

Abstract

Decarboxylative halogenation, or halodecarboxylation, represents one of the fundamental key methods for the synthesis of ubiquitous organic halides. The method is based on conversion of carboxylic acids to the corresponding organic halides via selective cleavage of a carbon–carbon bond between the skeleton of the molecule and the carboxylic group and the liberation of carbon dioxide. In this review, we discuss and analyze major approaches for the conversion of alkanoic, alkenoic, acetylenic, and (hetero)aromatic acids to the corresponding alkyl, alkenyl, alkynyl, and (hetero)aryl halides. These methods include the preparation of families of valuable organic iodides, bromides, chlorides, and fluorides. The historic and modern methods for halodecarboxylation reactions are broadly discussed, including analysis of their advantages and drawbacks. We critically address the features, reaction selectivity, substrate scopes, and limitations of the approaches. In the available cases, mechanistic details of the reactions are presented, and the generality and uniqueness of the different mechanistic pathways are highlighted. The challenges, opportunities, and future directions in the field of decarboxylative halogenation are provided.
1. Introduction
The development of useful methods for the chemo-, regio-, and stereoselective cleavage and formation of chemical bonds for the construction of functional materials has engaged chemists for more than a century. Recent progress in our understanding of selective carbon–hydrogen and carbon–carbon bond activation provides an optimistic hope for revolutionary approaches to the direct introduction of the desired functionality in nonfunctionalized substrates, which would have a transformable influence on our economics and environment. While these emerging approaches are in their infancy, the selective conversion of available functional substrates to valuable materials via functional group interconversion constitutes an essential core of organic chemistry. Many such conversions are based on the ability to selectively activate the C–C bond in functional groups, which has opened a door to a wide range of tremendously useful applications in organic synthesis (described in this issue of Chemical Reviews).
Organic halides are one of the most widely used precursors or intermediates for numerous organic transformations. Unlike other functionalities, halogenated compounds can be considered, from a retrosynthetic point of view, as a synthetic equivalent for cationic, radical, or anionic carbon-based synthons (Scheme 1). Polarization of the carbon–halogen bond and the good leaving abilities of the halides enable these compounds to efficiently undergo nucleophilic substitution and elimination reactions. Furthermore, in the presence of a suitable reducing agent, organohalides could act as radical precursors or can be converted to organometallic species. Additionally, oxidation furnishes valuable hypervalent halogen reagents, which act as powerful oxidants or versatile reagents with their own rich and unique chemistry. The reactivity of halogenated compounds is strongly influenced by the hybridization of the carbon atom in the C–X bond as well as the halogen itself.
Scheme 1. Retrosynthetic Disconnections of the Organohalide Moieties.

Along with their great synthetic utility, halogenated organic compounds have found wide use in numerous valuable commercial products. One of the major industrially employed halogen-containing product–vinyl chloride–is estimated to be produced on an about 51-million-ton scale in 2020,1 being almost exclusively used for the production of polyvinyl chloride. Among others, organohalides are extensively used as solvents, plasticizers, fire retardants, organic light-emitting devises, heat transfer, and hydraulic fluids. Often, halogenation can significantly alter the biological properties of the molecule, rendering the use of these compounds as drugs, agrochemicals, biocides, etc. Moreover, while a huge number of halogenated compounds are anthropogenic, the amount of isolated naturally occurring organohalides has exceeded 5000.2 Since most of these natural compounds have a specific role in living organisms, their structural features often serve as an inspiration in the search for new classes of biologically active compounds. All these indispensable applications of organic halides highlight the importance of the targeted incorporation of the halogen atom at the desired position in the molecule.3
On a par with classical laboratory methods for the preparation of organic halides from alcohols or unsaturated compounds, carboxylic acids represent an important alternative. The attractiveness of the use of acids can be attributed to their wide abundance. A large number of carboxylic acids are readily commercially available, since many of them are obtained from renewable sources or are prepared on an industrial scale. Generally air-stable and easy to handle, they can also be synthesized on demand on a laboratory scale by a wide range of well-established methods.
The acids are mainly converted to the corresponding halides in two principal ways. The first method represents a separate combination of two well-studied methods: reduction of the acid to the alcohol, with further conversion to the halide. The second approach involves cleavage of the carbon–carbon bond next to the carboxylic group by inducing a decarboxylation process and trapping the intermediate with a suitable halogen source. The process is known as decarboxylative halogenation or the halodecarboxylation reaction (Scheme 2).
Scheme 2. Halodecarboxylation Reaction.

Being a conceptually distinct transformation, the halodecarboxylation reaction has several important features, which make it of great synthetic utility. It allows a one-pot conversion of relatively inexpensive and available carboxylic acids into valuable and highly useful organic halides. The best methods are applicable for both aliphatic and aromatic substrates. Moreover, the unreacted starting carboxylic acid can be facilely removed by aqueous basic workup.
Aromatic acids subjected to the halodecarboxylation reaction yield the corresponding aryl halides. This process not only serves as a useful alternative, but can also provide regioisomers that are only obtained in negligible amounts by direct aromatic halogenation, especially if the substituents on the aromatic ring do not direct the electrophilic substitution to the desired position. Remarkably, the carboxyl group on the aromatic ring is selectively obtained by a simple oxidation of the corresponding methyl (or alkyl) group of benzene derivatives. Such derivatives constitute a basic chemical feedstock, and their oxidation to the corresponding carboxylic acids is a well-established process.
In the case of aliphatic carboxylic acids, the alkyl chain of the halodecarboxylation product will be one carbon shorter than the parent acid. This feature can play a significant role when considering the preparation of materials, which are tediously synthesized by other means. For example, if one looks at the prices of n-alkyl bromides, it is clearly seen that products with an even number of carbon atoms are significantly cheaper than their odd-numbered homologues. This interesting observation can be attributed to the facile production of alkyl halides, bearing an even-numbered carbon chain, from natural fatty acids (which also possess an even number of carbons in their skeleton) by reduction-bromination approach (Table 1). However, it is impossible to utilize the same approach to prepare alkyl halides, bearing an odd number of carbons. Halodecarboxylation would be an ideal alternative.
Table 1. Prices for Alkyl Bromides with Even and Odd Number of Carbon Atoms.

Prices from Sigma-Aldrich Israel as of February 2020.
The concept of a halodecarboxylation was developed about 80 years ago. However, there are only a few comprehensive reviews, which cover a few methods in the field of decarboxylative halogenation.4−13 Since then, numerous alternative halodecarboxylation approaches have been developed, although these methods have been mentioned only sporadically in a few nonthematic reviews, as a small subdivision. In this review, we aim to summarize the long history of the halodecarboxylation reaction, describing its scope and limitations, and presenting mechanistic insights into the methods described in the literature up to June 2020. In the first section, we will discuss pioneering approaches. Since these reactions have already been summarized elsewhere, no full details will be given. Further, the discussion will separately outline the preparation of iodo-, bromo-, and chloro-organics from aliphatic and aromatic acids. Because of the similarities in the mechanistic details, the transformations of alkenylic and alkynilic acids will be described in one chapter. As a final topic, we will outline the achievements in a challenging decarboxylative fluorination process.
2. Pioneering and Classical Methods
2.1. The Hunsdiecker–Borodin Reaction
The first report of the halodecarboxylation reaction was by Alexander Borodin in 1861.14 In his seminal work, solid silver salts of valeric and butyric acids were treated with bromine vapors to give the monobrominated acid derivatives. However, attempts to obtain bromoacetic acid by a similar approach failed, producing silver bromide and a mixture of gases, which were identified as carbon dioxide and methyl bromide. Since then, a very few sporadic examples were reported, mentioning the introduction of the halogen function, via a decarboxylation in the very specific substrates.15−19 These included a first halodecarboxylation of aromatic acids, which was observed by Pope and Wood upon halogenation of hydroxybenzoic acids. They subjected 4-hydroxybenzoic acid in cold sulfuric acid to bromine in an attempt to synthesize 2,6-dibromophenol, but the major isolated product from the reaction was 2,4,6-tribromophenol.20 Later, Krishna and Pope21 during their analysis of salicylic acid by the iodometric determination of acids observed significantly lower amounts of liberated iodine than expected from reaction 1:
| 1 |
Evolution of carbon dioxide and iodine was observed during the reaction. However, the iodine color vanished with time, accompanied by the formation of a precipitate, which was identified as 2,4,6-triiodophenol. The reaction was shown to proceed in a similar manner for some 2- and 4-hydroxybenzoic acids, such as 2 and 3, giving the same triiodinated product 1 (Scheme 3). Further mechanistic studies22,23 of this type of transformations showed that after excessive electrophilic halogenation, carboxyphenol 4 underwent an additional halogenation at ipso to carboxyl group position, followed by decarboxylation.
Scheme 3. Halodecarboxylation of Phenolcarboxylic Acids.

Only at the end of the 1930s did Hans Hunsdiecker24,25 realize the power of this transformation, which laid the foundation for the development of halodecarboxylation as a useful synthetic method for the preparation of alkyl haildes.5,6 Because of his vast contribution to the study of this transformation, it is often referred to as the Hunsdiecker reaction, or Hunsdiecker–Borodin, paying a tribute to the pioneer of this transformation.14
Hunsdiecker systematically showed that suspensions of aliphatic carboxylic acid salts in carbon tetrachloride are efficiently transformed to the corresponding alkyl bromides upon treatment with bromine under reflux conditions (Scheme 4). While most of his work was dedicated to silver salts, other metal derivatives, such as mercury(I/II) and thallium(I) salts, were claimed to be efficient.25 Potassium salts could also be employed, although the yield of the product in this case was substantially lower. Various alkyl bromides were prepared in good yields, independently of the length of the alkyl chain. Interestingly, the reaction could be applied to a variety of alpha-substituted acids. As such, silver salts of α-bromo, and α-keto acids gave geminal dibromide 20 and acyl bromide 13, respectively. The salts of α-hydroxy (14) and amino- (16) acids are also suitable substrates for the reaction, delivering the aldehydes 15 and 17, respectively, upon hydrolysis. Chlorinated analogues also proved to be attainable if elemental chlorine is used.
Scheme 4. Examples of Bromodecarboxylation by Hunsdiecker.

It should be noted that an interesting observation was made by Bockemüller and Hoffmann, independently from Hunsdiecker, during their studies on acyl hypohalites.26 The authors reported on the decomposition of acyl hypobromites in the presence of silver(I) and Br2 with the release of CO2 and the formation of alkyl bromides as the main products. Methyl- and propyl bromide were obtained in synthetically useful 69% and 61% yields, respectively.
2.1.1. Scope of the Reaction
After the initial work of Hunsdiecker, the area of halodecarboxylation was extensively explored by others. The most studied halogen in this transformation was bromine; yet, a considerable amount of work was dedicated to the employment of chlorine and iodine for the preparation of the corresponding alkyl halides. While the highest yields were obtained for the alkyl bromides, the chloride analogues usually had significantly lower yields due to the radical chlorination of both the starting acid and the product.27 Alkyl iodides could be obtained in a similar manner, although the outcome of the reaction significantly depended on the reaction conditions. The reaction of silver carboxylates with I2 was studied at the end of the 19th century as a method for the preparation of esters and is known as the Simonini reaction.28,29 In this transformation, 2 equiv of a silver salt are reacted with one equivalent of I2 (Reaction 2). As was shown, the utilization of a 3:2 ratio of salt to iodine produced equimolar amounts of ester and alkyl iodide (Reaction 3).30 Increasing the proportion of the I2/substrate to 1:1 (or even higher) was recommended for obtaining the desired alkyl iodide in a good yield, although special experimental precautions should be taken to minimize the formation of the ester. Bromodecarboxylation also produces esters as a side product, but in smaller amounts, while no esters were isolated for the chlorodecarboxylation process.27
 |
2 |
With respect to the scope, the Hunsdiecker reaction can be efficiently applied to primary and secondary aliphatic acids (Scheme 5). Yields of the primary alkyl bromides vary in a range of 60–90%, while secondary carboxylic acids are less efficient (the yield is 30–70%). The synthesis of tertiary alkyl bromides is problematic, furnishing only traces of the product, unless the carboxylic group is attached to a bridgehead carbon. Substituents elsewhere in the aliphatic chain, but in the α-position, do not significantly affect the yield, unless these substituents are incompatible with the intermediate acyl hypohalite. Carboxylic acids bearing electron-rich aromatics suffer from an electrophilic halogenation of the ring instead of the desired reaction. For example, subjecting silver 3-methoxyhydrocinnamate 42 to the bromodecarboxylation conditions resulted in the bromoaryl product 43 (Scheme 6). Alkene or alkyne-decorated acids usually do not deliver the desired product, unless they are in conjugation with the carboxylic group. For compounds, bearing isolated double bonds, lactonization is one of the possible outcomes (Scheme 6). If an excess of halogen is avoided, α,β-unsaturated acids undergo halodecarboxylation, providing the haloalkynes (34) and haloalkenes (35). The N-protection of amino acids allowed the isolation of moisture-sensitive α-bromoamines 31 in a 28–40% yield (Scheme 5).31 Halogenated substrates are also tolerated, making the method suitable for the preparation of polyhalogenated compounds. Thus, a number of mono-, di-, and trihalogenated acetic acids were successfully converted to the corresponding polyhalogenated methanes in good yields (compounds 36–38). Perfluorinated acids are also easily converted to the desired halides, including the relatively problematic iodides, with no formation of ester byproducts (39–41).
Scheme 5. Selected Examples of the Aliphatic Hunsdiecker–Borodin Halodecarboxylation Reaction5,30−45.

Cl2 instead of Br2.
I2 instead of Br2.
Scheme 6. Examples of Unsuccessful Bromodecarboxylation Reactions46,47.

The one-pot bromodecarboxylation of dicarboxylic acids was studied.25,44,46,48,49 The outcome of the reaction mainly depends on the relative position of the two carboxylic groups. α,ω-Dicarboxylic acids with alkyl chains longer than that of 1,7-heptandicarboxylic acid are efficiently converted to the corresponding dibromides with yields of 55–80% (Scheme 7). For adipic acid, the yield of dibromide 46 is relatively high (58%), although a substantial amount of δ-valerolactone is formed (26%). γ-Butyrolactone 53 becomes the almost exclusive product when glutaric acid or its derivatives are employed, providing a yield of the dibromide 52 in the range of 5–10%. For succinic acid, the yield of the product 54 rises to 37%. Silver salts of ethylmalonic acid undergo double decarboxylation to give the geminal dibromide 55 in a poor yield (28%), and a significant amount of 1,1,1-tribromopropane 56 is isolated from the reaction mixture.
Scheme 7. Examples of Double Bromodecarboxylation of Aliphatic Diacids25,44,46,48,49.

Substantial efforts were made to develop an aromatic version of the reaction due to the enormous practical importance of aromatic halides. However, the Hunsdiecker reaction of aromatic carboxylic acids proved to be less general compared to that of the aliphatic counterparts, and reports on the product yields were sometimes contradictory.26,44,45,50,51 For instance, the yields of bromobenzene in the bromodecarboxylation of silver benzoate varied from 0 to 80%.26 Such a drastic difference may be attributed to variations in the reaction conditions, purity of the reagents, and amounts of water present in the silver salts. In general, silver benzoates bearing electron-withdrawing groups gave synthetically useful yields of the corresponding aryl bromides (Scheme 8). However, electron-neutral and especially electron-rich benzoic acids were inefficient since the electrophilic bromination of the aromatic ring was the predominant process. A number of pyridine-based carboxylic acids were reported to undergo bromodecarboxylation, although the yield was low or unstated.52,53
Scheme 8. Selected Examples of Aromatic Bromodecarboxylation26,44,45,50,51.

Asterisk indicates a bromine introduced via electrophilic bromination.
2.1.2. Mechanism of the Reaction
Most of the mechanistic studies were conducted on the bromodecarboxylation reaction. While the suggested mechanism is also applicable for chlorodecarboxylation, the iodination process is, likely, more complicated and is influenced by the reagents ratio and the experimental protocol. The mechanistic aspects of the iododecarboxylation will be discussed later in the text.
The Hunsdiecker–Borodin reaction represents a sequential combination of two elemental steps (Scheme 9). In the first step, a silver salt of a carboxylic acid produces an acyl hypohalite in the reaction with halogen. The obtained intermediate possesses a poor stability and decomposes, with extrusion of carbon dioxide, to provide the targeted alkyl halide. The first step of the sequence was extensively studied previously.7 Even though the acyl hypohalite cannot be isolated in the pure form, and can be handled only in solution, there is spectroscopic evidence for its existence. In addition, this unstable intermediate has been chemically trapped with alkenes to form the corresponding haloesters 68 or diesters 69 (Scheme 10).6
Scheme 9. General Mechanism of the Hunsdiecker Bromodecarboxylation Reaction.

Scheme 10. Reaction of Silver Salts with Iodine in the Presence of Double Bond (Prevost Reaction).

There is solid experimental evidence that the initiation step–decomposition of an acyl hypobromite 65– proceeds via homolytic cleavage of the oxygen-halogen bond to give acyloxy and halogen radicals. The rate of this process depends on the stability of the hypohalites. Moreover, in the presence of light, the decomposition of 65 is accelerated by 1–2 orders of magnitude.26 The resulting unstable acyloxy radical 66 liberates carbon dioxide to form an alkyl radical 67, which further abstracts a halogen atom from another molecule of either the acyl hypobromite or bromine, or recombines with a free bromine radical (Scheme 9).
As indicated in numerous studies, this final step of carbon–halogen bond formation involves the intermediacy of alkyl radicals rather than carbocations. As such, some optically active carboxylic acids (70, 72) were subjected to a bromodecarboxylation reaction, furnishing racemic products 71 and 73 (Scheme 11).37,54 Racemization of these alkyl halides in the presence of silver halide was shown to be possible; yet, the rate of the racemization is too slow to be the cause of the complete loss of enantioselectivity.
Scheme 11. Halodecarboxylation of Optically Active Acids.

Another supporting experiment for the radical mechanism is the reaction of silver tert-butyl acetate 74 in bromodecarboxylation (Scheme 12).55 If the reaction involves the carbocation intermediate, then the neopentyl cation should undergo a 1,2-methyl shift to give the more stable tert-amyl cation and, correspondingly, tert-amyl bromide 76 as one of the products. However, the reaction proceeds with the formation of neopentyl bromide 75 as the sole product. Additionally, the reaction of 2-methyl-2-phenylpropionic acid 77 furnishes a mixture of 2-phenyl-2,2-dimethylbromoethane 78 and 1-phenyl-2-methyl-2-bromopropane 79.56 The presence of product 79 suggests the intermediacy of a neophyl radical, which is prone to a 1,2-phenyl shift. Product 78 is stable under the reaction conditions and does not undergo rearrangement in the presence of AgBr. Furthermore, the absence of ring contraction in the case of the halodecarboxylation of cyclopentyl and cyclohexylacetic acids precludes a carbocation mechanism.39,57
Scheme 12. Mechanistic Insights of the Reaction Intermediates.

A chain process was questioned by Cristol and co-workers during studies of the mechanism of the reaction.58 Subjecting silver salts of endo and exo isomers of the acid 79a and 79b to the reaction with Br2 in dark resulted in a mixture of bromides endo-80a and exo-80b in a 69:31 ratio regardless of the configuration of the starting acid. On the other hand, decomposition of the acylperoxides 81a and 81b in refluxing carbon tetrabromide led to the formation of solely exo bromide 80b without traces of endo-80a (Scheme 13). This difference in product distribution was attributed to different reaction mechanisms. While formation of a significant amount of exo bromide during decomposition of peroxides is explained by a free radical chain reaction mechanism, the prevalent endo isomer in a bromodecarboxylation process is more consistent with geminate recombination of an alkyl and bromine radicals within a solvent cage.
Scheme 13. Bromodecarboxylation of Isomer Acids.

Details of the bromodecarboxylation reaction of aromatic silver salts are also consistent with a radical pathway. During the reaction of silver benzoate 82 with Br2 in CCl4, along with bromobenzene 57 (53%), some chlorobenzene 83 was detected (5%), accompanied by 6.7% of BrCCl3 (Scheme 14).45,59 The presence of these products can be rationalized by the reaction of the phenyl radical intermediate with CCl4 to give chlorobenzene and the trichloromethyl radical, which abstracts a Br atom from the benzoyl hypobromite.
Scheme 14. Side Products during Bromodecarboxylation of Silver Benzoate.

As aforementioned, the outcome of the reaction of silver salts of carboxylic acids with I2 is mainly influenced by the ratio of the reagents. Similarly to the reaction with Br2, the treatment of a carboxylic acid 84 with iodine initially forms an acyl hypoiodite 85. However, decomposition of this intermediate to form an alkyl iodide 86 is a rather slow process, and reaction with another equivalent of the silver salt to give a 1:1 complex 87 takes place (Scheme 15).60 The intermediate 87 can be isolated in some cases.61 The complex 87 undergoes further decomposition to acyloxy radicals 88 with the concomitant formation of an ester 89 (Scheme 15, path A). When the ratio between the silver salt and I2 is kept to 3:2, the reaction gives rise to the formation of iodine triacyl 90 as the major intermediate (Scheme 15, path B). Decomposition of 90 leads to an ester 89, accompanied by an alkyl iodide 86. Presumably, the iodine complex 90 undergoes homolytic cleavage to give acyl radicals and acyl hypoiodite, which decompose, in turn, to give the Hunsdiecker product. However, if the decomposition of 90 proceeds in the presence of excess iodine, the alkyl iodide 86 is formed in high yields.
Scheme 15. Proposed Reaction Sequence of Silver Carboxylates with Iodine.

The presence of carbocation intermediates was assumed in some cases. The most notable example involves the reaction of the silver salt of cyclobutylcarboxylic acid 91 with I2 (Scheme 16).62 Among other products, a mixture of esters was isolated and determined to consist of cyclobutyl-, cyclopropylcarbinyl-, and allylcarbinyl cyclobutylacetates (92, 93, and 94, respectively). The presence of these side products is attributed to a rearrangement, characteristic for cyclobutyl cations. One of the possible pathways of carbocation formation involves the reaction between cyclobutyl iodide and silver carboxylate with concomitant rearrangement and reaction with the carboxylate.
Scheme 16. Mechanistic Insights on Reaction of Silver Carboxylates with Iodine.

2.1.3. Limitations and Features
The pioneering influence of the Hunsdiecker–Borodin process in the development of the field of decarboxylative halogenation is truly invaluable. However, a number of limitations currently preclude a broad employment of this reaction. The main disadvantage of the method is its poor tolerance toward moisture. Silver salts are usually prepared in an aqueous medium followed by thorough drying. Even traces of moisture lead to a significant decrease in the reaction yield and are responsible for inconsistencies in the reported yields. In order to obtain the maximum efficacy, not only the silver salts, but also the solvent, halogen, and glassware must be scrupulously dry. Otherwise, a significant amount of the starting acid is regenerated due to hydrolysis of the reaction intermediates. Additionally, the sensitivity of silver salts to light poses another hurdle to their preparation in a pure form. The preferred solvent for the reaction is carbon tetrachloride, which is industrially unacceptable due to its toxicity and environmental concerns. Other solvents, such as chloroform, dichloromethane, dichloroethane, carbon disulfide, chlorobenzene, and nitrobenzene, among others, might be useful to some extent, although the yields are inferior to those using carbon tetrachloride. Moreover, the reaction employs strong oxidants in the form of elemental halides, which limits the method to rather simple substrates. The small number of chlorodecarboxylation examples is dictated by the inconvenience of using gaseous chlorine as well as the extensive radical chlorination of the materials, which is not suppressed even at −70 °C.27 Mainly, the reaction is performed under ambient conditions, although the employment of an inert atmosphere may be beneficial in some cases.
2.2. Modifications of the Hunsdiecker–Borodin Reaction
The work of Hunsdiecker stimulated an investigation into alternatives to the reaction, mainly due to the high sensitivity of the method toward moisture. In an analogy to the known process, salts of other metals were tested in an attempt to replace silver. Thus, salts of mercury(I), mercury(II), and thallium gave good results, although the yields were reduced, compared to those of silver. Salts of alkali metals such as sodium and potassium also showed encouraging results, but the lower reactivity of these salts required longer reaction times and higher temperatures, leaving room for side reactions to occur. Attempts to use salts of barium, copper, calcium, zinc, cadmium, magnesium, aluminum, and nickel resulted in the desired product, although the yields were synthetically irrelevant in many cases.
An interesting approach was developed by Rice in order to overcome the problem of sensitivity to moisture.63 Since the main problem is the presence of moisture in the silver salt, it was proposed to prepare the salt in situ by reacting intrinsically dry acid chloride with silver oxide in the presence of bromine.63 Such an approach was effectively applied to the bromodecarboxylation of gluconic acid pentaacetate 95, to give the bromide 96 in 86% isolated yield (Scheme 17). This approach was further extended toward aromatic acid chlorides, such as benzoyl, nitrobenzoyl, and naphthoyl chlorides.64 While nearly quantitative yields, based on the evolved carbon dioxide, were estimated, no isolated yields were given. The mechanism of this transformation is unclear, although the authors proposed the formation of a silver salt in situ, with further decomposition by bromine as in the classical Hunsdiecker reaction. However, some skepticism was expressed regarding such a scenario, since only traces of the silver carboxylate were obtained by subjecting the acid chloride to silver oxide under the reaction conditions in the absence of Br2. Noticeably, attempts to isolate an aryl bromide upon re-examination of the reaction by other researchers resulted in only trace amounts of the product.65
Scheme 17. Decarboxylation of an Acid via Acyl Chloride with the Aid of Silver Oxide.

2.2.1. The Cristol–Firth Modification
During the mechanistic studies of the Hunsdiecker reaction, Cristol and Firth attempted a different approach to the intermediate acyl hypobromites.66 By the treatment of a carboxylic acid and a slurry of red mercury(II) oxide in CCl4 with equimolar amounts of Br2 in the dark, a smooth formation of the alkyl bromide took place. Thus, stearic acid was converted to the corresponding bromide 97 in a 93% crude yield. Other compounds, such as bromocyclopropane 98 and 9-bromo-9,10-dihydro-9,10-ethanoanthracene 99 were prepared in fair to excellent yields; yet, the values were not specified (Scheme 18). The reaction of glutaric acid yielded 1,3-dibromopropane in a low yield, but contrary to the reaction of the corresponding silver salt, the formation of γ-butyrolactone was not observed. Performing the reaction in the presence of light caused excessive radical bromination of the aliphatic chain.
Scheme 18. Examples of Cristol–Firth Modification of the Hunsdiecker Reaction.

The reaction formally proceeds according to eq 4, meaning that one mole of water is formed during the process. However, contrary to the extremely moisture-sensitive Hunsdiecker reaction, the formed water does not interfere with the reaction progress. When an acid 79c is subjected to the Cristol–Firth reaction, the same ratio (80a: 80b 71:29) of bromides is obtained as in the case of silver salts (Scheme 13), suggesting that the reaction might proceed via the same acyl hypobromite intermediate.67 The authors have also assumed the intermediacy of some other positive bromine species to explain the higher tolerance toward moisture compared to the reaction of the corresponding silver salts.
| 4 |
2.2.1.1. Scope of the Reaction
Despite the absence of comprehensive studies on the scope of the reaction and its comparison to the original process using silver, the Cristol–Firth modification was extensively used as a method for the selective introduction of a halogen. The reaction was used almost exclusively to introduce bromine, and very few examples of the corresponding iododecarboxylation are reported.66,68−71 Examples of the chlorination reaction were surprisingly absent, likely due to the inconvenience of using chlorine gas.
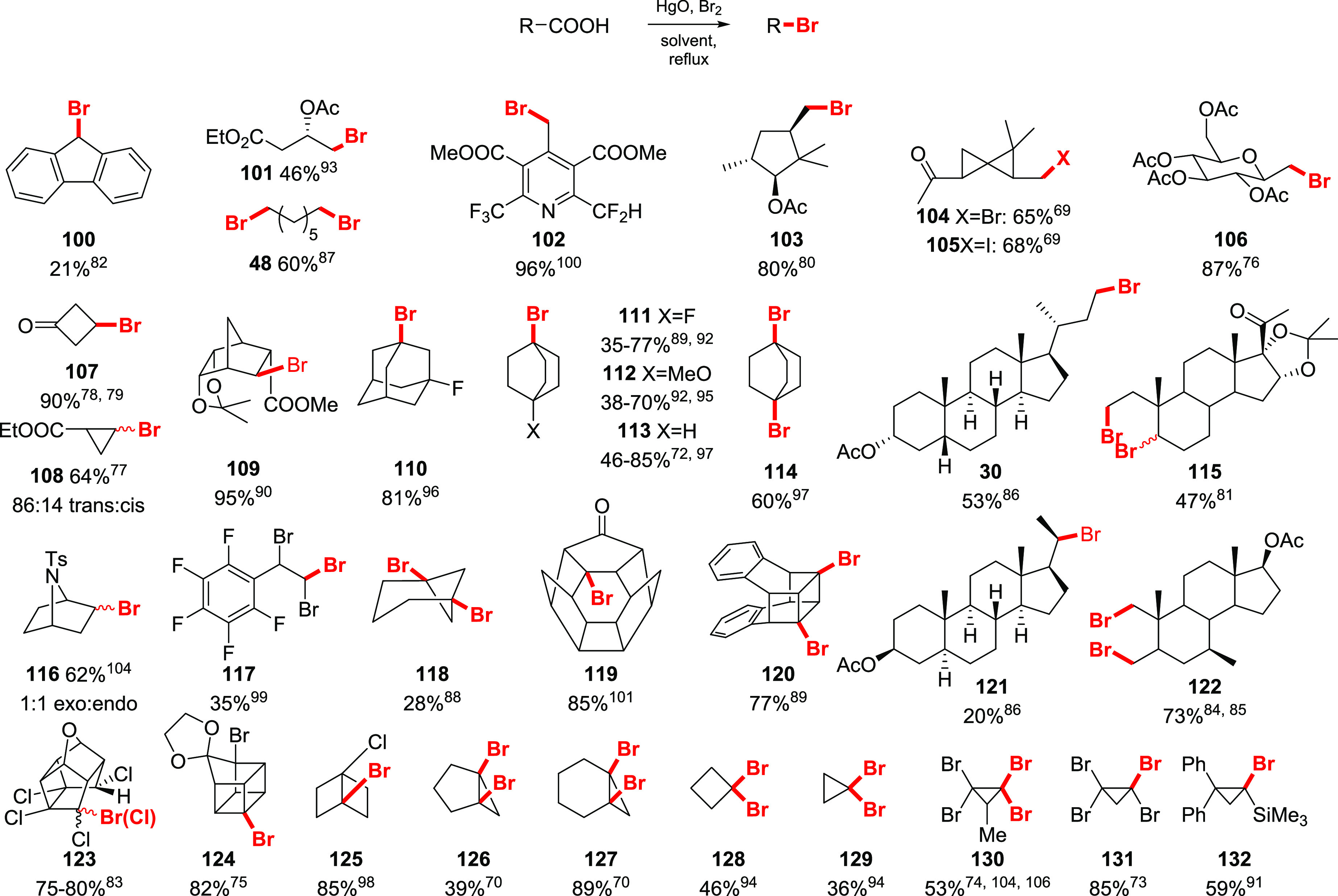
In Scheme 18 and Scheme 19, the scope of the Cristol–Firth reaction, reported throughout the literature, is combined. The scope of the bromodecarboxylation of aliphatic acids is similar to that of the classical Hunsdiecker reaction. Notably, the primary acids were observed to be less involved in radical side reactions, providing the highest yields of alkyl bromides, in a range 70–95% (Scheme 19). The bromodecarboxylation yields of secondary carboxylic acids are lower, compared to the primary counterparts. Tertiary bromides are usually not formed in more than a trace amount since rapid decomposition in the presence of metal salts was observed.27 Only the rigid caged systems deliver corresponding tertiary bromides in good to excellent yields (e.g., 110, 111–114). Unlike the reaction of silver salts, attempts to introduce iodine in the bridgehead position via this approach usually led to the formation of the Simonini ester as a sole product, and no alkyl iodide was observed.72 The reaction demonstrated a broad functional group compatibility. Dicarboxylic acids furnished dibromides as the main product, although, unlike under the Hunsdiecker conditions, they are less prone to form a lactone, even if δ- or γ-lactone is the estimated side product.
Scheme 19. Selected Examples of Cristol–Firth Halodecarboxylation of Aliphatic Acids69,70,72−106.
Aromatic bromodecarboxylation proved to be more problematic in this approach, compared to the classical Hunsdiecker reaction. Despite the work of Davis,65 who claimed good reactivity of both electron-rich and electron-poor aromatic acids (he based the yields on the evolved CO2 rather than on the obtained aryl bromide), later reports revealed that isolated amounts of the aromatic bromides were unacceptably low.66,107 The only aromatic acids that could furnish synthetically useful amounts of the product were nitro-substituted benzoic acids (Scheme 20). Benzoic and p-toluic acids furnished negligible amounts of the corresponding bromides 57 and 60 in 11 and 14% yield respectively, while 4-chlorobenzoic acid failed to produce any product at all. The poor yields were attributed to the sluggish solubility of carboxylates in the reaction medium.107,108 Light irradiation significantly improved the reaction rates and yields of aromatic bromides.109 Thus, various electron-poor acids underwent the reaction in good to excellent yields. Electron-rich substrates, as well as naphthoic acid, gave only polybrominated products. Some heteroaromatic carboxylic acids, such as nicotinic acid, were found to be suitable substrates for decarboxylative halogenation.110 In general, the Cristol–Firth modification provides slightly higher yields than the original Hunsdiecker reaction for electron-poor substrates, although the performance of electron-neutral and especially electron-rich acids is significantly inferior.
Scheme 20. Selected Examples of the Halodecarboxylation of Aromatic Acids by the Cristol–Firth Modification.

Reaction performed in nitrobenzene at 180 °C; hv–reaction was irradiated with light.
2.2.1.2. Mechanism of the Reaction
There were a number of speculations about the reaction mechanism. While the process seems to behave similarly to the original Hunsdiecker–Borodin reaction, the intermediacy of mercury salts was initially questioned, due to Hunsdiecker’s report on the inferiority of mercury salts compared to silver.24,25 Since the intermediacy of an acyl halide was anticipated, its formation was expected to take place in a different way. The reaction of mercuric oxide with Br2 was anticipated to give rise to the formation of some “positive bromine species”, which are responsible for the bromodecarboxylation.66 Bromine(I) dioxide was proposed in the role of these species (Scheme 21, pathway 1).66,111 The plausibility of this assumption was supported by Jennings and Ziebarth, who isolated the bromine dioxide and performed the degradation of pentanoic acid by means of Br2O, to butyl bromide, in a 30% yield.111 Despite the observation of the product in this reaction, some inconsistencies were noted. The first disagreement comes from the outcome of the reaction at different temperatures. Performing the Cristol–Firth reaction at ambient conditions generally results in sluggish yields of the desired bromides, while elevated temperatures lead to maximum yields. This behavior is counterintuitive since the intermediate Br2O possesses poor stability and slowly decomposes, even at −50 °C. Following this assumption, the iododecarboxylation reaction should proceed, in turn, through iodine(I) oxide—an even more unstable and elusive intermediate. Additionally, while bromodecarboxylation proceeded quickly and was essentially quantitative, the reaction between HgO and Br2 to give Br2O was very slow and incomplete and required low temperatures to obtain a noticeable amount of the oxide.
Scheme 21. Possible Reaction Pathways in the Cristol–Firth Bromodecarboxylation.

Another plausible and well-accepted pathway includes the in situ formation of a mercury salt of the acid followed by reaction with Br2 to give the acyl hypobromite (Scheme 21, pathway 2). Many reports indicate that the mercury salt is likely an intermediate in the Cristol–Firth method.107 Following the salt formation, the reaction mechanism is anticipated to be identical to that of the classical Hunsdiecker reaction, meaning formation of the acyl hypohalite and its further transformation to the alkyl bromide.
The solubility of the salt in the reaction medium was considered as one of the main factors responsible for a good reaction. Most of the aliphatic acids are suitable substrates for the transformation and upon reaction with mercuric oxide provide salts of the type (RCOO)2Hg, which were denoted as “normal”.107 However, some acids, such as phenylacetic acid, failed to produce the organobromide, having a very poor solubility in boiling CCl4. Other, mainly aromatic acids, with poor reactivity, were found to be insoluble in the reaction medium. It was found that these acids form complex salts of general structure (RCOO)2Hg·HgO with an uncontrollable acid/HgO ratio, which possess poor solubility in the reaction media.
Even though in the initial work Cristol and Firth reported excessive chain bromination if the reaction is not carried out in the dark, in many instances additional light irradiation was found to be beneficial for the course of the reaction.109 Irradiation with a tungsten lamp allowed the preparation of both aryl and alkyl bromides. While the effect of light has not been systematically studied, it was widely applied in this transformation.
Despite the formation of water upon the reaction of HgO with an acid, the reaction tolerates moisture and, contrary to the original Hunsdiecker reaction, provides the desired product in high yields in the presence of H2O. Because of their poor solubility, silver salts mainly react heterogeneously, while the reaction between (RCO2)2Hg and bromine likely proceeds homogeneously. This leads to the formation of an acylhypohalite in the anhydrous body of the solvent, which continues to the desired alkyl bromide. In the case of silver salts, the acyl hypohalite is formed on the border between the phases and is more likely to be hydrolyzed by water, which is immiscible with the solvent.
This hypothesis was supported by control experiments, comparing the outcome of the reaction of silver and mercury salts of valeric acid versus mercuric oxide/acid mixture, with Br2, in dry or “wet” solvents possessing different miscibility with water, at different temperatures (Table 2). The bromodecarboxylation reaction, employing the mercuric oxide/acid system, proved to be unaffected by the presence of water under different reaction conditions. At reflux temperatures in CCl4, the reaction provides bromobutane in a good yield, while at ambient conditions the reaction performance drops significantly. The reaction in acetonitrile proceeds poorly in any conditions, being a result of the miscibility of water with the reaction medium, which apparently results in a faster hydrolysis of the intermediates. The reaction of the silver salt with Br2, in turn, significantly deteriorated in the presence of moisture. The reactivity of mercuric salts resembles the performance of their silver counterparts in dry solvents, while in wet conditions the yields are similar to those of mercuric oxide/acid combinations.
Table 2. Influence of Water on the Bromodecarboxylation of Valeric Acid and Its Salts.

