size was 36 nm (Figure 3D). A mixture of TEOS and cyclohexane resulted in smaller droplets because the TEOS is prone to rapid hydrolysis at the droplet interface, which then affects the droplet surface energy and hence stabilizes smaller droplets. Furthermore, DLS measurements were also performed for spherical 92 nm nanoparticle clusters in different reaction mixture compositions, but not for chainlike or disclike particles, because DLS can only be used reliably for measurements of spherical nanoparticles (Figure 3E). The hydrodynamic size distributions of the aqueous spherical nanoparticle cluster suspensions were significantly affected by the addition of the different reaction mixture components (i.e., base catalyst, CTAB, and cyclohexane) (Figure 3E). The suspension pH was increased from 6.5 to 9.9 when the general amount of the base catalyst TEOA was added to the suspension, and this pH change increased the absolute value of the zeta potential from −39.1 to −45.6 mV at the reaction temperature of 65 °C (Figure 3H). Improved colloidal stability of the nanoparticles at high pH (9.9) compared to low pH (6.5) resulted in decreased mean hydrodynamic size, from 163 to 133 nm (Figure 3E). The reaction mixtures composed of nanoparticle suspensions and CTAB, or cyclohexane and CTAB, increased the mean hydrodynamic sizes of the nanoparticles to 296 and 332 nm, respectively. CTAB micelles and nanoscale droplets of the organic phase had positive zeta-potentials of above ∼45 mV (Figure 3F,G), and they were therefore strongly associated with negatively charged nanoparticles (−45.6 mV) at 65 °C (general procedure reaction temperature) (Figure 3H). This strong contact interaction between positively charged soft-template components and the negatively charged nanoparticle surface is the reason for the increased nanoparticles hydrodynamic size. Indeed, the zeta-potential of cyclohexane-based soft-templates (75.3 mV) was significantly higher compared to that of toluene-based soft templates (45.2 mV), which suggested stronger contact interactions between positively charged soft-template components and negatively charged nanoparticles for the cyclohexane-based system (Figure 3G). Furthermore, the outermost surfaces of the micelles and nanoscale droplets were composed of identical surfactant polar heads, which provided sufficient electrostatic repulsion between the soft-template components and therefore resulted in good colloidal stability of the reaction system. The nature of the surfactant polar head is crucial to achieve the attractive contact interaction between the surface of the core particles and the soft-template components and to be able to deposit the silica precursors on the surface of the micelles and nanoscale droplets. The soft-template components need to form attractive electrostatic interactions with the primary, thin, and nonporous silica surface of core particles, which can be achieved as the positive polar heads of the surfactant provides positively charged micelles and nanoscale droplets (Figure 3F,G). The strong affinity between the soft-template components and the silica precursors originates from the zeta-potential of the silica, which is negative at the alkaline pH at which the deposition of silica oligomers usually takes place.37 For example, replacing positively charged CTAB with equimolar amounts of negatively charged SDS while retaining the other reaction conditions of the general procedure showed no changes and did not lead to the formation of additional silica shells on the nanochain surfaces (Figure S2).
The pore size distribution from the gas adsorption–desorption analysis showed two peaks, which confirmed a bimodal distribution that most likely originated from the small CTAB micelles and the larger nanoscale droplets of the organic solvent, with mean pore diameters of 3.4 ± 0.4 and 17 ± 5 nm, respectively (Figure 2E). The silica obtained by using CTAB without an organic solvent was compact, which suggested a requirement for organic solvents for the formation of very large mesopores (Figure 4E). It was anticipated that thin silica walls, which support radially aligned silica pore channels, contain small and uniform holes (∼3–4 nm) that are generated by uniform micelles, while large radially aligned pores are obtained by silica precursor deposition onto the surface of the nanoscale droplets of the organic solvent (Figure 1B). This is in agreement with the hydrodynamic sizes of the nanoscale droplets of the organic solvent cyclohexane, which were between 10 and 25 nm. We believe that the silica pore growth direction is limited to the perpendicular according to the core particle surface because other growth directions are spatially restricted by the dense cover of the electrostatically attached nanoscale droplets on the surface of the core particles (Figure 1B). A relatively broad pore size distribution linked to the large nanoscale droplets of the organic solvent corresponds to a conical shape of the silica pores. In addition, the nitrogen adsorption–desorption isotherm of the sample produced by following the general procedure showed a typical type IV curve with a narrow capillary condensation step at a relative pressure of ∼0.65 to 0.85, which indicated cylindrical pores (Figure 2F). Data extraction from the adsorption branch indicated that the samples produced by following the general procedure had a high mean specific surface area of 577 m2 g–1 and a total pore volume of 1.817 cm3 g–1, which are the highest reported values for anisotropic magnetically guidable core–shell nanomaterials.7,26
Figure 4.

Representative transmission electron microscopy micrographs of the core–shell magnetic nanochains with diverse silica-shell morphologies that resulted from the organic solvents that were used as pore expanders. (A–D) Changes in the volume of cyclohexane from the general procedure, where raspberry-like mesoporous silica was synthesized with one-sixth the cyclohexane (A), while radially aligned silica pores were synthesized in the general procedure (B) and with 3-fold (C) and 6-fold (D) the cyclohexane volumes. (E) A dense silica shell was obtained when no organic solvent was used. (F–H) Core–shell nanochains with radially aligned pores were prepared by replacing the cyclohexane of the general procedure with the same volume of n-hexane (F), dibenzyl ether (G), and toluene (H). Scale bars: 100 nm.
Surfactant as a Soft-Template Agent
Surfactants are the key element in the synthesis of mesoporous silica because they form supramolecular soft-template components, such as the micelles and small droplets of organic solvents that are crucial for pore structure organization. Heterogeneously deposited silica embraces the soft-template components and permanently preserves the mesoporous structure, while the soft-template components can be removed easily once the deposited silica is solidified and therefore self-supporting. The concentration of the surfactant has an important role for efficient template assembly, which is a prerequisite for the formation of well-defined mesoporous silica and to avoid the formation of irregular supramolecular structures.38 The CTAB concentration that was 2-fold that used for the general procedure resulted in the formation of undesired supramolecular assemblies, which supported the silica deposition and hence the formation of planar silica sheets with holes (Figure S3). Indeed, such a relatively high CTAB concentration increased the viscosity of the reaction mixture, which led to ineffective mixing and therefore poor transport of the reagents. With the CTAB concentration reduced by a factor of 3 from the general procedure, this resulted in mixed products that contained well-formed radially aligned silica pores on nanochains as well as undesired formless silica deposits (Figure S4). Furthermore, no silica was deposited on the nanochains when the CTAB concentration was reduced by a factor of 20. When the positively charged surfactant CTAB was replaced with the negatively charged SDS, there was no formation of additional silica shells on the nanochain surfaces due to the repulsive electrostatic interactions among the negatively charged species of soft-template components, the silica precursors, and nanochains surfaces (Figure S2).39−41
Silica Source
Tetraethyl orthosilicate was used as the silica precursor across all of the experiments. The silica shell thickness was proportional to the amount of silica precursor when the TEOS concentration was reduced from the general procedure. For example, when the TEOS concentration was increased by 3-fold, there was formation of undesired silica deposits (Figure S5 and Table S1). Moreover, the 9-fold increase in TEOS concentration resulted in less of a decrease in the silica shell thickness compared to that for the reduction of the TEOS concentration by a factor of 3, while there was also a substantial fraction of monodispersed mesoporous silica nanoparticles with mean diameter of ∼60 nm (Figure S6 and Table S1). These data can be explained by the instantaneous formation of a large number of small silica nuclei that grew to mature silica nanoparticles by consuming a substantial part of the hydrolyzed silica precursors that would otherwise be used for synthesis of thicker silica shells.28,42,43 Additionally, the formation of undesired extra silica nanoparticles with TEOS concentration 3-fold the general procedure was reduced from ∼5% to ∼2% by an increase in the stirring rate from 600 to 900 rpm, which enhances the transport of the silica precursors in the viscous reaction mixture (Table S1). Interestingly, with the 9-fold increase in the TEOS concentration with the other reaction conditions unchanged, there was a decrease in the pore size. This effect was gradual and ranged from 18 nm with the TEOS concentration reduced by a factor of 3 to 12 nm for the 9-fold TEOS concentration (compared to the general procedure; Table S1). Hence, it appears that the presence of TEOS in cyclohexane affected the soft-template droplet size, which then corresponds to the silica pore size (Table S1). Indeed, although TEOS is fully miscible with cyclohexane, it is significantly less hydrophobic, which most probably affects the formation of the smaller droplets of the inner emulsion phase, therefore producing a smaller pore size.
Organic Solvent as a Pore Expander
The organic solvent is a crucial component of the reaction mixture as it affects the mean diameter of the silica pores; thus, it can be used as a pore expander. The organic solvent that contained TEOS represents the inner phase of the emulsion system, where these nanoscale droplets associate with the surface of the oppositely charged external particles in the water phase. This process is known as interfacial coassembly because the nanoscale droplets of the organic solvent occupy the core particle surfaces electrostatically, while the silica is then deposited on the interface between the organic phase and the outer aqueous phase. The data here show that both the volume and type of organic solvents have important roles in the formation of diverse silica pores (Table S2). Cyclohexane is recognized as an efficient pore expander, and different volumes were initially examined for the preparation of silica shells with mesopores (Figure 4A–D). A 3-fold increase in the cyclohexane volume, compared to the general procedure, resulted in slightly thinner silica shells, with a thickness of 83 nm and a mean pore size of 14 nm (Figure 4C). Then, a 6-fold increase in the cyclohexane volume resulted in even thinner silica shells, of 72 nm, while the mean pore size was greatly expanded, to 40 nm (Figure 4D). Reduced amounts of cyclohexane, such as by a factor of 6 from the general procedure volume, resulted in raspberry-like, 5 nm thick silica pores, with a silica shell thickness of ∼25 nm (Figure 4A). Moreover, the complete absence of the solvent resulted in a compact silica shell of 25 nm with negligible porosity (Figure 4E). This provides direct evidence to confirm that a minimal volume of an organic solvent is required for effective pore expansion. Both 3-fold and 6-fold increases in the cyclohexane volume compared to the general procedure showed a small extent of the silica precursors in the form of extra silica deposits (Figure S7). It appears that very large volumes of the organic phase form droplets that are too large, and these are temporarily not stable enough for interfacial coassembly to take place.