Three equiv of water added.
On the basis of the evidence, Bunce107 suggested three factors for a successful outcome of the reaction: (1) the substrate acid should form a salt, soluble in the reaction medium; (2) water-immiscible solvents are preferred; and (3) elevated temperatures are beneficial for a good yield.
2.2.1.3. Limitations and Features
The Cristol–Firth reaction represents a certain modification of the original Hunsdiecker reaction. However, from a synthetic standpoint, both transformations have similar intrinsic limitations: incompatibility with (a) multiple C–C bonds in the skeleton not conjugated with a carboxylic group, and (b) electron-rich aromatics. The moieties that are sensitive toward strong oxidants are also not compatible with the transformation. Usually, the yields of the Cristol–Firth modification are slightly lower compared to the original modification. Nevertheless, the Cristol–Firth modification represents a significant advance in the development of the halodecarboxylation reaction since it overcomes the severe moisture sensitivity of silver salts. An additional removal of the moisture by distillation or the addition of a desiccant helps to further improve the yield, which makes the Cristol–Firth modification synthetically attractive in comparison to the original Hunsdiecker reaction.70,89,97,108 Also, the possibility of performing double halodecarboxylation on dicarboxylic acids, without the formation of a lactone, is a substantial advantage.
The major disadvantage of the Cristol–Firth method is the employment of highly toxic mercury oxide, which precludes this reaction form use in modern synthesis of drugs and bioactive candidates. While red mercury oxide is usually employed, the yellow form has proved to be equally effective. Attempts to employ oxides of less toxic metals (silver, cadmium) led to significantly inferior results.65 CCl4 was established as the most useful solvent for primary or secondary aliphatic acids. For the preparation of bridgehead tertiary bromides, brominated solvents, such as dibromomethane or 1,2-dibromoethane, are preferred in order to avoid possible Cl-radical transfer from the CCl4.72,112 Although the reaction can be performed under air, an inert atmosphere may be beneficial for the reaction outcome. In some cases, light must be excluded in order to suppress extensive chain bromination; however, for electron-poor aromatic acids, additional light irradiation is important to achieve acceptable yields of aryl bromide. The success of the double halodecarboxylation of 1,2-bridgehead dicarboxylic acid also depends on efficient light irradiation.70,102,103
2.2.2. The Kochi Reaction
Despite the extensive development of the Hunsdiecker–Borodin and Cristol–Firth halodecarboxylation reactions, the synthesis of chlorinated materials was underrepresented. The use of gaseous chlorine and excessive radical chlorination forced researchers to seek alternatives. The problem was solved in 1965 by Kochi, who was the first to employ an ionic source of a halogen in the Hunsdiecker-type transformation.113,114 Upon treating the carboxylic acids with lead tetraacetate and lithium chloride in refluxing benzene a number of short-chain primary, secondary and tertiary alkyl chlorides were obtained in good to excellent yields (Scheme 22). Benzoic acid gave only 8% of chlorobenzene.
Scheme 22. First Examples of Chlorodecarboxylation by Kochi.

3–6 equiv of acetic acid were added; GC yields based on amounts of Pb(OAc)4.
Even though the employment of Pb(OAc)4 in a halodecarboxylation reaction was largely developed by Kochi, an early example by Barton of the use of lead salt in such a transformation should be noted.115,116 Inspired by the work of Wettstein on the preparation of alcohol hypoiodites with lead tetraacetate and iodine,117 it was suggested that treatment of an acid with Pb(OAc)4 and I2 might result in the desired acyl hypoiodite, which can further degrade to an organoiodide. Indeed, upon light irradiation, the reaction between a carboxylic acid with a nearly equimolar amount of Pb(OAc)4 and iodine proceeded smoothly, to give primary and secondary alkyl- and aryl iodides in good yields (Scheme 23). Comparable yields might be achieved with silver or mercury acetates, although more than a 3-fold excess of the metal salt and iodine is required.116
Scheme 23. Early Examples of the Employment of Lead Tetraacetate in the Iododecarboxylation Reaction by Barton.

2.2.2.1. Modifications of the Kochi Reaction
The original Kochi procedure is suitable for the preparation of primary and secondary alkyl chlorides. Tertiary alkyl chlorides are obtained in significantly lower yields, especially on a large scale.118 The problem with the preparation of tertiary chlorides was addressed by Grob and co-workers.118 The use of N-chlorosuccinimide (NCS) as a source of the chlorine atom instead of LiCl, and applying a DMF/acetic acid solvent system, made the preparation of various non-bridgehead tertiary alkyl chlorides possible. It allowed the chlorodecarboxylation to be performed at lower temperatures (compared to the original process), enabling the synthesis of a number of heat-sensitive tertiary chlorides in reasonable yields (Scheme 24). Secondary carboxylic acids are also converted to the chlorides in good yields, although primary alkyl chlorides are obtained only in poor yields. Aromatic acids do not react at all (compound 147).
Scheme 24. Examples of the Grob Modification of the Kochi Reaction.

2.2.2.2. Scope of the Reaction
The Kochi reaction is suitable for the preparation of a wide variety of structure-independent alkyl chlorides. In many cases, a combination of lead tetraacetate and lithium chloride gives satisfactory yields of the desired chloroalkane (Scheme 22 and Scheme 24). The synthesis of the corresponding alkyl bromides leads to significantly lower yields and is accompanied by substantial amounts of bromine. Employment of the iodide salt in this transformation furnishes trace amounts of alkyl iodides, producing the ester as a main product, along with considerable amounts of iodine. Iodides could be efficiently prepared in reasonable yields by the Barton modification, utilizing I2 instead of an iodide salt (Scheme 23 and Scheme 25).
Scheme 25. Selected Examples of the Kochi Reaction and Its Modifications97,119−143.

Grob modification, DMF/AcOH 5:1 as a solvent.
Barton modification.
Acetate protecting group was partially removed.
THF as a solvent.
Pyridine as a solvent.
The Kochi reaction is capable of transforming primary secondary and tertiary carboxylic acid to the corresponding alkyl chlorides (Scheme 25). Unlike the Hunsdiecker and Cristol–Firth reactions, the process is not restricted to bridgehead tertiary carboxylic acids, although the yields of tertiary chlorides are relatively low.118 The aforementioned Grob modification allows the preparation of sensitive non-bridgehead tertiary alkyl chlorides in good to excellent yields. The Barton approach can furnish tertiary alkyl iodides, although in very low yields. The Kochi reaction possesses a substantial functional group tolerance. Ethers, esters, ketones, secondary and tertiary amides, among others, are compatible with the reaction conditions. Silyl-,144 formyl-, and acetoxy-protecting groups are well tolerated; however, the acetoxy protective group is cleaved under the Grob reaction conditions.119 Noticeably, the unsaturated moieties are tolerated (176, 184, 185), unless the double bond is favorably located toward intramolecular radical cyclization. Interestingly, suitably located unprotected hydroxy groups do not affect the course of the reaction (179).127 The chlorodecarboxylation of alpha-substituted carboxylic acids was shown to proceed in good yields. Thus, α-chloroamide137169 and 2-chloro-oxetane125170 derivatives were successfully prepared. The synthesis of germinal iodo,chloro- and iodo,bromoalkanes was attempted via iododecarboxylation of α-chloro and α-bromopropionic acids under Barton conditions.141 However, the yields of these products were substantially low (186–188). Some dichloroalkanes were prepared from the corresponding diacids in moderate yield (165, 174, 175); however, the chlorodecarboxylation of 1,2-dicarboxylic acids usually furnished the products in low yields.122,135,140 Interestingly, even quadruple, one-pot chlorodecarboxylation was achieved in 20% yield (182).122
Very few results of the halodecarboxylation of aromatic acids by the Kochi reaction and its modifications are reported. Some examples of the iododecarboxylation of aromatic acids via the Barton approach, resulting in moderate yields, are presented in Scheme 26.145,146 Attempts to perform the chlorodecarboxylation of aromatic acids by either the Kochi or Grob approach failed or provided negligible yields.113,114,118,147 Therefore, we can conclude that this reaction is, most likely, not a method of choice for the preparation of aryl halides.
Scheme 26. Examples of Aromatic Halodecarboxylation by Kochi Reaction and Its Modifications.

Conditions: Pb(OAc)4, LiCl, benzene, reflux.
Pb(OAc)4, NCS, DMF/AcOH 5:1, 40 °C.
Pb(OAc)4, I2, benzene, reflux.
in the presence of 3 mol % AIBN, CCL4 as solvent.
Br2 instead of I2.
2.2.2.3. The Reaction Mechanism
Comprehensive studies by Kochi have shed light on the mechanism of the reaction.113,114 The reaction conditions, behavior and functional group compatibility offer the proposition of a mechanism that is different from the Hunsdiecker reaction. For instance, as apparent from the scope of the Kochi reaction, alkene groups are well tolerated in the Kochi procedure, presuming the absence of an acylhypohalite as the key reaction intermediate. Additionally, contrary to the Hunsdiecker reaction, the Kochi process is readily quenched in the presence of oxygen, indicating another important mechanistic difference of the reaction.
The extensive experimental data provide strong evidence that the chlorodecarboxylation reaction proceeds via a radical mechanism.148,128,149,150 In light of this, Kochi proposed the mechanism depicted in Scheme 27. Initially, the reaction mixture undergoes pre-equilibration, with exchange of the carboxylate ligands on the Pb center, producing a mixed lead carboxylate 191. Further coordination of the halide ion produces the unstable -ate complex 192. Two alternative decomposition routes of 192 are possible. In the first pathway, the main chain sequence is initiated by decomposition of the complex 192 with the evolution of carbon dioxide to give the alkyl radical and the intermediate complex 193. Such type of decomposition of (RCO2)4Pb, accelerated by halide coordination, is well documented.151,152 The carbon-centered radials are prone to react with an additional molecule of 192, furnishing the final alkyl halide and generating the chain-propagating Pb(III)-species 194. The chain process may be terminated upon the reaction of a carbon-centered radical with 193. Alternatively, a homolytic cleavage of the lead-halogen bond in the complex 192 might take place, giving rise to a halogen radical and complex 194. The halogen radical may initiate chain propagation by a reaction with 192, giving the lead salt 194 and the elemental halogen.
Scheme 27. Proposed Mechanism of the Kochi Reaction.

Fragmentation of the plumbate 192 depends on the nature of the carboxylate ligands and the nature of the halide. The lead complex carrying a chloride ligand predominantly undergoes fragmentation of the Pb-carboxylate bond. In the case of iodides, extensive fragmentation of the Pb–I bond might represent a significant side process, with the concurrent formation of the elemental halogen. This assumption rationalizes the formation of nearly quantitative amounts of iodine (which remains intact until the end of the reaction) and only traces of the alkyl iodide upon attempts of a Kochi-type iododecarboxylation. Significant amounts of Br2 are also observed upon employment of the bromine salts, although this disappears during the reaction, presumably by radical bromination of the solvent, acid, etc. Small amounts of Cl2 could also be detected during the reaction of chlorides. The fragmentation of the carboxyl moiety is believed to occur via concerted double bond cleavage, generating an alkyl radical and carbon dioxide. The selectivity of the fragmentation in the carboxylates series follows the order of the carbon-centered radical stability: phenyl < methyl < primary < secondary < tertiary.
The mechanism of the Barton approach is postulated to be similar to the halide-induced reaction (Scheme 28). Since the reaction involves the use of elemental iodine, it was envisioned to be initiated by the light-induced dissociation of I2 to the iodine radicals. Reaction of the I-radical with lead(IV) carboxylate 191 results in the formation of the acyl hypoiodite 85 and the unstable lead(III) intermediate 196, which, in turn, decomposes, with the production of the alkyl radical. Scavenging of the alkyl radical with iodine provides the target alkyl iodide and the iodine radical that propagates the chain.
Scheme 28. Proposed Reaction Pathway of the Barton Approach.

2.2.2.4. Limitations and Features
The major drawback of the Kochi reaction is its use of the highly toxic lead salt as a key reagent. However, the Kochi reaction exhibits significant distinctions from the original Hunsdiecker method. Unlike the Hunsdiecker and Cristol–Firth reactions, the Kochi approach is synthetically useful for the preparation of a variety of alkyl chlorides. While the requirement for a corrosive Cl2 gas was overcome, Pb(OAc)4, used in the reaction as a key reagent, also possesses strong oxidative properties.
Although LiCl is used as a convenient source of halide, the requirement for a high excess has a deleterious effect on the reaction outcome. A large excess of chloride boosts the formation of elemental chlorine. For example, the use of 1.05 equiv of LiCl (with respect to Pb(OAc)4) in the chlorodecarboxylation of valeric acid provides butyl chloride in a 93% yield. With 2.3 equiv of LiCl, the yield drops to 63%, while with 4 equiv, the evolution of copious amounts of Cl2 is observed, and the product is obtained in only a 4% yield.113 The observed phenomenon is general for various acids, but the effect is more pronounced for the reaction of primary acids rather than for the secondary or tertiary counterparts. LiCl is not the only salt that can be used in the reaction. Tetramethylammonium, sodium, potassium, and calcium chlorides give acceptable yields of the corresponding alkyl chloride. While benzene is considered to be the most suitable solvent for the original Kochi reaction, other solvents such acetonitrile, pyridine, and THF could be used in some specific cases.131,140,126
Notably, minor amounts of oxygen readily halt the process, so the reaction must be conducted under an inert atmosphere. Although the reaction does not have to be scrupulously dry, moisture-free solvents were preferred in many cases. The halide salt also does not need to undergo excessive drying; yet, in some cases additional drying of the salt was performed.134
2.3. Metal-free Methods for Halodecarboxylation
The Hunsdiecker reaction and its modifications provided a valuable pioneering input for establishing the field of halodecarboxylation. Detailed studies of the reaction mechanism shed light on possible reaction intermediates and the role of the metal in these transformations. In the above-discussed methods, the role of the metal could be elucidated as (i) a precursor to the key acylhypohalite intermediate (silver, mercury, etc.) or as (ii) a strong oxidant that facilitates the concerted fragmentation with the evolution of CO2 to generate the carbon-centered radical (Pb(IV)). The use of metals dictates a number of operational and mechanistic limitations. The major, severe operational limitation is associated with the high toxicity of the employed heavy metals. Contamination of a product with traces of these heavy metals, as well as the problem of waste disposal, especially on a large scale, significantly limits the application of these methods in modern synthesis. An initial heterogeneous reaction medium and/or the formation of a heavy precipitate could affect the yield of the reaction and create difficulties in scaling up the process. Even though the Hunsdiecker reaction proceeds via a free radical mechanism, characteristic carbocation rearrangement products were observed in many cases with sensitive substrates due to the interaction of the product with the metal species. Furthermore, attempts to prepare sensitive substrates such as non-bridgehead tertiary alkyl halides generally failed, while the formation of esters, or decomposition of the product, was observed, due to the presence of the metals. All these factors led researchers to investigate alternative metal-free approaches for the halodecarboxylation reaction.
The first substrate independent, metal-free, approach for iododecarboxylation was developed by Barton. Working on iododecarboxylation of the carboxylic acids with lead(IV) acetate and iodine, Barton and co-workers came up with an alternative way of preparing acyl hypohalites, with no need for heavy metal precursors.116 It was shown that the treatment of a carboxylic acid with tert-butylhypoiodite, prepared from potassium tert-butoxide and iodine, accompanied by light irradiation resulted in the formation of an alkyl iodide. Acyl hypohalite is anticipated as an intermediate in the reaction, which decomposes under irradiation to give the alkyl iodide. Notably, the yields of the products are comparable, and in some cases are even superior, to those of the Pb(OAc)4/I2 system (Scheme 29). Thus, the yields of primary and secondary alkyl iodides are slightly lower, while the iododecarboxylation of pivalic, glutaric, and adipic acids is significantly better than the previously reported Pb-based approach. In the cases of poor acid solubility in the reaction medium, sulpholane or small amounts of DMF were found to be useful cosolvents, while DSMO or pyridine destroyed the reagent. Substrates bearing unprotected hydroxy groups are not suitable, since tert-butylhypoiodite readily reacts with free alcohols. Alkene groups are mainly unaffected under the reaction conditions.
Scheme 29. Selected Examples of the Barton Iododecarboxylation with t-BuOI.

Reaction performed in a benzene–sulpholane mixture at 55–80 °C.
Isolated yields. Yields based on the evolved CO2 are given in parentheses.116,153−155
Because of its low stability, tert-butylhypoiodite cannot be obtained in a pure form, so it is usually prepared in situ prior to use. It was observed that the structure and reactivity of the reagent are significantly influenced by the method of preparation.156 Goosen and co-workers during their studies of the light-assisted iododecarboxylation reaction prepared tert-butylhypoiodite by treating tert-butylhypochlorite with iodine, and the major product of the reaction was the alkyl chloride.157 A series of 4-substituted phenylacetic acids was subjected to the reaction conditions to provide the corresponding benzyl chlorides in excellent yields, accompanied by minor amounts of esters (Scheme 30). Investigation of this unexpected exchange indicated that the initially formed benzyl iodide product reacts with iodine monochloride (ICl), formed in the course of the reaction, to give the corresponding benzyl chloride. Cycloalkylcarboxylic acids of various ring sizes also react smoothly to give cycloalkyl chlorides in usually good yields. The reaction rates were found to be in the order CH3 > H > I > Cl > Br > NO2 for the aryl substituent, indicating that radical stability is not the major factor controlling the reaction rate. The ρ value of −1.09 corresponds to a free radical process, and correlation with σ+ suggests that the reaction rates are influenced by a polar effect. The decomposition of acylhypoiodites was concluded to be a combination of two elemental steps, namely, homolytic cleavage of O–I, followed by the evolution of CO2 to form an alkyl radical.
Scheme 30. Examples of Iododecarboxylation with t-BuOCl + I2.

2.3.1. Suarez Halodecarboxylation
Suarez and co-workers discovered that the Pb(OAc)4/I2 system can be effectively substituted with phenyliodine diacetate/I2 without compromising the yield of the reaction.158,159 Indeed, irradiation of carboxylic acids with nearly equimolar amounts of phenyliodine diacetate (PIDA) and iodine, under reflux, gave very good yields of primary and secondary steroid-derived alkyl iodides (Scheme 31). Interestingly, utilization of lithium chloride instead of iodine makes it possible to perform the chlorodecarboxylation reaction, although in very small yields (compound 206, 15%). Increasing the amounts of PIDA and LiCl to 5 equiv raises the yield of the reaction to 40%. Tertiary carboxylic acids gave mixture of alkenes, and the desired iodide was not detected.
Scheme 31. First Examples of Suarez Iododecarboxylation Reaction.

One equiv of LiCl instead of I2, benzene as solvent.
Five equiv of PhI(OAc)2 and LiCl instead of I2, benzene as solvent.
2.3.1.1. Scope of the Reaction
The Suarez halodecarboxylation reaction proved to be a useful method, mainly for the preparation of aliphatic iodides (Scheme 32). Alkyl bromides could also be prepared by a similar approach, although a large excess of PIDA and Br2 is required, and yields are inferior, compared to those of the corresponding iodides.160 Very few examples of chlorodecarboxylation, using a combination of PIDA/LiCl, were reported, although the yields were low.159 The yields of primary alkyl iodides are generally higher than secondary iodides, while the iododecarboxylation of tertiary carboxylic acids results in a mixture of alkenes rather than in the desired products. Similar to other radical halodecarboxylation reactions, the preparation of bridgehead alkyl iodides is attainable in high yields. Such functional groups as alkenes, alkynes, esters, ethers, ketones, and azides tolerated the reaction conditions. Alcohols and amines must be protected. The one-pot iododecarboxylation of di- and tetracarboxylic acids was achieved (compounds 220, 221, 223). The reaction was also applied to prepare iodinated single-wall carbon nanotubes.161
Scheme 32. Selected Examples of Suarez Iododecarboxylation70,160,162−181.

Recently Uchiyama and co-workers applied PIDA in combination with KBr under fluorescent light irradiation to perform a bromodecarboxylation reaction. Additionally, (difluoroiodo)benzene can be used to increase the yields of some aliphatic bromides. Even though the reaction is quenched in the presence of radical scavengers, implying a radical nature of the process, bromomethylcyclopropane was obtained in 56% yield. The authors suggest that the bromination of an alkyl radical is a significantly faster process than the opening of the cycolopropylmethyl radical (kabs(37 °C) = 1.2 × 108 s–1).
The halodecarboxylation of aromatic acids can be achieved using a PIDA/halogen mixture (Scheme 33). The reaction is limited to electron-neutral and electron-poor substrates. Low yields of electron-rich aryl iodides were obtained, while bromides failed to give any product at all. Notably, the reaction tolerates unprotected aromatic aldehydes and alkynes (229, 227). The double iododecarboxylation of 1,8-naphthalenedicarboxylic acid proceeds smoothly without the formation of an anhydride (see compound 232). Remarkably, the iododecarboxylation of electron-rich aromatic acids could be achieved employing LiI instead of I2, in trifluoroethanol or hexafluoroisopropanol as the solvent (230, 85).182 Although the reaction is not selective, and excessive ring halogenation is observed, quenching the reaction in the early stages allows the isolation of the iododecarboxylation products in useful yields. Interestingly, the iododecarboxylation of meta-methoxy or electron-poor carboxylic acids does not take place. The preparation of electron-rich bromoaromatics is possible, although ring bromination precedes the bromodecarboxylation step, making this side process unavoidable under the current reaction conditions.
Scheme 33. Examples of Halodecarboxylation of Aromatic Acids by the Suarez Approach160,182,183.

Reaction with PhI(OAc)2 and LiX in HFIP.
Reaction with PhI(OAc)2 and Br2 in dibromomethane; asterisk indicates an iodine introduced via electrophilic iodination.
2.3.1.2. Mechanism of the Reaction
It is proposed that the Suarez reaction proceeds via a radical pathway. PIDA is known to possess poor thermal stability, forming iodobenzene and acyloxy radicals upon decomposition. In a pre-equilibration step, one or two acetoxy groups on PIDA undergo exchange with the substrate acid, and the new λ3-iodane consequently undergoes thermal or photolytic decomposition (Scheme 34). Depending on their nature, acyloxy radicals produce either carbon-centered radicals by the rapid extrusion of carbon dioxide or, in the case of aromatic acids, react with iodine to produce acyl hypoiodite.7 Acyl hypoiodite can be indicated as an intermediate in the reaction by 1H NMR.184 The obtained carbon radicals are quenched with iodine, furnishing alkyl iodides as the final product. The adduct of carbon-centered radicals to a spin-trap was observed by ESR spectroscopy.185
Scheme 34. Plausible Mechanism of the Suarez Reaction.

In the case of the employment of halide, the mechanism is not reported. We can assume that the reaction would involve a pre-equilibration to (halo)(acetoxy)iodobenzene 236 (Scheme 34). Its decomposition in a radical manner gives the acyloxy species with the concomitant formation of a carbon-centered radical and iodine(II) intermediate 237. Abstraction of a halogen from 237 or 236 by a carbon-centered radical provides the targeted organohalide.
2.3.1.3. Features and Limitations
The facile experimental setup and absence of heavy metals have made the Suarez reaction one of the most convenient approaches for the preparation of organoiodides via the decarboxylation approach. Despite the intermediacy of an acyl hypoiodite, suitably located double bonds in the substrate backbone are well tolerated since a large excess of reagents is avoided and hypoiodites are readily decomposed under thermal conditions and strong light irradiation. However, alkene motifs are likely to be affected during the bromodecarboxylation variant due to the employment of large excess of the reagents. Unprotected hydroxy and amino groups do not tolerate the reaction. Secondary protected amines are compatible with the reaction conditions, although the yields are lower than in the case of the corresponding tertiary amines. The major side products are acetates and elimination products.159 For the preparation of nonpolar compounds, the presence of iodobenzene as a coproduct must be taken into account since the purification and separation of the product from PhI might be troublesome. The common solvents used for the reaction are CCl4, DCM, cyclohexane, or benzene. Acetonitrile was shown to be preferable when a poor solubility of the starting acid was encountered.174 On the other hand, DMF and methanol proved to be a poor choice of solvent. For the bromodecarboxylation reaction, dibromomethane was employed, to avoid the bromination of benzene as the predominant process.160 α,α,α-Trifluorotoluene was also found to be a useful solvent for the bromodecarboxylation.186 In some cases, iodinated products were isolated during the bromodecarboxylation reaction, arising from the abstraction of an iodine atom from the iodobenzene.160 Usually, the use of inert atmosphere and dry solvents is not required, although in many protocols the reaction was performed under nitrogen and in a dry solvent.
2.3.2. Barton Halodecarboxylation
A significant improvement in the halodecarboxylation process was achieved by Barton. While most of the investigations were focused on alternative ways to prepare an acyl hypohalite, Barton’s synthesis involved a novel approach for the generation of alkyl radicals from carboxylic acids, followed by their scavenging with an appropriate halogen donor. During their work on a reductive decarboxylation of N-acyloxy-2-pyridinethione with tributyltin hydride, Barton and co-workers discovered that this reaction proceeded via a free radical chain process with the tributyltin radical being a propagating species.116 It was anticipated that the employment of a reagent, which can serve as both a halogen donor and a source of the propagating radical species, would lead to the development of a halodecarboxylation reaction. Initially, such a reagent was found to be CCl4 or BrCCl3. Thermal decomposition of N-acyloxy-2-pyridinethione in these solvents led to the smooth formation of alkyl chlorides and bromides, respectively, in very high yields.
According to the Barton protocol, the starting carboxylic acid is converted to the acyl chloride, which reacts, in turn, with N-hydroxypyridine-2-thione 238 to furnish the corresponding N-acyloxy-2-pyridinethione 239 (Scheme 35). Barton and co-workers showed the general applicability of the designed reaction to prepare primary, secondary, and tertiary alkyl chlorides, bromides, and iodides, in high to excellent yields.187−189 Moreover, the bromodecarboxylation of aromatic and α,β-unsaturated carboxylic acids was achieved with minor modifications of the reaction conditions.
Scheme 35. Selected Examples of Barton Halodecarboxylation Reaction of Aliphatic Acids187,190−210.

2.3.2.1. Scope of the Reaction
The halodecarboxylation of N-acyloxy-2-pyridinethiones 239, so-called Barton esters, proved to be one of the mildest approaches to the preparation of organohalides from carboxylic acids. Wide functional-group tolerance and the absence of strong oxidants and transition metals resulted in the widespread application of the reaction as a valuable synthetic tool. Being developed almost 40 years ago, the reaction still finds a wide application in the multistep synthesis of natural products.
With respect to halides, maximum yields are obtained for the preparation of brominated products (Scheme 35). Corresponding chlorides and iodides can generally be obtained in lower yet useful yields. Primary, secondary, tertiary acyclic, cyclic, and bridgehead aliphatic carboxylic acids efficiently undergo the halodecarboxylation reaction. Although possessing a poor stability, sterically congested secondary and tertiary alkyl bromides and chlorides could be prepared in good yields.190 Numerous functionalities, such as esters, ketones, azides, skipped dienes, α,β-unsaturated ketones, protected amines and alcohols, thioethers, epoxides, and dioxaborolanes, among others, remain unaffected under the reaction conditions. Notably, unprotected β-hydroxy carboxylic acids undergo the halodecarboxylation reaction, although in moderate yields (264, 265, 273).191 Halodecarboxylation in proximity to the functional group or heteroatom was successfully achieved. For examples, α-bromoesters (271), 2-chloro-oxetane (71) and germinal fluorobromo and fluoroiodocyclopropanes (249, 250) were efficiently prepared.
The Barton halodecarboxylation can also be efficiently applied to aromatic acids (Scheme 36). Again, the highest yields are achieved in the preparation of aryl bromides, while aryl chlorides and iodides are obtained in lower yields. A wide variety of aromatic carboxylic acids are suitable substrates for the halodecarboxylation reaction. In contrast to other modifications, the absence of highly electrophilic reagents and intermediates allowed for the preparation of both electron-poor and electron-rich aryl halides. For example, eudesmic acid can be efficiently converted to the corresponding bromo or iodo derivative 280 and 281 in 62–40% yield. While deep analysis of the compatibility of the types of aromatic substituents in this reaction is not available, halogens, ester, and nitro functions are well tolerated. As for the substitution patterns, ortho-, meta-, and para-substituted benzoic acids perform well in the reaction. Even 2,6-disubstituted aryl bromide 289 can be obtained in a good yield. Derivatives of 2- and 3-bromopyridines 292 and 293 were successfully prepared via a double bromodecarboxylation reaction, although in rather poor yields. Interestingly, the approach is applicable to α,β-unsaturated acids to provide vinyl halides in very good yields (35, 294–297). The mildness of the reaction conditions allowed the preparation of the sensitive triene 296 in an acceptable yield.
Scheme 36. Selected Examples of Barton Halodecarboxylation Reaction of Aromatic Acids188,189,211−220.

2.3.2.2. The Reaction Mechanism
The success of the reaction can be attributed to its simple mechanism (Scheme 37). Initially, the acid is converted to the Barton ester in a variety of ways, followed by decomposition. As was anticipated by Barton and co-workers, N-acyloxy-2-pyridinethione 239 possesses a weak oxygen–nitrogen bond, which is prone to homolytic cleavage. This process is initiated either thermally or with the aid of light irradiation. The produced acyloxy radical undergoes degradation, with the evolution of carbon dioxide, to give a carbon-centered radical. Further reaction with an appropriate halogen donor (CCl4, BrCCl3, etc.) gives an organohalide and a stabilized chain-carrying radical species 298. Propagation of the chain is achieved by the reaction of 298 with another molecule of N-acyloxy-2-pyridinethione 239 by attack on a sulfur atom, facilitating its homolytic fragmentation. The adduct 299 was isolated in many reactions supporting the proposed mechanism. The quantum yield of the reaction is greater than 1 (Φ = 6–55), which is in line with a chain pathway.221
Scheme 37. Mechanism of the Barton Halodecarboxylation Reaction.

The efficacy of the reaction depends on the selectivity of the trapping of the radical 67 by the halogen source. Otherwise, this radical might be scavenged by the Barton ester producing 300 as a side product. Indeed, 300 is a major product when the reaction is performed in the absence of a halide source. The rate constant for the scavenging of an alkyl radical 67 with thione 239 is 2.3 × 106 M–1·s–1 (octyl radical, 50 °C, benzene).222 This implies that the rate constant for the halogen abstraction must be a few orders of magnitude higher, in order to avoid a large excess of a halogen source. For example, abstraction of bromine form BrCCl3 proceeds with rate of 2.8 × 108 M–1·s–1 (butyl radical, 80 °C, BrCCl3);223 therefore, only a slight excess of a few equivalents of BrCCl3 in an inert solvent is needed to achieve a good selectivity. On the other hand, the chlorodecarboxylation reaction has to be performed in neat halogen donor (e.g., CCl4, FCCl3) and in relatively dilute solutions since the rate of chlorine abstraction from CCl4 is 2.4 × 104 M–1·s–1 (octyl radical, 50 °C, CCl4).224 Even in this case, significant amounts of the adduct 300 are observed.
The halodecarboxylation of aromatic and α,β-unsaturated acids proved to be a more problematic process. While the chain radical process is essentially the same for both aliphatic and aromatic Barton esters, the aromatic halodecarboxylation reaction resulted in poor yields of aromatic bromides when using the conditions for the aliphatic counterparts. The main product of the reaction was identified as the acid anhydride accompanied by the disulfide 302. The process of N–O bond scission was estimated to be significantly slower in the series of aromatic esters 239, compared to the aliphatic counterparts. An alternative degradation pathway was proposed, leading to an anhydride (Scheme 38). Bimolecular decomposition of 239 presumably gives an arylcarboxylate and disulfide 301, followed by the concomitant attack of a carboxylate ion to result in the acid anhydride and disulfide 302. The decomposition of the Barton ester by the desired radical pathway was facilitated by adjusting the experiment setup. Slow addition of the acid chloride to the pyrithione sodium salt slowed down the ester formation, while an increase in the reaction temperature facilitated the unimolecular decomposition of the Barton ester. Moreover, addition of an external initiator, such as azobis(isobutyronitrile) (AIBN), was found to have a beneficial effect on the reaction (Scheme 39). Thermal decomposition of AIBN resulted in the formation of isobutyronitrile radicals, which, in turn, abstracts a halogen from the solvent (BrCCl3) giving an effective rise to the chain-carrying trichloromethyl radicals (Scheme 39).
Scheme 38. Decomposition of Barton Ester of Aromatic Acids.

Scheme 39. Generation of a Chain-Carrying Trichloromethyl Radical by Addition of AIBN.

A similar activation mechanism was proposed for a sonochemically activated reaction.225 The Barton ester itself was shown to be stable upon sonication in benzene solutions, even in the presence of AIBN, although a rapid reaction was observed in aliphatic halogenated solvents. It was found that CCl4 undergoes decomposition by sonication, giving chlorine and trichloromethyl radicals that initiate the reaction by addition to the Barton ester 239.
2.3.2.3. Features and Limitations
Even though the Barton halodecarboxylation reaction is highly appealing in terms of scope, it has some weaknesses that may limit its application. As might be expected from the reaction mechanism, the preparation of some materials might possess intrinsic problems. For example, the halodecarboxylation of acids that produce highly stabilized or electrophilic radicals, such as perfluoroalkyl, will result in an unproductive scavenging of these radicals with the Barton ester rather than with the halogen donor.226
A number of delicate issues are associated with the experimental setup. First, the method requires the preparation of the Barton ester prior to the reaction, which represents a major drawback of the approach. In many cases, these esters possess poor stability and have to be prepared with the exclusion of light. The low stability precludes their isolation and purification by convenient methods. Therefore, such esters are usually prepared in situ, and the overall yields of halodecarboxylation reaction depend on the selected esterification approach. Although various esterification conditions were developed, there are no general methods suitable for all acids. While the Steglich-type esterification of acids (DCC, DIC,227 EDC,207,216 etc. and catalytic amounts of DMAP) with pyrithione 303 generally provides the highest yields, in some cases, the preparation via acid chlorides with sodium pyrithione 304 was beneficial (Figure 1).208 Additionally, prefunctionalization of the acid with an alkyl chloroformate228 and CDI proved to be useful.228 Other successful esterification techniques include the use of pyrithione derivatives, such as 305,195,229−231306,232 and disulfide dipyrithione 307 (in combination with tributylphosphine).233 The purity of reagent 305 was found to have a profound effect on the reaction yield.230
Figure 1.

Precursors to Barton-type esters and their analogues.
When the multiple-step Barton process (ester preparation, isolation and concomitant radical degradation) is performed in a one-pot manner, it increases the difficulty of purification of the target product from the reaction mixture. In some cases, the presence of coproduct of type 299 made the isolation of a pure final product impossible, and additional synthetic manipulations were required to remove impurities, or even to disregard the method. The issue of impurities might be addressed by employing other efficient reagents from the thiohydroxamic acid family (308–311)234−236 or by using polymer-supported reagents.237 Moreover, the esters derived from reagents 308–311 possess higher thermal and/or photostability than the original 239. Usually, the yields of the alkyl halides are not significantly diminished, and hence utilization of these esters should be considered in the case of problematic ester stability.
Three approaches to initiate a decarboxylation sequence were found. The most general method is thermal decomposition. Heating the reaction mixture to moderate or high temperature (also by means of microwave irradiation238) initiates the desired decomposition of the Barton ester. Some of these esters are poorly stable even at low temperatures.218 A light-assisted activation enables the reaction of sensitive substrates at low temperatures (ca. 0–20 °C). The effect of a light source was studied.239 The Barton ester of type 239 has two absorption bands with maxima at 301 and 374 nm. It was found that the wavelength of the light is not important, unless it is absorbed by the ester (275–430 nm). The model reaction finishes in 15–40 s upon irradiation with sunlight, in 2 min in a Rayonet reactor (350 nm) or in about 5 min in the case of tungsten lamp. The reactions are often performed by combining thermal and photoirradiation. The third approach is based on the addition of an external source of radicals to initiate decomposition of the Barton ester. This was achieved by sonication of the reaction mixture in a chlorinated solvent225 or, most commonly, by the addition of a radical initiator, such as AIBN. While the presence of AIBN additive is important for the success of reaction in the case of aromatic Barton esters, in the aliphatic series the radical initiator reduces the reaction time rather than has a significant effect on the yield.238 The sonication-induced reaction yields were greatly influenced by the reaction concentration. Moreover, the formation of chlorine and acid chloride was observed in this case.
Various halogen sources were utilized in the Barton reaction. In order to estimate the applicability of a compound as an efficient halogen donor, general selection criteria were proposed.240 According to these criteria, the compound should (1) readily donate a halogen radical, (2) give rise to a relatively stable localized carbon-centered radical, (3) be easily removable from the reaction mixture, and (4) be stable under the reaction conditions. A number of halogenated aliphatic compounds meet these requirements. The most commonly used chlorine source is neat CCl4, which hampers the application of this method due to the incompatibility of poisonous CCl4 with industrial regulations. When an easily removable solvent is needed, 1,1,1-trichloro-2,2,2-trifluoroethane or fluorotrichloroethane might be employed, although performing of the reaction in a sealed vessel might be required in order to reach effective reaction temperatures.194 The bromodecarboxylation reaction usually employs a few equivalents of bromotrichloromethane, although 1-bromo-1-chloro-2,2,2-trifluoroethane (halothane) was found to be a useful substitute.201,241,242 Several types of iodine donors were used in the iododecarboxylation approach. The utilization of elemental iodine usually results in poor yields due to the poor ability of the iodine radical to serve as a chain-carrying species. Iodoform and especially 1-iodo-2,2,2-trifluoroethane217,240,243,244 are of the highest utility. Diethyl iodo(methyl)malonate was shown to be an extremely good iodine donor.222 Equimolar amounts of this reagent led to a nearly quantitative yield with barely detectable amounts of the side product 300. Similar reagents, such as iodoacetonitrile or ethyl iodoacetate, could also be utilized, although a larger amount of 300 was observed. Analogous bromides could be employed to perform the bromodecarboxylation.222,245
2.3.3. Other Photoactive Esters (Including Some Recent Approaches)
Barton’s seminal discovery of photoactive esters has had a great impact on the synthetic community. The ability to generate a carbon-centered radical under mild reaction conditions has allowed numerous decarboxylative transformations of carboxylic acids.8,10,246 Attempts were dedicated toward the development of alternative photosensitive moieties that can efficiently deliver carbon-centered radicals. As a common feature, these moieties possess a weak N–O bond that undergoes cleavage under light irradiation.
Soon after, Barton, Hasebe and Tsuchiya demonstrated the possibility of generating aryl radicals from benzophenone oxime esters (Scheme 40).247 The irradiation of (hetero)aromatic and aliphatic esters of type 312 with a high-pressure mercury lamp, equipped with a Pyrex filter, in CCl4, at room temperature, provided the corresponding organochlorides in good to excellent yields.248 Thus, the oxime esters of primary, secondary and bridgehead aliphatic acids smoothly underwent chlorodecarboxylation. Ketones, protected amides, and free alcohol functionalities tolerated the reaction conditions. In the aromatic series para-substituted electron-poor aryl chlorides, naphthalenes, pyridines, (iso)quinolines, and quinaxolines were obtained in very good to nearly quantitative yields. The preparation of electron-rich aryl chlorides via the designed approach was not reported.
Scheme 40. Photo-Assisted Chlorodecarboxylation of Benzophenone Oxime Esters.