The effects of different organic solvents on the morphology of the mesoporous silica were also examined (Figure 4F–H). n-Hexane resulted in formation of thin silica shells (thickness ∼ 40 nm) and smaller pores (diameter ∼ 14 nm) (Figure 4F) compared to cyclohexane in the general procedure (Figure 4B). The largest molecular size used as a pore expander was dibenzyl ether (Figure 4G). Here, the pores showed a diameter of 18 nm, which was not significantly different than those obtained by using the same volume of cyclohexane (Figure 4B and Table S2). However, although toluene is a smaller molecule than dibenzyl ether, it resulted in formation of ∼90 nm thick shells and the largest radially aligned pores seen in this study with a diameter of ∼41 nm (Figure 4H and Table S2). We can therefore conclude that there are no clear relationships between the molecular size of the solvent and the silica pore size. Also, as demonstrated by Shen et al., solvent hydrophobicity is an important feature because it dictates the rapid diffusion of the hydrolyzed silica precursor onto the nanoscale droplet interface, thereby stabilizing the smaller pores.44 The present study confirms this finding because the smallest pores were seen by using dibenzyl ether, which was the most nonpolar of these pore expanders. Interestingly, ethyl acetate has also been used, although the nanochain colloidal stability was disrupted because ethyl acetate is partially miscible with the water phase. As a result of this instability, there was no formation of mesoporous silica. To ensure better colloidal stability of the nanochains, the volume of ethyl acetate was also reduced by a factor of 6 from the general procedure with cyclohexane. However, the nanochains remained stable, and the silica obtained was compact and hence similar to that obtained by reduction of the volume of cyclohexane by a factor of 6, and also to the system without organic solvent added, which indicated the weak pore expanding behavior of ethyl acetate.
A Base as a Reaction Catalyst
The base used as the catalyst for TEOS hydrolysis and nucleation represents another reaction system component that can affect the final silica morphology. The kinetics of TEOS hydrolysis directly govern the evolution rate of the silica precursor in the reaction system, which is then deposited either as a silica shell on the surface of the core particles or as undesired extra silica nanoparticles or 2D silica sheetlike deposits (Figures S5 and S6). We hypothesized that the stronger base accelerated TEOS hydrolysis, which rapidly generated numerous small silica nuclei that led to undesired extra silica nanoparticles. As a result of this extra silica consumption, only part of the silica precursors produced was used for silica shell growth on the core particles, and therefore a thinner silica shell was formed. To confirm this hypothesis, the effects of different strengths (i.e., alkalinities) of five bases were studied in terms of the mesoporous silica morphology, with all of these bases used at the same concentration (Figure 5). The bases used were as follows: TEOA (pKa, 7.76), TRIS (pKa, 8.07), ammonia solution (pKa, 9.25), TEA (pKa, 10.78), and NaOH (completely dissociated).45 Here, the silica shell thickness decreased gradually from 95 to 20 nm with the use of bases with increased alkalinity (Table S3). A compact, 20 nm thick, nonporous silica shell was formed when NaOH was used as catalyst, which represented the strongest base in the series (Figure 5H). Substantial amounts of extra silica nanoparticles and silica deposits were generated with TEA and ammonia solution, which resulted in thinner silica shells, with pore diameters of 38 and 45 nm, respectively. On the other hand, both TEOA (Figure 5A–D) and TRIS (Figure 5E) favored the formation of mesoporous silica shells on the core nanochains with thicker silica shells for mean shell thicknesses of 95 and 86 nm, respectively. Interestingly, TEOA and TRIS resulted in well-defined silica coatings with similar structures, porosities, and thicknesses, which can be attributed to their relatively similar chemical structures and pKa values.
Figure 5.

Representative transmission electron microscopy micrographs of the core–shell magnetic nanochains with diverse silica shell morphologies that resulted from the bases used as reaction catalysts. (A–D) Changes in the amounts of TEOA added from the general procedure, where core–shell nanochains with radially aligned silica pores were synthesized with one-third the TEOA (A), with the general procedure with TEOA (B), and with 3-fold the TEOA (C). A 6-fold increase of the TEOA resulted in disordered pores with pore sizes of 31 nm (D). (E–H) Core–shell nanochains with radially aligned pores with pore sizes of 17, 21, and 19 nm were prepared by replacing TEOA with the same amounts of TRIS (E), ammonia solution (F), and TEA (G), respectively. A compact nonporous silica shell was formed when NaOH was used as the catalyst (H). Scale bars: 100 nm.
Furthermore, the effects of different amounts of TEOA on the mesoporous silica were examined. Increases in the TEOA above the general procedure led to a gradual decrease in the silica shell thickness (Table S4). This behavior was in general agreement with the trend observed by using a series of increased base strengths. Finally, larger amounts of TEOA generated more alkaline conditions, which was similar to replacing TEOA with a stronger base. The high rate of silica precursors generation with the 6-fold increase in TEOA led to less defined silica pores, although they showed a larger pore size of 31 nm (Figure 5D).
Effects of Temperature, Reaction Time, and Stirring Rate
As well as the reaction components discussed above, the reaction parameters are important for the fine-tuning of the mesoporous silica properties, such as temperature, reaction time, and stirring rate. The reaction temperature influences the rate of silica precursor hydrolysis and condensation, where both of these processes were accelerated by increased temperature. Moreover, the reaction mixture viscosity was decreased by increased temperature, which enhanced the mass transport of the reaction components as well as affecting the behavior of the soft-template components. However, the reaction temperature was limited by the solvent boiling point. We examined reaction temperatures from 20 to 90 °C, and the results are given in Table S5. It was determined experimentally that the reaction temperature of 65 °C resulted in the desired well-formed silica pores on the core particles, with no extra silica nanoparticles detected. The reaction at room temperature was far from optimal because the CTAB did not dissolve completely before the reaction took place, even though it was stirred overnight, and therefore undesired extra silica nanoparticles and poorly ordered compact silica shells were formed (Figure S8). Similarly, the reaction temperature of 40 °C had the same issues as those for room temperature. On the other hand, the reaction temperature of 75 °C, which is close to the boiling point of cyclohexane, generated moderate mixture foaming, where the silica formed had a similar thickness to and smaller pores than (∼10 nm) that prepared at 65 °C. A further reaction temperature increase to 90 °C resulted in excessive boiling and foaming, where the silica was formed was raspberry-like, with small pores (∼5 nm) (Figure S8E).
As the TEOS is admixed into the organic solvent, while the base is added into the outer aqueous phase, the formation of silica precursors is a relatively slow process that takes place only at the nanoscale droplet interface. Our synthesis product was analyzed 90 min after addition of the base when it was noted that the silica had just barely started to form on the surface of the core nanochains. Typically, the silica was condensed completely in 3 h if the base and TEOS were both added into the aqueous or the water/ethanol solutions.33 However, it appeared that there was an additional way for us to control the silica shell thickness, simply by immediate termination of the reaction. This can be achieved by rapid cooling of the reaction mixture and isolation of the particles from the mixture by magnetic separation.
Finally, the stirring intensity also has a role in the formation of mesoporous silica. Yue et al. studied the effects of different stirring rates on the structures of mesoporous silica shells, and they reported that pore size can be controlled by tuning the stirring intensity.46 They thus showed that stirring at 170 rpm generated pores of 5 nm, at 250 rpm generated pores of 9 nm, and at 500 rpm generated a bimodal size distribution with mean pore sizes of 8.8 and 18.8 nm. Interestingly, Yue et al. also detected rougher surfaces when the stirring rate was increased.46 Here, an intense stirring rate of 900 rpm was used to put extra energy into the relatively viscous reaction system, which might result in better transport of the reagents to reduce the formation of undesired silica deposits and silica nanoparticles. Although an intense stirring rate of 900 rpm diminished the formation of undesired silica nanoparticles when TEOS concentration was increased by 3-fold (Table S1), such a high stirring rate generated too intense foaming. When a stirring rate was reduced from 600 to 300 rpm, the reaction mixture in the general procedure appeared inhomogeneous due to visible separation of cyclohexane phase from the water phase. These changes in stirring rates showed no effect on the structures of mesoporous silica shells. Hence, for the general procedure, a stirring rate of 600 rpm was optimal to reduce foam generation, while the formation of extra silica nanoparticles was also completely avoided.
Versatility of the Method
The scientific community is urgently looking for robust and fully transferrable methods for mesoporous silica coatings on the surfaces of diverse core nanoparticles. Here, we have demonstrated the broad applicability of the general procedure to other nanomaterials with different magnetic properties, shapes, and dimensions (Figure 6A–C). First, the general procedure without any modifications was applied to spherical, superparamagnetic (saturation magnetization MS ∼ 64 emu g–1), and small (10.4 ± 1.6 nm) iron oxide nanoparticles (Figure S9). A silica shell with large radially aligned pores with silica thickness of ∼36 nm and mean diameter of ∼19 nm was produced. Furthermore, the versatility was also shown for spherical, superparamagnetic (saturation magnetization MS ∼ 54 emu g–1), iron oxide nanoparticle clusters (92 ± 16 nm) (Figure 6D–F), and highly anisotropic, platelet-like, ferromagnetic (coercivity HC ∼ 142 kA m–1; saturation magnetization MS ∼ 40 emu g–1), barium hexaferrite nanoplatelets (Figure 6G–I), with a thickness of 3.8 nm and a mean cross-sectional diameter of 52 ± 14 nm (Figure S9). The nanoparticle clusters and hexaferrite nanoplatelets used as the core nanoparticles had silica shells with radially aligned (stellate) pores with silica shell thicknesses of 90 and 55 nm, respectively, and mean pore sizes of 17 and 22 nm, respectively. Therefore, the good applicability of the silica-coating method is shown here for diverse core nanoparticles.