Irradiation with a 400 W high-pressure mercury lamp with a Pyrex filter.
The decomposition of a benzophenone oxime ester is likely to proceed from an excited triplet state. Upon excitation, the molecule 312 undergoes homolytic scission of the N–O bond to give acyloxy and iminyl radicals (Scheme 41). Decarboxylation of an acyloxy radical gives a carbon-centered radical, which abstracts halogen from the solvent (or other halogen donor). The iminyl radical undergoes recombination or reacts with the solvent to give benzophenone azine 319 and N-chloroimine 320 (hydrolyzes to benzophenone upon workup). The trichloromethyl radical recombines with itself to give hexachloroethane. All these mentioned compounds were isolated from the reaction mixture. The oxime 312 cannot be activated thermally or by an external radical initiator, such as AIBN. This observation suggests the non-chain mechanism of the reaction. The ester 312 (R = Ph) exhibits two strong absorption lines with λmax = 263 and 280 nm (ε = 21 900 and 14 100 M–1·cm–1, respectively), but does not absorb light beyond 350 nm, making the use of a high-pressure mercury lamp inevitable.249 Less energetic irradiation could be employed for the decomposition of an oxime ester by means of a suitable photosensitizer. It was shown that the decomposition could be efficiently induced by irradiation with light at λ > 350 nm, employing benzophenone as a photosensitizer without affecting the product distribution.249
Scheme 41. Proposed Pathway for the Chlorodecarboxylation of Oxime Esters.

Recently, oxime esters as a source of aryl radicals were addressed by Glorius et al.250 A minor modification in the oxime backbone and the use of an iridium photocatalyst 321 ([Ir(dF(CF3)ppy)2(dtbbpy)]PF6) enabled a reaction upon irradiation with a 400 nm LED light. The reaction proved viable for the preparation of aromatic bromides and iodides by employing BrCCl3 or ICCl3 respectively as the halogen source (Scheme 42). The performed calculations suggest that the fragmentation of the oxime ester do not involve the intermediacy of an acyloxy radical but proceeds via concerted scission of N–O and C–C bonds, producing CO2, aryl, and iminyl radicals.
Scheme 42. Glorius Approach to Photoassisted Halodecarboxylation of Oxime Esters.

Reaction performed in EtOAc:CCl4 1:1.
“X” source: ICH2Cl, BrCCl3, or CCl4.
Another example of a photoactive ester was introduced by Okada and Oda.251 In their work, aliphatic carboxylic acids were converted to photoactive N-acyloxyphthalimides 340. Irradiation with a high-pressure mercury lamp (Pyrex filter) in the presence of DABCO (1,4-diazabicyclo[2.2.2]octane) in tert-BuOH/CCl4/H2O (85:10:5) as the solvent gave corresponding alkyl chlorides in moderate to high yields. The chlorodecarboxylation of primary, secondary, and bridgehead carboxylic acids was achieved by this process (Scheme 43). The presence of both water and DABCO was important to obtain an efficient reaction outcome.
Scheme 43. Conversion of N-Acyloxyphthalimides to Alkyl Chlorides.

No DABCO added.
No water added.
Irradiation with a 100 W high-pressure mercury lamp (Pyrex filter); 1–3 equiv of DABCO.
The reaction mechanism is proposed in Scheme 44. Upon excitation of N-acyloxyphthalimide to a triplet state with UV light, 340* is reduced with DABCO to give an anion-radical 341. Protonation with water gives a radical 342, which in turn, undergoes fragmentation to produce phthalimide, CO2, and an alkyl radical. The latter species abstracts chlorine from the CCl4 to result in the alkyl chloride. Similarly to oxime esters, the decomposition of 340 might be induced by less energetic radiation if an appropriate photosensitizer is applied.252 In the last five years, the functionalization of carboxylic acids via N-acyloxyphthalimides has become a popular approach to induce various decarboxylative transformations.253,254
Scheme 44. Anticipated Mechanism of the Chlorodecarboxylation of N-Acyoxyphthalimides.

3. Recent Methods for the Halodecarboxylation of Aliphatic Acids
In this chapter, we will discuss some recent approaches to the halodecarboxylation of aliphatic carboxylic acids. Since the discovery of the reactivity of N-acyoxyphthalimides in 1988, the field of halodecarboxylation has remained static in terms of fundamentally new approaches. In 2011, we presented a novel, one-pot, metal-free method for iododecarboxylation.255 We envisioned that N-iodoamides could be used as a dual-purpose reagent for both initiation of the reaction and iodine donation. Commercially available 1,3-diiodo-5,5-dimethylhydantoin (DIH) was found to be the N-iodoamide of choice. While N-iodosuccinimide (NIS) can also be used in numerous cases, it generally provides lower yields. Refluxing a carboxylic acid with DIH under visible light irradiation furnishes a wide range of primary, secondary, and tertiary bridgehead alkyl iodides in good to excellent yields (Scheme 45). Notably, for the first time, the reaction allowed the efficient preparation of various geminal dihaloalkanes (345, 346, 349, 350) and perfluorinated iodides (351).256−258 Lactones, N-Boc, N-Cbz, acetyl, and formyl groups remain unaffected during the reaction. Double decarboxylation of remote and 1,1-dicarboxylic acid proceeds smoothly in high yields. Aromatic iododecarboxylation is also a feasible process under the current conditions; yet, the scope is limited (see Section 4, Scheme 64).
Scheme 45. Selected Examples of Iododecarboxylation with 1,3-Diiodo-3,3-dimethylhydantoin.

Scheme 64. Iododecarboxylation of Benzoic Acids with DIH.

The important aspects of this approach are its simple experimental setup and facile isolation of the desired product. A straightforward aqueous acid–base workup of the reaction mixture is sufficient to obtain an essentially pure organoiodine product without the need for column chromatography. It enables the preparation of sensitive and barely stable geminal (iodo)haloalkanes.
It is hypothesized that the reaction is initiated by a homolytic cleavage of the N–I bond, producing iodine and an amide radicals (Scheme 46). The amide radical can reversibly abstract a hydrogen atom from the carboxylic group, producing an acyloxy radical. Rapid emission of carbon dioxide produces a carbon-centered radical, which, in turn, abstracts an iodine atom from the N-iodoamide. The presence of 10% TEMPO significantly retards the reaction, suggesting a chain radical process.
Scheme 46. Proposed Mechanism for Iododecarboxylation with 1,3-Diiodo-3,3-dimethylhydantoin.

Togo and co-workers used a similar approach for the one-pot transformation of aliphatic carboxylic acids to N-alkyl succinimides, with the intermediacy of alkyl iodides.259N-Iodosuccinimide was used for the iododecarboxylation reaction, under thermal conditions, with light exclusion (Scheme 47). The addition of elemental iodine slightly increased the yield of the alkyl iodides. The reaction was studied only for primary aliphatic acids. Aromatic acids were mainly unreactive, and only poor yields of the corresponding aryl iodides were obtained with both electron-rich and electron-poor benzoic acids. Additionally, the in situ preparation of NIS from N-chlorosuccinimide and sodium iodide was utilized in a one-pot preparation of N-alkyl succinimides. The proposed reaction mechanism is essentially the same as that depicted on Scheme 46.
Scheme 47. Iododecarboxylation with NIS and I2.

A significant advance in the area of halodecarboxylation was made by Li’s group.260 The choice of a suitable ligand and halogenating agent allowed for a first general catalytic approach toward aliphatic chloro- and bromodecarboxylation reactions. The use of a silver-phenanthroline complex, and tert-butylhypochlorite as both an oxidant and a chlorine donor, provided various alkyl chlorides in good to excellent yields (Scheme 48). Utilization of dibromoisocyanuric acid, in turn, gave access to the corresponding bromides.261 The halodecarboxylation reaction of primary, secondary, and tertiary aliphatic acids can be achieved under nearly identical conditions. Various alkyl chlorides, bearing benzyl- (350, 351), allyl- (348, 349), and protected α-oxy- (357) and α-amino (358) groups can be prepared in high yields. Moreover, ketones, esters, tertiary amides, halides, nitro-, terminal alkynes, and electron-poor alkenes do not interfere with the reaction. Aromatic chlorides and bromides cannot be obtained by the designed approach, and the starting acid is recovered. The reaction times increases in the order benzyl ≈ tertiary < secondary < primary acids. The difference in reactivity has enabled chemoselective decarboxylation. For example, tertiary acids can be selectively converted to a chloride in the presence of a primary acid (356), while phenylacetic acids are selectively decarboxylated in the presence of aromatic carboxylic groups (355).
Scheme 48. Examples of Silver-Catalyzed Aliphatic Chloro- and Bromodecarboxylation.

As suggested by a number of control experiments, the reaction is likely to proceed via a radical mechanism. It is hypothesized that tert-butylhypochlorite oxidizes the Ag(I) species to the dinuclear Ag(II) complex 359, followed by a ligand exchange with a carboxylate ion to give complex 360 (Scheme 49). The carboxylate ion is oxidized by the Ag(II) species to produce a carboxyl radical, which emits CO2 to form an alkyl radical. The alkyl radical then abstracts a chlorine atom from the Ag(II)-Cl species 361 to afford an alkyl chloride and regenerate the catalyst. Attempts to use I2, Br2, or NXS (X = I, Br, Cl) as halogenating agents (instead of working with tert-BuOCl or DBI) were completely unsuccessful, which can be explained by their low oxidation potential and inability to oxidize Ag(I)(Phen)2 to Ag(II)(Phen)2. The order of reactivity of the acids suggests that the rate-limiting step is the oxidation of the carboxyl ion to the acyloxy radical. A computational study of the reaction mechanism supports the outlined pathway.262
Scheme 49. Proposed Mechanism for Ag(I)-Catalyzed Chlorodecarboxylation.

Glorius and co-workers introduced an interesting photocatalytic method for the halodecarboxylation reaction.263 Irradiation of a carboxylic acid in the presence of cesium carbonate, Ir-based photocatalyst 321 ([Ir(dF(CF3)ppy)2(dtbbpy)]PF6) and a suitable halogen donor, provided access to primary, secondary, and tertiary (bridgehead and acyclic) alkyl halides (Scheme 50). The yields of primary alkyl halides are generally lower than those of their secondary and tertiary counterparts. In the halide series, the yields typically decrease in the order chlorides > bromides > iodides. Functional groups, such as esters, silyl- and phenyl ethers, protected tertiary amines and aliphatic bromides tolerate the reaction conditions. For the preparation of alkyl chlorides and iodides, N-halosuccinimide proved to be the halogen source of choice. For the bromodecarboxylation process, employment of NBS led to decomposition of the product; therefore, diethylbromomalonate was employed as an alternative bromine source.
Scheme 50. Light-Assisted Halodecarboxylation of Aliphatic Acids.

The product was isolated as a mixture of alkenes.
On the basis of studies of the bromodecarboxylation process, a reaction mechanism was proposed (Scheme 51). Upon excitation, the iridium photocatalyst 321 oxidizes the cesium salt of the carboxylic acid 377 to produce an alkyl radical with the liberation of CO2. The alkyl radical abstracts bromine from the diethyl bromomalonate 362 to give the product of the reaction and a malonyl radical 378. Reduction of the malonyl radical 378 with an Ir(II) species gives a malonyl anion 379 and regenerates the photocatalyst. However, the quantum yield of the reaction Φ = 2.7 indicates a chain process, in addition (or as an alternative) to the proposed one. An additional possible pathway was suggested after examining the outcome of the control experiments. Diethyl bromomalonate 362 was found to decompose upon light irradiation in the presence of a photocatalyst to give bromine (among other products). On the basis of these observations, the Hunsdiecker-type reaction between cesium carboxylate and Br2, involving the intermediacy of an acyl hypobromite as well as Br-radical or Br2·– as a chain-carrying species, was proposed.
Scheme 51. Proposed Mechanism for the Photocatalytic Bromodecarboxylation Reaction.

A striking approach for an enantioselective chlorodecarboxylation was developed by Shibatomi and co-workers.264 β-Ketoacids delivered α-chloroketones in very good yields and good-to-excellent enantioselectivities upon treatment with NCS in the presence of a chiral amine catalyst 391 (Scheme 52). The ee of the product depended on the backbone of the starting material. In the case of tertiary α-chloroketones, excellent values were obtained for benzannulated cyclic tertiary derivatives (382–384), whereas in acyclic motifs, a better enantioselectivity was achieved for secondary chlorides (387–389).
Scheme 52. Enantioselective Chlorodecarboxylation of β-Ketoacids.

Unlike other halodecarboxylation reactions, this process proceeds via an ionic mechanism (Scheme 53). β-Ketoacid 392 is deprotonated with a chiral amine catalyst 391 to produce a carboxylate ion. Further decarboxylation provides an enolate anion 393, which is tightly bound to a chiral cation. Rapid chlorination of these species with NCS furnishes the reaction product 394.
Scheme 53. Proposed Mechanism for the Enantioselective Chlorodecarboxylation of β-Ketoacids.

A similar approach was utilized to prepare α-chloro-α-fluoroketones from α-fluoro-β-oxocarboxylic acids using an amine catalyst (Scheme 54).265 DABCO and quinuclidine were found to be the optimal bases. Very high yields of chloro-fluoroketones are generally obtained for acyclic backbones (395–397). Cyclohexanone-based β-ketoacid gives only moderate yields of the corresponding chloride (398). The reaction was also performed in an enantioselective manner employing catalytic amounts of a chiral amine 391.266 Generally, high yields were secured for this transformation; however, the enantioselectivity of the product was found to be significantly dependent on an acid backbone. Very high ee’s were obtained for benzannulated six-membered cyclic ketones (399); however, the values for corresponding five-membered cyclic or acyclic ketones are rather poor (400–405).
Scheme 54. Examples of α-Chloro-α-Fluoroketones Prepared from α-Fluoro-β-oxocarboxylic Acids.

Asterisks indicate the reaction performed with 391 as a catalyst instead of DABCO and −20 °C – 0 °C instead of room temperature.
4. Additional Methods for the Halodecarboxylation of Aromatic Carboxylic Acids
In general, the halodecarboxylation of aromatic acids is more challenging than that of aliphatic substrates. We have discussed some approaches in the previous sections. Thus, classical Ag-, Hg-, or Pb-based methods for the conversion of aryl carboxylic acids to aryl halides are presented in Scheme 8 and Scheme 20. Typical metal-free methods are summarized in Scheme 33, Scheme 36, Scheme 40, and Scheme 42. As can be seen from these examples, the decarboxylative halogenation of aromatic acids suffers from intrinsic problems and a lack of general trends. The development of an operationally simple protocol, applicable for a broad range of substrates bearing both electron-donating and electron-withdrawing groups on the aromatic ring, and capable of installing Cl, Br, or I, represents a substantial challenge, to which the solutions are yet to be found. In this chapter, we discuss some representative examples of recent efforts in this direction to complete the contemporary state-of-the-art.
The halodecarboxylation reactions of aromatic acids possess their own mechanistic features and complexity. If for the halodecarboxylation of alkanoic acids a radial intermediacy is generally recognized, the mechanism for aromatic acids is somewhat ambiguous. This is due to the high stability of the benzyloxy radical (407, Scheme 55), which inclines to intra- or inter-H-abstraction more than to homolytic decay, with the formation of the aryl radical (408).176 Special forcing reaction conditions are required to promote the decarboxylation of 407 to generate the aryl radical. Examples of these triggers may be transition metals, light- or MW-irradiation, or radical initiators.
Scheme 55. Plausible Mechanisms for the Halodecarboxylation of Aromatic Acids and Possible Side-Reactions.

An alternative pathway is possible which does not involve a radical species and implies that the preformed aroylhypohalide (406) undergoes a concerted direct ipso substitution to the desired aryl halide 410 via transition state 409. An additional possible channel for side-reactions—the electrophilic halogenation of the aromatic ring, both in the starting acid and in the product 410—adds to the complexity of the process.
Lee and co-workers attempted a metal-free halodecarboxylation of substituted benzoic acids. The reaction is based on the application of a strong oxidizer, potassium hydrogen persulfate (KHSO5, commercially available as Oxone), in combination with NaBr and Na2CO3 base (Scheme 56).267
Scheme 56. Bromodecarboxylation of Benzoic Acids with Oxone.

Asterisks indicate a halogen introduced via electrophilic halogenation.
Unfortunately, the reaction worked only for benzoic acids bearing MeO- or AcNH-electron-donating substituents at the ortho- and para-positions. Moreover, it was accompanied by a side process of aromatic electrophilic bromination. As a result, 1,3-dibromoarenes were obtained in good yields using 2 equiv of the Oxone-NaBr system. The corresponding desired monobromoarenes were selectively obtained for acids bearing a meta-substituent, such as 3-chloro-4-methoxy- and 3-chloro-4-acetamidobenzoic acids (products 412 and 413).
Liu et al. demonstrated the applicability of the iodine pentoxide-KX system for the preparation of mono-, di-, and even trihalides in the presence of various methoxy or amino-groups (Scheme 57).268 However, additional electron-withdrawing groups, such as Cl and Br, reduced the product yield or made the reaction impossible, as was observed for 2,6-dimethoxy-3-bromobenzoic acid (423).
Scheme 57. Halodecarboxylation of Arylcarboxylic Acids with I2O5–KX System.

Asterisks indicate a halogen introduced via electrophilic halogenation.
A poor iododecarboxylation yield was noted for 2,4,6-trimethylbenzoic acid (424), while no bromodecarboxylation occurred at all (425). In contrast, the 5,6-dibromobenzo[d][1,3]dioxole (427) was obtained in a moderate yield (45%), although the corresponding diiodide (426) was found only in trace amounts.
Similar results were obtained for the synthesis of iodo- and bromoarenes with another oxidizing reagent, PhI(OAc)2, in combination with LiX (Scheme 58).182 Again, the procedure was limited to acids, bearing at least two electron-donating groups (RO-, NH2-) on the ring, and led to dihaloarenes due to the concurrent undesired electrophilic halogenation.
Scheme 58. Selected Examples of the Halodecarboxylation of Methoxy Substituted Benzoic Acids with a Hypervalent Iodine(III)-LiX System.

Asterisks indicate a halogen introduced via electrophilic halogenation
The Larrosa group found that aromatic acids bearing electron-donating groups can be effectively converted to the corresponding monoiodoarenes by simply using molecular iodine and K3PO4 as a base (Scheme 59).269 The conversion was not effective with other bases, such as Li2CO3,Na2CO3, K2CO3, and Cs2CO3, and with molecular bromine, or for the Ag-salt of the starting acid. High yields of monoiodides were achieved in the presence of methoxy- or amino-activating substituents. The position of the activating aromatic substituent barely affected the product yield. The only exception was 3-methoxybenzoic acid, which was unreactive under the developed conditions. As was suggested by the authors, this indicated that the ipso-position of the decarboxylation must be sufficiently nucleophilic. Additional electron-withdrawing groups (halogens or CF3) significantly reduced the yields. However, in the absence of the activating groups, acids bearing strong electron-withdrawing groups, such as NO2, CF3, or CN, were unreactive or gave the monoiodide in a poor yield (<13%). Surprisingly, polyfluorobenzoic acids gave the corresponding polyfluoroiodobenzenes in excellent yields.
Scheme 59. Halodecarboxylation of Arylcarboxylic Acids with a Molecular Iodine-K3PO4 System.

Asterisks indicate the position of an electrophilic halogenation in the main side product. Isolated yields. NMR yields are given in square brackets.
The procedure was also applied to a range of heteroaromatic and cinnamic acids (Scheme 60). Furyl-2-carboxylic (446) and thienyl-2-carboxylic (448) acids gave the corresponding iodides in good yields, although the presence of a strong acceptor substituent, such as a NO2 group (447), drastically reduced the product yield. Iodo-substituted indole- (455, 456), benzothiophenes (451, 453), and benzofuranes (452, 454) were efficiently prepared by this method. However, the reactions were less selective for benzo[b]thiophene carboxylic acids, lacking a substituent in the 2- or in a 3-position, since the subsequent iodination led to the formation of diiodides. 1-Methyl-1H-pyrazole-4-carboxylic acid, 4-oxo-4H-chromene-3-carboxylic acid and nicotinic acids, bearing substituents in the potentially active sites led to the corresponding iodides (450, 458, 459, 460 respectively) in a selective mode. It was found that cinnamic acids—representative of the α,β-unsaturated acids family—were also active under the developed conditions and furnished the corresponding iodides (461, 462) in reasonable yields.
Scheme 60. Halodecarboxylation of Heteroaromatic Carboxylic and Cinnamic Acids with an I2–K3PO4 System.

Asterisks indicate the position of an electrophilic halogenation in the main side product; isolated yields.
Experimental and computational studies of this interesting reaction suggest that its mechanism involves the concerted conversion of an aroylhypohalide 463 to the final iodoarene through the transition state 464 rather than via a Hunsdiecker-type radical pathway (Scheme 61).269
Scheme 61. Proposed Mechanism for Iododecarboxylation with an I2–K2CO3 System.

Using the density functional theory (DFT), a barrier to 463, namely, the key transition state, was found to be of 27.6 kcal/mol (Scheme 61). This value is reasonable for a reaction proceeding at temperatures as high as 100 °C. Moreover, computational studies and the Hammett plot analysis showed the substantial buildup of positive charge in the transition state 464, which might explain the positive effect of the electron-donating group on the efficiency of the iododecarboxylation.
Recently, the same group found that Bu4NBr3, in combination with K3PO4, could effectively induce the bromodecarboxylation of electron-rich aromatic acids to the corresponding monobromides (Scheme 62).270 The behavior and scope of the substrate acids were observed to be similar to those in the iododecarboxylation.269 Contrary to the three previously described methods, based on the strong oxidizer/halide ion combination, this method does not lead to undesired electrophilic halogenation and aromatic dihalides as side products. Exceptions were naphthoic acids and 2,3,4,5-tetrafluorobenzoic acids, where the corresponding dibromides were formed in a low 5–7%.
Scheme 62. Bromodecarboxylation of Arylcarboxylic Acids with a Bu4NBr3–K3PO4 System.

Asterisks indicates the position of an electrophilic halogenation in the side product. Isolated yields. NMR yields are given in square brackets.
Other brominating agents, such as molecular bromine, N-bromosuccinimide (NBS), 1,3-dibromo-4,4-dimethylhydantoin (DBH), and pyridinium tribromide (PTB) were less effective and led to mixtures of mono- and dibromobenzenes, as well as causing bromination of the starting acids. As in the iodination approach, the reaction likely proceeds by a concerted mechanism via a 4-membered transition state (ΔG= 19.2 kcal·mol–1) as depicted in Scheme 61.
The procedure was also effective for heteroaromatic acids, furnishing the corresponding bromides of thiophenes (476), furanes (477), thiazoles (478), pyrazoles (479), indoles (480), benzothiophenes (481, 482, 484), benzofuranes (483, 485), pyridines (486), and 4-oxo-4H-chromene (487) in good yields (Scheme 63). However, in contrast to the iododecarboxylation, the reaction with benzofuryl- and benzothienyl carboxylic acids, nonsubstituted at the C3-position, was much less selective and was accompanied by a substantial amount of dibrominated products. The bromodecarboxylation of trans-4-methylcinnamic acid proceeded with significant isomerization at the double bond (488).
Scheme 63. Bromodecarboxylation of Aryl Carboxylic Acids with a Bu4NBr3–K3PO4 System.

Asterisks indicate the position of an electrophilic halogenation in the side product. Figures in brackets indicate monobromide:dibromide ratios.
Our group has developed the iododecarboxylation of aromatic acids by a photoinduced reaction with DIH as the sole reagent (Scheme 64).255 Electron-poor and electron-neutral iodobenzenes, bearing halogen, phenyl, or ester substituents, could be obtained in fair yields. Electron-rich aromatic acids mainly suffer from ring iodination, and the desired products are obtained in poor yields.
Using this approach, McDonald and co-workers converted ortho-iodobenzoic acids to the corresponding 1,2-di- and 1,2,3-triiodides using NIS (Scheme 65).271 In general, 1,2,3-trihaloarenes were obtained in good to excellent yields (>75%) for acids bearing electron-withdrawing groups (halogens and esters) in the para-position to the carboxylic group. At the same time, a halogen in the ortho-position and electron-neutral substituents (e.g., Me) in the ortho- or para-positions slightly reduced the product yield.
Scheme 65. Iododecarboxylaton of Benzoic Acids with NIS.

Recently, a successful example of a visible light photocatalytic iododecarboxylation has been disclosed.272 Benzoic and naphthoic acids bearing both electron-donating and electron-withdrawing substituents, as well as thiophene-2-carboxylic acid, were converted to iodoarenes with the photocatalytic assistance of [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 (321; Scheme 66). The employment of I2 as an additive, together with an excess of N-iodosuccinimide and Cs2CO3, proved necessary for provision of the products in moderate to good yields.
Scheme 66. Photocatalytic I2-Assisted Iododecarboxylation with NIS.

The reaction did not work without visible light irradiation, a photocatalyst, or a base. The addition of substoichiometric amounts of I2 played an important role in the process. The product yield deteriorated drastically without the additive, while increasing the amount of I2 to 20 mol % favored the process of iododecarboxylation of the less active substrates, namely, MeCO2-, CN-, or NO2-substituted benzoic acids. However, iodine alone, without NIS, led to the desired product in a poor yield. Under optimal conditions, the nature of the substituents and their position in the aromatic ring had no significant effect on the product. The reaction was effective even in the case of heterocyclic acids with a pronounced aromatic character, such as nicotinic acid or thienyl-2-carboxylic acid. The system NBS-Br2 was inefficient for the preparation of the corresponding aryl bromides.
Elegant mechanistic studies allowed the authors to propose a detailed radical pathway for this reaction (Scheme 67). The acid substrate is first deprotonated by Cs2CO3 to the carboxylate 511, which is converted to the benzoyloxy radical 512 as a result of SET between the photoexcited Ir(III) catalyst and 511. The reaction of the radical with I2 afforded the iodine radical and aroylhypoiodide 513, which undergoes the photocatalyst-assisted decarboxylation to form the aryl radical 514 and an iodide anion. The elusive radical 514 reacts with I2 to furnish the final aryl iodide and an I-radical, which undergoes recombination to molecular iodine. The role of NIS is likely in the oxidation of the formed iodide anion to I2, which is regenerated for the further catalytic cycles. It should be noted that this mechanism is in accordance with the data obtained by Glorius for the in situ photocatalytic formation of elusive aryl radicals from the corresponding carboxylic acids.273
Scheme 67. Proposed Mechanism of the Photocatalytic I2-Assisted Iododecarboxylation with NIS.

The application of a Pd-based catalyst enables the performance of iododecarboxylation in nonirradiative conditions.274 It was found that aryl carboxylic acids bearing ortho-hydroxy- or alkoxy substituents easily react with N-iodosuccinimide in the presence of catalytic amounts of Pd(II), to afford monoiodides in good yields (Scheme 68).
Scheme 68. Pd(OAc)2-Catalyzed Iododecarboxylation of Aryl Carboxylic Acids with NIS.

Yields of diiodides are given in brackets. Asterisks indicate an iodine atom introduced via electrophilic iodination.
Thus, under the optimized conditions, 1-iodo-2,6-dialkoxybenzenes (515, 516) and 1-iodo-2-alkoxynaphthalenes (520, 521) were obtained in 80–91% isolated yields. Bulky alkoxy groups in the ortho-position slightly reduced the product yields to 70–78%. The ortho-positions must be substituted in order to prevent a competitive Pd-catalyzed C–H iodination, directed by the carboxylic group.271,275 Notably, the amount of diiodides (the second iodination is a result of electrophilic aromatic substitution) did not exceed 2%. No reaction was observed for benzoic acid and for electron-deficient aromatic carboxylic acids bearing halogens or nitro-substituents (133, 519, 444).
Additionally, a range of heteroaromatic acids also underwent iododecarboxylation under the same conditions, affording the corresponding iodides in moderate to good yields (Scheme 68). The iododecarboxylation of 3-methylthienyl-2-carboxylic acids led to the 2,5-diiodide rather than to the target monoiodide (524). The same reaction with benzo[b]thiophene-2-carboxylic and 3-methylbenzo[b]furan-2-carboxylic acids gave the monoiodides (453, 522, 513) only for 3-substituted acids; otherwise, the diiodide was obtained (524).
Mechanistic studies reveal that the reaction proceeds most likely via ipso-iododecarboxylation in the coordination sphere of Pd rather than via a radical-type mechanism (Scheme 69). It was proposed that a ligand exchange in Pd(OAc)2 leads to the formation of the acyloxy-Pd species 525, which undergo concerted Pd-assisted ipso-decarboxylation. The resulting Pd(II)-aryl intermediate 526 is oxidized with NIS to provide the Pd(IV) species 527, which is prone to C–I reductive elimination to form the desired iodoarene and regenerate the Pd(II) catalyst.
Scheme 69. Proposed Mechanism for Pd(OAc)2-Catalyzed Iododecarboxylation with NIS.

Wu and co-workers found that CuBr2 and CuCl2 could be used as a source of halogen for the halodecarboxylation of benzoic acids bearing an ortho-nitro group, and of 1-methylindole, if Ag2CO3 was used as the catalyst, which obviated the use of the Pd-species (Scheme 70).276
Scheme 70. Ag-Catalyzed O2-Promoted Halodecarboxylation of 2-Nitrobenzoic Acids with CuX2.

The reaction takes place only in the presence of oxygen. This fact allowed authors to propose a radical mechanism for the transformation. Other salts, such as LiCl, NaCl, or tetrabutylammonium chloride, promoted the reaction even in the presence of catalytic amounts of Cu- and Ag-salts. The nitro group in the ortho-position was critical for the success of the transformation, which implies a serious limitation to this method. Benzoic acids with other substituents (MeO, CN, or CF3) in the ortho-position were inert under the developed conditions. Interestingly, chlorodecarboxylation proceeded in higher yields than bromodecarboxylation.
Recently, it was found that a similar Ag-catalyzed iododecarboxylation can be performed by the employment of I2 as the halogen source, and potassium peroxydisulfate, K2S2O8, as the oxidant (Scheme 71).277 Attempts to use KI and NaI instead of I2 were ineffective.
Scheme 71. Ag-Catalyzed Peroxydisulfate-Assisted Iododecarboxylation of Benzoic Acids with I2.

Asterisks indicate the position of an electrophilic halogenation in the main side product.
The reaction furnished excellent yields (>90%) of iodoarenes in the case of benzoic acids bearing electron-withdrawing substituents (halogens, nitro, and CF3) in the ortho- and meta- positions, although the yields of para-substituted iodoarenes were lower (65–75%). Good yields were also reached for iodonaphthalenes and disubstituted iodobenzenes. Strong electron-donating groups, such as MeO, promoted the formation of diiodides as a result of electrophilic substitution in the activated aromatic ring (e.g. 428). The presence of the ortho-nitro group inhibited the undesired electrophilic iodination and gave the monoiodide 548 in a good yield (85%). 5-Bromonicotinic acid was converted to the corresponding dihalopyridine 549 in a 63% yield. Unfortunately, attempts to carry out chloro- and bromodecarboxylation using Br2, NBS, or NCS failed. The corresponding chlorides and bromides were obtained in poor yields (20–40%).
The proposed radical mechanism of this reaction is depicted in Scheme 72. In the first step, a single electron transfer between the carboxyl anion and Ag(II), generated in situ by the oxidation of Ag(I) with S2O82–, forms the acyloxy radical 550, which is decarboxylated under MW irradiation to an aryl radical 551. When the reaction is performed in the presence of CD3CN, monodeuterated benzene (552) is obtained, which supports the plausible formation of the aryl radical 551. In the presence of iodine, the aryl radical is transformed into the desired iodobenzene.
Scheme 72. Proposed Mechanism for Ag-Catalyzed Silver Peroxydisulfate-Assisted Iododecarboxylation of Benzoic Acids with I2.

Liu and co-workers found that the reaction can be done without a strong oxidant (like K2S2O8 in the previous case). However, a stoichiometric amount of the Ag(I) salt should be used. The process is catalyzed by PdCl2, and CuBr2 or CuCl2 are used as the halogen sources (Scheme 73).278 Electron-rich aryl carboxylic acids showed significantly better reactivity than their counterparts bearing electron-poor aryls, which indicates that the process involves an ionic mechanism with a concerted direct ipso-substitution rather than a radical pathway. Unfortunately, in several cases the reaction was accompanied by the formation of dihaloarenes as a result of an electrophilic substitution on the electron-enriched aromatic ring. This circumstance led to moderate isolated yields in a number of substrates, especially for chlorobenzenes.
Scheme 73. Selected Examples of the Ag-Promoted Pd-Catalyzed Halodecarboxylation of 2-Methoxybenzoic Acids with CuX2.

The authors suggested that the reaction proceeds via the aryl palladium complex 563 (likely obtained via concerted direct ipso-palladation of the corresponding acyloxy-Pd), which undergoes halogen-transmetalation to result in the aryl palladium halide 564 (Scheme 74). The consequent reductive elimination affords the product and Pd(0) species, which is oxidized by Ag(I) to regenerate the active catalyst.
Scheme 74. Proposed Mechanism for the Ag-Promoted Pd-Catalyzed Halodecarboxylation of 2-Methoxybenzoic Acid with CuX2.

Interestingly, Cai and co-workers revealed that a simple combination of CuI and oxygen is sufficient for the iododecarboxylation of ortho-nitrobenzoic acids and heteroaromatic acids in reasonable yields (Scheme 75).279 Other sources of iodine, such as LiI, KI, NH4I, or the system CuO + KI, gave only negligible yields of the iodoarenes. These reaction conditions are unsuitable for the iododecarboxylation of electron-rich benzoic acids. However, the addition of catalytic amounts of Pd(OAc)2 enabled the reaction with ortho-methoxy substituted electron-rich aromatic acids. Additionally, this bimetallic system made possible the iododecarboxylation of indol-3-carboxylic acids. However, the reaction was nonselective and provided a mixture of mono- and diiodides because of an additional electrophilic iodination.
Scheme 75. Oxygen-Promoted Iododecarboxylation of 2-Nitrobenzoic and Heteroaromatic Acids with CuI.

Reaction in the presence of 10 mol % Pd(OAc)2.
A yield of the diiodinated product is given in parentheses. Asterisks indicates the position of an additional electrophilic iodination.
Later, the same group developed a Cu/Ag catalyzed reaction, which allowed the preparation of iodo-, bromo-, and chloroarenes by a single approach.280 Sodium halide (NaI, NaBr, or NaCl), with catalytic amounts of Ag2SO4 and Cu(OAc)2, in the presence of the 2,9-dimethyl-1,10-phenanthroline (2,9-DMP) ligand were able to convert ortho-nitrobenzoic acids into the corresponding 1-halo-2-nitrobenzenes (Scheme 76).
Scheme 76. Examples of the Oxygen-Promoted Ag-Catalyzed Halodecarboxylation of Benzoic Acids with NaX, in the Presence of 2,9-Dimethyl-1,10-phenanthroline.

Figures in parentheses are yields for CuI, CuI–Pd(OAc)2 or for CuI–CuX systems.
As a special case of halodecarboxylation, we should point out a method in which metalorganic species can be prepared by decarboxylation and isolated, following by concomitant halogenation. Larrosa’s group succeeded in the preparation of haloarenes and heterohaloarenes from the corresponding stable gold(I) aryl compounds (Scheme 77).281 The intermediate Ar–Au(I) could be isolated in more than 75% yield. The authors combined these two processes to directly prepare bromo- and iodoarenes, as well as halopyridines, from carboxylic acids in high yields.
Scheme 77. Halodecarboxylation of Benzoic Acids via Intermediate Au-Complexes.

NMR studies demonstrated that the formation of the gold(I)-aryl compounds likely proceeds via transmetalation from the initially formed Ag-salts 585 to t-Bu3AuCl (Scheme 78). The generated Au-complex 586 undergoes decarboxylation to the aryl-gold species 587, which in turn converts to the desired aryl halide upon reaction with NXS. It should be noted that a related approach was reported a while ago, in which permercurated aromatic systems, isolated after reaction of benzoic acid and Hg(OCOCF3)2, were converted to perhalobenzenes with an appropriate halogen source.282
Scheme 78. Proposed Mechanism of Halodecarboxylation of Benzoic Acids via Intermediate Au-Complexes.

We can conclude that the decarboxylation of the aromatic acids suffers from a lack of generality, and methods applicable to a wide range of acids, bearing different electronic natures of the aromatic rings, are practically absent. However, some trends can be generalized, so the suitability of the substrate to the selected decarboxylation approach will be fitted to the reaction mechanism. Acids bearing electron-donating substituents on the aromatic ring proved efficient in halodecarboxylation reactions proceeding via a concerted electrophilic ipso substitution in hypohalites or acyloxy metal intermediates (RCO2X or RCO2M, correspondinly). In such reactions, electron-rich aromatics promote a nucleophilic attack in this key mechanistic step. Electron-poor aromatic acids are usually inefficient in such transformations, although they are suitable for radical-type halodecarboxylation reactions. This is because in radical transformations, the halogenating agent, which is utilized, can be active in concurrent, undesired electrophilic aromatic substitution. Electron-poor aromatic acids are less active in such undesired processes and can selectively undergo halodecarboxylation.
5. Additional Miscellaneous Examples of the Halodecarboxylation of Heteroaromatic Carboxylic Acids
Heteroaromatic moieties are common motifs in biologically active compounds. One of the most useful ways of their installation in the complex skeleton is by a cross-coupling reaction of the corresponding halo-heterocycle. Although the preparation of a heteroaromatic halide from carboxylic acid seems an attractive approach, reliable procedures, applicable to a wide range of substrates, remain elusive, and commonly are heterocycle-specific. In the previous chapter, we described methods for the decarboxylative halogenation of aromatic acids, pointing out their applicability to heteroaromatic counterparts in suitable cases. In this chapter, we provide number of interesting miscellaneous halodecarboxylation methods, which were explored for some specific heteroaromatic acids. In contrast to alkyl and aryl carboxylic acids, the halodecarboxylation of heteroaromatic acids mainly proceeds via an ipso-substitution mechanism.
Back in early works, it was found that heating a solution of pyrrole-2-carboxylic acid and NaHCO3 with a mixture of I2/KI led to the corresponding iodopyrroles in good yields (Scheme 79).283 Some derivatives of the related indole-2-carboxylic acid may undergo iododecarboxylation under similar reaction conditions.284
Scheme 79. Selected Examples of the Iododecarboxylation of Pyrrole-2 Carboxylic Acids with I2/KI283,285−291.