Figure 6.

Representative transmission electron microscopy micrographs with increasing magnification of three different types of core–shell magnetic nanoparticles with radially aligned silica pores: superparamagnetic iron oxide nanocrystals (A–C), superparamagnetic nanocrystal clusters (D–F), and hard-magnetic barium hexaferrite nanoplatelets (G–I), generated after the coating process by using the general procedure. Scale bars: 200 nm (A, D, G); 100 nm (B, E, H); 50 nm (C, F, I).
Finally, the mechanical stability of the silica shells on the core particles was estimated by exposure of the suspensions to strong, 1 s long ultrasound pulses (3 min; volume = 10 mL; amplitude = 40%; Sonics Vibra-Cell VC505) followed by overnight drying at 80 °C. The TEM analysis confirmed good stability of the silica coating, with its fully preserved mesoporous morphology (Figure S10).
Conclusions
In summary, we have developed here a versatile, simple, soft-template-based, and low-cost synthesis of hierarchical dual-mesoporous silica shells on diverse core nanoparticles with a specific set of applicable pore morphologies. Manipulation of the reaction parameters resulted in synthesis of a wide variety of highly desired silica-shell morphologies with hierarchical dual pore sizes from ∼3 to ∼40 nm, with centro-radial or stellate and raspberry-like geometries. This study reveals the effects of each individual synthesis parameter on the growth of the mesoporous silica shells on the surface of magnetic nanochains, in terms of surfactant concentration, type and amount of catalyst and organic solvent, amount of silica precursor, reaction temperature, reaction time, and stirring intensity. Precise characterizations of the core nanoparticles with mesoporous silica coating revealed the mechanisms involved and provided information to allow the further and better design of diverse core–shell nanostructures. The general procedure defined here was successfully applied to spherical small nanoparticles and nanoparticle clusters as well as to ferromagnetic hexaferrite nanoplatelets. The method is therefore fully versatile and easy to apply to a plethora of diverse core nanoparticles.
Acknowledgments
The authors acknowledge the financial support from the Slovenian Research Agency (ARRS) for Young Researcher Scheme (S.N.; 1000-18-0106), Research Core Funding (P2-0089), and ARRS Projects. The authors thank Hermina Hudelja for assistance with the gas adsorption–desorption measurement, and Patricija Hribar Boštjančič for assistance with the zeta-potential measurements. We acknowledge the CENN Nanocenter (Ljubljana, Slovenia) for the use of the electron microscope (TEM 2100).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsami.0c17863.
Demonstration of magnetic responsiveness, hydrodynamic size measurements, TEM characterization, synthesis parameters data (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Shi J. On the Synergetic Catalytic Effect in Heterogeneous Nanocomposite Catalysts. Chem. Rev. 2013, 113, 2139–2181. 10.1021/cr3002752. [DOI] [PubMed] [Google Scholar]
- Li W.; Liu J.; Zhao D. Mesoporous Materials for Energy Conversion and Storage Devices. Nat. Rev. Mater. 2016, 1, 1–17. 10.1038/natrevmats.2016.23. [DOI] [Google Scholar]
- Li Y.; Shi J. Hollow-Structured Mesoporous Materials: Chemical Synthesis, Functionalization and Applications. Adv. Mater. 2014, 26, 3176–3205. 10.1002/adma.201305319. [DOI] [PubMed] [Google Scholar]
- Pan P.; Zhang T.; Yue Q.; Elzatahry A. A.; Alghamdi A.; Cheng X.; Deng Y. Interface Coassembly and Polymerization on Magnetic Colloids: Toward Core–Shell Functional Mesoporous Polymer Microspheres and Their Carbon Derivatives. Adv. Sci. 2020, 7, 2000443. 10.1002/advs.202000443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells C.; Vollin-Bringel O.; Fiegel V.; Harlepp S.; Van der Schueren B.; Bégin-Colin S.; Bégin D.; Mertz D. Engineering of Mesoporous Silica Coated Carbon-Based Materials Optimized for an Ultrahigh Doxorubicin Payload and a Drug Release Activated by pH, T, and NIR-Light. Adv. Funct. Mater. 2018, 28, 1–13. 10.1002/adfm.201706996. [DOI] [Google Scholar]
- Schacht S.; Huo Q.; Voigt-Martin I. G.; Stucky G. D.; Schuth F. Oil-Water Interface Templating of Mesoporous Macroscale Structures. Science 1996, 273, 768–771. 10.1126/science.273.5276.768. [DOI] [PubMed] [Google Scholar]
- Wan Y.; Zhao D. On the Controllable Soft-Templating Approach to Mesoporous Silicates. Chem. Rev. 2007, 107, 2821–2860. 10.1021/cr068020s. [DOI] [PubMed] [Google Scholar]
- Qu Q.; Min Y.; Zhang L.; Xu Q.; Yin Y. Silica Microspheres with Fibrous Shells: Synthesis and Application in HPLC. Anal. Chem. 2015, 87, 9631–9638. 10.1021/acs.analchem.5b02511. [DOI] [PubMed] [Google Scholar]
- Zhao T.; Elzatahry A.; Li X.; Zhao D. Single-Micelle-Directed Synthesis of Mesoporous Materials. Nat. Rev. Mater. 2019, 4, 775–791. 10.1038/s41578-019-0144-x. [DOI] [Google Scholar]
- Jana S. K.; Nishida R.; Shindo K.; Kugita T.; Namba S. Pore Size Control of Mesoporous Molecular Sieves Using Different Organic Auxiliary Chemicals. Microporous Mesoporous Mater. 2004, 68, 133–142. 10.1016/j.micromeso.2003.12.010. [DOI] [Google Scholar]
- Deng Y.; Yue Q.; Sun J.; Kang Y. Recent Advance in Interfacial Assembly Growth of Mesoporous Silica on Magnetite Particles. Angew. Chem., Int. Ed. 2020, 59, 15804. 10.1002/anie.201911690. [DOI] [PubMed] [Google Scholar]
- Narayan R.; Nayak U.; Raichur A.; Garg S. Mesoporous Silica Nanoparticles: A Comprehensive Review on Synthesis and Recent Advances. Pharmaceutics 2018, 10, 118. 10.3390/pharmaceutics10030118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge J.; Zhang Q.; Zhang T.; Yin Y. Core-Satellite Nanocomposite Catalysts Protected by a Porous Silica Shell: Controllable Reactivity, High Stability, and Magnetic Recyclability. Angew. Chem. 2008, 120, 9056–9060. 10.1002/ange.200803968. [DOI] [PubMed] [Google Scholar]
- Zhang Q.; Zhang T.; Ge J.; Yin Y. Permeable Silica Shell through Surface-Protected Etching. Nano Lett. 2008, 8, 2867–2871. 10.1021/nl8016187. [DOI] [PubMed] [Google Scholar]
- He Q.; Shi J. Mesoporous Silica Nanoparticle Based Nano Drug Delivery Systems: Synthesis, Controlled Drug Release and Delivery, Pharmacokinetics and Biocompatibility. J. Mater. Chem. 2011, 21, 5845–5855. 10.1039/c0jm03851b. [DOI] [Google Scholar]
- Kim J.; Kim H. S.; Lee N.; Kim T.; Kim H.; Yu T.; Song I. C.; Moon W. K.; Hyeon T. Multifunctional Uniform Nanoparticles Composed of a Magnetite Nanocrystal Core and a Mesoporous Silica Shell for Magnetic Resonance and Fluorescence Imaging and for Drug Delivery. Angew. Chem., Int. Ed. 2008, 47, 8438–8441. 10.1002/anie.200802469. [DOI] [PubMed] [Google Scholar]
- Nerantzaki M.; Michel A.; Briot E.; Siaugue J. M.; Menager C.; Wilhelm C.; Griffete N. Controlled Drug Delivery for Cancer Cell Treatment via Magnetic Doxorubicin Imprinted Silica Nanoparticles. Chem. Commun. 2020, 56, 10255–10258. 10.1039/D0CC01325K. [DOI] [PubMed] [Google Scholar]
- Perton F.; Tasso M.; Muñoz Medina G. A.; Ménard M.; Blanco-Andujar C.; Portiansky E.; van Raap M. B. F.; Bégin D.; Meyer F.; Begin-Colin S.; Mertz D. Fluorescent and Magnetic Stellate Mesoporous Silica for Bimodal Imaging and Magnetic Hyperthermia. Appl. Mater. Today 2019, 16, 301–314. 10.1016/j.apmt.2019.06.006. [DOI] [Google Scholar]