Similarly, the bromodecarboxylation of pyrrole-2-carboxylic acid has been performed using molecular bromine in absolute methanol (Scheme 80).283
Scheme 80. Examples of the Bromodecarboxylation of Pyrrol-2 Carboxylic Acids with Br2.

In a series of works,292−295 Miki et al. investigated the halodecarboxylation of N-protected indole mono- and dicarboxylic acids with a bromonium ion, generated in situ by the oxidation of a bromide anion. The employed oxidants were Oxone, cerium ammonium nitrate (CAN), m-chloroperbenzoic acid (m-CPBA), and PhI(OAc)2. The method was attempted for the synthesis of polybromoindoles, which serve as precursors for the preparation of biologically active compounds.294
The PhI(OAc)2-LiBr system (4 + 4 equiv) was effective for the halodecarboxylation of indolecarboxylic acids, which could be converted to 2,3-dibromoindoles 599 or 3,3-dibromo-2-oxoindole 600, depending on the type of N-protective group (Scheme 81). The formation of oxoindole of type 602 was explained by the formation of intermediate 601, which is the result of an electrophilic attack of the halodecarboxylated indole ring on the halonium ion. This intermediate underwent hydrolysis during workup to form oxindole 602, which is claimed to be an attractive synthon in indole-based alkaloid synthesis.293 For example, synthesis of the alkaloid Kalbretorine was performed via the corresponding diiodoindole (604), which was prepared by iododecarboxylation of the corresponding diacid 603.294
Scheme 81. Bromodecarboxylation of N-Protected Pyrrolecarboxylic Acids, Proposed Mechanism for the Formation of Oxoindoles and Application Towards Synthesis of Kalbretorine.

Exploring indazoles as a potential scaffold for an in-house kinase inhibitor, Henderson and Honey found that indazole-, indole-, azaindole-, and azaindazol-3-carboxylic acids can be converted into the corresponding 3-bromo heterocycles by simple treatment with an equimolar amount of NBS in DMF, providing products in good to excellent yields (Scheme 82).296 The process was successful only in DMF. The authors assumed that the reaction proceeds through a nonradical pathway, as occurs in the absence of light.
Scheme 82. Bromodecarboxylation of Indazole-3-carboxylic Acids.

Halopyridines constitute an extremely important building block in organic synthesis. A notable study revealed that 2-halopyridine derivatives can be prepared by the halodecarboxylation of 2-picolinic acids with dihalomethanes, using tert-BuOCl as an oxidant, under heating in air (Scheme 83).297 In the same way, quinoline-2- and isoquinoline-1-carboxylic acids were converted to the corresponding 2-haloquinoline and 1-haloisoquinoline, respectively.
Scheme 83. Halodecarboxylation of 2-Picolinic Acids with t-BuOCl.

Figures in parentheses indicate the amount of dibromide.
It was found that under standard conditions the reaction did not proceed without t-BuOCl. No reaction was observed in the presence of the radical scavenger TEMPO. The proposed mechanism involves an initial homolytic cleavage of t-BuOCl to t-BuO and a Cl radicals (Scheme 84). The latter is oxidized by oxygen to the chloronium ion, which in turn attacks the nitrogen atom of the pyridine ring in the Na-salt of the acid 626 to result in species 627. The formed positive charge in the ammonium intermediate 627 promotes the decarboxylation leading to the ylide 628/carbene 629 species. The reaction of the carbene with dihalomethane affords the desired 2-halopyridine and bromochloromethane, which was detected by GC-MS.
Scheme 84. Proposed Mechanism of Halodecarboxylation of 2-Picolinic Acids.

The bromodecarboxylation of quinoline-4-carboxylic acids can be accomplished by a simple treatment with NBS; however, the reaction is highly limited to precursors bearing hydroxy- or amino-substituents in the 3-position (Scheme 85).298 This fact was explained by the presence of an enol or an enamine fragment in the 3-hydroxy- and 3-amino-derivatives, which make these compounds prone to electrophilic attack of the bromonium ion at the ipso position of the aromatic ring, to form bromo-derivative 634. This intermediate undergoes decarboxylation affording the desired 2-bromo-3-hydroxyquinolines.
Scheme 85. Bromodecarboxylation of 3-Amino- and 3-Hydroxyquinoline-4-carboxylic Acids and Proposed Mechanism.

The microwave-assisted iodo- and bromodecarboxylation of isoxazole-4-carboxylic acids with halosuccinimides, in the presence of K3PO4, was recently described by the Batra group (Scheme 86).299
Scheme 86. Examples of Microwave-Assisted iodo- and Bromodecarboxylation of Isoxazole-4-carboxylic Acids with N-Halosuccinimides.

Both iodo- and bromodecarboxylation were efficient with a broad scope of substrates. Surprisingly, the exceptions were acids bearing the CN substituent on the aromatic ring or the amino or hydrazino group in the isoxazole ring. The mechanism of this process is unclear. While the reaction furnished a complex mixture of products in the presence of TEMPO, the radical clock experiment269,270 did not lead to the typical radical-assisted rearranged products. On the basis of this data, the authors assumed that both radical and electrophilic ipso-substitution paths could be concurrently operative (Scheme 87).
Scheme 87. Proposed Scheme for the Halodecarboxylation of Isoxazole-4-carboxylic Acids.

Thiophene-based carboxylic acids comprise an additional object of interest for the preparation of valuable halo-thiophenes. Some examples are provided in section 4. In Scheme 88, a specific approach to the conversion of a trithiophene-based diacid to the corresponding 2,6-dibromodithieno[3,2-b;2′,3′-d]thiophene (644) is presented.300 It was found that treatment of the Li-salt of 643 with an excess of solid NBS at room temperature led to 644 in an 81% yield. The proposed mechanism implies the electrophilic attack of a bromonium ion, derived from NBS, at the ipso position of the thiophene ring, followed by decarboxylation to afford the corresponding 2-bromothiophene derivative (Scheme 88).
Scheme 88. Bromodecarboxylation of Dithieno[3,2-b;2′,3′-d]thiophene-2,6-dicarboxylic Acid with NBS and the Proposed Mechanism.

6. Halodecarboxylation of α,β-Unsaturated Carboxylic Acids
The decarboxylative halogenation of unsaturated carboxylic acids has a significant synthetic importance for the preparation of halogen-containing olefins and acetylenes (Scheme 89).
Scheme 89. Synthesis of Haloalkenes and Haloalkynes via the Halodecarboxylation of Unsaturated Carboxylic Acids.

6.1. Halodecarboxylation of Substituted α,β-Alkenoic Acids
6.1.1. Halodecarboxylation with Molecular Halogens
The original Hunsdiecker procedure has undergone versatile modifications, and various methods are available nowadays for the halodecarboxylation of organic carboxylic acids. Unfortunately, these approaches rarely provide good results when applied to α,β-unsaturated carboxylic acids. For instance, the modified Hunsdiecker-type bromodecarboxylation of both trans- and cis-cinnamic acids gave β-bromostyrene in very poor yields (Scheme 90).5
Scheme 90. Hunsdiecker Bromodecarboxylation of Cinnamic Acid.

Attempts to replace RCO2Ag with Na analogues were unsuccessful. In aqueous solutions, the bromination of sodium cis-cinnamate with Br2 gave a 53% yield of cis-α-bromostyrene 650 in a mixture with the starting acid; meanwhile, the bromination of sodium trans-cinnamate 652 afforded the corresponding trans-α-bromostyrene 35 in only 30% yield, along with tolane (654, 20%) and 21% of the parent acid (Scheme 91).301
Scheme 91. Halodecarboxylation of Cinnamate Ions with Molecular Bromine and Chlorine.

An attempt to perform a direct chlorodecarboxylation of the trans-cinnamate ion with an excess of chlorine led to a mixture of products (Scheme 91).302 The similar bromination of cinnamate ion derivatives with Br2 in water or methanol resulted in a series of products (658–662), the ratios and stereospecificities between them depending on the nature of the R-substituent, the solvent and the presence of NaBr (Scheme 92).303 Notably, under any conditions, the desired vinyl bromide 658 was not obtained selectively.
Scheme 92. Oxidative Bromination of Substituted Cinnaate Ion with Molecular Bromine in Aqueous Methanol.

Later, the efficiency of the bromodecarboxylation with molecular bromine was improved by altering the solvent. It was found that in 10% acetic acid in dichloromethane the reaction of some cinnamic acids proceeded selectively and led to (E)-β-bromostyrenes in moderate to good yields (Scheme 93).304 Although optimization of the reaction conditions and the use of a pyridine-HBr3 complex instead of Br2 could improve the selectivity for the bromodecarboxylation of some cinnamic and acrylic acids, the method suffers from a highly limited scope, providing mixtures of products for many substrates.
Scheme 93. Bromodecarboxylation of Substituted Cinnamic and Acrylic Acids with Br2 in DCM-AcOH.

Yield for PyHBr3 instead of Br2.
A significant improvement in the efficiency of the preparation of (E)-vinyl bromides from (E)-cinnamic acids was achieved by the treatment of anti-3-aryl-2,3-dibromopropanoic acids 669 with AgOAc in AcOH. The (E)-β-arylvinyl bromides 670–673 were obtained stereoselectively in high yields, from the corresponding acids bearing both electron-donating and electron-withdrawing substituents on the aromatic ring (Scheme 94).305 Microwave irradiation allowed completion of the reaction in minutes.
Scheme 94. Microwave-Assisted Bromodecarboxylation-Debromination of anti-3-Aryl-2,3-dibromopropanoic Acids.

The starting anti-dibromopropionic acids 669 were obtained stereoselectively by the direct bromination of cinnamic acids with Br2.306 It is noteworthy that this two-step procedure can be carried out in one pot. Thus, (E)-1-bromo-4-(2-bromovinyl)benzene 676 was obtained directly from (E)-4-bromocinnamic acid 675 in an overall yield of 91% (E/Z > 98/2). Direct treatment of the acid 674 with bromine/AcOH, at 55 °C for 2 h, followed by the microwave-assisted treatment of the reaction mixture with 1.2 equiv AgOAc for 1 min led to the product 676 (Scheme 95).305
Scheme 95. Microwave-Assisted One-Pot Preparation of (E)-1-Bromo-4-(2-bromovinyl)benzene from (E)-4-Bromo Cinnamic Acid.

Interestingly, when this type of reaction is halted after 5 s of MW-irradiation, the product mixture consists of the desired vinyl bromide 670 and lactone 678, which was exemplified on anti-3-(4-methoxycarbonylphenyl)-2,3-dibromopropanoic acid 677 (Scheme 96).305
Scheme 96. Composition of Reaction Products after Short Microwave-Irradiation of anti-3-(4-Methoxycarbonylphenyl)-2,3-dibromopropanoic Acid.

This result led to speculations that the reaction might proceed via two possible pathways, presented in Scheme 97.305
Scheme 97. Proposed Mechanism for the Bromodecarboxylation of anti-3-(4-Methoxycarbonylphenyl)-2,3-dibromopropanoic Acids.

The starting acid 669 undergoes an Ag-promoted debromination to give the zwitterionic intermediate 679, which can eliminate CO2 to result in (E)-vinyl bromide. In parallel, the bromonium ion 680 can be formed, which would decompose to α-bromo-β-lactone 682. AcOH-assisted elimination of CO2 from the lactone 682 occurs with a retention configuration to give (E)-vinyl bromide. The authors supposed that in most cases the two pathways coexist. Thus, for acids with strong electron-donating groups on the aromatic ring, the pathway involving the zwitterion 681, is preferred. Acids possessing electron-poor aromatic rings should prefer the mechanistic scenario of the formation of α-bromo-β-lactone.
The dehalogenation-decarboxylation of anti-dibromopropionic acids was used for the stereoselective preparation of (Z)-vinyl halides from the corresponding (E)-cinnamic acids. It was found that (Z)-β-bromostyrenes could be selectively prepared from anti-3-aryl-2,3-dibromopropanoic acids by treatment with a Et3N/DMF system without Ag-salts. In turn, 2,3-dibromopropanoic acids could be obtained by the bromination of (E)-cinnamic acid with Br2 (Scheme 98).307
Scheme 98. Synthesis of (Z)-Vinyl Halides from (E)-Cinnamic Acids.

Later, this procedure was significantly improved with regard to shortening the reaction time and increasing the yields and stereoselectivity, by using additional microwave irradiation.308 A high stereospecificity was observed for both anti- and syn-dibromides.
Scheme 99. Microwave-Assisted Debromination-Bromodecarboxylation of anti-3-Aryl-2,3-dibromopropanoic Acids.

The high stereoselectivity of the reaction for most substrates was explained by the simultaneous loss of CO2 and a bromide ion during the trans-β-elimination (Scheme 100). However, in the case of 3-(4-methoxyphenyl)-2,3-dibromopropanoic acid, the presence of a strong electron-donating group provided stabilization of the carbocations 694 and 695, which after decarboxylation, gave (Z)- and (E)-vinyl bromides, respectively, with a preferential formation of the (Z)-isomer.
Scheme 100. Proposed Mechanism for the Bromodecarboxylation-Debromination of anti-3-Aryl-2,3-dibromopropanoic Acids.

Recently, it was demonstrated that (E)-cinnamic acids can be directly converted to (Z)-vinyl bromides by their treatment with Br2 and 1,8-diazabicyclo[5.5.0]undec-7-ene (DBU; Scheme 101).309 While the convenient one-pot procedure was successfully implemented, in some cases (where R is not a strong electron donating group), bromolactone intermediates 682 were isolated.
Scheme 101. One-Pot Synthesis of (Z)-Bromostyrenes from (E)-Cinnamic Acids in the Presence of DBU.

Unfortunately, an exclusive stereoselectivity was only observed for cinnamic acids bearing weak electron-donating or strong electron-withdrawing groups on the aromatic ring. A methoxy group, especially in the ortho-position, induced strong isomerization and afforded mixtures of (Z)- and (E)-isomers. The mechanism of this process is shown in Scheme 102.
Scheme 102. Proposed Mechanism for the Synthesis of (Z)-Bromostylbenes from (E)-Cinnamic Acids.

The selective formation of the Z-isomer cannot be explained by the thermal or concerted decarboxylation of bromolactone 698. Such a scenario should lead to a more stable E-isomer. It was proposed that after the deprotonation of the acidic α-proton with DBU in 698, the resulting enolate 699 undergoes stereospecific β-elimination by a E1cB mechanism to give the carboxylate 700 with a fixed Z-configuration (pathway 1). The latter underwent decarboxylation relatively easily, since the resulting 1-bromovinyl carbanion 701 is stabilized by the bromine substituent. In the case of a strong electron-donating group on the aromatic ring (2- or 4-MeO) it was suggested this mechanism is accompanied by the alternative pathway 2. The initially formed bromonium ion 702 exists in equilibrium with a stabilized benzylic carbocation 703, which is subjected to rotation before cyclization in β-lactone, to give a mixture of both diastereomers. Thus, after the decarboxylation, both the (Z)-and (E)-vinyl bromides were formed.
Treatment of the dithioketals of monoethyl esters of 2-oxo-3-oxomethylidenesuccinic acid 704 or the dithioketals of 2,4-dioxo-3- oxomethylidenepentanoic acid 705 with iodine and NaHCO3 in aqueous ethanol produced the corresponding monoiodide 706 and 707 in a high yield (Scheme 103).310
Scheme 103. Iododecarboxylation of Dithioketals of 2,4-Dioxo-3-oxomethylidenepentanoic Acid and Esters with Iodine.

The iododecarboxylation of the dithioketals of 2-oxo-3-oxomethylidenesuccinic acid 708 with iodine in aqueous ethanol gave the corresponding diiodide in excellent yields (Scheme 103). Under the same conditions, the iododecarboxylation of cyclic dithioketals 710 also provided the monoiodides in high to excellent yields (Scheme 104).311
Scheme 104. Iododecarboxylation of α-Carboxylate, α-Aroyl Ketene Cyclic Dithioketals.

The mechanism of this reaction involves the electrophilic addition of iodine on the double bond to form the carbocation 715, which is stabilized by the adjacent sulfur atoms due to the delocalization of the positive charge (Scheme 105).
Scheme 105. Proposed Mechanism for the Iododecarboxylation of α-Carboxylate, α-Aroyl Ketene Cyclic Dithioketals.

Both the strong nucleophilicity of dithioketals and the stability of the carbocation benefit this step. The release of CO2 from this intermediate produces the monoiodide product 716. In the case of R = OH, the second iododecarboxylation takes place in a similar way.
6.1.2. Halodecarboxylation with N-Halosuccinimides
Attempts of the halodecarboxylation of α,β-unsaturated carboxylic acids by reaction with N-halosuccinimides, in the absence of additives, usually lead to a mixture of products.312 In the last two decades, extensive studies have been conducted to understand the factors affecting the yields and selectivity of the reactions with NXS, as well as to elucidate a plausible mechanism.
In pioneering works, Roy and co-workers revealed that manganese and alkali metal acetates could serve as good catalysts for the conversion of trans-α,β-unsaturated carboxylic acids to β-haloalkenes (Scheme 106).313,314 Among the alkali metal acetates (Na, K, or Cs acetate), lithium acetate was beneficial. Mn(OAc)2 gave similar results in the bromodecarboxylation, but the reaction was sensitive to the amount of water and NBS, providing halohydrines as a side- or even in some cases a major product.
Scheme 106. LiOAc-Promoted Halodecarboxylation of Cinnamic Acids with Halosuccinimides.

Starting acid E/Z = 87:13.
On the basis of semiempirical calculations, a plausible ionic pathway for the halodecarboxylation of α,β-unsaturated carboxylic acids in the presence of lithium acetate was suggested (Scheme 107).315,316 Indeed, the reaction was unaffected by the presence of a radical trap, which supports the ionic mechanism. It was stated that the role of the catalyst was to form a tight ion pair 724 in organic solvents. This anion participates in the reaction with NXS faster than the acid itself.
Scheme 107. Proposed Mechanism for the LiOAc-Catalyzed Bromodecarboxylation of Cinnamic Acid.

A nucleophilic attack of 724 on NBS leads to the formation of the bromonium ion 725. This species could proceed to an α-bromo-β-lactone 682 or open up to the carbocation 695, depending on electronic and steric factors. The regioselectivity in 695 could be explained by the fact that the α-carbon of the double bond of the carboxylate anion 724 bears a greater negative charge (−0.164 eV) compared to the β-carbon (−0.074 eV) in these species. The formation of the lactone 682 under the conditions of the halodecarboxylation was observed previously.303 These compounds were also isolated in preparative yields in the case of α-substituted cinnamic acid and alkylvinylcarboxylic acids.315,317
It was found that MW irradiation substantially accelerates the LiOAc-promoted halodecarboxylation of substituted cinnamic acids, reducing the reaction time to 1–2 min (Scheme 108).316,318,319
Scheme 108. MW-Assisted Halodecarboxylation of Cinnamic Acids with the NBS/LiOAc System.

In continuation of these studies, the Roy group demonstrated that tetrabutylammonium trifluoroacetate (TBATFA) can be employed as a catalyst instead of LiOAc, with a reaction outcome on a par with the alkali metal acetates (Scheme 109).320−322
Scheme 109. Tetrabutylammonium Trifluoroacetate (TBATFA)-Catalyzed Bromodecarboxylation of α,β-Unsaturated Carboxylic Acids with N-Halosuccinimides.

At −40 °C.
E:Z = 1:1.
E:Z = 89:11.
Startingacid E:Z = 87:13.
The proposed mechanism for the TBATFA-catalyzed process was similar to that of the alkali metal acetates.321,322 The formation of a tight ion-pair of carboxylate anion–quaternary ammonium cation is believed to be crucial, since it has a beneficial reactivity with NXS compared to the starting carboxylic acid (Scheme 110).
Scheme 110. Proposed Mechanism for the TBATFA-Catalyzed Bromodecarboxylation of Cinnamic Acid.

Semiempirical calculations (AM1) of the intermediates 736 and 737 provided interesting information. They show that the electron charge on the α-carbon atom (−0.202 eV) in 736 is much higher than that on the β-carbon (−0.026 eV) and that the Cα-Br bond (1.96 Å, close to a Br–Csp3 bond) in 737 is significantly shorter than the Cβ–Br bond (2.74 Å). The Cα–COO bond in 737 is significantly longer in comparison to that in 736. Therefore, the bromonium ion attack was believed to be directed to the α-carbon atom, while the weakening of the Cα–COO bond in 737 promoted the elimination of CO2. Such a mechanism also explains the positive effect on the product yield of electron-donating substituents on the aromatic ring and on the double bond due to their stabilization of the cation 737.
Later, it was discovered that Et3N was also a powerful catalyst, both in dichloromethane and in aqueous acetonitrile (Scheme 111).323 MW irradiation shortened the reaction time and increased the product yield in the case of nonactivated cinnamic acids.
Scheme 111. The Et3N-Catalyzed Halodecarboxylation of α,β-Unsaturated Carboxylic Acids with NXS.

Yields of the reaction in CH3CN/H2O (97:3) under 260 W MW irradiation.
A comparative spectroscopic UV–vis kinetic study of the LiOAc-, TBATFA-, and triethylamine-catalyzed halodecarboxylation of aryl-substituted cinnamic acid in various solvents revealed that the efficiency of lithium acetate and Et3N in acetonitrile–water (97:3) was 1 order of magnitude higher than that of TBATFA in DCM or acetonitrile–water (Table 3).323
Table 3. Rate Data for Different Catalysts and Solvent Combinations.
| catalyst | solvent | Kobs × 105 (s–1) | K+ (rel. rate) (s–1) |
|---|---|---|---|
| Et3N | DCM | 42 | 4.6 |
| Et3N | CH3CN | 21 | 2.3 |
| Et3N | CH3CN-H2O (97:3) | 192 | 21.3 |
| LiOAc | CH3CN-H2O (97:3) | 209 | 23.2 |
| TBATFA | DCM | 13 | 1.4 |
| TBATFA | CH3CN | 9 | 1 |
| TBATFA | CH3CN-H2O (97:3) | 44 | 4.8 |
It was found that halodecarboxylation involving N-halosuccinimides could be stereospecific in the presence of potassium acetate. Thus, the slow addition of N-bromo-, chloro-, or iodosuccinimide to hot solutions of E-5-(2-carboxyvinyl)-2′-deoxyuredine 739 and potassium acetate induced an immediate evolution of gas and resulted in the stereoselective formation of the potent antiherpes virus agents, E-5-(2-halovinyl)-2′-deoxyuredines halides 740–742 in 69%, 39%, and 43% yield, respectively (Scheme 112).324 This procedure was further used for the synthesis of other halovinyl analogs of the carbocyclic nucleosides 743 and 744 (Scheme 112).325,326
Scheme 112. Synthesis of Potent Anti-Herpes Virus Agents via the AcOK-Promoted Halodecarboxylation of Potassium α,β-Unsaturated Carboxylate.

The high activity of both quaternary ammonium and alkali metal catalysts allowed their application in a one-pot halodecarboxylation-cross coupling strategy, which was first demonstrated in an example of the bromodecarboxylation-Heck reaction.321,322 TBATFA proved to be the best catalyst for the one-pot synthesis of 5-aryl-2,4-pentadienoic acids, esters, and amides from cinnamic and acrylic acids.
A similar one-pot strategy was also utilized in a bromodecarboxylation-Suzuki cross-coupling sequence to prepare various highly conjugated styrenes from in situ synthesized cinnamyl bromides with phenylboronic esters.327
Additional catalysts for the halodecarboxylation with NXS have been further developed. It was found that iodosobenzene and (diacetoxyiodo)benzene promoted stereospecific transformation of 3-arylprop-2-enoic acids to the corresponding (2-halovinyl)benzenes (Scheme 113).
Scheme 113. Halodecarboxylation of Cinnamic Acids with NXS in the Presence of Polyvalent Iodine.

E/Z ratio.
The reaction with NBS proceeds in good yields (60–89%) for most of the (E)- and (Z)-cinnamic acid derivatives. The stereoselectivity of the conversion of (E)-cinnamic acids to (E)-vinyl bromides was remarkably high, whereas the (Z)-counterpart furnished a mixture of isomeric products (compounds 686, 745, and 746). However, substituents in the α-position in the substrate significantly reduced the yield of bromides (compound 728 and 747). Satisfactory yields of chloro- and iodovinylbenzenes in the analogous reaction with NCS or NIS were observed only for cinnamic acids bearing a strong electron-donating group on the aromatic ring.
Computational analysis suggests that the reaction starts by the formation of a hypervalent iodine-based intermediate 748, followed by electrophilic attack to form the bromonium ion 749 (Scheme 114). This species undergoes fragmentation to release CO2, the desired bromoalkene 750, and a polyvalent iodine intermediate 751, which might be considered as the active catalyst.
Scheme 114. Proposed Mechanism for Bromodecarboxylation of α,β-Unssaturated Acids with PIDA.

An additional interesting approach to the NXS-based halodecarboxylation employs a micellar catalysis, namely, the use of surfactants, such as sodium dodecyl sulfate, hexadecyltrimethylammonium bromide (CTAB), or Triton X-100.328 As it turned out, it could be considered as a powerful and universal method for the conversion of para-substituted (E)-cinnamic and acrylic acids to the corresponding β-halovinyls derivatives.328 In a typical procedure, refluxing a suspension of the halosuccinimide (1.5 equivalents), substrate, and 5% mol of CTAB afforded the corresponding haloalkenes in good to excellent yields (65–95%; Scheme 115).
Scheme 115. Halodecarboxylation of α,β-Unsaturated Acids with N-Halosuccinimide in the Presence of a Micellar Catalyst.

Notably, high yields of haloalkenes were observed for cinnamic acids, bearing electron-poor and electron-rich substituents on the aromatic ring, 3-phenylcrotonic acids 753–755, and metacrylic acids 756–758, which were usually inefficient in reactions with NXS. It was proposed that micelles enhance the reaction rates by their solubilization capability, which could increase the reactant concentration within the hydrophilic or hydrophobic area of the micellar clusters, thus facilitating the reaction. Novates and Pastre used this method in the total synthesis of the natural product actinoranone, a potential antitumor agent.329
6.1.3. Halodecarboxylation with an Alternative Source of Halonium Ions
Following the idea to employ N-halogen compounds as halogenating agents, few alternatives for N-halosuccinimides were investigated. The use of N-chlorobenzotriazole (NCBT) as the chlorinating agent was demonstrated for the preparation of (Z)-β-vinyl chlorides from the corresponding (Z)-cinnamic acids.330 Microwave irradiation (5–10 min, 200 W) of the acid, NCBT, and catalytic LiOAc gives the β-vinyl chlorides with complete retention of the stereochemistry in high yields. An α-substitution deteriorates stereochemistry of the reaction. Thus, chlorodecarboxylation of (Z)-2-methyl-3-phenylacrylic acid led to a mixture of (Z)-and (E)-(2-chloroprop-1-en-1-yl)benzene 725 (Scheme 116).
Scheme 116. Microwave-Assisted Chlorodecarboxylation of α,β-Unsaturated Acids with N-Chlorobenzotriazole.

Recently N,N-dibromo-p-toluene sulfonamide (TsNBr2) was suggested as an effective reagent for the bromodecarboxylation of α,β-unsaturated carboxylic acids. In the presence of a base, a fast, selective reaction occurred, affording the corresponding bromoalkenes in good to excellent yields331 The reaction proceeded stereospecifically, forming only E-isomer.
Scheme 117. Bromodecarboxylation of α,β-Unsaturated Acids with N,N-Dibromo-p-toluene Sulfonamide.

In the last two decades, alternative methodologies for the generation of halonium ions were developed, with special attention to hypervalent iodine reagents. Such reagents were used for the generation of the Br+ ion from quaternary ammonium salts. Thus, PhI(OAc)2 in combination with tetraethylammonium bromide (TEBA) induced the bromodecarboxylation of various substituted (E)-cinnamic and acrylic acids, in excellent yields. Remarkably, the reaction is efficient regardless of the nature of the substituents on the aromatic ring or double bond (Scheme 118).332 Saturated aliphatic acids (e.g., butanoic, pentanoic, 2-(phenyl)-propionic acids) did not undergo this bromodecarboxylation protocol.
Scheme 118. Bromodecarboxylation of α,β-Unsaturated Acids with PIDA/TEBA-System.

Other conceptually similar hypervalent reagents were investigated and proved to be useful in the bromodecarboxylation with TEBA. Thus, o-iodoxy benzoic acid333 and Dess-Martin periodinane334 in combination with TEBA convert various alkenoic acids to the corresponding bromoalkenes in practical yields.
Another interesting iodine reagent, diphosphorous tetraiodide, in combination with TEBA, demonstrated a high efficiency in the bromodecarboxylation of (E)-cinnamic and (E)-acrylic acids in anhydrous carbon disulfide (Scheme 119).335
Scheme 119. Bromodecarboxylation of α,β-Unsaturated Acids with Diphosphorous Tetraiodide.

The reaction is applicable to a wide variety of substrates having substituents with various electronic characteristics and provides the (E)-isomers of the final bromides. A remarkable observation was noted regarding the stereoselectivity of this process: if the pure Z-isomer, or a mixture of the Z,E-isomers of the acids, were used as substrates, the (E)-isomer of vinyl bromide was obtained exclusively. It was concluded that in the case of (Z)-α,β-unsaturated carboxylic acids, inversion took place, while the (E)-counterparts react with retention of their configuration to provide thermodynamically stable product in both cases (Scheme 120). It should be noted that the P2I4/TEBA combination was inefficient for the halodecarboxylation of saturated alkanoic carboxylic acids.
Scheme 120. Stereoselectivity of Bromodecarboxylation of α,β-Unsaturated Acids with Diphosphorous Tetraiodide.

Following the idea of the in situ generation of a positive halogen species for the halodecarboxylation, Rousseau and co-workers found that bis(sym-collidine)bromine(I) hexafluorophosphate and bis(sym-collidine)iodine(I) hexafluorophosphate can be used to convert some cinnamic and acrylic acids to the corresponding haloalkenes in satisfactory yields (60–93%; Scheme 121).336
Scheme 121. Halodecarboxylation of α,β-Unsaturated Acids with Bis(sym-collidine)halogen(I) Hexafluorophosphate.

In toluene at 65 °C.
Starting acid E/Z = 3:97.
E/Z-ratio.
The reactions were stereoselective for trans-cinnamic acids, providing the E-isomer almost exclusively. For the cis-cinnamic acids and acrylic acids, a notable isomerization was observed. The iododecarboxylation of acrylic acid gave similar results, although the reaction with cinnamic acids was noneffective (yield of 28% for trans-cinnamic acid).
Recently, it was demonstrated that ionic liquids based on imidazolium tribromide could be useful for the bromodecarboxylation of sodium salts of α,β-unsaturated acids.337,338 These brominating agents were used both as solvents and reagents. For instance, stoichiometric amounts of ethylenebis(N-methylimidazolium) ditribromide (EBMIDTB) react with α,β-unsaturated acids in the presence of K2CO3, to furnish the corresponding vinyl bromide in reasonable yields (Scheme 122). However, the stereoselectivity of the resulting bromoalkenes was highly substrate-dependent.
Scheme 122. Bromodecarboxylation of α,β-Unsaturated Acids with Ethylenebis(N-methylimidazolium) Ditribromide.

We previously described the utilization of simple alkali salts of halides as a source of halogen in the presence of relatively strong oxidizers, for the synthesis of aryl halides. A similar approach, employing different oxidizing agents with sodium halides, proved efficient for the halodecarboxylation of α,β-unsaturated acids.339−344 Here, we would like to discuss some representative examples.
Stoichiometric combinations of Oxone (KHSO5)/NaCl, NaBr or NaI in the presence of Na2CO3, afforded (E)-β-halostyrenes (Scheme 123a) and 2-aryl-1-bromophosphonates (Scheme 123b) from the corresponding (E)-α,β-unsaturated acids.339,340 While bromo- and chloroalkenes were obtained in useful yields, iododecarboxylation by this approach was less effective (yields 0–45%). The stereoselectivity varies from high to moderate, which might impose certain limitations on the method.
Scheme 123. (a, b) Oxone-Promoted Halodecarboxylation of α,β-Unsaturated Acids with Alkali Halides.

A surprisingly high efficiency was observed for a 48% solution of HBr in the process of bromodecarboxylation of (E)-α,β-unsaturated carboxylic acids, when the reaction was performed in the presence of catalytic amounts of sodium nitrite and atmospheric oxygen (Scheme 124).343 Notably, the resulting bromides were obtained as (E)-isomers in excellent yields (87–91%) regardless of the substituents on the aromatic ring and on the double bond.
Scheme 124. Sodium Nitrite-Catalyzed Bromodecarboxylation of α,β-Unsaturated Acids with 48%HBr.

The role of oxygen and the bromine source was critical. In the absence of O2, the reaction with cinnamic acid proceeded very slowly (48 h) and gave β-bromostyrene in poor yields (5%). No reaction was observed when HBr was replaced with TEBA or NBS. The proposed mechanism of the process comprises a series of redox reactions (Scheme 125). The authors assumed that in situ formed molecular bromine was responsible for the electrophilic attack on the double bond of the acid, followed by decarboxylation. However, the electrophilic attack of NOBr on the double bond to form a bromonium ion cannot be ruled out.
Scheme 125. Proposed Mechanistic Steps of Bromodecarboxylation with HBr/NaNO2/O2.

Because of the simplicity of the procedure, this approach can be considered as one of the simplest and economic methods for the preparation of (E)-haloalkenes. The use of highly concentrated HBr is a certain drawback of the process.
Another possible way to oxidize KBr to the bromonium cation is by the employment of Selectfluor (Scheme 126a).345
Scheme 126. (a) Bromodcarboxylation of Cinnamic Acids with a Selectfluor/KBr System and (b) Plausible Mechanism.

E/Z-ratio.
Isolated yields for the E-isomer.
Interestingly, no fluorodecarboxylation products were observed in the reaction mixture, which was explained by the fast oxidation of KBr with Selectfluor to afford the Br+ ion, followed by its addition to the double bond (Scheme 126b). However, in the presence of a base, Selectfluor induced the fluorodecarboxylation of cinnamic acids in moderate yields and poor stereoselectivity (Scheme 127a).346
Scheme 127. (a) Fluorodecarboxylation of α,β-Unsaturated Acids with Selectfluor in the Presence of a Base and (b) Proposed Mechanism.

E/Z ratio.
Other alkali-metal bases (NaF, KF and CsF, Na2CO3) can effectively promote the reaction. Unfortunately, cinnamic acids with electron-withdrawing groups on the aromatic ring, and heteroaromatic acrylic acids, did not react under the developed conditions. No product formed in the presence of the radical scavenger TEMPO and the low stereoselectivity of the reaction hinted at a radical-based mechanism (Scheme 127b). Despite the above-mentioned drawbacks, this work is, to the best of our knowledge, the only example of the fluorodecarboxylation of α,β-unsaturated carboxylic acids. We will provide a detailed discussion of fluorodecarboxylation methods in the next chapter.
Another oxidative method is based on the use of sodium hypochlorite (NaClO, bleach) for the generation of a positively charged chlorine species. Thus, an excess of bleach was employed for the decarboxylative chlorination of α,β-unsaturated aromatic and heteroaromatic acids (Scheme 128).344
Scheme 128. Examples of Chlorodecarboxylation with Bleach.

The corresponding (E)-chloroalkenes were obtained in satisfactory yields. The formation of chlorides from saturated acids, alkyl acrylic acid, and (E)-β-(2-pyridyl)-acrylic acid was not observed. In the presence of molecular iodine or bromide, mixtures of the haloalkenes were formed. No oxidation or epoxidation of the double bond, oxidation of methyl groups, or aromatic chlorination were observed under the reaction conditions. It should be noted that this is one of the rare approaches which allows the preparation of vinyl chlorides from the carboxylic acids in practical yield.
A series of works was devoted to the generation of Br+ from an alkali halide using transition metal peroxo-complexes.347−349 Thus, a “green variant of the Hunsdiecker reaction” was suggested involving a Na2MoO4/H2O2 agent for generation of the bromonium ion from KBr (Scheme 129).347 Unfortunately, satisfactory yields of β-bromostyrenes were reached only for nonsubstituted 4-methoxy and 4-chlorocinnamic acids (65 and 85%, respectively).
Scheme 129. Na2MoO4-Promoted Bromodecarboxylation of Cinnamic Acids with the KBr/H2O2 System.

Later, Roy improved the scope of this reaction by using sodium tungstate (Scheme 130).348 As in the previous case, the reaction is limited to acids with electron-donating substituents. Acids bearing electron-withdrawing groups, such as nitrocinnamic acids and phenacyl acrylic acids, as well as aliphatic α,β-unsaturated carboxylic acids (e.g., crotonic acid), remained intact under reaction conditions.
Scheme 130. Na2MoO4-Promoted Bromodecarboxylation of Cinnamic Acids with the KBr/H2O2 System.

The reaction mechanism is depicted in Scheme 131. Na2WO4 is oxidized in situ to form the tetraperoxotungstate 808, which simultaneously deprotonates the acid and oxidizes the bromide to form the W–O–Br motif in the coordination sphere of the complex. Carboxylates and esters are not active in this reaction, which clearly demonstrates the importance of the acidic proton. Nucleophilic attack of the double bond on the formal positive bromine leads to the formation of the cation 811 and species 810, which are further oxidized by H2O2 to regenerate an active catalyst. The formation of the intermediate 811 critically depends on the stabilization factors of the adjacent aromatic ring. The presence of an electron-withdrawing group on the aromatic ring destabilizes the carbocation, which explains the inertness of the corresponding precursors in this reaction.
Scheme 131. Proposed Mechanism of the Na2MoO4-Promoted Bromodecarboxylation of Cinnamic Acids with the KBr/H2O2 System.

Later, it was found that such type of reactions can be performed utilizing the powerful and common oxidizer ceric(IV) ammonium nitrate [Ce(NH4)2(NO3)6; CAN]. CAN was able to oxidize LiBr or LiCl in the course of the reaction with α,β- unsaturated carboxylic acids to provide β-halostyrenes (Scheme 132).350 Notably, both chloro- and bromodecarboxylation was efficient in this approach.
Scheme 132. Ceric(IV) Ammonium Nitrate-Promoted Halodecarboxylation with LiBr and LiCl.

Interestingly, a solvent-free solid-phase version of this reaction was recently reported, using CAN for the halodecarboxylation of unsaturated acids.351
One of the challenges in halodecarboxylation chemistry is the development of processes which do not use halogenated solvents and corrosive and toxic molecular halogens. In this respect, stable and inexpensive trichloro- (TCCA) and tribromoisocyanuric acids (TBCA) constitute an attractive alternative (Scheme 133).352 Both of these reagents promoted the halodecarboxylation of substituted cinnamic acids to result in 2-bromostyrenes, in satisfactory yields, when tested in the presence of a base in a biphasic medium (diethyl ether-water). Substrates bearing electron-withdrawing groups were not reactive under the reaction conditions.
Scheme 133. Halodecarboxylation of Sodium Cinnamates with Trihaloisocyanuric Acids.

Electrochemical organic synthesis has lately received a great deal of attention. Recently, an anodic oxidation of ammonium bromide to the bromonium ion on platinum electrodes was employed to promote the stereoselective halodecarboxylation of substituted (E)-cinnamic acids and (E)-β-(2-thienyl)acrylic acids into the corresponding β-bromoalkenes (Scheme 134).353 The reaction trends are similar to those found in the previous examples of Br–/Br+ oxidation. The reaction works mainly with substrates in which a carbocation/bromonium intermediate can be stabilized by electron-donating substituents. Experimental data supported an ionic rather than a radical mechanism.
Scheme 134. Bromodecarboxylation of α,β-Unsaturated Acids under Anodic Oxidation with Ammonium Bromide.

6.2. Halodecarboxylation of Acetylenic Carboxylic Acids
In general, the halodecarboxylation of acetylenic carboxylic acids possesses some similarities with the reactions of olefinic acids. In this section, we will describe representative methods for the synthesis of haloalkynes from alkynoic acids.
In the classical Hunsdiecker reaction, the Ag-salt of phenylpropiolic acid was converted to iodophenylacetylene 34 in an almost quantitative yield (Scheme 135).5 We have discussed the considerable drawbacks of the Hunsdiecker reaction, and its modifications in the previous chapters. These limitations prompted chemists to search for alternative efficient and facile approaches.
Scheme 135. Iododecarboxylation of Ag-Salt of Phenylpropiolic Acid.

One of the practical alternatives to the Hunsdiecker reaction is the utilization of halide salts as a source of halogen, in the presence of an oxidizer to generate the halonium cation in situ. We described a number of conceptually similar methods based on such an approach in the section on the halodecarboxylation of α,β-unsaturated acids. Here, we summarize the related representative examples for acetylenic acids.
It was reported that the combination of Oxone with NaBr, in the presence of a base, selectively converts various arylpropiolic acids to the corresponding β-bromoalkynes in excellent yields (Scheme 136).354 The reaction did not work for (4-nitrophenyl)propiolic acid which bears a strong electron-withdrawing aromatic ring substituent. Moreover, propiolic acid, substituted with a heteroaromatic ring, results in the desired product in a substantially lower yield.
Scheme 136. Halodecarboxylation of Acetylenic Acids with the Oxone–NaBr System.

Other combinations, based on such an oxidative principle, can be used for this transformation. Thus, Na2WO4/H2O2/KBr was used for the bromodecarboxylation of propiolic acid,348 whereas I2O5/I2 was applied for the corresponding iododecarboxylation of phenylpropiolic and 2-hexynoic acids.355
Another conceptual approach is to employ the reagents which can be considered as potential carriers of the positively charged halonium ion. For example, bis(sym-collidine)bromine hexafluorophosphate and bis(sym-collidine)iodine hexafluorophosphate converted acetylenic acids to the corresponding mono- and dihaloacetylenes in moderate to good yields (Scheme 137).336
Scheme 137. Bromodecarboxylation of Acetylenic Acids with Bis(sym-collidine)halogen (I) Hexafluorophosphates.

An additional route is to use N-haloamides as the source of halogen, in the presence of catalytic amounts of base. For instance, N-bromo- and N-iodosuccinimides in the presence of tetrabutylammonium trifluoroacetate converted substituted propiolic acids to the β-haloalkynes at room temperature (Scheme 138).356
Scheme 138. Halodecarboxylation of Propiolic Acids with N-Halosuccinimides.

The yield of 1-iodoacetylenes was usually higher than that of 1-bromoacetylenes. Both halodecarboxylations were effective with acids bearing enyne and diyne motifs. The reaction of phenylpropiolic acid with NBS gave the corresponding bromoalkyne in less than a 10% yield in the absence of the catalyst. The reaction was unaffected by the presence of a nitrone radical trap. On the basis of experimental and computational studies, the authors proposed an ionic pathway for the reaction (Scheme 139).
Scheme 139. Proposed Mechanism of the TBATFA-Catalyzed Bromodecarboxylation of Phenylpropiolic Acid with NBS.

The calculations revealed that the energies of formation of the intermediates 836, 837, and 838 were about the same. The authors believed that the open vinyl cation 837 is the preferred one based on its stereoelectronic features.
It was found that triethylamine was a more effective catalyst in the halodecarboxylation with N-bromo- and iodosuccinimides (Scheme 140).323
Scheme 140. TEA-Catalyzed Halodecarboxylation of Propiolic Acid.

An improvement to the Et3N-catalysis resulted in the ability to perform both bromo- and iododecarboxylation in an efficient manner to produce β-haloalkynes in nearly similar high yields.
The combination of N,N-dibromo-p-toluene sulfonamide (TsNBr2) with DBU was effective for the bromodecarboxylation of propiolic acids.331 The reaction instantaneously led to 1-bromoalkynes in good to excellent yields at room temperature (Scheme 141).
Scheme 141. Bromodecarboxylation of Acetylenic Acids with N,N-Dibromo-p-toluene Sulfonamide.

The utilization of inexpensive and stable trihaloisocyanuric acids as halogenating agents was also explored. Interestingly, an attempt at the chlorodecarboxylation of arylpropiolic acids, bearing neutral or electron-withdrawing substituents, with TCCA afforded 1,1,1-trichloromethyl phenyl ketones in moderate to good yields rather than the corresponding chloroacetylenes (Scheme 142).357
Scheme 142. Synthesis of 1,1,1-Trichloromethyl Phenyl Ketones via the Chlorodecarboxylation of Arylpropiolic Acids with TCCA.

A radical scavenger (2,6-di-tert-butyl-4-methylphenol) did not halt the reaction, thus supporting the electrophilic mechanism. Experiments with 18O-labeled water indicate that the carbonyl oxygen in the final product originated from the carboxyl group rather than from H218O. The proposed mechanism is presented in Scheme 143.
Scheme 143. Proposed Mechanism for the Formation of 1,1,1-Trichloromethyl Phenyl Ketones.

The bromodecarboxylation of acetylenic acids with dibromoisocyanuric acid (DBCA) may result in number of products depending on the catalytic additives. The reaction of DBCA with propiolic acid in the presence of 10 mol % of AgOAc in an aqueous solvent selectively led to 1,1,1-tribromomethylketones (Scheme 144).358
Scheme 144. Synthesis of 1,1,1-Bromomethyl Phenyl Ketones via the Bromodecarboxylation of Propiolic Acids with DBCA.

At the same time, the presence of 10% of a radical scavenger TEMPO led to the exclusive formation of 1,2,2-tribromostyrene, whereas 1,1,1-tribromomethyl ketones were not obtained at all (Scheme 145).
Scheme 145. Synthesis of 1,2,2-Tribromostyrenes via Bromodecarboxylation of Propiolic Acids with DBCA.

Experimental mechanistic studies enabled the proposal of a possible reaction pathway (Scheme 146).
Scheme 146. Proposed Mechanism for the Reactions of Phenylpropiolic Acid with DBCA.

It is well-documented that DBCA generates Br2 and hypobromous acid (HOBr) in the presence of water.359 The formation of tribromo ketones proceeds by path I, which involves both DBCA and HOBr. DBCA presumably forms 2-bromophenylacetylene via the bromonium ion 836. HOBr reacts with 814 to form intermediate 870, which is hydroxylated, and the resulting species 871 converts to the final tribromoketone by an additional bromination with DBCA. 1,2,2-Tribromostyrenes, in turn, are obtained due to the electrophilic addition of Br2, formed in situ, to 2-bromophenylacetylene 814 via path II. It was assumed that the Br2 is scavenged by AgOAc, negating the feasibility of path II. In contrast, TEMPO can easily reduce HOBr,360 thus preventing path I.
The iododecarboxylation of some acetylenic acids can be performed by their reaction with N,N-diiodo-3,3-dimethylhydantoin (DIH). Thus, iodophenyl acetylene was prepared from propiolic acid and DIH, in a 91% isolated yield (Scheme 147).361 Interestingly, when such an iododecarboxylation was performed in the presence of a fluorine source, 2,2-diiodo-1-fluoro-alkenes were isolated as the reaction products (Scheme 148a).
Scheme 147. Iododecarboxylation of Propiolic Acid.

Scheme 148. Fluorination-Iododecarboxylation of Propiolic Acids with N,N-Diiodo-3,3-dimethylhydantoin in the Presence of the Py·HF/AgOAc System.

It was proposed that the reaction proceeds via the formation of 2-iodoacetylenes (34, Scheme 147), which further reacts with DIH to provide an iodonium ion intermediate. The latter species is nucleophilically opened by the fluoride ion. Indeed, subjecting the isolated iodoalkyne 34 to the reaction conditions leads to the formation of 1-fluoro-2,2-iodostyrene (872) in 86% isolated yield (Scheme 148b).
7. The Fluorodecarboxylation Reaction
Fluoro-organic compounds occupy a special place among the organic halides and have attracted enormous attention within the chemical community. Because of their unique physical, chemical and biological properties, organic fluorides have found wide use in numerous valuable commercial products and industrial applications.362 The incorporation of fluorine is a key tool used in the pharmaceutical and agrochemical industries for altering the activity, bioavailability, and metabolic stability of the organic compound. Therefore, reliable and selective methods for the fluorination of organic molecules are highly desirable for discovery of drugs and agrochemicals. Undoubtedly, an attractive approach would be the direct exchange of a carboxylic group with fluorine. Although a number of reactions for fluorodecarboxylation exist, an operationally simple, economical and general method still presents a considerable challenge. In general, the existing reactions mechanistically differ from the approaches described by us for other halodecarboxylations. All these factors prompted us to discuss the fluorodecarboxylation chemistry in a separate chapter.
7.1. Early Examples
In analogy to other halodecarboxylation reactions, the initial attempts to introduce a fluorine atom in place of a carboxylic group were performed using the elemental halogen. However, because of the severe reactivity of a fluorine gas, the reaction usually resulted in the nonselective fluorination of the organic substrate. The first successful results were obtained for intrinsically inert perfluorinated substrates, so a selective reaction on the carboxyl group could be achieved.363,364 Vapors of trifluoroacetic 879 or pentafluoropropionic acids were reacted with a fluorine gas to yield perfluoroacyl hypofluorite 880 (Scheme 149a). These hypofluorites were isolated and shown to decompose slowly at room temperature via a free-radical pathway to yield carbon tetrafluoride and hexafluoroethane, respectively.
Scheme 149. (a–c) Early Examples of the Fluorodecarboxylation Reaction with Fluorine Gas.

Grakauskas isolated alkyl fluorides after the treatment of cold aqueous solutions of sodium α,ω-dicarboxylates with fluorine gas.365 The main product of the fluorination of sebacic acid was ω-fluorocarboxylic acid 883. 1,8-Difluorooctane 884 was also obtained, although the samples were contaminated with significant amounts of randomly fluorinated octanes 885 (Scheme 149b). The formation of polyfluorooctane 885 was proposed to occur via the fluorination of 884 since no signs of the fluorination of 8-fluorooctanoic acid were observed. Monobasic aliphatic acids also underwent fluorodecarboxylation, but reaction conversions were low and substantial amounts of undesired fluorinated products were observed. Salts of aromatic acids generally failed to provide the corresponding aryl fluorides, and only a dark tar was obtained after the attempted reactions. However, 1-fluoro-4-nitrobenzene 887 was isolated in 4% yield under similar conditions. In contrast to the reaction of trifluoroacetic acid 879, in this case it was proposed that the process proceeds via the formation of an acyl hypofluorite 889, which decomposes via an ionic solvent-cage SNi mechanism to form the alkyl fluoride (Scheme 149c).
Other strong oxidizing reagents were investigated in addition to elemental fluorine. Successful results were obtained for mercury(II) fluoride366 and bromine trifluoride367 in reactions with specific substrates (Scheme 150).
Scheme 150. Use of HgF2 and BrF3 in a Fluorodecarboxylation Reaction.

7.2. Fluorodecarboxylation with Xenon Difluoride
7.2.1. Scope of the Reaction
Patrick and co-workers demonstrated that among other strong oxidizing fluorinated reagents, xenon difluoride can be employed as a versatile reagent in the fluorodecarboxylation reaction.368,369 Primary alkyl fluorides are generally prepared in good yields (Scheme 151). Various activated fluorides, such as benzyl, allyl, and aryloxymethyl fluorides, were obtained by this method. The fluorodecarboxylation of secondary acids usually provides very poor yields of the corresponding fluorides. For example, cyclopentyl fluoride was obtained in only 10% yield, while norbornyl-2-carboxylic acid failed to deliver the product at all.369 Tertiary acids without β-hydrogens, such as triphenylacetic (891) and Mosher’s acids (904), underwent fluorodecarboxylation in excellent yields. The outcome of the reaction of tertiary bridgehead carboxylic acids depends on the size of the backbone. Camphanic and 1-bicyclo[n.1.1]alkyl carboxylic acids (906) failed to deliver the corresponding fluorides, while the protodecarboxylation product was predominant.243,370,371 The fluorodecarboxylation of 1-adamanthyl and lesser strained bicyclic carboxylic acids provided the desired fluorides in high yields (905); yet, significant amounts of hydrogen atom abstraction from the solvent were observed in many cases.243,369 Employment of hexafluorobenzene as the solvent helped to overcome this problem.372 Fluorobenzene cannot be obtained by treatment of benzoic acids with xenon difluoride, and some amounts of benzoyl fluoride were obtained as the major product. No fluorodecarboxylation was observed in the presence of unprotected hydroxy, amino, and thio groups.
Scheme 151. Selected Examples of Fluorodecarboxylation with XeF2.

Benzoic acid as the starting material
Sammis, Paquin, and co-workers used XeF2 to prepare a range of di- and trifluoromethoxyarenes via the challenging fluorodecarboxylation of mono- and difluoroaryloxyacetic acids under ambient conditions (Scheme 152).373 Generally, good yields of trifluoromethoxyarenes are obtained for substrates bearing both electron-donating and electron-withdrawing substituents on the aryl ring in very short reaction times. However, acids possessing poor solubility, bearing alkoxy or naphthyl substituents, or having benzylic protons, provide poor yields of the corresponding fluorodecarboxylation products. Additionally, difluoromethyl(aryl) ethers can be prepared via a similar approach, using aryloxy(fluoro)acetic acids. The yields of the corresponding difluoromethoxy arenes are somewhat lower than for the analogous trifluoromethoxy derivatives due to the more facile ring fluorination. The yields of the trifluoromethoxyarenes are on a par with those obtained by the method, reported by Hartwig et al., which uses the AgF2/AgF system (vide infra).374
Scheme 152. Preparation of Trifluoromethoxy- and Difluoromethoxyarenes Using XeF2.

7.2.2. The Reaction Mechanism
The first step of the reaction is likely to be the formation of a xenon ester as the key intermediate. The ester 916 was isolated when trifluoroacetic acid 879 was used as the substrate (Scheme 153).375 There is evidence for both radical and ionic pathways of the xenon esters decomposition. The first indication of a radical intermediacy was presented by DesMarteau, who isolated and characterized xenon(II) trifluoroacetate 916.375 Thermal decomposition of the xenon ester led to the formation of hexafluoroethane 917 as the main product. Additionally, the treatment of trifluoroacetic or phenylacetic acids 919 with XeF2, in benzene as the solvent, resulted in the formation of trifluoromethylbenzene 918 and diphenylmethane 920, which is in line with a radical pathway. The presence of the protodehalogenated product 892 during the fluorodecarboxylation of bridgehead carboxylic acid 921 also suggests the involvement of carbon-centered radicals. The reaction of 6-heptenoic acid 924 furnished a mixture of 1-fluoro-5-hexene 925 and (fluoromethyl)cyclopentane 926 in 75% and 25% yields, respectively. The observation of product 926 clearly indicates a radical-involved mechanism.
Scheme 153. Evidence for a Radical Pathway of the Fluorodecarboxylation Reaction with XeF2.

On the other hand, there is experimental evidence that indicates the possible involvement of an ionic route. A strong indication was obtained in the reaction of 4-bromobutanoic acid 927 with XeF2 in the presence of an equimolar amount of labeled 18F-tetrabutylammonium fluoride (Scheme 154).369 A complete exchange of the 18F– with XeF2, to produce [18FXe19F], would give a maximum 50% radiochemical yield of the product. However, 1-fluoro-3-bromopropane 928 was obtained in a 78% radiochemical yield, indicating an ionic reaction pathway. Moreover, the recovered XeF2 showed no radioactivity, indicating no fluoride exchange between the 18F– and XeF2. In an additional experiment, the fluorodecarboxylation of 3,3-diphenylpropionic acid 929 did not provide any 1-fluoro-1,2-diphenylethane 931, which would be expected to form upon radical rearrangement. Moreover, the product of an internal radical cyclization 933 was not observed during the reaction of 4-phenylbutyric acid 932.
Scheme 154. Evidence for an Ionic Pathway in the Fluorodecarboxylation Reaction with XeF2.

RCY – radiochemical yield.
Considering these experimental data, two competitive reaction pathways were proposed (Scheme 155). In the first pathway, the xenon ester 935 undergoes nucleophilic attack by a fluoride anion at the α-position to provide a decarboxylative fluorination. Alternatively, a homolytic cleavage of the oxygen–xenon bond furnishes an acyloxy radical 937 with the concomitant formation of the alkyl radical 938. The carbon-centered radical, in turn, either abstracts a fluorine atom from XeF2 (or ·XeF) or is oxidized to the carbocation 939, followed by further association with a fluoride anion.
Scheme 155. Proposed Mechanisms for Fluorodecarboxylation with XeF2.

Additionally, the reaction outcome was found to be dependent on the reaction vessel material. The desired product of a fluorodecarboxylation reaction was observed in PTFE or quartz vessel, while the reaction in a borosilicate glass vessel provided a completely different set of products.376
7.3. Radical Fluorodecarboxylation Reactions
7.3.1. Metal-free Fluorodecarboxylation Reaction
The renaissance in the radical fluorination chemistry was launched by Sammis, Paquin, and co-workers in 2011. In their seminal work, the authors demonstrated the feasibility of the utilization of electrophilic fluorinating agents as a fluorine atom donor upon interaction with alkyl radicals.377 As a proof of concept, an alkyl radical, generated by the decomposition of an aliphatic acid peroxide under thermal or photoassisted conditions, was able to abstract a fluorine atom from N-fluorobenzenesulfonimide (NFSI), to yield an alkyl fluoride (Scheme 156). While primary alkyl fluorides are obtained in poor yields, secondary and tertiary peroxoesters provide the corresponding alkyl fluorides in nearly quantitative yields. Benzylic, α-oxy, and α-amino stabilized radicals can deliver alkyl fluorides in the 40–60% yield range (compounds 943–945).
Scheme 156. Examples of Alkyl Radical Fluorination with NFSI.

In 2018, Renaud and co-workers developed a family of N-fluoro-N-aryl sulfonamides, which has weaker nitrogen–fluorine bond by 30–45 kJ/mol than that in NFSI. This important phenomenon allowed a more facile fluorination of alkyl radicals.378 As a comparison, the new fluorinating reagent 948 exhibited a superior fluorine atom transfer than NFSI under similar reaction conditions (Scheme 157).
Scheme 157. Fluorination of an Alkyl Radical with a New Family of N-Fluoro-N-arylsulfonamides.

Further Sammis, Paquin, and co-workers expanded this concept into a synthetically useful approach for the preparation of aryl(fluoromethyl) ethers from carboxylic acids. The fluorodecarboxylation process was achieved by treating aqueous solutions of sodium 2-aryloxyacetates with Selectfluor in the presence of light irradiation (Scheme 158). The reaction could be performed under irradiation with energetic UV light source of hv = 300 nm,379 or with the aid of a tungsten lamp in the presence of a ruthenium photocatalyst.380 The scopes of the reactions obtained by both methods are intrinsically the same with only minor differences. Electron-neutral aryloxyacetic acids undergo fluorodecarboxylation in high yields. The reaction performance diminishes with a decrease of the electron density on the aromatic ring. Phenylacetic and phenyloxy(fluoro)acetic acids proved to be viable substrates for the fluorodecarboxylation reaction under UV irradiation (894, 955). The reaction protocol without a photocatalyst seems to be preferential for electron-poor substrates (951), while the modification with the photocatalyst proved to be superior in the case of methyl- and phenyl-substituted aryloxyacetic acids (950 and 952). Electron-rich substrates, such as naphthyl- or methoxyphenyl ethers suffer from electrophilic ring fluorination, producing the desired fluoromethyl ethers in very low yields.
Scheme 158. Selected Examples of the Fluorodecarboxylation of 2-Aryloxyacetic Acids.

The problem of the preparation of electron-rich aryl(fluoromethyl) ethers was later addressed by the same groups.381 Since a ring fluorination with Selectfluor was the predominant side reaction, it was envisioned that the utilization of a less electrophilic fluorinating reagent, NFSI, would allow for a broader substrate scope. The reaction between aryloxyacetic acid and NFSI, in the presence of 2,6-di-tert-butyl-4-methylpyridine (956) as the base, under UV irradiation, provided the model 4-fluorophenyloxy(fluoromethyl) ether (920) in a high yield (Scheme 159). Interestingly, it was found that the reaction was significantly accelerated when acetone was used as the solvent, or by the addition of catalytic amounts of benzophenone. The ketone additive acts both as a single-electron oxidant and as a base in its triplet state. Implementation of these conditions significantly improved the yields and allowed for the preparation of previously unobtainable electron-rich aromatic fluoromethyl ethers. 2-Aryloxyacetic acids, bearing a methoxy group in the para- position still provide a complex mixture, without the presence of the desired fluoride.
Scheme 159. The Photosensitized Fluorodecarboxylation of Electron-rich 2-Aryloxyacetic Acids with NFSI.

Reaction with Selectfluor and NaOH in CH3CN.
In these three protocols (described in Scheme 158 and Scheme 159), the reaction proceeds via a similar mechanism, involving the oxidation of the carboxylate anion 963 to the zwitterionic radical 965 (Scheme 160). Decarboxylation of the radical 965 produces a stabilized carbon-centered radical 966 that further abstracts a fluorine atom from the Selectfluor or NFSI. However, in all three cases, the oxidant species that are responsible for the oxidation of 963 to 965 are believed to be different. In the protocol using the Selectfluor-UV combination, the anion 963 is excited to its triplet state 964, which is oxidized by Selectfluor to give the intermediate 965. In the case of the ruthenium photocatalyzed reaction, the excited triplet state of the ruthenium photocatalyst 960 is oxidized by Selectfluor to give the Ru(bpy)33+ species 961, which is responsible for the oxidation of the carboxylate 963 to the intermediate 965. Lastly, in the photosensitized reaction with NFSI, the triplet state of acetone or benzophenone 962 simultaneously acts as a single electron oxidant and a base, to produce 961. Importantly, the reaction does not involve direct oxidation of the carboxylate; therefore, the presence of the easily oxidizable 2-phenyloxy motif in the substrate is crucial for the reaction feasibility.
Scheme 160. Proposed Mechanism for the Fluorodecarboxylation Reaction of 2-Aryloxyacetic Acids.

The MacMillan group made a significant improvement in the field of light-assisted fluorodecarboxylations.382 The employment of a photocatalyst with a higher oxidative potential allowed for overcoming the requirement of an α-alkoxy substituent in the carboxylic acid. Iridium photocatalyst Ir[dF(CF3)ppy]2(dtbbpy) 321 in combination with Selectfluor was able to induce the decarboxylation by direct oxidation of the carboxylate anion to an acyloxy radical by a transient Ir(IV) species. This approach enabled the preparation of tertiary, secondary, and even primary alkyl fluorides in high yields and with notable functional group tolerance (Scheme 161).
Scheme 161. Selected Examples of the Photocatalyzed Fluorodecarboxylation Reaction of Nonactivated Carboxylic Acids.

The proposed reaction mechanism is based on the photoexcitation of Ir(III) catalyst, which undergoes an oxidation to Ir(IV) from the excited state (Scheme 162). The transiently formed Ir(IV) react with the in situ formed alkyl carboxylate with the thermodynamically favorable single electron transfer (SET) to give Ir(III) ground state species and a carboxylate radical. The latter immediately liberates CO2 to result in an alkyl radical, which, in turn, abstracts F-radical from Selectfluor to furnish the product.
Scheme 162. Proposed Mechanism for the Photocatalyzed Fluorodecarboxylation of Nonactivated Acids.

The Ye group presented a similar approach for the fluorodecarboxylation involving an organic photocatalyst based on an acridinium core (Scheme 163).383 The mesityl-acridinium photocatalyst 980 in the excited state possesses a high oxidation potential (*E1/2red = +2.08 V vs SCE), which presumably enables the direct oxidation of the carboxylate by such an excited species. The yields of the fluorides are generally good, although somewhat lower compared to those obtained with the Ir-photocatalyst 321. Interestingly, chemoselective fluorodecarboxylation of a secondary acid in the presence of a primary acid was achieved (978).
Scheme 163. Fluorodecarboxylation with Acridinium-Based Organic Photocatalyst.

In 2018, Gilmore and co-workers developed a continuous-flow, heterogeneous, photoassisted fluorodecarboxylation reaction.384 The use of a modified carbon nitride (CMB-C3N4) as the photocatalyst allowed the performance of the reaction with a light irradiation of 420 nm (Scheme 164). Additional dosed gas feeding into the flow reactor forms a segmented gas–liquid stream that helps to prevent the settling of solids and the clogging of tubing, thus significantly enhancing the reproducibility of the reaction. This modification offers generally high to nearly quantitative yields for the fluorodecarboxylation of 2-phenyloxyacetic acids. Phenylacetic acid derivatives deliver the corresponding benzyl fluorides in good yields, although benzylic fluorination is a competitive side reaction. Notably, the reaction can be performed in a nondegassed solvent without a significant effect on the yield.
Scheme 164. Light-Assisted In-Flow Fluorodecarboxylation of Aryloxyacetic Acids.

Tarantino and Hammond revealed that titanium oxide in combination with Selectfluor can serve as an effective photocatalyst for fluorodecarboxylation reaction in aqueous media.385 The reaction gives excellent yields for tertiary water-soluble carboxylic acids, while yields drop for the secondary and primary counterparts (Scheme 165a). The addition of acetone helps to solubilize the hydrophobic acids, although a significant decrease in the reaction yield is observed. Interestingly, tertiary acids can be selectively decarboxylated in the presence of primary acids by this method.
Scheme 165. Scope (a) and the Mechanism (b) of the Titania-Photocatalyzed Fluorodecarboxylation Reaction.

Reaction carried in a water–acetone mixture as a solvent.
As to the mechanism, the irradiation of titania with a xenon ark lamp induces the formation of electron–hole pairs (Scheme 165b). The formed positive holes possess a sufficient oxidation potential to abstract an electron from the carboxylate anion to produce the acyloxy radical. Decarboxylation provides a carbon-centered radical that abstracts a fluorine atom from the Selectfluor to give the target alkyl fluoride and TEDA radical 990, that takes an electron from the titania and closes the catalytic cycle. The turnover frequency of TiO2 was calculated to be TOF = 1050 ± 70 h–1. The apparent photon efficiency is estimated to be ξ = 3.5 ± 0.5, indicating that the light acts as a catalyst in the reaction.
7.3.2. Silver-Catalyzed and Silver-Mediated Fluorodecarboxylation Reactions
In 2012, the Li group made a significant contribution to the area of fluorination reactions by developing a synthetically useful catalytic method for the fluorodecarboxylation of aliphatic carboxylic acids.386 In a manner similar to their previously reported approach to chlorodecarboxylation, the current method also employed a silver-based catalysis. However, bidentate silver complexes, which proved to be crucial for the chlorodecarboxylation process, failed to provide any fluorinated product. Surprisingly, a simple water-ligated silver(I) complex gave the desired products. The reaction of a carboxylic acid with Selectfluor in the presence of catalytic amounts of silver nitrate in aqueous solution provided access to a range of primary, secondary, and tertiary alkyl fluorides in good to excellent yields (Scheme 166). Amides, esters, ethers, ketones, and arene functionalities were compatible with the reaction conditions. Activated substrates, such as 2-oxy and 2-amino acids, are also amenable substrates. The yields of primary alkyl fluorides are generally lower than those of the secondary and tertiary counterparts. Aromatic acids were completely unreactive in this reaction. The reactivity of acids decreases in the order tertiary > secondary > primary ≫ aromatic. This pattern allows for the selective fluorodecarboxylation of tertiary acids in the presence of primary carboxylic groups. Free aromatic acids do not interfere with the reaction progress and may be present in unprotected form during the reaction.
Scheme 166. Examples of the AgNO3-Catalyzed Fluorodecarboxylation of Aliphatic Acids.

Comprehensive studies by Patel and Flowers addressed the mechanistic details of this Ag-catalyzed fluorodecarboxylation.387 The obtained results revealed the intermediacy of silver(II) species, which are responsible for the decarboxylation process, while Selectfluor acts as a fluorine atom donor (Scheme 167). Additionally, the studies showed the presence of an induction period, which was explained by the formation of a silver carboxylate as a resting state of the catalyst. The reaction exhibits an inverse order in the carboxylic acid, meaning an inhibition of the reaction by a substrate via the formation of the Ag(carboxylate)2– species. Oxidation of Ag(I) to Ag(II) by Selectfluor was found to be a rate-limiting step of the catalysis. The use of sodium persulfate as an external oxidant resulted in an 8-fold acceleration of the reaction rate, without compromising the yield.387 The absence of reaction in the case of NFSI was explained by the inability of this fluorinating reagent to oxidize Ag(I) to Ag(II). However, the use of NFSI in the presence of persulfate produced the alkyl fluoride in a moderate yield. The presence of water in at least 10 vol % was crucial, likely for the dissolution of all the reaction components, as well as to serve as a labile ligand for the silver.
Scheme 167. Proposed Mechanism of the Silver-Catalyzed Fluorodecarboxylation Reaction.

A computational investigation of the reaction mechanism agrees with the proposal of Flowers.262 Oxidation of the Ag(I) species was found to be feasible process with both Selectfluor and a TEDA radical cation to form the Ag(II) intermediate, while the formation of AgF(II) and AgF(III) is an unattainable process at room temperature. The process of CO2 elimination from the acyloxy radical and the abstraction of fluorine from Selectfluor by an alkyl radical were proposed to proceed without an activation barrier since the transition state was not located. In a different computational study on decarboxylation of acyloxy radicals, the barrier was estimated to be about 5 kcal/mol for the acetyloxy radical.388
Inspired by the seminal work of Li, a field of silver-catalyzed fluorodecarboxylation emerged, with numerous notable applications. Recently, Chen and co-workers presented an interesting example of a selective fluorodecarboxylation of disubstituted malonic acids.389 The choice of the base and the solvent had a significant influence on the reaction outcome (Scheme 168). The utilization of sodium benzoate in a tricomponent solvent system of acetonitrile, water, and hexane, led to a smooth double fluorodecarboxylation, furnishing a variety of geminal difluoro compounds. On the other hand, K2HPO4 in a cyclopentyl methyl ether (CPME)-water system led to a selective monofluorodecarboxylation, providing the α-fluorocarboxylic acid as the predominant product. Despite the conceptual similarity between the two processes, differences in the reaction conditions slightly affect the functional group tolerance. Thus, both protocols tolerate arylhalides, phthalimides, esters, ethers, sulfones, cyanides, tertiary amides, and ketones. The presence of aliphatic alcohols and alkynes does not interfere with the selective monofluorodecarboxylation, although the conditions for double fluorodecarboxylation cause a significant oxidation of alcohols to ketones and preclude the use of alkynes and alkenes. The fluorodecarboxylation of monosubstituted malonic acids proceeds in poor selectivities and yields (1003, 1004). Both approaches failed to deliver any product in the case of α-heteroatom substituted malonic acids. The presence of an aromatic ring or a bulky substituent in the α-position of a malonic acid has a deleterious effect on the yield and selectivity of the monofluorodecarboxylation, making a double reaction preferable.
Scheme 168. Selective Mono- and Double Fluorodecarboxylation of Malonic Acids.

Gouverneur et al. elegantly applied the Ag-catalyzed approach to the synthesis of trifluoromethyl- and difluoromethylarenes (Scheme 169).390 The reaction provided access to electron-rich trifluoromethylarenes, while a significant decrease in the yield was observed for substrates bearing electron-withdrawing substituents. Importantly, this method was employed for the synthesis of 18F labeled compounds, using [18F]Selectfluor.
Scheme 169. Synthesis of Trifluoromethyl- and Difluoromethylarenes via a Silver-Catalyzed Fluorodecarboxylation Reaction.

[18F]Selectfluor bis(triflate) was used.
RCY – radiochemical yield.
Hu disclosed a method for the preparation of trifluoro(thio)methoxy arenes via the fluorodecarboxylation of the corresponding acids (Scheme 170).391 While attempts to apply photocatalytic fluorodecarboxylation methods to such precursors resulted in traces of the desired fluorinated products, the use of a silver salt and Selectfluor II, in the presence of a strong acid, provided access to the products in moderate to high yields. Silver nitrate and silver iodide proved to be the most efficient silver catalysts. Interestingly, the use of Selectfluor II—a weaker fluorinating agent—was crucial for the success of the reaction. For example, product 913 was obtained in only 4% yield when Selectfluor was used, while a 42% yield was obtained with Selectfluor II. The reaction generally performs better with electron-poor substrates, and an increase in the electron density on the aryloxy moiety decreases the yield. Esters, ketones, and halides are well tolerated, while methoxy-substituted arenes fail to deliver the product at all. Later, Prakash presented a similar approach to create the same type of substrates, utilizing Selectfluor as a fluorine source and a different strong acid.392 The yields of this modification are generally lower.
Scheme 170. Synthesis of Trifluoro(thio)methoxy Arenes via Silver-Catalyzed Fluorodecarboxylation.

Reaction with AgOTf, Selectfluor and CF3COOH.
Additionally, the silver-catalyzed decarboxylative fluorination approach allowed for a straightforward preparation of 3-fluorobicyclo[1,1,1]pentane-1-carboxylic acid 1032 (Scheme 171).393,394 The applied method significantly shorten the synthesis of 1032 from 8−11 steps to only one step. The reaction of 1028 with XeF2, as well as the fluorodecarboxylation of 1027 according to the aforementioned protocols, resulted in poor yields and complex reaction mixtures. However, the silver catalyzed approach furnished the desired product 1032 in 50–63% yield.
Scheme 171. Preparation of 3-Fluoro-bicyclo[1,1,1]pentane-1-carboxylic Acid.

Soorukram and co-workers have prepared β-fluoro-γ-butyrolactones and -lactams via silver mediated fluorodecarboxylation (Scheme 172).395,396 Unfortunately, the use of catalytic amounts of the silver catalyst provided the desired compounds in rather poor yields, accompanied by a low conversion. In order to achieve synthetically useful yields, equimolar amounts of silver nitrate were used. The reactions of 5-substituted lactones and lactams gave rise to a mixture of diastereomeric fluorides. Generally, the diastereomeric ratio varies in a range from 2:1 to 5:1, and only for specific lactams does the dr ratio reach 13:1. The reaction yield depends on the substitution pattern and varies in the range of 50–80%. N-Aryl-β-fluoro-γ-lactams are generally obtained in poor yields, due to a competitive fluorination of aryl ring, unless ortho-substituents on the aryl ring is present (1043 vs 1044). For substrates bearing a long alkyl-chain substituent in the γ-position, a radical intermediate suffers from a competitive 1,5-hydrogen atom transfer with the concomitant formation of an alkyl fluoride 1051 or 1052. The authors suggested the presence of a fluorinated silver species that act as a fluorine atom donor, hence justifying the need for the equimolar loading of the silver salt.
Scheme 172. Silver-Mediated Fluorodecarboxylation of β-Carboxyl-γ-butyrolactones and -Lactams.

Ratio of diastereomers is given in parentheses.
Fluorodecarboxylation was employed as the final step in the synthesis of a fluorinated corticosteroid fluticasone propionate. After minor fine-tuning of the reaction conditions, the compound 1053 was obtained in 92.7% yield on a 100-g scale (Scheme 173).
Scheme 173. Synthesis of Fluticasone Propionate.

Some additional applications should be noted. Thus, the Ag-catalyzed fluorination approach developed by Li was applied for the modification of poly(methyl)acrylic acids in aqueous solutions.397 The fluorine atoms are incorporated into the polymer backbone in a predictable manner in approximately 80% yield, based on the amount of Selectfluor used. Fluorodecarboxylation takes place in a well-distributed manner, furnishing a random copolymer. Additionally, the synthesis of a fluorinated graphene oxide was performed via fluorodecarboxylation of the carboxylated graphene oxide.398
Hartwig and co-workers envisioned the use of silver(II) fluoride as an oxidant for the synthesis of trifluoromethoxyarenes via a challenging fluorodecarboxylation of aryloxy(difluoro)acetic acids.374 AgF2 possesses a sufficient oxidation potential to oxidize the substrate and initiate the decarboxylation process (Scheme 174). Using only AgF2 as both oxidant and a fluorine atom donor, the model aryl trifluoromethyl ether was obtained in 49% yield. Attempts to improve the yield by the addition of an electrophilic fluorinating agent proved deleterious. However, the addition of a pyridine ligand and substoichiometric amounts of silver(I) fluoride drastically improved the reaction yield. The designed approach allowed for the synthesis of electron-rich and electron-poor trifluoromethoxyarenes in 50–80% yield. Esters, nitriles, ketones, secondary and tertiary amides, diaryl ethers, and benzylic protons are well tolerated under the reaction conditions. In the case of brominated substrates, a competitive abstraction of bromine was observed (1017). Substrates bearing an alkoxy substituent failed to deliver the desired product due to predominant side reactions. Notably, the reaction is applicable for the preparation of aryl fluoromethyl and difluoromethyl ethers 910 and 944 via fluorodecarboxylation of the corresponding acetic acid derivatives. The presence of radical intermediates was detected in the course of the reaction.
Scheme 174. Fluorodecarboxylation with the AgF2/AgF System.