- Ménard M.; Meyer F.; Parkhomenko K.; Leuvrey C.; Francius G.; Bégin-Colin S.; Mertz D. Mesoporous Silica Templated-Albumin Nanoparticles with High Doxorubicin Payload for Drug Delivery Assessed with a 3-D Tumor Cell Model. Biochim. Biophys. Acta, Gen. Subj. 2019, 1863, 332–341. 10.1016/j.bbagen.2018.10.020. [DOI] [PubMed] [Google Scholar]
- Deng Y.; Cai Y.; Sun Z.; Liu J.; Liu C.; Wei J.; Li W.; Liu C.; Wang Y.; Zhao D. Multifunctional Mesoporous Composite Microspheres with Well-Designed Nanostructure: A Highly Integrated Catalyst System. J. Am. Chem. Soc. 2010, 132, 8466–8473. 10.1021/ja1025744. [DOI] [PubMed] [Google Scholar]
- Cazares-Cortes E.; Nerantzaki M.; Fresnais J.; Wilhelm C.; Griffete N.; Menager C. Magnetic Nanoparticles Create Hot Spots in Polymer Matrix for Controlled Drug Release. Nanomaterials 2018, 8, 850. 10.3390/nano8100850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisjak D.; Mertelj A. Anisotropic Magnetic Nanoparticles: A Review of Their Properties, Syntheses and Potential Applications. Prog. Mater. Sci. 2018, 95, 286–328. 10.1016/j.pmatsci.2018.03.003. [DOI] [Google Scholar]
- Kralj S.; Potrc T.; Kocbek P.; Marchesan S.; Makovec D. Design and Fabrication of Magnetically Responsive Nanocarriers for Drug Delivery. Curr. Med. Chem. 2017, 24, 454–469. 10.2174/0929867323666160813211736. [DOI] [PubMed] [Google Scholar]
- Wan L.; Song H.; Chen X.; Zhang Y.; Yue Q.; Pan P.; Su J.; Elzatahry A. A.; Deng Y. A Magnetic-Field Guided Interface Coassembly Approach to Magnetic Mesoporous Silica Nanochains for Osteoclast-Targeted Inhibition and Heterogeneous Nanocatalysis. Adv. Mater. 2018, 30, 1–9. 10.1002/adma.201707515. [DOI] [PubMed] [Google Scholar]
- Yue Q.; Li J.; Luo W.; Zhang Y.; Elzatahry A. A.; Wang X.; Wang C.; Li W.; Cheng X.; Alghamdi A.; Abdullah A. M.; Deng Y.; Zhao D. An Interface Coassembly in Biliquid Phase: Toward Core-Shell Magnetic Mesoporous Silica Microspheres with Tunable Pore Size. J. Am. Chem. Soc. 2015, 137, 13282–13289. 10.1021/jacs.5b05619. [DOI] [PubMed] [Google Scholar]
- Kwon D.; Cha B. G.; Cho Y.; Min J.; Park E. B.; Kang S. J.; Kim J. Extra-Large Pore Mesoporous Silica Nanoparticles for Directing in Vivo M2Macrophage Polarization by Delivering IL-4. Nano Lett. 2017, 17, 2747–2756. 10.1021/acs.nanolett.6b04130. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Yue Q.; Zagho M. M.; Zhang J.; Elzatahry A. A.; Jiang Y.; Deng Y. Core-Shell Magnetic Mesoporous Silica Microspheres with Large Mesopores for Enzyme Immobilization in Biocatalysis. ACS Appl. Mater. Interfaces 2019, 11, 10356–10363. 10.1021/acsami.8b18721. [DOI] [PubMed] [Google Scholar]
- Ding H. L.; Zhang Y. X.; Wang S.; Xu J. M.; Xu S. C.; Li G. H. Fe3O4@SiO2 Core/Shell Nanoparticles: The Silica Coating Regulations with a Single Core for Different Core Sizes and Shell Thicknesses. Chem. Mater. 2012, 24, 4572–4580. 10.1021/cm302828d. [DOI] [Google Scholar]
- Kralj S.; Makovec D. Magnetic Assembly of Superparamagnetic Iron Oxide Nanoparticle Clusters into Nanochains and Nanobundles. ACS Nano 2015, 9, 9700–9707. 10.1021/acsnano.5b02328. [DOI] [PubMed] [Google Scholar]
- Tadic M.; Kralj S.; Jagodic M.; Hanzel D.; Makovec D. Magnetic Properties of Novel Superparamagnetic Iron Oxide Nanoclusters and Their Peculiarity under Annealing Treatment. Appl. Surf. Sci. 2014, 322, 255–264. 10.1016/j.apsusc.2014.09.181. [DOI] [Google Scholar]
- Drofenik M.; Kristl M.; Žnidaršič A.; Hanžel D.; Lisjak D. Hydrothermal Synthesis of Ba-Hexaferrite Nanoparticles. J. Am. Ceram. Soc. 2007, 90, 2057–2061. 10.1111/j.1551-2916.2007.01740.x. [DOI] [Google Scholar]
- Lisjak D.; Drofenik M. Chemical Substitution—An Alternative Strategy for Controlling the Particle Size of Barium Ferrite. Cryst. Growth Des. 2012, 12, 5174–5179. 10.1021/cg301227r. [DOI] [Google Scholar]