Variations of the fluorodecarboxylation reaction involving a heterogeneous silver catalyst were investigated. Hammond and co-workers employed titania-supported silver oxide, obtained via sol immobilization (1 wt % Ag/TiO2).399 While the obtained heterogeneous catalyst performed in a manner similar to homogeneous silver nitrate in terms of the yield, the conversions were achieved in shorter time periods with the same catalyst loadings (Scheme 175). No leaching of the catalyst into the solution was observed, indicating the true heterogeneous nature of the catalyst. The catalyst was shown to convert primary and tertiary acids to the corresponding fluorides with turnover numbers of TON = 3–18, depending on the type of substrate. It was noted that fine-tuning of the acid–base ratios is important, in order to achieve maximum efficiency. The catalyst can be recycled at least five times without a significant drop in the catalytic activity.
Scheme 175. Heterogeneous Silver-Catalyzed Fluorodecarboxylation.

1Ag/TiO2 – mechanically mixed Ag2O and TiO2 containing 1 wt % of Ag; TON – turnover number.
The Truong group showed that silver ferrite nanoparticles (50–70 nm) can be used as an effective heterogeneous catalyst for the decarboxylative fluorination reaction (Scheme 176). Compared to the initial homogeneous variation, an AgFeO2 catalyst allows the reaction to be performed at room temperature with significantly reduced reaction times. The catalyst was shown to be truly heterogeneous with <5 ppm of silver present in the noncatalytically active supernatant solution. The catalyst could be recycled at least 7 times, retaining 93% of its activity. Interestingly, a solution of silver(I) nitrate and iron(III) nitrate shows an identical performance, in terms of the reaction kinetics and yield, to those of the heterogeneous catalyst. While a direct comparison with original Li procedure is not comprehensive due to the lack of similar substrates, a silver ferrite catalyst seems to perform with similar efficacy.
Scheme 176. Fluorodecarboxylation Reaction Catalyzed by AgFeO2 50–70 nm Nanoparticles.

7.4. Fluorodecarboxylation Reaction with an Anionic Fluorination Source
The first example of a fluorodecarboxylation reaction involving an anionic fluorine source was disclosed by Wang and Mallouk.400 The treatment of specific easily oxidized carboxylic acids with silver fluoride and titanium dioxide (rutile), under irradiation with a mercury–xenon lamp, resulted in the formation of alkyl fluorides (Scheme 177). Only a few carboxylic acids were reported to deliver the fluorinated product. The substrate reactivity was correlated to their oxidation potentials, setting a threshold level of ca. +1.3 V (vs SCE) required to observe a fluorodecarboxylation product. This limitation was attributed to the valence-band potential of titania in the reaction medium.
Scheme 177. Fluorodecarboxylation with an Anionic Fluorine Source.

3,3,3-Triphenylpropionic acid as the starting material.
AgF·HF used instead of AgF and KF.
A plausible mechanism for the reaction involves the direct oxidation of the acid to an acyloxy radical with the concomitant evolution of carbon dioxide. The generated carbon-centered radical undergoes an additional oxidation to a carbocation, followed by a nucleophilic attack by a fluoride anion. The presence of 1-fluoro-1,1,2-triphenylethane 1069 upon the fluorodecarboxylation of 3,3,3-triphenylpropionic acid supports the presence of a radical intermediate.
Further mechanistic studies were in line with this assumption.401 Laser flash-photolysis experiments on triphenylacetic acid revealed the intermediacy of carbon-centered radical Ph3C•, which was stable in the presence of a fluoride anion. On the other hand, the signal from the carbocation Ph3C+, whose presence was also observed in the reaction mixture, is efficiently quenched by a fluoride anion. Additionally, the transient absorbance of the radical Ph3C• has a linear dependence on the laser pulse power, while the absorbance of the carbocation Ph3C+ increases with the square of the intensity. This observation implies that the formation of the carbocation Ph3C+ is a two-photon process. The reaction rate of the fluorodecarboxylation was also in a square dependency of the light intensity, being consistent with the formation of R-F from R+. Interestingly, it was found that utilization of the AgF·HF complex allowed an increase in the range of suitable substrates by increasing the oxidizing power of the semiconductor by preventing the absorption of free fluorides on the titania surface.
Another striking example was reported by Groves and co-workers.402 The fluorodecarboxylation process was enabled by means of catalytic amounts of the manganese-porphyrin complex 1082 in the presence of iodosobenzene, using NEt3·3HF as the fluorine source. The designed approach is capable of converting benzylic and aryloxy carboxylic acids into the corresponding fluorides in 50–70% yields (Scheme 178). Secondary and tertiary bridgehead carboxylic acids are also viable substrates, although the yields are significantly lower. Primary acids provide only traces of the final fluorides. The reaction exhibits great functional group tolerance. Thus, alkynes, ketones, esters, nitriles, aliphatic bromides, and heterocycles, such as pyridines and thiophenes, are well tolerated. Notably, electron-rich aromatics were compatible with the reaction conditions, and no ring fluorination was observed. Additionally, no epoxidation or allylic C–H activation was detected, despite the well-documented reactivity of the employed catalytic system in such reactions.403 Short reaction times allow the utilization of this approach toward 18F labeling, providing the radiofluorides in 25–50% radiochemical conversions.
Scheme 178. A Fluorodecarboxylation Reaction with NEt3·HF Catalyzed by a Manganese Complex.

RCC – radiochemical conversion.
The reaction is assumed to involve the intermediacy of fluoromanganese(IV) species, acting as a fluorine donor for the alkyl radical. There are two possible pathways to obtain this intermediate. The first one involves oxidation of the manganese(III)-porphyrin complex 1088 to a fluoromanganese(IV) 1090 intermediate following by decarboxylation (Scheme 179c, path 1). The second pathway proceeds via the formation of oxomanganese(V) complex 1089 followed by a hydrogen atom abstraction from the carboxylic group and a ligand exchange to give 1090 (path B). It was suggested that path A is likely to be operative, since (diacetoxyiodo)benzene is also an efficient oxidant in this fluorodecarboxylation reaction. The estimated barrier for the oxidation step of 1083 to form the F-Mn(IV)-F species 1090 by (acetoxy)(fluoro)iodobenzene is 18.2 kcal·mol–1. The intermediacy of carbon-centered radicals was suggested by using an alkyl radical trap. Subjecting the acid 1083 to the reaction conditions in the presence of bromotrichloromethane gave rise to the formation of a bromide 1085, although a significant amount of a fluorinated product 1084 was observed (Scheme 179a). This implies that the rate of the abstraction of a fluorine atom from the manganese complex 1091 by a carbon-centered radical is competitive to the bromine transfer from BrCCl3, which is around 108 m–1 s–1.223 Additionally, when a chiral Mn-salen complex 1087 was employed, a product 931 was obtained in 11% ee, supporting the involvement of a Mn–F intermediate in the C–F formation step (Scheme 179b).
Scheme 179. (a–c) Proposed Mechanism for the Manganese-Catalyzed Fluorodecarboxylation.

Gouverneur et al. addressed a feasibility of manganese porphyrin-based catalysis to the synthesis of [18F]-labeled difluoromethylarenes from carboxylic acids. Treatment of an excess of aryl(fluoro)acetic acids with a substoichiometric amounts of tetrabutylammonium fluoride resulted in the formation of the desired difluoromethylarene in a 50% yield (based on the consumed fluoride). On the other hand, the preliminary conversion of the acid to an iodobenzene dicarboxylate, followed by addition of the catalyst and the fluoride source, resulted in a quantitative incorporation of the fluoride into the desired molecule (Scheme 180). These modifications allowed the preparation of various [18F]difluoromethyl arenes. Generally, the highest radiochemical conversions were obtained for aryl(fluoro)acetic acids bearing electron-rich substituents on the aromatic ring. Interestingly, a λ3-iodane derived from an arylacetic acid, does not provide any fluorinated product under the same conditions.
Scheme 180. Preparation of [18F]-Labeled Difluoroarenes via Mn-Catalyzed Fluorodecarboxylation.

Recently Baran and co-workers developed an excellent method for the electrochemical synthesis of hindered dialkyl ethers.404 This method is based on an electrochemical oxidation of a carboxylic acid to a carbocation in an undivided cell, in the presence of a slight excess of an alcohol. It was demonstrated that the reaction is not limited to an alcohol as the nucleophile and, among others, fluorides can be utilized in this transformation, providing access to alkyl fluorides. A few examples of decarboxylative fluorination were presented, including the introduction of a fluorine atom at the benzylic 1099, bridgehead 989, tertiary cyclic 1100, and α-to-imide positions 945 (Scheme 181). The silver additive played the role of a sacrificial oxidant, rather than taking place in the decarboxylation process.
Scheme 181. First Example of Electrochemical Fluorodecarboxylation.

A similar approach was employed by Waldvogel and co-workers in the electrochemical fluorodecarboxylation of aryloxyacetic acids.405 Like in the previous example, the reaction was performed in an undivided cell with graphite electrodes using 2,4,6-collidine as the base (Scheme 182). The highest performance of the reaction was achieved using NEt3·5HF as the fluorine source. Interestingly, no product was observed when NEt3·3HF was tried. Additionally, the reaction takes place only with graphite electrodes; reactions performed with platinum, boron-doped diamond or glassy carbon electrodes failed to deliver any product. Aryl fluoromethyl ethers are generally obtained in 40–70% yields. Aryloxyacetic acids, bearing electron-rich and electron-poor substituents, are suitable substrates for this method. For nitro- and cyano-substituted derivatives (1104 and 1108), yields are low. Pyridine and pyrimidine-acetic acids are also viable substrates for the transformation (1106). The fluorodecarboxylation of α,α-dimethyl(aryloxy)acetic acid failed to deliver a fluorinated product.
Scheme 182. Electrochemical Fluorodecarboxylation of Aryloxyacetic Acids.

Very recently, Doyle et al. developed a method for a redox-neutral nucleophilic fluorodecarboxylation of N-hydroxyphthalimides by means of a photoactive catalyst.406 In the designed approach, the decomposition of a redox-active ester produces an alkyl radical, which is oxidized to a carbocation by a photocatalyst, followed by nucleophilic attack of a fluoride (Scheme 183). The reaction delivers primary, secondary, and tertiary benzyl fluorides, as well as unactivated tertiary fluorides, in good to excellent yields. However, electron-poor primary benzylic phthalimide afforded the benzylic fluoride 1111 in a poor yield, while a similar secondary benzylic derivative 1112 was highly efficient. The fluorodecarboxylation of α-oxy and α-thio carboxylic acids is also feasible by this protocol. A wide variety of functional groups, such as ketones, cyanides, iodides, thioethers, secondary amides, and azides are tolerated under the reaction conditions. Fluorodecarboxylation in the presence of heterocycles, such as pyridine and thiophene, proved to be efficient. Additionally, the reaction can be applied for the preparation of [18F]-labeled fluorinated motifs.
Scheme 183. Photocatalyzed Nucleophilic Fluorodecarboxylation of N-Phthalimide Esters.

7.5. Fluorodecarboxylation of β-Keto- and Related Acids
A few examples of the decarboxylative fluorination of β-keto acids are presented in the literature. A general feature of the fluorodecarboxylation of this type of substrates is the intermediacy of an anionic species that undergoes electrophilic fluorination. Depending on the conditions, the reaction can proceed via two distinct pathways (Scheme 184). The first possibility involves the initial decarboxylation of the acid, to form the enolate 1122, which undergoes electrophilic fluorination to give the α-fluoroketone 1123. Alternatively, the enol form of the acid undergoes electrophilic fluorination to give the α-fluoro-β-keto acid 1120. Further deprotonation and liberation of CO2, followed by protonation, gives the product 1123, while interception of the fluoroenolate with a fluorinating agent provides the difluoroketone 1121.
Scheme 184. Possible Pathways for the Fluorodecarboxylation of β-Ketoacids.

Deng and co-workers showed that 3-phenyl-3-oxopropionic acids upon treatment with Selectfluor in the presence of the phase transfer catalyst triethylbenzylammonium bromide (TEBAB) and potassium carbonate as the base can be converted to the α-fluoroacetophenone in excellent yields (Scheme 185).407 The reaction proceeds in high yields with electron-rich groups on the phenyl ring, while no reaction is observed for the analogues with electron-withdrawing substituents (e.g., 1129). An additional alkyl substituent in the α-position significantly decreases the reaction yield (e.g., 1130), and a large amount of difluorinated ketone is observed for electron-poor 3-aryl-β-ketoacids. In an attempt to perform asymmetric fluorination, N-benzylcinchonidinium chloride was used as the catalyst, although the product 1126 was obtained in a fairly racemic fashion.
Scheme 185. Preparation of α-Fluoroacetophenones via the Fluorodecarboxylation of β-Ketoacids.

TEBAB – triethylbenzylammonium bromide.
The same transformation can be achieved using a combination of NFSI and cesium carbonate in an acetonitrile–water mixture.408 Under these conditions, the yields are generally lower, although the nature of the substituents does not have a significant impact on the reaction outcome, and both electron-poor and electron-rich α-fluoroacetophenones can be prepared. However, this modification did not overcome the problem of the substituent in the α-position of the acids, and the target fluoride was obtained in a very poor yield. Tertiary β-ketoacid failed to deliver the fluorinated product at all.
In continuation, Deng expanded the use of 3-aryl-β-ketoacids for the preparation of α,α-difluoroacetophenones with minor modification of the reaction conditions.409 While they were the major undesired product in previous works, performing the reaction with higher loadings of Selectfluor in nitromethane without a base allowed for the creation of difluoroketones as the major product. The reaction has intrinsically the same limitations as the initial work,407 namely, the sluggish reactivity of the electron-poor 3-aryl-β-ketoacids and the drastic drop in the yield for acyclic α-substituted acids (Scheme 186).
Scheme 186. Preparation of α,α-Difluoroacetophenones via the Fluorodecarboxylation of β-Ketoacids.

Tertiary β-ketoacids were found to undergo a smooth fluorodecarboxylation upon treatment with Selectfluor in the absence of a base, in DMF. β-Ketoacids, based on various aromatic, aliphatic cyclic, and acyclic backbones, proved to be viable substrates for the reaction and delivered the target fluorides in useful yields (Scheme 187).410 Protodecarboxylation was detected as the main side reaction.
Scheme 187. Fluorodecarboxylation of Tertiary β-Ketoacids.

2-Pirydyl acetic acids, similarly to β-ketoacids, are known to undergo thermal decarboxylation to produce protonated product. Shibatomi et al. envisioned that interception of the 2-picolyl anion with an electrophilic fluorinating agent would provide 2-(fluoroalkyl)pyridines (Scheme 188a).411 The reaction of lithium (2-pirydyl)α,α-dialkylacetic acetates 1143 with Selectfluor provided the corresponding fluorides in high to nearly quantitative yields. On the other hand, the reaction of mono-α-substituted 2-pyridylacetates provided a mixture of mono- and difluoroalkylpyridines, with the difluorinated compound as the major product in almost all cases.
Scheme 188. Selected Examples of the Fluorodecarboxylation of 2-Pyridylacetic Acids and the Proposed Mechanistic Pathway.

It is proposed that the reaction involves the fluorination of the pyridine nitrogen as the first step (Scheme 188b). The formation of the N-fluoropyridinium species facilitates a decarboxylation process to give the intermediate 1156. A further rearrangement of 1156 leads to the target fluoride 1157. To support this pathway, 3- and 4-pyridylacetates were subjected to the reaction. As expected, no 3-(fluoroalkyl)pyridine 1158 was formed, while 4-(fluoroalkyl) pyridine 1159 was obtained in a 60% yield. This observation is in line with the stabilization of the carbanion intermediate by the pyridine moiety.
7.6. Aromatic Fluorodecarboxylation
The fluorodecarboxylation reaction is deemed to be an attractive methodology for the selective introduction of a fluorine atom into an aromatic backbone. However, unlike the other aforementioned halodecarboxylation processes, the fluorodecarboxylation of aromatic carboxylic acids proved to be an extremely challenging reaction. Despite numerous attempts, a scarce number of successful approaches were reported. For the discovered transformations, substrates are limited to either heteroaromatic carboxylic acids with a low degree of aromaticity or easily oxidizable 2,4-dihydroxybenzoic acids. A general approach for aromatic decarboxylative fluorination is yet to be discovered.
The only example of an aryl fluorodecarboxylation reaction was shown by Grakauskas in 1969.365 The reaction of an aqueous solution of sodium 4-nitrobenzoate and elemental fluorine provided 4-fluoronitrobenzene in only 4% yield. It was proposed that the process involves an acylhypofluorite intermediate that undergoes rearrangement, with the evolution of carbon dioxide, to give the arylfluoride (Scheme 149).
It was found that furan-412 and pyrrole-2-carboxylic413 acids can give the desired decarboxylative fluorination products upon treatment with Selectfluor in a biphasic mixture of CH2Cl2 and aqueous sodium carbonate, albeit in moderate yields (Scheme 189). Later, the method underwent a minor improvement and was extended to benzofuranes, (benzo)thiophenes, pyrazoles, isoxazoles, indazoles, and indoles.414 Electron-rich and electron-poor 2-heteroaryl fluorides were obtained in moderate to good yields. For the ultimate performance, the pyrazole ring must be fully substituted; otherwise, a 4,4′-dimer of type 1166 is formed as the major product. The same outcome is observed for indoles lacking a substituent in the 3-position. The reaction mechanism was not studied in depth. Unlike the Selectfluor-mediated fluorodecarboxylation of cinnamic acids346 (see Scheme 127, section 6.1.3), this reaction is barely influenced by 5 equiv of TEMPO. However, the biphasic conditions of the reaction do not allow ruling out a radical pathway.
Scheme 189. Fluorodecarboxylation of Heteroaromatic Acids.

In 2018, Liu and co-workers showed the possibility of the fluorodecarboxylation of ortho-hydroxynaphthoic and related acids.415 Treatment with Selectfluor under basic conditions resulted in a rapid reaction, providing ortho-hydroxyfluoronaphthalenes in good yields (Scheme 190a). The nature and position of substituents on the ring did not have a strong influence on the reaction outcome. 3-Hydroxy- and 3-aminoquinoline-4-carboxylic acids are also amenable substrates for the reaction (product 1171). Only 2,4-dihydroxy (1169) and 2-hydroxy-4-methoxybenzoic acid gave the corresponding fluorides in good yields, while traces of aryl fluorides were detected in the case of salicylic acid.
Scheme 190. (a, b) Fluorodecarboxylation of ortho-Hydroxynaphthoic Acids and the Proposed Reaction Mechanism.