- Kralj S.; Makovec D.; Čampelj S.; Drofenik M. Producing Ultra-Thin Silica Coatings on Iron-Oxide Nanoparticles to Improve Their Surface Reactivity. J. Magn. Magn. Mater. 2010, 322, 1847–1853. 10.1016/j.jmmm.2009.12.038. [DOI] [Google Scholar]
- Schindelin J.; Arganda-Carreras I.; Frise E.; Kaynig V.; Longair M.; Pietzsch T.; Preibisch S.; Rueden C.; Saalfeld S.; Schmid B.; Tinevez J.-Y.; White D. J.; Hartenstein V.; Eliceiri K.; Tomancak P.; Cardona A. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadic M.; Kralj S.; Kopanja L. Synthesis, Particle Shape Characterization, Magnetic Properties and Surface Modification of Superparamagnetic Iron Oxide Nanochains. Mater. Charact. 2019, 148, 123–133. 10.1016/j.matchar.2018.12.014. [DOI] [Google Scholar]
- Li W.; Zhang M.; Zhang J.; Han Y. Self-Assembly of Cetyl Trimethylammonium Bromide in Ethanol-Water Mixtures. Front. Chem. China 2006, 1, 438–442. 10.1007/s11458-006-0069-y. [DOI] [Google Scholar]
- Kralj S.; Drofenik M.; Makovec D. Controlled Surface Functionalization of Silica-Coated Magnetic Nanoparticles with Terminal Amino and Carboxyl Groups. J. Nanopart. Res. 2011, 13, 2829–2841. 10.1007/s11051-010-0171-4. [DOI] [Google Scholar]
- Beck J. S.; Vartuli J. C.; Roth W. J.; Leonowicz M. E.; Kresge C. T.; Schmitt K. D.; Chu C. T. W.; Olson D. H.; Sheppard E. W.; McCullen S. B.; Higgins J. B.; Schlenker J. L. A New Family of Mesoporous Molecular Sieves Prepared with Liquid Crystal Templates. J. Am. Chem. Soc. 1992, 114, 10834–10843. 10.1021/ja00053a020. [DOI] [Google Scholar]
- Lv X.; Zhang L.; Xing F.; Lin H. Controlled Synthesis of Monodispersed Mesoporous Silica Nanoparticles: Particle Size Tuning and Formation Mechanism Investigation. Microporous Mesoporous Mater. 2016, 225, 238–244. 10.1016/j.micromeso.2015.12.024. [DOI] [Google Scholar]
- Chen B. C.; Lin H. P.; Chao M. C.; Mou C. Y.; Tang C. Y. Mesoporous Silica Platelets with Perpendicular Nanochannels via a Ternary Surfactant System. Adv. Mater. 2004, 16, 1657–1661. 10.1002/adma.200306327. [DOI] [Google Scholar]
- Gao C.; Qiu H.; Zeng W.; Sakamoto Y.; Terasaki O.; Sakamoto K.; Chen Q.; Che S. Formation Mechanism of Anionic Surfactant-Templated Mesoporous Silica. Chem. Mater. 2006, 18, 3904–3914. 10.1021/cm061107+. [DOI] [Google Scholar]
- Zainal N. A.; Shukor S. R. A.; Wab H. A. A.; Razak K. A. Study on the Effect of Synthesis Parameters of Silica Nanoparticles Entrapped with Rifampicin. Chem. Eng. Trans. 2013, 32, 2245–2250. [Google Scholar]
- Yuan H.; Gao F.; Zhang Z.; Miao L.; Yu R.; Zhao H.; Lan M. Study on Controllable Preparation of Silica Nanoparticles with Multi-Sizes and Their Size-Dependent Cytotoxicity in Pheochromocytoma Cells and Human Embryonic Kidney Cells. J. Health Sci. 2010, 56, 632–640. 10.1248/jhs.56.632. [DOI] [Google Scholar]
- Shen D.; Yang J.; Li X.; Zhou L.; Zhang R.; Li W.; Chen L.; Wang R.; Zhang F.; Zhao D. Biphase Stratification Approach to Three-Dimensional Dendritic Biodegradable Mesoporous Silica Nanospheres. Nano Lett. 2014, 14, 923–932. 10.1021/nl404316v. [DOI] [PubMed] [Google Scholar]
- Kim S.; Chen J.; Cheng T.; Gindulyte A.; He J.; He S.; Li Q.; Shoemaker B. A.; Thiessen P. A.; Yu B.; Zaslavsky L.; Zhang J.; Bolton E. E. PubChem 2019 Update: Improved Access to Chemical Data. Nucleic Acids Res. 2019, 47, D1102–D1109. 10.1093/nar/gky1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Q.; Li J.; Luo W.; Zhang Y.; Elzatahry A. A.; Wang X.; Wang C.; Li W.; Cheng X.; Alghamdi A.; Abdullah A. M.; Deng Y.; Zhao D. An Interface Coassembly in Biliquid Phase: Toward Core-Shell Magnetic Mesoporous Silica Microspheres with Tunable Pore Size. J. Am. Chem. Soc. 2015, 137, 13282–13289. 10.1021/jacs.5b05619. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.