Mechanistic insights showed that the reaction likely involves a radical species since the process is inhibited in the presence of a radical scavenger. Thus, the assumed reaction pathway launches with the oxidation of a phenolate by Selectfluor to a phenyloxy radical 1173 (Scheme 190b). This radical can easily undergo dearomatization to its carbon-centered tautomeric form 1174, which is fluorinated to give an unstable α-fluoro-β-ketoketoacid 1175. A facile decarboxylation of the intermediate 1175 furnishes the target aryl fluoride.
8. Conclusions and Outlook
The chemistry presented in this review clearly shows that the decarboxylative halogenation presents a highly attractive approach for the synthesis of ubiquitous organic halides. The method possesses a number of advantages: (1) carboxylic acids are widely available and relatively inexpensive substrates; (2) the acids are either natural products or can be prepared by a variety of well-established routes; (3) due to the nature of the transformation, carboxylic acids can be considered as traceless activating groups; (4) the protocols for direct carboxylic group conversion to the corresponding organic halides meet the atom-economical requirements of modern organic synthesis.
In this review, major known approaches for the conversion of alkanoic, alkenoic, acetylenic, and (hetero)aromatic acids to the corresponding alkyl, alkenyl, alkynyl, and (hetero)aryl halides were collected and comprehensively analyzed. These methods encompass the preparation of families of valuable organic iodides, bromides, chlorides, and fluorides. Historic and modern methods, including analysis of their advantages and drawbacks, were broadly discussed. Features, substrate scopes, and limitations of the approaches were also critically addressed. In the available cases, mechanistic details of the reactions were provided, highlighting the generality and uniqueness of the different mechanistic pathways.
The halodecarboxylation reaction is an established, well-studied process, which has found numerous prolific applications in organic synthesis. However, substantial challenges and opportunities still remain. Thus, a number of the halodecarboxylation protocols are based on stepwise processes, employ heavy or poisonous metals and highly toxic, corrosive or strongly oxidizing reagents, use halogenated environmentally unfriendly solvents, and include tedious separation of the final organic halides. Moreover, the methods usually lack generality. Namely, the same approach usually cannot be used for the aliphatic and aromatic acids, or applied for installation of different halogens. As to the substrate scope, one should carefully match the halodecarboxylation approach with the functional groups in the substrate. For example, many methods, described here, are based on the participation of halonium-based reagents and intermediates. Therefore, they could be problematic when applied to the acids bearing unsaturated bonds in the skeleton not conjugated to the CO2H group. The same is applied to the substrates bearing easily oxidizable functions (e.g., some heterocycles). In general, aromatic and heteroaromatic decarboxylative halogenation has been more challenging than that for the aliphatic counterparts.
Although some examples of aromatic chlorodecarboxylation are reported, a reliable and practical approach, which can be applied to a broad scope of aromatic carboxylic acids, is still lacking. Strikingly, to the best of our knowledge, the corresponding decarboxylative fluorination of benzoic acid derivatives is a completely unresolved reaction. One of the reasons could be a problematic step of F atom abstraction by an aryl radical. No doubt, a process that could directly convert aromatic acids to valuable organic fluorides would find a great resonance in the chemical community, and the wide application of such process for the preparation of existing or potential pharmaceuticals or agrochemicals can easily be envisioned.
Since organic halides are often used as intermediates in the multistage synthesis, their in situ preparation by halodecarboxylation and direct employment with no separation could significantly streamline the synthesis. An example of such a tandem approach could be an application of in situ prepared aryl iodides in cross-coupling reactions.270,321,327
Finding practical solutions to these formidable, yet intriguing, problems would broaden the applicability of halodecarboxylation reactions and open the door to the facile and efficient preparation of numerous organohalides for pharmaceutics, agrochemicals, polymers, and organic electronic materials, among many others.
Acknowledgments
We are thankful to Israel Innovation Authority, KAMIN Project 69747, for financial support.
Biographies
Andrii Varenikov received his B.Sc. degree (2012) and M.Sc. in Chemistry from the Kharkiv National University in 2013 (under supervision of Prof. Alexander Roshal). In 2014, he joined the group of Prof. Mark Gandelman as a Ph.D. student and is working on the enantioselective construction of trifluoromethyl-substituted stereogenic centers via cross-coupling reactions, as well as utilization of organotitanium nucleophiles in these transformations.
Evgeny Shapiro received his Ph.D. and Doc. Sci. from Zelinskii Institute of Organic Chemistry, Russian Academy of Science, Moscow. He worked as a group leader at the same institute from 1987–1992. In 1992, he was a research associate in the Department of Organic Chemistry at the Trinity University, Texas. From 1994 to 2016, he served as a senior researcher in IMI (TAMI) Institute for Research and Development, Israel. From 2017, he joined the Gandelman group as a research associate.
Mark Gandelman is Professor of Chemistry at the Technion-Israel Institute of Technology. Mark was born and raised in Moldova, and at the age of 18 he emigrated to Israel. He received a B.Sc. degree in chemistry in 1995 from Tel Aviv University, and a Ph.D. degree in 2003 from the Weizmann Institute of Science under the supervision of Prof. David Milstein. Holding the Rothschild fellowship, Mark performed his postdoctoral studies at Harvard University in the group of Prof. Eric N. Jacobsen. In 2005, he joined the Schulich Faculty of Chemistry in the Technion as an assistant professor and became a full professor in 2020. His fields of interests encompass organic and organometallic chemistry, ligand design, catalysis, and halogenation chemistry.
The authors declare no competing financial interest.
References
- Global Demand for PVC to Rise by About 3.2%/Year to 2021. Addit. Polym. 2014, 2014, 10–11. [Google Scholar]
- Zhdankin V. V.; Gribble G. W. Newly Discovered Naturally Occurring Organohalogens. ARKIVOC 2019, 2018, 372–410. 10.24820/ark.5550190.p010.610. [DOI] [Google Scholar]
- Lin R.; Amrute A. P.; Perez-Ramirez J. Halogen-Mediated Conversion of Hydrocarbons to Commodities. Chem. Rev. 2017, 117, 4182–4247. 10.1021/acs.chemrev.6b00551. [DOI] [PubMed] [Google Scholar]
- Sheldon R. A.; Kochi J. K. Oxidative Decarboxylation of Acids by Lead Tetraacetate. In Org. React. 1972, 19, 279–422. [Google Scholar]
- Johnson R. G.; Ingham R. K. The Degradation of Carboxylic Acid Salts by Means of Halogen - the Hunsdiecker Reaction. Chem. Rev. 1956, 56, 219–269. 10.1021/cr50008a002. [DOI] [Google Scholar]
- Wilson C. V. The Reaction of Halogens with Silver Salts of Carboxylic Acids. In Org. React. 1957, 9, 332–387. [Google Scholar]
- Tanner D. D.; Bunce N. J.. The Acyl Hypohalites. In Acyl Halides; Patai S., Ed.; Wiley, 1972; pp 455–500. [Google Scholar]
- Crich D.; Quintero L. Radical Chemistry Associated with the Thiocarbonyl Group. Chem. Rev. 1989, 89, 1413–1432. 10.1021/cr00097a001. [DOI] [Google Scholar]
- Tius M. A. Xenon Difluoride in Synthesis. Tetrahedron 1995, 51, 6605–6634. 10.1016/0040-4020(95)00362-C. [DOI] [Google Scholar]
- Saraiva M. F.; Couri M. R. C.; Le Hyaric M.; de Almeida M. V. The Barton Ester Free-Radical Reaction: A Brief Review of Applications. Tetrahedron 2009, 65, 3563–3572. 10.1016/j.tet.2009.01.103. [DOI] [Google Scholar]
- Crich D.; Sasaki K.. 7.27 the Hunsdiecker and Related Reactions. In Comprehensive Organic Synthesis II; Knochel P., Molander G., Eds.; Elsevier: Amsterdam, 2014; pp 818–836. [Google Scholar]
- Chatalova-Sazepin C.; Hemelaere R.; Paquin J. F.; Sammis G. M. Recent Advances in Radical Fluorination. Synthesis 2015, 47, 2554–2569. 10.1055/s-0034-1378824. [DOI] [Google Scholar]
- Schwarz J.; König B. Decarboxylative Reactions with and without Light – a Comparison. Green Chem. 2018, 20, 323–361. 10.1039/C7GC02949G. [DOI] [Google Scholar]
- Borodine A. Ueber Bromvaleriansäure Und Brombuttersäure. Liebigs Ann. 1861, 119, 121–123. 10.1002/jlac.18611190113. [DOI] [Google Scholar]
- Chapman E. T.; Smith M. H. XIII.—on Some Decomposition of the Acids of the Acetic Series. J. Chem. Soc. 1869, 22, 185–191. 10.1039/JS8692200185. [DOI] [Google Scholar]
- E. Blaise L. P. Action De Chlorures Des Acides a-Alcoxylés Sur Les Dérivés Organo Métalliques Mixtes Du Zinc (II). Ann. Chim. Phys. 1912, 258–288. [Google Scholar]
- Jones M. A Novel, Convenient Synthesis of Chloromethyl Methyl Ether. Synthesis 1984, 1984, 727–727. 10.1055/s-1984-30946. [DOI] [Google Scholar]
- Stadlwieser J. Notiz Zur Herstellung Von Chloromethylmethylether Aus Methoxyessigsäure. Synthesis 1985, 1985, 490–490. 10.1055/s-1985-31240. [DOI] [Google Scholar]
- Bunge N. Einwirkung Der Haloide Auf Mettalderivate Einiger Kohlenstoff Verbindungen. Liebigs Ann. Suppl. 1870, 7, 117–124. [Google Scholar]
- Pope F. G.; Wood A. S. CXCIII.—the Bromination of Phenol. 2:4- and 2:6-Dibromophenol. J. Chem. Soc., Trans. 1912, 101, 1823–1829. 10.1039/CT9120101823. [DOI] [Google Scholar]
- Krishna S.; Pope F. G. LXXXIX.—the Action of Potassium Iodide and Iodate on Some Hydroxy-Acids. J. Chem. Soc., Trans. 1922, 121, 798–800. 10.1039/CT9222100798. [DOI] [Google Scholar]
- Grovenstein E.; Henderson U. V. Decarboxylation. III. The Kinetics and Mechanism of Bromodecarboxylation of 3,5-Dibromo-2-Hydroxy- and 3,5-Dibromo-4-Hydroxybenzoic Acids1,2. J. Am. Chem. Soc. 1956, 78, 569–578. 10.1021/ja01584a017. [DOI] [Google Scholar]
- Grovenstein E.; Ropp G. A. Carbon-13 Isotope Fractionation as a Criterion of the Mechanism of Bromodecarboxylation of 3,5-Dibromo-4-Hydroxybenzoic Acid. J. Am. Chem. Soc. 1956, 78, 2560–2562. 10.1021/ja01592a062. [DOI] [Google Scholar]
- Hunsdiecker H.; Hunsdiecker C. Über Den Abbau Der Salze Aliphatischer Säuren Durch Brom. Ber. Dtsch. Chem. Ges. B 1942, 75, 291–297. 10.1002/cber.19420750309. [DOI] [Google Scholar]
- Hunsdiecker H.; Hunsdiecker C.; Vogt E.. Halogen-Containing Organic Compounds. US2176181, 1939.
- Bockemüller W.; Hoffmann F. W. Über Carbonsäure-Hypohalogenite. Liebigs Ann. 1935, 519, 165–192. 10.1002/jlac.19355190115. [DOI] [Google Scholar]
- Bunce N. J.; Murray N. G. On the Relationship between the Hunsdiecker and Simonini Reactions. Tetrahedron 1971, 27, 5323–5335. 10.1016/S0040-4020(01)91697-7. [DOI] [Google Scholar]
- Simonini A. Über Den Abbau Der Fetten Säuren Zu Kohlenstoffärmeren Alkoholen. Monatsh. Chem. 1892, 13, 320–325. 10.1007/BF01523646. [DOI] [Google Scholar]
- Simonini A. Über Den Abbau Der Fetten Säuren Zu Kohlenstoffärmeren Alkoholen. Monatsh. Chem. 1893, 14, 81–92. 10.1007/BF01517859. [DOI] [Google Scholar]
- Oldham J. W. H.; Ubbelohde A. R. 71. Some Acyl Derivatives of Iodine. J. Chem. Soc. 1941, 368–375. 10.1039/jr9410000368. [DOI] [Google Scholar]
- Heyns K.; Stange K. Uber Abbaureaktionen an Aminosäuren Und Polypeptiden. Z. Naturforsch., B: J. Chem. Sci. 1955, 10b, 129–140. 10.1515/znb-1955-0302. [DOI] [Google Scholar]
- Hauptschein M.; Stokes C. S.; Grosse A. V. The Properties and Reactions of Perfluorobutyrolactone. J. Am. Chem. Soc. 1952, 74, 1974–1976. 10.1021/ja01128a030. [DOI] [Google Scholar]
- Haszeldine R. N. 124. The Reactions of Metallic Salts of Acids with Halogens. Part I. The Reaction of Metal Trifluoroacetates with Iodine, Bromine, and Chlorine. J. Chem. Soc. 1951, 584–587. 10.1039/jr9510000584. [DOI] [Google Scholar]
- Haszeldine R. N. 817. The Reactions of Metallic Salts of Acids with Halogens. Part Iii. Some Reactions of Salts of Fluorohalogenoacetates and of Perfluoro-Acids. J. Chem. Soc. 1952, 4259–4268. 10.1039/jr9520004259. [DOI] [Google Scholar]
- Wieland H.; Gottwalt Fischer F. Über Das Auftreten Freier Radikale Bei Chemischen Reaktionen Iv. Die Umsetzung Der Silbersalze Von Carbonsäuren Mit Jod. Liebigs Ann. 1926, 446, 49–76. 10.1002/jlac.19264460105. [DOI] [Google Scholar]
- Bergström S.; Rottenberg M.; Voltz J.; Schonfeldt E.; Steensgaard I.; Rosenberg T. The Preparation of Some Carboxylabelled Bile Acids. Bile Acids and Steroids 2. Acta Chem. Scand. 1953, 7, 481–484. 10.3891/acta.chem.scand.07-0481. [DOI] [Google Scholar]
- Heintzeler M. Über Den Mechanismus Des Abbaus Von Silbersalzen Von Carbonsäuren Zu Alkyl-Halogeniden. Liebigs Ann. 1950, 569, 102–116. 10.1002/jlac.19505690204. [DOI] [Google Scholar]
- Buchman E. R.; Deutsch D. H.; Fujimoto G. I. Spiro [3.5] Nonane1. J. Am. Chem. Soc. 1953, 75, 6228–6230. 10.1021/ja01120a035. [DOI] [Google Scholar]
- Hunsdiecker H.; Hunsdiecker C.; Vogt E.. Verfahren Zur Herstellung Von Isocyclischen Gesaettigten Chlor- Und Bromverbindungen. DEDE000000730410A, 1935.
- Mehta T. N.; Mehta V. S.; Thosar V. B. Halogenation of Fatty Acids. Ii. Reaction between Halogens and Metallic Salts of Higher Fatty Acids. J. Indian Chem. Soc., Ind. Ed. 1940, 3, 166–173. [Google Scholar]
- Bergström S.; Pääbo K.; Rottenberg M. The Preparation of 8,11-Heptadecadienoic (nor-Linoleic) Acid. Acta Chem. Scand. 1953, 7, 1001–1006. 10.3891/acta.chem.scand.07-1001. [DOI] [Google Scholar]
- Anker H. S. On the Mechanism of Fatty Acid Synthesis in Vivo. J. Biol. Chem. 1952, 194, 177–183. [PubMed] [Google Scholar]
- Cope A. C.; Synerholm M. E. Cyclic Polyolefins. XI. Carbonyl-Bridged Compounds Derived from the Adduct of α-Carbethoxycyclohexanone and Acrolein. J. Am. Chem. Soc. 1950, 72, 5228–5232. 10.1021/ja01167a123. [DOI] [Google Scholar]
- Oldham J. W. H. 22. A Study of the Action of Bromine on the Silver Salts of Organic Acids. J. Chem. Soc. 1950, 100–108. 10.1039/jr9500000100. [DOI] [Google Scholar]
- Barnes R. A.; Prochaska R. J. The Reaction of Bromine with Silver Salts of Aromatic Acids1. J. Am. Chem. Soc. 1950, 72, 3188–3191. 10.1021/ja01163a106. [DOI] [Google Scholar]
- Conly J. C. Applications of the Hunsdiecker Silver Salt Degradation. The Preparation of Dibromides and Tribromides1. J. Am. Chem. Soc. 1953, 75, 1148–1150. 10.1021/ja01101a041. [DOI] [Google Scholar]
- Papa D.; Schwenk E.; Klingsberg E. X-Ray Diagnostics. III. Iodinated Alkoxyaryl Aliphatic Acids and Ethyl Esters1. J. Am. Chem. Soc. 1950, 72, 2623–2626. 10.1021/ja01162a078. [DOI] [Google Scholar]
- Lüttringhaus A.; Schade D. Über Den Abbau Fettsaurer Silbersalze Zu Alkylbromiden. Ber. Dtsch. Chem. Ges. B 1941, 74, 1565–1568. 10.1002/cber.19410740916. [DOI] [Google Scholar]
- Schmid H. Über Thiophanverbindungen III. Helv. Chim. Acta 1944, 27, 127–142. 10.1002/hlca.19440270116. [DOI] [PubMed] [Google Scholar]
- Goncalves R.; Brown E. V. Preparation of Thiophanthrenequinones. III. The Thenoylnitrobenzoic Esters. J. Org. Chem. 1954, 19, 4–10. 10.1021/jo01366a002. [DOI] [Google Scholar]
- Lodi S. S. M. N. K.; Ahmed M.-D. Reaction of Bromine with Metallic Salts of Aromatic Acids. Pakistan J. Sci. Res. 1954, 6, 92–94. [Google Scholar]
- Kuffner F.; Russo C. Versuche Über Den Verlauf Des Silbersalzabbaues Mittels Brom Bei Den Pyridin-Monocarbonsüuren. Monatsh. Chem. 1954, 85, 1097–1103. 10.1007/BF00899858. [DOI] [Google Scholar]
- Mariella R. P.; Belcher E. P. A-Oxygenated Pyridines. IIII. The Reaction of N-Bromosuccinimide with Some Pyridine Derivatives1. J. Am. Chem. Soc. 1952, 74, 1916–1919. 10.1021/ja01128a013. [DOI] [Google Scholar]
- Bell F.; Smyth I. F. B. The Interaction of the Silver Salts of Optically Active Acids with Bromine - Notes. J. Chem. Soc. 1949, 2372–2373. [Google Scholar]
- Smith W. T.; Hull R. L. The Silver Salt Degradation of T-Butylacetic Acid. J. Am. Chem. Soc. 1950, 72, 3309–3309. 10.1021/ja01163a540. [DOI] [Google Scholar]
- Winstein S. Neighboring Groups in Displacements and Rearrangements. Bull. Soc. Chim. Fr. 1951, 5, C55–C61. [Google Scholar]
- Parker W.; Raphael R. A. The Conversion of Cyclohexanone into 1:1-Disubstituted Cyclohexanes. J. Chem. Soc. 1955, 1723–1727. 10.1039/jr9550001723. [DOI] [Google Scholar]
- Cristol S. J.; Douglass J. R.; Firth W. C.; Krall R. E. Bridged Polycyclic Compounds. XII. A Mechanism for the Hunsdiecker Reaction1,2. J. Am. Chem. Soc. 1960, 82, 1829–1830. 10.1021/ja01492a067. [DOI] [Google Scholar]
- Dauben W. G.; Tilles H. The Reaction of Bromine with the Silver Salts of Aromatic Acids. J. Am. Chem. Soc. 1950, 72, 3185–3187. 10.1021/ja01163a105. [DOI] [Google Scholar]
- Beattie I. R.; Bryce-Smith D. Structure of Silver Iodide Dibenzoate. Nature 1957, 179, 577–578. 10.1038/179577a0. [DOI] [Google Scholar]
- Bunce N. J.; Hadley M. On the Constitutions of Silver Iodine Dibenzoates (Simonini Complexes). Can. J. Chem. 1976, 54, 2612–2616. 10.1139/v76-370. [DOI] [Google Scholar]
- Roberts J. D.; Mazur R. H. Small-Ring Compounds. IV. Interconversion Reactions of Cyclobutyl, Cyclopropylcarbinyl and Allylcarbinyl Derivatives. J. Am. Chem. Soc. 1951, 73, 2509–2520. 10.1021/ja01150a029. [DOI] [Google Scholar]
- Rice F. A. H. Decarboxylation Via the Acid Chloride of Penta-O-Acetyl-D-Gluconic Acid1a,1b. J. Am. Chem. Soc. 1956, 78, 3173–3175. 10.1021/ja01594a053. [DOI] [Google Scholar]
- Rice F. A. H.; Morganroth W. Reaction of the Acid Chlorides of Aromatic Acids with Bromine and Silver Oxide. J. Org. Chem. 1956, 21, 1388–1389. 10.1021/jo01118a015. [DOI] [Google Scholar]
- Davis J. A.; Herynk J.; Carroll S.; Bunds J.; Johnson D. Modifications of the Hunsdiecker Reaction1a. J. Org. Chem. 1965, 30, 415–417. 10.1021/jo01013a026. [DOI] [Google Scholar]
- Cristol S.; Firth W. Jr. A Convenient Synthesis of Alkyl Halides from Carboxylic Acids. J. Org. Chem. 1961, 26, 280–280. 10.1021/jo01060a628. [DOI] [Google Scholar]
- Cristol S. J.; Douglass J. R.; Firth W. C.; Krall R. E. Bridged Polycyclic Compounds. XVII. The Stereochemistry of Conversions of Some Substituted Norbornanecarboxylic Acids to Substituted Norbornyl Bromides1. J. Org. Chem. 1962, 27, 2711–2715. 10.1021/jo01055a001. [DOI] [Google Scholar]
- Cristol S. J.; Gaston L. K.; Tiedeman T. Bridged Polycyclic Compounds. XXIV. The Halogenation of Substituted Norbornyl Radicals1. J. Org. Chem. 1964, 29, 1279–1282. 10.1021/jo01029a001. [DOI] [Google Scholar]
- Wolk J. L.; Goldschmidt Z.; Dunkelblum E. A Short Stereoselective Synthesis of (+)-Cis-Planococcyl Acetate, Sex Pheromone of the Citrus Mealybugplanococcus Citri(Risso). Synthesis 1986, 1986, 347–348. 10.1055/s-1986-31613. [DOI] [Google Scholar]
- Wiberg K. B.; Artis D. R.; Bonneville G. 1,2-Bridged Cyclopropenes. J. Am. Chem. Soc. 1991, 113, 7969–7979. 10.1021/ja00021a024. [DOI] [Google Scholar]
- Davies D. I.; Mason P. Free-Radical Reactions of Halogenated Bridged Polycyclic Compounds. Part XI. Application of the Cristol–Firth Reaction to Some Carboxylic Acids Containing a Chloronorbornene Ring System. J. Chem. Soc. C 1971, 0, 288–295. 10.1039/J39710000288. [DOI] [Google Scholar]
- Baker F. W.; Holtz H. D.; Stock L. M. The Brominative Decarboxylation of Bicyclo[2.2.2]Octane-1-Carboxylic Acid in Halogenated Solvents1. J. Org. Chem. 1963, 28, 514–516. 10.1021/jo01037a062. [DOI] [Google Scholar]
- Al Dulayymi A. R.; Al Dulayymi J. a. R.; Baird M. S.; Gerrard M. E.; Koza G.; Harkins S. D.; Roberts E. 1,2,2-Tribromocyclopropanecarboxylic Acid and Derivatives—Valuable Intermediates for Four Carbon Cyclopropane and Cyclopropene Synthons. Tetrahedron 1996, 52, 3409–3424. 10.1016/0040-4020(95)01123-4. [DOI] [Google Scholar]
- Al Dulayymi J. a. R.; Baird M. S.; Fitton H. L. The Stereochemistry of Ring-Opening of 3-Alkyl-1,1-Dihalocyclopropenes to Vinylcarbenes at Ambient Temperature. Tetrahedron Lett. 1992, 33, 4803–4806. 10.1016/S0040-4039(00)61290-X. [DOI] [Google Scholar]
- Klunder A. J. H.; Zwanenburg B. Chemistry of Strained Polycyclic Compounds—III. Tetrahedron 1972, 28, 4131–4138. 10.1016/S0040-4020(01)93644-0. [DOI] [Google Scholar]
- Hanessian S.; Pernet A. G. Carbanions in Carbohydrate Chemistry: A New Synthesis of C-Glycosyl Compounds. J. Chem. Soc. D 1971, 755–756. 10.1039/c29710000755. [DOI] [Google Scholar]
- Kaiser C.; Tedeschi D. H.; Fowler P. J.; Pavloff A. M.; Lester B. M.; Zirkle C. L. Analogs of Phenothiazines. 3. Synthesis and Potential Antidepressant Activity of Some Phenothiazine Derivatives and Related Compounds Containing a Carbocyclic Basic Side Chain. J. Med. Chem. 1971, 14, 179–186. 10.1021/jm00285a001. [DOI] [PubMed] [Google Scholar]
- Sieja J. B. Synthesis of Cyclobutenone. J. Am. Chem. Soc. 1971, 93, 2481–2483. 10.1021/ja00739a020. [DOI] [Google Scholar]
- Roggen M.; Carreira E. M. Preparation of Cyclobutenone. Org. Synth. 2012, 89, 491–500. 10.15227/orgsyn.089.0491. [DOI] [Google Scholar]
- Collet A.; Jacques J.; Chion B.; Lajzerowicz J. Configuration Absolue Et Dichroïsme Circulaire Du Nitroxyde De La Tetramethyl-2,2,5,5 Hydroxy-3 Pyrrolidine. Etude Comparee De La Pentamethyl-2,2,3,5,5 Cyclopentanone. Tetrahedron 1975, 31, 2243–2246. 10.1016/0040-4020(75)80222-5. [DOI] [Google Scholar]
- Cimarusti C. M.; Giarrusso F. F.; Grabowich P.; Levine S. D. An α-nor-3-Thia-5β-Pregnane. Steroids 1975, 26, 359–362. 10.1016/0039-128X(75)90080-X. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Kinson P. L. Perturbed [12]Annulenes. Derivatives of Dibenzo[cd,gh]Pentalene. J. Am. Chem. Soc. 1975, 97, 2438–2449. 10.1021/ja00842a022. [DOI] [Google Scholar]
- Marchand A. P.; Chou T.-C.; Ekstrand J. D.; Van der Helm D. Synthesis of Tetracyclo[6.3.0.04,11.05,9]Undeca-2,6-Diene, 10-Oxatetracyclo[6.3.0.04,11.05,9]Undeca-2,6-Diene, and Hexacyclo[5.4.0.02,6.04,11.05,9.08,10]Undecane. Structure of Syn-4, Syn-7-Diiodopentacyclo[6.3.0.02,6.03,10.05,9]Undecane. Synthesis and Rhodium(I)-Promoted Rearrangement. J. Org. Chem. 1976, 41, 1438–1444. 10.1021/jo00870a033. [DOI] [Google Scholar]
- Chiu W. H.; Wolff M. E. Syntheses and Biological Activities of 7 Beta-Methyl Steroids. J. Med. Chem. 1979, 22, 1257–1260. 10.1021/jm00196a020. [DOI] [PubMed] [Google Scholar]
- Chiu W. H.; Klein T. H.; Wolff M. E. Apparent Bioisosteric Replacement of--S--by Ncn: Synthesis of N-Cyano-2-Aza-A-nor-5α-Androstan-17β-Ol Acetate, an Aza Steroid Androgen. J. Med. Chem. 1979, 22, 119–120. 10.1021/jm00187a028. [DOI] [PubMed] [Google Scholar]
- Knapp F. Steroid Synthesis. The Modified Hunsdiecker Degradation of Bile Acids and Related Compounds. Steroids 1979, 33, 245–249. 10.1016/0039-128X(79)90001-1. [DOI] [PubMed] [Google Scholar]
- Halim H.; Locksley H. D.; Memon J. J. Vesicant Principles of Poison Ivy and Related Plants: Synthesis of the Urushiols, 1,2- Dihydroxy-3-[(Z)-Pentadec-8-Enyl]Benzene and 1,2 Dihydroxy-3-Pentadecylbenzene. J. Chem. Soc., Perkin Trans. 1 1980, 1, 2331–2337. 10.1039/p19800002331. [DOI] [Google Scholar]
- Gassman P. G.; Proehl G. S. [3.1.1]Propellane. J. Am. Chem. Soc. 1980, 102, 6862–6863. 10.1021/ja00542a040. [DOI] [Google Scholar]
- Adcock W.; Abeywickrema A. N. Synthesis of 4-Substituted Bicyclo[2.2.2]oct-1-yl Fluorides. J. Org. Chem. 1982, 47, 2951–2957. 10.1021/jo00136a028. [DOI] [Google Scholar]
- Bin Sadikun A.; Davies D. I.; Kenyon R. F. An Approach to the Synthesis of Cyclopentane Analogues of the Lyxosyl C-Nucleosides. J. Chem. Soc., Perkin Trans. 1 1981, 1, 2299–2305. 10.1039/p19810002299. [DOI] [Google Scholar]
- Paquette L. A.; Uchida T.; Gallucci J. C. Silanes in Organic Synthesis. 21. Configurational Stability of the 2,2-Diphenyl-1-(Trimethylsilyl)Cyclopropyl Carbanion and Free Radical. Absolute Stereochemical Assignments to Select Silylcyclopropanes. J. Am. Chem. Soc. 1984, 106, 335–340. 10.1021/ja00314a014. [DOI] [Google Scholar]
- Adcock W.; Abeywickrema A. N.; Kok G. B. Transmission of Polar Substituent Effects in Bicycloalkane Systems. Synthesis and Nuclear Magnetic Resonance Study (Carbon-13 and Fluorine-19) of 4-Substituted Bicyclo[2.2.1]hept-1-yl Fluorides. J. Org. Chem. 1984, 49, 1387–1397. 10.1021/jo00182a014. [DOI] [Google Scholar]
- Gopalan A. S.; Sih C. J. Bifunctional Chiral Synthons Via Biochemical Methods. 5. Preparation of (S)-Ethyl Hydrogen-3-Hydroxyglutarate, Key Intermediate to (R)-4-Amino-3-Hydroxybutyric Acid and L-Carnitine. Tetrahedron Lett. 1984, 25, 5235–5238. 10.1016/S0040-4039(01)81572-0. [DOI] [Google Scholar]
- Blankenship C.; Paquette L. A. Double Hunsdiecker Reactions. Convenient Preparation of 1,1-Dibromocyclopropane and 1,1-Dibromocyclobutane. Synth. Commun. 1984, 14, 983–987. 10.1080/00397918408059624. [DOI] [Google Scholar]
- Adcock W.; Gangodawila H.; Kok G. B.; Iyer V. S.; Kitching W.; Drew G. M.; Young D. Polar Substituent Effects on Nmr Chemical Shifts of Group 14 Elements: Synthesis and Nmr (Carbon-13, Silicon-29, Tin-119, and Lead-207) Substituent Chemical Shifts (SCS) of 4-Substituted Bicyclo[2.2.2]Oct-1-ylbutanes, -Trimethylsilanes, -Trimethylstannanes, and -Trimethyl Plumbanes (M(CH3)3, M = Carbon, Silicon, Tin, and Lead) and 4-Substituted Bicyclo[2.2.1]hept-1-yltrimethylstannanes. Organometallics 1987, 6, 156–166. 10.1021/om00144a029. [DOI] [Google Scholar]
- Adcock W.; Kok G. B. Transmission of Polar Substituent Effects in Saturated Systems: Synthesis and Fluorine-19 Nmr Study of 3-Substituted Adamant-1-Yl Fluorides. J. Org. Chem. 1987, 52, 356–364. 10.1021/jo00379a008. [DOI] [Google Scholar]
- Honegger E.; Heilbronner E.; Heß N.; Martin H.-D. Synthesis and Pe Spectra of 1-Ethynyl- and 1,4-Diethynylbicyclo[2.2.2]octane, and of Related 1,4-Dihalobicyclo[2.2.2]octanes. Chem. Ber. 1987, 120, 187–193. 10.1002/cber.19871200210. [DOI] [Google Scholar]
- Wiberg K. B.; Matturro M. G.; Okarma P. J.; Jason M. E.; Dailey W. P.; Burgmaier G. J.; Bailey W. F.; Warner P. Bicyclo[2.2.0]hex-1(4)-ene. Tetrahedron 1986, 42, 1895–1902. 10.1016/S0040-4020(01)87609-2. [DOI] [Google Scholar]
- Brooke G. M.; Mawson S. D. Oligomeric Polyparatetrafluorophenylene Vinylenes: A New Synthesis Based on the Nucleophilic Substitution of the Para Fluorine During the Reaction of (E)-2-(pentafluorophenyl)ethenyl Lithium with Hexafluorobenzene. J. Fluorine Chem. 1990, 50, 111–122. 10.1016/S0022-1139(00)82183-1. [DOI] [Google Scholar]
- Chupp J. P.; Molyneaux J. M. Derivation of Fluorine-Containing Pyridine Dicarboxylates.II. Elaboration at the 4-Position. J. Heterocycl. Chem. 1989, 26, 645–653. 10.1002/jhet.5570260325. [DOI] [Google Scholar]
- Mehta G.; Padma S. Synthetic Studies Towards Prismanes: 1,4-Bishomo-[6]-Prismane (“Garudane”). Tetrahedron 1991, 47, 7807–7820. 10.1016/S0040-4020(01)81937-2. [DOI] [Google Scholar]
- Sims C. G.; Wege D. Cyclopropa-Fusion to Seven-Membered Unsaturated Rings. The Synthesis of 4-Bromo-8,8-Dimethylbicyclo[5.1.0]octa-1,4,6-trien-3-one, a Cyclopropa-Fused Tropone. Aust. J. Chem. 1995, 48, 469–490. 10.1071/CH9950469. [DOI] [Google Scholar]
- Chenier P. J.; Bauer M. J.; Southard D. A. Attempted Synthesis of Tricyclo[3.3.2.024]Dec-2(4)-Ene. Synth. Commun. 1996, 26, 1199–1207. 10.1080/00397919608003729. [DOI] [Google Scholar]
- Armstrong A.; Bhonoah Y.; Shanahan S. E. Aza-Prins-Pinacol Approach to 7-Azabicyclo[2.2.1]Heptanes: Syntheses of (±)-Epibatidine and (±)-Epiboxidine. J. Org. Chem. 2007, 72, 8019–8024. 10.1021/jo701536a. [DOI] [PubMed] [Google Scholar]
- Al Dulayymi J. a. R.; Baird M. S.; Fitton H. L.; Rajaram L. 1,2-Dibromoalk-2-enylidenes by Ring-Opening of 1,2-Dibromo-3-alkylcyclopropenes at Ambient Temperature. J. Chem. Soc., Perkin Trans. 1 1994, 1, 1633–1641. 10.1039/p19940001633. [DOI] [Google Scholar]
- Baird M. S.; Fitton H. L.; Clegg W.; McCamley A. (R)-1,3-Dimethylcyclopropene—One Isomer of the Smallest Chiral Hydrocarbon. J. Chem. Soc., Perkin Trans. 1 1993, 1, 321–326. 10.1039/P19930000321. [DOI] [Google Scholar]
- Bunce N. J. Mercury Salt Pathway for the Degradation of Carboxylic Acids to Alkyl Halides Using Halogen and Mercuric Oxide. J. Org. Chem. 1972, 37, 664–669. 10.1021/jo00969a030. [DOI] [Google Scholar]
- Cason J.; Walba D. M. Reaction Pathway in the Modified Hunsdiecker Reaction. J. Org. Chem. 1972, 37, 669–671. 10.1021/jo00969a031. [DOI] [Google Scholar]
- Meyers A. I.; Fleming M. P. Photoassisted Cristol-Firth-Hunsdiecker Reaction. J. Org. Chem. 1979, 44, 3405–3406. 10.1021/jo01333a029. [DOI] [Google Scholar]
- Uemura S.; Tanaka S.; Okano M.; Hamana M. Decarboxylative Ipso Halogenation of Mercury(II) Pyridinecarboxylates. Facile Formation of 3-Iodo- and 3-Bromopyridines. J. Org. Chem. 1983, 48, 3297–3301. 10.1021/jo00167a027. [DOI] [Google Scholar]
- Jennings P. W.; Ziebarth T. D. Mechanism of the Modified Hunsdiecker Reaction. J. Org. Chem. 1969, 34, 3216–3217. 10.1021/jo01262a101. [DOI] [Google Scholar]
- Tabushi I.; Aoyama Y.; Kojo S.; Hamuro J.; Yoshida Z. Free-Radical Halogenation of Adamantane. Selectivity and Relative Lifetime of 1- and 2-Adamantyl Radicals. J. Am. Chem. Soc. 1972, 94, 1177–1183. 10.1021/ja00759a024. [DOI] [Google Scholar]
- Kochi J. K. Formation of Alkyl Halides from Acids by Decarboxylation with Lead(IV) Acetate and Halide Salts. J. Org. Chem. 1965, 30, 3265–3271. 10.1021/jo01021a002. [DOI] [Google Scholar]
- Kochi J. K. A New Method for Halodecarboxylation of Acids Using Lead(IV) Acetate. J. Am. Chem. Soc. 1965, 87, 2500–2502. 10.1021/ja01089a041. [DOI] [Google Scholar]
- Barton D. H. R.; Serebryakov E. P. Proceedings of the Chemical Society. Proc. Chem. Soc. 1962, 309–309. September 1962. [Google Scholar]
- Barton D. H. R.; Faro H. P.; Serebryakov E. P.; Woolsey N. F. 445. Photochemical Transformations. Part XVII. Improved Methods for the Decarboxylation of Acids. J. Chem. Soc. 1965, 2438–2444. 10.1039/jr9650002438. [DOI] [Google Scholar]
- Meystre C. H.; Heusler K.; Kalvoda J.; Wieland P.; Anner G.; Wettstein A. Reaktionen Von Steroid-Hypojoditen II. Über Die Herstellung 18-Oxygenierter Pregnanverbindungen. Über Steroide, 187. Mitteilung. Helv. Chim. Acta 1962, 45, 1317–1343. 10.1002/hlca.19620450431. [DOI] [Google Scholar]
- Becker K.; Geisel M.; Grob C.; Kuhnen F. Improved Preparation of Tertiary Chlorides by Halodecarboxylation. Synthesis 1973, 1973, 493–494. 10.1055/s-1973-22243. [DOI] [Google Scholar]
- Martin J. D.; Palazon J. M.; Perez C.; Ravelo J. L. Syntheses of Marine Molecules. Pure Appl. Chem. 1986, 58, 395–406. 10.1351/pac198658030395. [DOI] [Google Scholar]
- Wender P. A.; Wolanin D. J. Total Synthesis of Quadrone and Terrecyclic Acid A. J. Org. Chem. 1985, 50, 4418–4420. 10.1021/jo00222a055. [DOI] [Google Scholar]
- Falkenberg-Andersen C.; Ranganayakulu K.; Schmitz L. R.; Sorensen T. S. Cis-1-Methylcyclopropylcarbinyl Cation. Preparation and Facile Interconversion with the 1-Ethylallyl Cation. J. Am. Chem. Soc. 1984, 106, 178–182. 10.1021/ja00313a036. [DOI] [Google Scholar]
- Hutmacher H.-M.; Fritz H.-G.; Musso H. Tetraasterane, Pentacyclo[6.4.0.02,7.04,11.05,10]Dodecane. Angew. Chem., Int. Ed. Engl. 1975, 14, 180–181. 10.1002/anie.197501801. [DOI] [Google Scholar]
- Schwab D.; Thom H.; Heinze J.; Kurz G. 3α,7α,12α-Trihydroxy-24-nor-5β-cholan-23-sulfonate: Synthesis and Suitability for the Study of Cholate Transport. J. Lipid Res. 1996, 37, 1045–1056. [PubMed] [Google Scholar]
- Wu Z.; Grubbs R. H. Preparation of Alternating Copolymers from the Ring-Opening Metathesis Polymerization of 3-Methylcyclobutene and 3,3-Dimethylcyclobutene. Macromolecules 1995, 28, 3502–3508. 10.1021/ma00114a002. [DOI] [Google Scholar]
- Wang Y.; Fleet G. W. J.; Storer R.; Myers P. L.; Wallis C. J.; Doherty O.; Watkin D. J.; Vogt K.; Witty D. R.; Wilson F. X.; et al. Synthesis of the Potent Antiviral Oxetane Nucleoside Epinoroxetanocin from D-Lyxonolactone. Tetrahedron: Asymmetry 1990, 1, 527–530. 10.1016/S0957-4166(00)80541-8. [DOI] [Google Scholar]
- Dupont A. C.; Audia V. H.; Waid P. P.; Carter J. P. A Convenient Large-Scale Synthesis of Cyclobutyl Halides. Synth. Commun. 1990, 20, 1011–1021. 10.1080/00397919008052804. [DOI] [Google Scholar]
- Di Maio G.; Migneco L. M.; Vecchi E. Studies on the Stereochemistry of Reduction Reactions on 10-R Substituted Trans Decal-2-ones. Tetrahedron 1990, 46, 6053–6060. 10.1016/S0040-4020(01)87929-1. [DOI] [Google Scholar]
- Gream G. E. The 9-Decalyl and Related Cations. I. Generation of the 9-Decalyl Cation by Solvolysis. Aust. J. Chem. 1972, 25, 1051–1079. 10.1071/CH9721051. [DOI] [Google Scholar]
- Stevenson D. E.; Akhtar M.; Gani D. Structural and Stereochemical Studies of Methiomine Decarboxylase from Dryopteris Felix-Mas. Tetrahedron Lett. 1986, 27, 5661–5664. 10.1016/S0040-4039(00)85294-6. [DOI] [Google Scholar]
- Ando M.; Asao T.; Hiratsuka N.; Takase K.; Nozoe T. The Total Syntheses of Chamaecynone, Isochamaecynone, and Dihydroisochamaecynone. Bull. Chem. Soc. Jpn. 1980, 53, 1425–1434. 10.1246/bcsj.53.1425. [DOI] [Google Scholar]
- Shinmyozu T.; Inazu T.; Yoshino T. Synthesis of [3.3]Metacyclophane. Chem. Lett. 1976, 5, 1405–1406. 10.1246/cl.1976.1405. [DOI] [Google Scholar]
- Mihailovic M. L.; Bošnjak J.; Cekovic Ž. The Lead Tetraacetate Reaction of Alcohols Containing a Small Ring. Part II. Cyclobutane-Methanols and Cyclopropane-Ethanols. Helv. Chim. Acta 1976, 59, 475–486. 10.1002/hlca.19760590214. [DOI] [Google Scholar]
- Cassal J.-M.; Fürst A.; Meier W. Synthese Der Enantiomeren 2-Pyrrolidinessigsäuren. Helv. Chim. Acta 1976, 59, 1917–1924. 10.1002/hlca.19760590602. [DOI] [Google Scholar]
- Miller R. D.; Schneider M. The Synthesis and Characterization of Spiro[3.4]Octa-1,5,7-Triene. Tetrahedron Lett. 1975, 16, 1557–1560. 10.1016/S0040-4039(00)72196-4. [DOI] [Google Scholar]
- Miyamoto T.; Tanaka S.; Odaira Y. Synthesis of Tricarbonylcyclobuta[l]phenanthreneiron. J. Chem. Soc., Perkin Trans. 1 1973, 1, 138–140. 10.1039/p19730000138. [DOI] [Google Scholar]
- Ahmed S.; Alauddin M.; Caddy B.; Martin-Smith M.; Sidwell W. T. L.; Watson T. R. Bisquaternary Ammonium Salts Derived from 3α,12α-Diamino-5β-Cholane and 3α,12α-Diamino-24-nor-5β-Cholane. Aust. J. Chem. 1971, 24, 521–547. 10.1071/CH9710521. [DOI] [Google Scholar]
- Hall H. K.; Smith C. D.; Blanchard E. P.; Cherkofsky S. C.; Sieja J. B. Synthesis and Polymerization of Bridgehead-Substituted Bicyclobutanes. J. Am. Chem. Soc. 1971, 93, 121–130. 10.1021/ja00730a022. [DOI] [Google Scholar]
- Glily-Terry S.; Klein J. Metallation Reactions. Part X. Allylic Metallation in the Presence of Amide Groups: Long Range Interactions. J. Chem. Soc. C 1971, 3821–3827. 10.1039/j39710003821. [DOI] [Google Scholar]
- Perold G. W.; Ourisson G. Photolyse Des Dihydro-1,2-(6β,11α, et 11β)-Santonines. Tetrahedron Lett. 1969, 10, 3871–3873. 10.1016/S0040-4039(01)88534-8. [DOI] [PubMed] [Google Scholar]
- Reich H. J.; Cram D. J. Racemization, Ring Opening, and Ring Expansion of [2.2] Paracyclophane Nucleus through a Diradicial Intermediate. J. Am. Chem. Soc. 1967, 89, 3078–3080. 10.1021/ja00988a068. [DOI] [Google Scholar]
- Neuman R. C.; Rahm M. L. Synthesis and Nuclear Magnetic Resonance Spectra of Some 1-Halo-1-Iodoalkanes. J. Org. Chem. 1966, 31, 1857–1859. 10.1021/jo01344a041. [DOI] [Google Scholar]
- Huang P. Q.; Zhou W. S. Efficient Syntheses of a New Chiral Diene and a New Bridgehead Enone for a Diels-Alder Approach to Kaura-9(11)-16-Dien-19-oic Acid. Tetrahedron: Asymmetry 1991, 2, 875–878. 10.1016/S0957-4166(00)82199-0. [DOI] [Google Scholar]
- Lambert J. B.; Marko D. E. Factors Influencing Conformational Preferences in Cyclohexenes. J. Am. Chem. Soc. 1985, 107, 7978–7982. 10.1021/ja00312a030. [DOI] [Google Scholar]
- Drège E.; Tominiaux C.; Morgant G.; Desmaële D. Synthetic Studies on Cyathin Terpenoids: Enantioselective Synthesis of the Tricyclic Core of Cyathin through Intramolecular Heck Cyclisation. Eur. J. Org. Chem. 2006, 2006, 4825–4840. 10.1002/ejoc.200600429. [DOI] [Google Scholar]
- Kiely J. S.; Nelson L. L.; Boudjouk P. A Convenient Synthesis of 1-Bromo-8-Iodonaphthalene and 1,8-Dibromonaphthalene from 8-Bromo-1-Naphthoic Acid. J. Org. Chem. 1977, 42, 1480–1480. 10.1021/jo00428a055. [DOI] [Google Scholar]
- Almeida M.; Boman A.; Lundstedt T. Synthesis Of N-(2-Chloro-3,4-Dimethoxybenzylideneamino)guanidinium Acetate [α-14c]. J. Labelled Compd. Radiopharm. 2002, 45, 371–377. 10.1002/jlcr.559. [DOI] [Google Scholar]
- Nikishin G. I.; Sokova L. L.; Chizhov A. O.; Makhaev V. D.; Kapustina N. I. Solid-State Chlorodecarboxylation of Mono- and Dicarboxylic Acids with the Pb(OAc)4-MCl System. Russ. Chem. Bull. 2004, 53, 2200–2204. 10.1007/s11172-005-0099-5. [DOI] [Google Scholar]
- Kochi J. K. Mechanisms of Organic Oxidation and Reduction by Metal Complexes. Science 1967, 155, 415–424. 10.1126/science.155.3761.415. [DOI] [PubMed] [Google Scholar]
- Stolow R. D.; Giants T. W. Stereochemistry of the Kochi Reaction. Tetrahedron Lett. 1971, 12, 695–696. 10.1016/S0040-4039(01)96533-5. [DOI] [Google Scholar]
- Monnier M.; Aycard J.-P. Structure Et Réactivité. III., Evolutions Stéréochimiques et Chemins Réactionnels des Radicaux Cyclohexyles Substitués en 2 et Cyclohexényles Substitués en 3 (Réaction De Kochi). Can. J. Chem. 1979, 57, 1257–1261. 10.1139/v79-205. [DOI] [Google Scholar]
- Kochi J. K. The Mechanism of Oxidative Decarboxylation with Lead(IV) Acetate. J. Am. Chem. Soc. 1965, 87, 1811–1812. 10.1021/ja01086a046. [DOI] [Google Scholar]
- Kochi J. K. Oxidation with Lead(IV). I. Mechanism of the Decarboxylation of Pentanoic Acids. J. Am. Chem. Soc. 1965, 87, 3609–3619. 10.1021/ja01094a015. [DOI] [Google Scholar]
- Chiasson B. A.; Berchtold G. A. Synthesis of Trans-3,4-Dihydroxy-3,4-Dihydrobenzoic Acid. J. Am. Chem. Soc. 1974, 96, 2898–2901. 10.1021/ja00816a039. [DOI] [Google Scholar]
- Wiberg K. B.; Walker F. H.; Pratt W. E.; Michl J. [2.1.1.] Propellane Reaction of 1,4-Diiodobicyclo[2.1.1]Hexane with tert-Butyllithium and with Potassium Atoms. J. Am. Chem. Soc. 1983, 105, 3638–3641. 10.1021/ja00349a048. [DOI] [Google Scholar]
- Schäfer J.; Szeimies G. Reaction of 1,6-Dihalopentacyclo[5.2.0.02,6.03,9.05,8]Nonanes with Tert-Butyllithium: Is 1,6-Dehydrohomocubane Involved as a Reactive Intermediate?. Tetrahedron Lett. 1990, 31, 2263–2264. 10.1016/0040-4039(90)80201-V. [DOI] [Google Scholar]
- Tanner D. D.; Gidley G. C.; Das N.; Rowe J. E.; Potter A. On the Structure of tert-Butyl Hypoiodite. J. Am. Chem. Soc. 1984, 106, 5261–5267. 10.1021/ja00330a038. [DOI] [Google Scholar]
- Britten-Kelly M. R.; Goosen A.; Scheffer A. Kinetic Studies on the Photodecarboxylation Reactions of Acylhypoiodites. S. Afr. J. Chem. 1975, 28, 224–234. [Google Scholar]
- Concepción J. I.; Francisco C. G.; Hernández R.; Salazar J. A.; Suárez E. Intramolecular Hydrogen Abstraction. Iodosobenzene Diacetate, an Efficient and Convenient Reagent for Alkoxy Radical Generation. Tetrahedron Lett. 1984, 25, 1953–1956. 10.1016/S0040-4039(01)90085-1. [DOI] [Google Scholar]
- Concepcion J. I.; Francisco C. G.; Freire R.; Hernandez R.; Salazar J. A.; Suarez E. Iodosobenzene Diacetate, an Efficient Reagent for the Oxidative Decarboxylation of Carboxylic-Acids. J. Org. Chem. 1986, 51, 402–404. 10.1021/jo00353a026. [DOI] [Google Scholar]
- Camps P.; Lukach A. E.; Pujol X.; Vázquez S. Hunsdiecker-Type Bromodecarboxylation of Carboxylic Acids with Iodosobenzene Diacetate–Bromine. Tetrahedron 2000, 56, 2703–2707. 10.1016/S0040-4020(00)00169-1. [DOI] [Google Scholar]
- Coleman K. S.; Chakraborty A. K.; Bailey S. R.; Sloan J.; Alexander M. Iodination of Single-Walled Carbon Nanotubes. Chem. Mater. 2007, 19, 1076–1081. 10.1021/cm062730x. [DOI] [Google Scholar]
- Moriarty R. M.; Khosrowshahi J. S.; Dalecki T. M. Hypervalent Iodine Iodinative Decarboxylation of Cubyl and Homocubyl Carboxylic Acids. J. Chem. Soc., Chem. Commun. 1987, 675–676. 10.1039/c39870000675. [DOI] [Google Scholar]
- Ötvös L.; Béres J.; Sági G.; Tömösközi I.; Gruber L. The First Stereospecific Synthesis of (+)-(1R,2S,4R)-4-Amino-2-Hydroxy-1-Cyclopentanemethanol and (+)-Carbocyclic Thymidine. Tetrahedron Lett. 1987, 28, 6381–6384. 10.1016/S0040-4039(01)91379-6. [DOI] [Google Scholar]
- Gargiulo D.; Blizzard T. A.; Nakanishi K. Synthesis of Mosesin-4, a Naturally Occurring Steroid Saponin with Shark Repellent Activity, and Its Analog 7-β-Galactosyl Ethyl Cholate. Tetrahedron 1989, 45, 5423–5432. 10.1016/S0040-4020(01)89488-6. [DOI] [Google Scholar]
- Kotsuki H.; Nishikawa H.; Mori Y.; Ochi M. Highly Practical, Enantiospecific Synthesis of the Cyclohexyl Fragment of the Immunosuppressant FK-506. J. Org. Chem. 1992, 57, 5036–5040. 10.1021/jo00044a051. [DOI] [Google Scholar]
- Sun U J.; Seok Park H.; Gupta S.; Kun Cha J. A Convenient Synthesis of (−)-Methyl Epijasmonate. Synth. Commun. 1997, 27, 2931–2941. 10.1080/00397919708004999. [DOI] [Google Scholar]
- Eaton P. E.; Pramod K.; Emrick T.; Gilardi R. Building with Cubane-1,4-Diyl. Synthesis of Aryl-Substituted Cubanes, p-[n]Cubyls, and Cubane-Separated Bis(Arenes)1. J. Am. Chem. Soc. 1999, 121, 4111–4123. 10.1021/ja983441f. [DOI] [Google Scholar]
- Liang X.; Lohse A.; Bols M. Chemoenzymatic Synthesis of Isogalactofagomine. J. Org. Chem. 2000, 65, 7432–7437. 10.1021/jo000699r. [DOI] [PubMed] [Google Scholar]
- Bolster M. G.; Jansen B. J. M.; Groot A. d. The Synthesis of (−)-Ambrox® Starting from Labdanolic Acid. Tetrahedron 2001, 57, 5657–5662. 10.1016/S0040-4020(01)00493-8. [DOI] [Google Scholar]
- Camps P.; Lukach A. E.; Rossi R. A. Synthesis of Several Halobisnoradamantane Derivatives and Their Reactivity through the SRN1 Mechanism. J. Org. Chem. 2001, 66, 5366–5373. 10.1021/jo010159+. [DOI] [PubMed] [Google Scholar]
- Camps P.; Pujol X.; Vázquez S.; Pericàs M. A.; Puigjaner C.; Solà L. Cross-Coupling of a Functionalized Highly Pyramidalized Alkene: DSC and NMR Study of the [2 + 2] Retrocycloaddition of Cyclobutane Cross Products, Hyperstability and Pyramidalization of the Formed Dienes. Tetrahedron 2001, 57, 8511–8520. 10.1016/S0040-4020(01)00802-X. [DOI] [Google Scholar]
- Kitahara T.; Tachihara T.; Watanabe H. Enantioselective Synthesis of (−)-N-Boc-7-Azabicyclo[2.2.1]Heptan-2-One and (+)-N-Boc-4-Amino-2-Cyclohexen-1-One: Formal Total Synthesis of Both Enantiomers of Epibatidine. Heterocycles 2002, 57, 781–785. 10.3987/COM-02-9459. [DOI] [Google Scholar]
- Tachihara T.; Kitahara T. Total Synthesis of (+)-Epiepoformin, (+)-Epiepoxydon and (+)-Bromoxone Employing a Useful Chiral Building Block, Ethyl (1R,2S)-5,5-Ethylenedioxy-2-Hydroxycyclohexanecarboxylate. Tetrahedron 2003, 59, 1773–1780. 10.1016/S0040-4020(03)00119-4. [DOI] [Google Scholar]
- Ayats C.; Camps P.; Font-Bardia M.; Muñoz M. R.; Solans X.; Vázquez S. Alternative Syntheses of the D2d Symmetric 1,3,5,7-Tetraiodotricyclo[3.3.0.03,7]Octane. Tetrahedron 2006, 62, 7436–7444. 10.1016/j.tet.2006.05.015. [DOI] [Google Scholar]
- Krow G. R.; Huang Q.; Lin G.; Centafont R. A.; Thomas A. M.; Gandla D.; Debrosse C.; Carroll P. J. 5-Carboxy-2-Azabicyclo[2.1.1]Hexanes as Precursors of 5-Halo, Amino, Phenyl, and 2-Methoxycarbonylethyl Methanopyrrolidines. J. Org. Chem. 2006, 71, 2090–2098. 10.1021/jo052506b. [DOI] [PubMed] [Google Scholar]
- Yamauchi N.; Endoh S. Improved Isotopic Deuterium Labeling at the Diastereotopic Methyl Group of Leucine: A Synthetic Route to (4S)- and (4R)-[5-2H1]Leucine. Biosci., Biotechnol., Biochem. 2006, 70, 276–278. 10.1271/bbb.70.276. [DOI] [PubMed] [Google Scholar]
- Zhao R.-B.; Wu Y.-L. A Formal Synthesis of (−)-Patchouli Alcohol from (−)-Carvone. Chin. J. Chem. 1991, 9, 377–380. 10.1002/cjoc.19910090414. [DOI] [Google Scholar]
- Wang P. C.; Fang J. M.; Tsai K. C.; Wang S. Y.; Huang W. I.; Tseng Y. C.; Cheng Y. S.; Cheng T. J.; Wong C. H. Peramivir Phosphonate Derivatives as Influenza Neuraminidase Inhibitors. J. Med. Chem. 2016, 59, 5297–5310. 10.1021/acs.jmedchem.6b00029. [DOI] [PubMed] [Google Scholar]
- Hung K.; Condakes M. L.; Novaes L. F. T.; Harwood S. J.; Morikawa T.; Yang Z.; Maimone T. J. Development of a Terpene Feedstock-Based Oxidative Synthetic Approach to the Illicium Sesquiterpenes. J. Am. Chem. Soc. 2019, 141, 3083–3099. 10.1021/jacs.8b12247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao G.; Shao X.; Zhu D.; Yu B. Chemical Synthesis of Marine Saponins. Nat. Prod. Rep. 2019, 36, 769–787. 10.1039/C8NP00087E. [DOI] [PubMed] [Google Scholar]
- Moriarty R.; Khosrowshahi J.; Dalecki T.. Hypervalent Iodine Iodinative Decarboxylation of Cubyl and Homocubyl Carboxylic Acids. SPIE: 1988; Vol. 0872. [Google Scholar]
- Hamamoto H.; Hattori S.; Takemaru K.; Miki Y. Hypervalent Iodine(III)-LiX Combination in Fluoroalcohol Solvent for Aromatic Halogenation of Electron-Rich Arenecarboxylic Acids. Synlett 2011, 2011, 1563–1566. 10.1055/s-0030-1260791. [DOI] [Google Scholar]
- Singh R.; Just G. An Efficient Synthesis of Aromatic Iodides from Aromatic Carboxylic Acids. Synth. Commun. 1988, 18, 1327–1330. 10.1080/00397918808078799. [DOI] [Google Scholar]
- Courtneidge J. L.; Lusztyk J.; Pagé D. Alkoxyl Radicals from Alcohols. Spectroscopic Detection of Intermediate Alkyl and Acyl Hypoiodites in the Suárez and Beebe Reactions. Tetrahedron Lett. 1994, 35, 1003–1006. 10.1016/S0040-4039(00)79950-3. [DOI] [Google Scholar]
- Alberti A.; Benaglia M.; Vismara E. An ESR Approach to Hypervalent Iodine Induced Iododecarboxylation. Res. Chem. Intermed. 1989, 11, 117–126. 10.1163/156856789X00023. [DOI] [Google Scholar]
- Watanabe A.; Koyamada K.; Miyamoto K.; Kanazawa J.; Uchiyama M. Decarboxylative Bromination of Sterically Hindered Carboxylic Acids with Hypervalent Iodine(III) Reagents. Org. Process Res. Dev. 2020, 24, 1328–1334. 10.1021/acs.oprd.0c00130. [DOI] [Google Scholar]
- Barton D. H. R.; Crich D.; Motherwell W. B. A Practical Alternative to the Hunsdiecker Reaction. Tetrahedron Lett. 1983, 24, 4979–4982. 10.1016/S0040-4039(01)99826-0. [DOI] [Google Scholar]
- Barton D. H. R.; Lacher B.; Zard S. Z. Radical Decarboxylative Bromination of Aromatic-Acids. Tetrahedron Lett. 1985, 26, 5939–5942. 10.1016/S0040-4039(00)98266-2. [DOI] [Google Scholar]
- Barton D. H. R.; Lacher B.; Zard S. Z. The Invention of Radical Reactions. Tetrahedron 1987, 43, 4321–4328. 10.1016/S0040-4020(01)90307-2. [DOI] [Google Scholar]
- Stofer E.; Lion C. Décomposition Des Esters De La N-Hydroxythiopyridone-2. Synthèse De Quelques Nouveaux Halogènures Secondaires et Tertiaires Encombres. Bull. Soc. Chim. Belg. 1987, 96, 623–628. 10.1002/bscb.19870960809. [DOI] [Google Scholar]
- Hartung J.; Greb M. A New Regioselective Synthesis of Vicinal Bromohydrins Via Free Radical Decarboxylative Bromination of N-Hydroxycarboxylic Acids. Synlett 2004, 69–72. 10.1055/s-2003-43379. [DOI] [Google Scholar]
- Barton D. H. R.; Hervé Y.; Potier P.; Thierry J. Manipulation of the Cabboxyl Groups of α-Amino-Acids and Peptides Using Radical Chemistry Based on Esters of α-Hydroxy-2-Thiopyridone. Tetrahedron 1988, 44, 5479–5486. 10.1016/S0040-4020(01)86053-1. [DOI] [Google Scholar]
- Barton D. H. R.; Ozbalik N.; Schmitt M. Radical Chemistry Based on (+)-cis-Pinonic Acid. Tetrahedron Lett. 1989, 30, 3263–3266. 10.1016/S0040-4039(00)99217-7. [DOI] [Google Scholar]
- Della E. W.; Tsanaktsidis J. Synthesis of Bridgehead Halides by Barton Halodecarboxylation. Aust. J. Chem. 1989, 42, 61–69. 10.1071/CH9890061. [DOI] [Google Scholar]
- Kobayashi S.; Kamiyama K.; Ohno M. The First Enantioselective Synthesis of Fortamine, the 1,4-Diaminocyclitol Moiety of Fortimicin A, by Chemicoenzymatic Approach. J. Org. Chem. 1990, 55, 1169–1177. 10.1021/jo00291a014. [DOI] [Google Scholar]
- Wang Y.; Fleet G. W. J.; Wilson F. X.; Storer R.; Wallis C. J.; Doherty O.; Watkin D. J.; Vogt K.; Witty D. R.; Peach J. M. Oxetane Nucleosides with Fluorine and Azide Substitutents: Nucleophilic Displacements on an Oxetane Ring. Tetrahedron Lett. 1991, 32, 1675–1678. 10.1016/S0040-4039(00)74302-4. [DOI] [Google Scholar]
- Channing M. A.; Simpson N. Radiosynthesis of 1-[11C] Polyhomoallylic Fatty Acids. J. Labelled Compd. Radiopharm. 1993, 33, 541–546. 10.1002/jlcr.2580330611. [DOI] [Google Scholar]
- Della E. W.; Head N. J.; Janowski W. K.; Schiesser C. H. Toward an Understanding of the Cubyl and Related Caged Carbocations. J. Org. Chem. 1993, 58, 7876–7882. 10.1021/jo00079a037. [DOI] [Google Scholar]
- Zhu J.; Klunder A. J. H.; Zwanenburg B. Generation and Characterization of Tricyclodecatrienone, a Norbornene Annulated Cyclopentadienone. Tetrahedron Lett. 1993, 34, 3335–3338. 10.1016/S0040-4039(00)73697-5. [DOI] [Google Scholar]
- Kraus G. A.; Su Q. Bridgehead Intermediates in Organic Synthesis Construction of the Tetracyclic Skeleton of Leucothol A. Synlett 1994, 1994, 237–237. 10.1055/s-1994-22808. [DOI] [Google Scholar]
- Lukin K.; Eaton P. E. Dimerization of Cubene. 1-Iodoadamantane as a Probe for Radical Intermediates. J. Am. Chem. Soc. 1995, 117, 7652–7656. 10.1021/ja00134a009. [DOI] [Google Scholar]
- Zhu J.; Klunder A. J. H.; Zwanenburg B. Synthesis of 6-Functionalized Tricyclodecadienones Using Barton’s Radical Decarboxylation Reaction. Generation of Tricyclo[5.2.1.02,6]Decatrienone, a Norbornene Annulated Cyclopentadienone. Tetrahedron 1995, 51, 5099–5116. 10.1016/0040-4020(95)98707-O. [DOI] [Google Scholar]
- Graf C.-D.; Malan C.; Knochel P. New Chiral Ligands with Nonstereogenic Chirotopic Centers for Asymmetric Synthesis. Angew. Chem., Int. Ed. 1998, 37, 3014–3016. . [DOI] [PubMed] [Google Scholar]
- Scheumann K.; Sackers E.; Bertau M.; Leonhardt J.; Hunkler D.; Fritz H.; Wörth J.; Prinzbach H. Pentagonal Dodecahedranes. Novel Substitution Patterns— Ms Fragmentation and Unsaturation. J. Chem. Soc., Perkin Trans. 2 1998, 2, 1195–1210. 10.1039/a709107i. [DOI] [Google Scholar]
- Luithle J. E.; Pietruszka J. (2R,3R)-1,4-Dimethoxy-1,1,4,4-Tetraphenyl-2,3-Butanediol: Chiral Auxiliary and Efficient Protecting Group for Boronic Acids. J. Org. Chem. 2000, 65, 9194–9200. 10.1021/jo0056601. [DOI] [PubMed] [Google Scholar]
- Pornpakakul S.; Pritchard R. G.; Stoodley R. J. Asymmetric Synthesis of (−)-4-Epi-Shikimic Acid. Tetrahedron Lett. 2000, 41, 2691–2694. 10.1016/S0040-4039(00)00225-2. [DOI] [Google Scholar]
- Starr J. T.; Koch G.; Carreira E. M. Enantioselective Synthesis of the Cyclopentyl Core of the Axinellamines. J. Am. Chem. Soc. 2000, 122, 8793–8794. 10.1021/ja0019575. [DOI] [Google Scholar]
- Zhong S.; Sauter P. F.; Nieger M.; Brase S. Stereoselective Synthesis of Highly Functionalized Hydroindoles as Building Blocks for Rostratins B-D and Synthesis of the Pentacyclic Core of Rostratin C. Chem. - Eur. J. 2015, 21, 11219–11225. 10.1002/chem.201501199. [DOI] [PubMed] [Google Scholar]
- Shepherd E. D.; Hallside M. S.; Sutro J. L.; Thompson A.; Hutchings M.; Burton J. W.. Synthesis of the Cyclopentane Core of Pepluanin A. Tetrahedron 2020, 76.130981. 10.1016/j.tet.2020.130981 [DOI] [Google Scholar]
- Gawronska K.; Gawronski J.; Walborsky H. M. Formation of a Chiral 1-Fluoro-2,2-Diphenylcyclopropyl Radical in the Barton Decarboxylation Reaction. J. Org. Chem. 1991, 56, 2193–2197. 10.1021/jo00006a042. [DOI] [Google Scholar]
- Vogel E.; Schieb T.; Schulz W. H.; Schmidt K.; Schmickler H.; Lex J. Bridged[14]Annulenes with a Phenanthrene-Perimeter:Anti-1,6:7,12-Bismethano[14]Annulene. Angew. Chem., Int. Ed. Engl. 1986, 25, 723–725. 10.1002/anie.198607231. [DOI] [Google Scholar]
- Braish T. F.; Fox D. E. Synthesis of 2-Bromo-4, 5-Difluorobenzolc Acid. Synth. Commun. 1992, 22, 3067–3074. 10.1080/00397919209409255. [DOI] [Google Scholar]
- Inouye M.; Miyake T.; Furusyo M.; Nakazumi H. Molecular Recognition of β-Ribofuranosides by Synthetic Polypyridine-Macrocyclic Receptors. J. Am. Chem. Soc. 1995, 117, 12416–12425. 10.1021/ja00155a006. [DOI] [Google Scholar]
- Romero F. M.; Ziessel R. A Straightforward Synthesis of 5-Bromo and 5,5′-Dibromo-2,2′-Bipyridines. Tetrahedron Lett. 1995, 36, 6471–6474. 10.1016/0040-4039(95)01306-3. [DOI] [Google Scholar]
- Shi H.; Liu H.; Bloch R.; Mandville G. Enantioselective Synthesis of the Paf Antagonist Mk-287. Tetrahedron: Asymmetry 2002, 13, 1423–1428. 10.1016/S0957-4166(02)00334-8. [DOI] [Google Scholar]
- Toyota S.; Shimasaki T.; Tanifuji N.; Wakamatsu K. Experimental and Theoretical Investigations of Absolute Stereochemistry and Chiroptical Properties of Enantiopure 2,2′-Substituted 9,9′-Bianthryls. Tetrahedron: Asymmetry 2003, 14, 1623–1629. 10.1016/S0957-4166(03)00252-0. [DOI] [Google Scholar]
- Carbain B.; Collins P. J.; Callum L.; Martin S. R.; Hay A. J.; McCauley J.; Streicher H. Efficient Synthesis of Highly Active Phospha-Isosteres of the Influenza Neuraminidase Inhibitor Oseltamivir. ChemMedChem 2009, 4, 335–337. 10.1002/cmdc.200800379. [DOI] [PubMed] [Google Scholar]
- Catozzi N.; Edwards M. G.; Raw S. A.; Wasnaire P.; Taylor R. J. Synthesis of the Louisianin Alkaloid Family Via a 1,2,4-Triazine Inverse-Electron-Demand Diels-Alder Approach. J. Org. Chem. 2009, 74, 8343–8354. 10.1021/jo901761r. [DOI] [PubMed] [Google Scholar]
- Czeskis B. A.; Satonin D. K. Synthesis of C-14 Radiolabeled Glucagon Receptor Antagonist and Its Use in a Human Mass Balance Study. J. Labelled Compd. Radiopharm. 2017, 60, 110–115. 10.1002/jlcr.3477. [DOI] [PubMed] [Google Scholar]
- Cole C. J. F.; Fuentes L.; Snyder S. A. Asymmetric Pyrone Diels–Alder Reactions Enabled by Dienamine Catalysis. Chem. Sci. 2020, 11, 2175–2180. 10.1039/C9SC05738B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton D. H. R.; Blundell P.; Jaszberenyi J. C. Quantum Yields in the Photochemically Induced Radical Chemistry of Acyl Derivatives of Thiohydroxamic Acids. J. Am. Chem. Soc. 1991, 113, 6937–6942. 10.1021/ja00018a034. [DOI] [Google Scholar]
- Newcomb M.; Kaplan J. Rate Constants and Arrhenius Function for Scavenging of Octyl Radical by an N-Hydroxypyridine-2-Thione Ester. Tetrahedron Lett. 1987, 28, 1615–1618. 10.1016/S0040-4039(00)95373-5. [DOI] [Google Scholar]
- Mathew L.; Warkentin J. Rate Constants for Abstraction of Bromine from Bromotrichloromethane by Butyl, Cyclopropylmethyl, and Phenyl Radicals in Solution. Can. J. Chem. 1988, 66, 11–16. 10.1139/v88-002. [DOI] [Google Scholar]
- Newcomb M.; Sanchez R. M.; Kaplan J. Fast Halogen Abstractions from Alkyl Halides by Alkyl Radicals. Quantitation of the Processes Occurring in and a Caveat for Studies Employing Alkyl Halide Mechanistic Probes. J. Am. Chem. Soc. 1987, 109, 1195–1199. 10.1021/ja00238a031. [DOI] [Google Scholar]
- Dauben W. G.; Bridon D. P.; Kowalczyk B. A. Sonication-Induced Halogenative Decarboxylation of Thiohydroxamic Esters. J. Org. Chem. 1989, 54, 6101–6106. 10.1021/jo00287a023. [DOI] [Google Scholar]
- Barton D. H. R.; Lacher B.; Zard S. Z. The Invention of New Radical Chain Reactions: Part XIII. Generation and Reactivity of Perfluoroalkyl Radicals from Thiohydroxamic Esters. Tetrahedron 1986, 42, 2325–2328. 10.1016/S0040-4020(01)90613-1. [DOI] [Google Scholar]
- Hartung J.; Greb M. A New Synthesis of the 2,2,3,5,6,6-Substituted Tetrahydropyran Aplysiapyranoid a and its 5-Epimer. Tetrahedron Lett. 2003, 44, 6091–6093. 10.1016/S0040-4039(03)01468-0. [DOI] [Google Scholar]
- Philippe N.; Rivron L.; De Bruin B.; Schofield J.; Roy S. Two New Convenient Syntheses of 14c-Squalene from Turbinaric Acid. J. Labelled Compd. Radiopharm. 2018, 61, 878–884. 10.1002/jlcr.3672. [DOI] [PubMed] [Google Scholar]
- Barton D. H. R.; Crich D.; Motherwell W. B. New and Improved Methods for the Radical Decarboxylation of Acids. J. Chem. Soc., Chem. Commun. 1983, 939–941. 10.1039/c39830000939. [DOI] [Google Scholar]
- Malachowski W. P.; Coward J. K. The Chemistry of Phosphapeptides: Investigations on the Synthesis of Phosphonamidate, Phosphonate, and Phosphinate Analogs of Glutamyl-γ-Glutamate. J. Org. Chem. 1994, 59, 7625–7634. 10.1021/jo00104a017. [DOI] [Google Scholar]
- Pound S. M.; Underwood S. J.; Douglas C. J. Studies Towards the Total Synthesis of Drimentine C. Preparation of the AB and CDEF Ring Fragments. Eur. J. Org. Chem. 2020, 2020, 2448–2453. 10.1002/ejoc.202000158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohn E.; Pietruszka J. Enantiomerically Pure Cyclopropane Building Blocks: Synthesis and Transformations of 2-Iodocyclopropylboronic Esters. Adv. Synth. Catal. 2004, 346, 863–866. 10.1002/adsc.200404055. [DOI] [Google Scholar]
- Camps P.; Pujol X.; Rossi R. A.; Vázquez S. Synthesis of Several 8-Halopentacyclo[6.4.0.02,10.03,7.04,9]Dodecane Derivatives. Synthesis 1999, 1999, 854–858. 10.1055/s-1999-3483. [DOI] [Google Scholar]
- Barton D. H. R.; Crich D.; Kretzschmar G. The Invention of New Radical Chain Reactions. Part 9. Further Radical Chemistry of Thiohydroxamic Esters; Formation of Carbon–Carbon Bonds. J. Chem. Soc., Perkin Trans. 1 1986, 1, 39–53. 10.1039/P19860000039. [DOI] [Google Scholar]
- Hua D. H.; Takasu K.; Huang X.; Millward G. S.; Chen Y.; Fan J. Palladium-Mediated Ring Closure Reactions. Facile Syntheses of Enantiopure Bicyclic and Tricyclic Alkenones. Tetrahedron 2000, 56, 7389–7398. 10.1016/S0040-4020(00)00658-X. [DOI] [Google Scholar]
- Nyfeler E.; Renaud P. Decarboxylative Radical Azidation Using MPDOC and MMOC Esters. Org. Lett. 2008, 10, 985–988. 10.1021/ol702832x. [DOI] [PubMed] [Google Scholar]
- De Luca L.; Giacomelli G.; Porcu G.; Taddei M. A New Supported Reagent for the Photochemical Generation of Radicals in Solution. Org. Lett. 2001, 3, 855–857. 10.1021/ol007032b. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Gunzner J. L.; Dirat O.; Rhee Y. H. Callipeltoside A: Total Synthesis, Assignment of the Absolute and Relative Configuration, and Evaluation of Synthetic Analogues. J. Am. Chem. Soc. 2002, 124, 10396–10415. 10.1021/ja0205232. [DOI] [PubMed] [Google Scholar]
- Dauben W. G.; Kowalczyk B. A.; Bridon D. P. Light Induced Halogenative Decarboxylation of Thiohydroxamic Esters. Tetrahedron Lett. 1989, 30, 2461–2464. 10.1016/S0040-4039(01)80425-1. [DOI] [Google Scholar]
- Tsanaktsidis J.; Eaton P. E. Synthesis of Iodocubanes by Decarboxylative Iodination. Tetrahedron Lett. 1989, 30, 6967–6968. 10.1016/S0040-4039(01)93399-4. [DOI] [Google Scholar]
- Della E. W.; Taylor D. K. Synthesis of Some Bridgehead Bridgehead-Disubstituted Bicyclo[1.1.1]Pentanes. J. Org. Chem. 1994, 59, 2986–2996. 10.1021/jo00090a015. [DOI] [Google Scholar]
- Wang J.; Ma D. 6-Methylenebicyclo[3.2.1]oct-1-en-3-one: A Twisted Olefin as Diels-Alder Dienophile for Expedited Syntheses of Four Kaurane Diterpenoids. Angew. Chem., Int. Ed. 2019, 58, 15731–15735. 10.1002/anie.201909349. [DOI] [PubMed] [Google Scholar]
- Della E. W.; Head N. J. Synthesis of Bridgehead Fluorides by Fluorodeiodination. J. Org. Chem. 1992, 57, 2850–2855. 10.1021/jo00036a018. [DOI] [Google Scholar]
- Carbain B.; Hitchcock P. B.; Streicher H. New Aspects of the Hunsdiecker–Barton Halodecarboxylation—Syntheses of Phospha-Shikimic Acid and Derivatives. Tetrahedron Lett. 2010, 51, 2717–2719. 10.1016/j.tetlet.2010.03.044. [DOI] [Google Scholar]
- Safronenko E. D.; Afanas’ev I. B. Reaction of Free Alkyl Radicals in the Liquid Phase Studied by a Competitive Addition Method. I. Reactions with Bromo-Substituted Derivatives of Carboxylic Acids. Zh. Org. Khim. 1968, 4, 2086. [Google Scholar]
- Barton D. H. R. The Invention of Chemical Reactions: The Last Five Years. Tetrahedron 1992, 48, 2529–2544. 10.1016/S0040-4020(01)88519-7. [DOI] [Google Scholar]
- Hasebe M.; Kogawa K.; Tsuchiya T. Photochemical Arylation by Oxime Esters in Benzene and Pyridine: Simple Synthesis of Biaryl Compounds. Tetrahedron Lett. 1984, 25, 3887–3890. 10.1016/S0040-4039(01)91195-5. [DOI] [Google Scholar]
- Hasebe M.; Tsuchiya T. Photodecarboxylative Chlorination of Carboxylic Acids Via Their Benzophenone Oxime Esters. Tetrahedron Lett. 1988, 29, 6287–6290. 10.1016/S0040-4039(00)82327-8. [DOI] [Google Scholar]
- Okada T.; Kawanisi M.; Nozaki H. Photolysis of Aromatic Oxime Benzoates. Bull. Chem. Soc. Jpn. 1969, 42, 2981–2983. 10.1246/bcsj.42.2981. [DOI] [Google Scholar]
- Patra T.; Mukherjee S.; Ma J.; Strieth-Kalthoff F.; Glorius F. Visible-Light-Photosensitized Aryl and Alkyl Decarboxylative Functionalization Reactions. Angew. Chem., Int. Ed. 2019, 58, 10514–10520. 10.1002/anie.201904671. [DOI] [PubMed] [Google Scholar]
- Okada K.; Okamoto K.; Oda M. Photochemical Chlorodecarboxylation Via an Electron-Transfer Mechanism. J. Chem. Soc., Chem. Commun. 1989, 1636–1637. 10.1039/c39890001636. [DOI] [Google Scholar]
- Okada K.; Okamoto K.; Oda M. A New and Practical Method of Decarboxylation: Photosensitized Decarboxylation of N-Acyloxyphthalimides Via Electron-Transfer Mechanism. J. Am. Chem. Soc. 1988, 110, 8736–8738. 10.1021/ja00234a047. [DOI] [Google Scholar]
- Mumtaz S.; Robertson M. J.; Oelgemöller M. Recent Advances in Photodecarboxylations Involving Phthalimides. Aust. J. Chem. 2018, 71, 634–648. 10.1071/CH18220. [DOI] [Google Scholar]
- Murarka S. N-(Acyloxy)Phthalimides as Redox-Active Esters in Cross-Coupling Reactions. Adv. Synth. Catal. 2018, 360, 1735–1753. 10.1002/adsc.201701615. [DOI] [Google Scholar]
- Kulbitski K.; Nisnevich G.; Gandelman M. Metal-Free Efficient, General and Facile Iododecarboxylation Method with Biodegradable Co-Products. Adv. Synth. Catal. 2011, 353, 1438–1442. 10.1002/adsc.201100145. [DOI] [Google Scholar]
- Jiang X.; Sakthivel S.; Kulbitski K.; Nisnevich G.; Gandelman M. Efficient Synthesis of Secondary Alkyl Fluorides Via Suzuki Cross-Coupling Reaction of 1-Halo-1-Fluoroalkanes. J. Am. Chem. Soc. 2014, 136, 9548–9551. 10.1021/ja504089y. [DOI] [PubMed] [Google Scholar]
- Jiang X.; Gandelman M. Enantioselective Suzuki Cross-Couplings of Unactivated 1-Fluoro-1-Haloalkanes: Synthesis of Chiral Beta-, Gamma-, Delta-, and Epsilon-Fluoroalkanes. J. Am. Chem. Soc. 2015, 137, 2542–2547. 10.1021/jacs.5b00473. [DOI] [PubMed] [Google Scholar]
- Jiang X.; Kulbitski K.; Nisnevich G.; Gandelman M. Enantioselective Assembly of Tertiary Stereocenters Via Multicomponent Chemoselective Cross-Coupling of Geminal Chloro(Iodo)Alkanes. Chem. Sci. 2016, 7, 2762–2767. 10.1039/C5SC04378F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai Y.; Moriyama K.; Togo H. One-Pot Transformation of Aliphatic Carboxylic Acids Inton-Alkylsuccinimides with Nis and Ncs/Nai. Eur. J. Org. Chem. 2016, 2016, 768–772. 10.1002/ejoc.201501315. [DOI] [Google Scholar]
- Wang Z.; Zhu L.; Yin F.; Su Z.; Li Z.; Li C. Silver-Catalyzed Decarboxylative Chlorination of Aliphatic Carboxylic Acids. J. Am. Chem. Soc. 2012, 134, 4258–4263. 10.1021/ja210361z. [DOI] [PubMed] [Google Scholar]
- Tan X.; Song T.; Wang Z.; Chen H.; Cui L.; Li C. Silver-Catalyzed Decarboxylative Bromination of Aliphatic Carboxylic Acids. Org. Lett. 2017, 19, 1634–1637. 10.1021/acs.orglett.7b00439. [DOI] [PubMed] [Google Scholar]
- Zhang X. Mechanism for Ag (I)-Catalyzed Decarboxylative Chlorination: A DFT Study. Theor. Chem. Acc. 2016, 135, 144. 10.1007/s00214-016-1903-z. [DOI] [Google Scholar]
- Candish L.; Standley E. A.; Gomez-Suarez A.; Mukherjee S.; Glorius F. Catalytic Access to Alkyl Bromides, Chlorides and Iodides Via Visible Light-Promoted Decarboxylative Halogenation. Chem. - Eur. J. 2016, 22, 9971–9974. 10.1002/chem.201602251. [DOI] [PubMed] [Google Scholar]
- Shibatomi K.; Kitahara K.; Sasaki N.; Kawasaki Y.; Fujisawa I.; Iwasa S. Enantioselective Decarboxylative Chlorination of Beta-Ketocarboxylic Acids. Nat. Commun. 2017, 8, 15600. 10.1038/ncomms15600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naruse A.; Kitahara K.; Iwasa S.; Shibatomi K. Synthesis of A-Fluoroenones by Elimination of α-Chloro-α-Fluoroketones. Asian J. Org. Chem. 2019, 8, 691–693. 10.1002/ajoc.201900072. [DOI] [Google Scholar]
- Kitahara K.; Mizutani H.; Iwasa S.; Shibatomi K. Asymmetric Synthesis of A-Chloro-A-Halo Ketones by Decarboxylative Chlorination of α-Halo-β-Ketocarboxylic Acids. Synthesis 2019, 51, 4385–4392. 10.1055/s-0039-1690009. [DOI] [Google Scholar]
- Koo B.-S.; Kim E.-H.; Lee K.-J. Bromodecarbonylation and Bromodecarboxylation of Electron-Rich Benzaldehydes and Benzoic Acids with Oxone® and Sodium Bromide. Synth. Commun. 2002, 32, 2275–2286. 10.1081/SCC-120005997. [DOI] [Google Scholar]
- Li Z. J.; Wang K. K.; Liu Z. Q. Transition-Metal-Free Hunsdiecker Reaction of Electron-Rich Arenecarboxylic Acids and Aryl Aldehydes in Water. Synlett 2014, 25, 2508–2512. 10.1055/s-0034-1378583. [DOI] [Google Scholar]
- Perry G. J. P.; Quibell J. M.; Panigrahi A.; Larrosa I. Transition-Metal-Free Decarboxylative Iodination: New Routes for Decarboxylative Oxidative Cross-Couplings. J. Am. Chem. Soc. 2017, 139, 11527–11536. 10.1021/jacs.7b05155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quibell J. M.; Perry G. J. P.; Cannas D. M.; Larrosa I. Transition-Metal-Free Decarboxylative Bromination of Aromatic Carboxylic Acids. Chem. Sci. 2018, 9, 3860–3865. 10.1039/C8SC01016A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Zoubi R. M.; Al-Mughaid H.; McDonald R. A Simple and Efficient Two-Step Synthesis of 1,2,3-Triiodoarenes Via Consecutive C–H Iodination/Ipso-Iododecarboxylation Strategy: A Potential Application Towards Ortho-Diiodoarenes by Regioselective Metal–Iodine Exchange Reaction. Aust. J. Chem. 2015, 68, 912–918. 10.1071/CH14386. [DOI] [Google Scholar]
- Jiang M.; Yang H. J.; Jin Y. H.; Ou L. Y.; Fu H. Visible-Light-Induced Decarboxylative Iodination of Aromatic Carboxylic Acids. Synlett 2018, 29, 1572–1577. 10.1055/s-0037-1610188. [DOI] [Google Scholar]
- Candish L.; Freitag M.; Gensch T.; Glorius F. Mild, Visible Light-Mediated Decarboxylation of Aryl Carboxylic Acids to Access Aryl Radicals. Chem. Sci. 2017, 8, 3618–3622. 10.1039/C6SC05533H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y.; Zhang L.; Deng G. J.; Gong H. Simple, Efficient and Controllable Synthesis of Iodo/Di-Iodoarenes Via Ipsoiododecarboxylation/Consecutive Iodination Strategy. Sci. Rep. 2017, 7, 40430. 10.1038/srep40430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Zoubi R. M.; Al-Mughaid H.; Al-Zoubi M. A.; Jaradat K. T.; McDonald R. Facile, One-Pot, and Gram-Scale Synthesis of 3,4,5-Triiodoanisole through a C-H Iodination/Ipso-Iododecarboxylation Strategy: Potential Application Towards 3,4,5-Trisubstituted Anisoles. Eur. J. Org. Chem. 2015, 2015, 5501–5508. 10.1002/ejoc.201500887. [DOI] [Google Scholar]
- Luo Y.; Pan X. L.; Wu J. Silver-Catalyzed Decarboxylative Halogenation of Carboxylic Acids. Tetrahedron Lett. 2010, 51, 6646–6648. 10.1016/j.tetlet.2010.10.054. [DOI] [Google Scholar]
- Zhan K.; Li Y. Microwave-Assisted Silver-Catalyzed Protodecarboxylation and Decarboxylative Iodination of Aromatic Carboxylic Acids. Catalysts 2017, 7, 314. 10.3390/catal7110314. [DOI] [Google Scholar]
- Peng X. F.; Shao X. F.; Liu Z. Q. Pd(II)-Catalyzed Bromo- and Chlorodecarboxylation of Electron-Rich Arenecarboxylic Acids. Tetrahedron Lett. 2013, 54, 3079–3081. 10.1016/j.tetlet.2013.03.135. [DOI] [Google Scholar]
- Fu Z.; Li Z.; Song Y.; Yang R.; Liu Y.; Cai H. Decarboxylative Halogenation and Cyanation of Electron-Deficient Aryl Carboxylic Acids Via Cu Mediator as Well as Electron-Rich Ones through Pd Catalyst under Aerobic Conditions. J. Org. Chem. 2016, 81, 2794–2803. 10.1021/acs.joc.5b02873. [DOI] [PubMed] [Google Scholar]
- Fu Z.; Jiang L.; Zuo Q.; Li Z.; Liu Y.; Wei Z.; Cai H. Inexpensive NaX (X = I, Br, Cl) as a Halogen Donor in the Practical Ag/Cu-Mediated Decarboxylative Halogenation of Aryl Carboxylic Acids under Aerobic Conditions. Org. Biomol. Chem. 2018, 16, 5416–5421. 10.1039/C8OB01095A. [DOI] [PubMed] [Google Scholar]
- Cornella J.; Rosillo-Lopez M.; Larrosa I. A Novel Mode of Reactivity for Gold(I): The Decarboxylative Activation of (Hetero) Aromatic Carboxylic Acids. Adv. Synth. Catal. 2011, 353, 1359–1366. 10.1002/adsc.201100109. [DOI] [Google Scholar]
- Deacon G. B.; Farquharson G. J. Permercurated Arenes. Iii. Syntheses of Periodoarenes and Perchloroarenes by Iodo- and Chloro-Demercuration of Some Permercurated Arenes. Aust. J. Chem. 1977, 30, 1701–1703. 10.1071/CH9771701. [DOI] [Google Scholar]
- Kleinspehn G. G.; Corwin A. H. A Proof of Structure for 2-Bromo-3-Methyl-4-Carbethoxypyrrole1. J. Am. Chem. Soc. 1953, 75, 5295–5298. 10.1021/ja01117a045. [DOI] [Google Scholar]
- Putey A.; Popowycz F.; Joseph B. A Non-Classical Route to 2,3-Diiodoindoles from Indole-2-Carboxylic Acids. Synlett 2007, 2007, 419–422. 10.1055/s-2007-967953. [DOI] [Google Scholar]
- Grigg R.; Johnson A. W.; Wasley J. W. F. 56. 2,2′-Bipyrroles. J. Chem. Soc. 1963, 0, 359–366. 10.1039/JR9630000359. [DOI] [Google Scholar]
- Grigg R.; Johnson A. W. 628. Bipyrroles. Part III. J. Chem. Soc. 1964, 3315–3322. 10.1039/jr9640003315. [DOI] [Google Scholar]
- Hayashi T.; Nakashima Y.; Ito K.; Ikegami T.; Aritome I.; Suzuki A.; Hisaeda Y. Synthesis, Structure, and Chemical Property of the First Fluorine-Containing Porphycene. Org. Lett. 2003, 5, 2845–2848. 10.1021/ol0348452. [DOI] [PubMed] [Google Scholar]
- Nonell S.; Bou N.; Borrell J.; Teixidó J.; Villanueva A.; Juarranz A.; Cañete M. Synthesis of 2,7,12,17-Tetraphenylporphycene (TPPO). First Aryl-Substituted Porphycene for the Photodynamic Therapy of Tumors. Tetrahedron Lett. 1995, 36, 3405–3408. 10.1016/0040-4039(95)00493-V. [DOI] [Google Scholar]
- Skowronek P.; Lightner D. A. Synthesis of a Chiral 3,3′-Di- Tert -Butyl-2,2′-Bipyrrole. Monatsh. Chem. 2003, 134, 889–899. 10.1007/s00706-002-0562-z. [DOI] [Google Scholar]
- Tu B.; Ghosh B.; Lightner D. A. A New Class of Linear Tetrapyrroles: Acetylenic 10,10a-Didehydro-10a-Homobilirubins. J. Org. Chem. 2003, 68, 8950–8963. 10.1021/jo030252t. [DOI] [PubMed] [Google Scholar]
- Jiao L.; Hao E.; Vicente M. G.; Smith K. M. Improved Synthesis of Functionalized 2,2′-Bipyrroles. J. Org. Chem. 2007, 72, 8119–8122. 10.1021/jo701310k. [DOI] [PubMed] [Google Scholar]
- Miki Y.; Umemoto H.; Umemoto M.; Ohta C.; Dohshita M.; Tanaka H.; Hattori S.; Hamamoto H. Decarboxylative Bromination of Indole-2,3-Dicarboxylic Acids Using Oxone® or CAN in the Presence of Lithium Bromide. Heterocycles 2009, 78, 2845–2850. 10.3987/COM-09-11802. [DOI] [Google Scholar]
- Hamamoto H.; Umemoto H.; Umemoto M.; Ohta C.; Dohshita M.; Miki Y. Hypervalent Iodine(III) Mediated Decarboxylative Halogenation of Indolecarboxylic Acids for the Synthesis of Haloindole Derivatives. Synlett 2010, 2010, 2593–2596. 10.1055/s-0030-1258585. [DOI] [Google Scholar]
- Miki Y.; Umemoto H.; Dohshita M.; Hamamoto H. Synthesis of Pyrrolophenanthridone Alkaloid Kalbretorine from Indolecarboxylic Acids Via Hypervalent Iodine(III) Mediated Halodecarboxylation and Reduction. Tetrahedron Lett. 2012, 53, 1924–1927. 10.1016/j.tetlet.2012.01.132. [DOI] [Google Scholar]
- Miki Y.; Hamamoto H.; Umemoto H.; Umemoto M.; Ohta C.; Fujita E.; Nakamura A.; Maegawa T. Decarboxylative Halogenation of Indolecarboxylic Acids Using Hypervalent Iodine(Iii) Reagent and Its Application to the Synthesis of Polybromoindoles. Heterocycles 2015, 91, 561–572. 10.3987/COM-14-13162. [DOI] [Google Scholar]
- Henderson S. H.; West R. A.; Ward S. E.; Honey M. A. Metal-Free Selective Mono-Halodecarboxylation of Heteroarenes under Mild Conditions. R. Soc. Open Sci. 2018, 5, 180333. 10.1098/rsos.180333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X. T.; Feng X. J.; Zhang H. X.; Yamamoto Y.; Bao M. Transition-Metal-Free Decarboxylative Halogenation of 2-Picolinic Acids with Dihalomethane under Oxygen Conditions. Green Chem. 2019, 21, 5565–5570. 10.1039/C9GC02407G. [DOI] [Google Scholar]
- Janz K.; Kaila N. Bromodecarboxylation of Quinoline Salicylic Acids: Increasing the Diversity of Accessible Substituted Quinolines. J. Org. Chem. 2009, 74, 8874–8877. 10.1021/jo9018232. [DOI] [PubMed] [Google Scholar]
- Barak D. S.; Dahatonde D. J.; Batra S. Microwave-Assisted Metal-Free Decarboxylative Iodination/Bromination of Isoxazole-4-Carboxylic Acids. Asian J. Org. Chem. 2019, 8, 2149–2154. 10.1002/ajoc.201900572. [DOI] [Google Scholar]
- Kwon T. H.; Armel V.; Nattestad A.; Macfarlane D. R.; Bach U.; Lind S. J.; Gordon K. C.; Tang W.; Jones D. J.; Holmes A. B. Dithienothiophene (DTT)-Based Dyes for Dye-Sensitized Solar Cells: Synthesis of 2,6-Dibromo-Dtt. J. Org. Chem. 2011, 76, 4088–4093. 10.1021/jo2001484. [DOI] [PubMed] [Google Scholar]
- Berman J. D.; Price C. C. The Stereochemistry and Mechanism of the Bromodecarboxylation of Unsaturated Carboxylate Ions1. J. Am. Chem. Soc. 1957, 79, 5474–5476. 10.1021/ja01577a039. [DOI] [Google Scholar]
- Cabaliero M. C.; Johnson M. D. Direct Chlorodecarboxylation of the Trans-Cinnamate Ion. Chem. Commun. 1965, 454–455. 10.1039/c19650000454. [DOI] [Google Scholar]
- Kingsbury C. A.; Max G. Substituent Effects on Bromodecarboxylation Reactions. J. Org. Chem. 1978, 43, 3131–3139. 10.1021/jo00410a008. [DOI] [Google Scholar]
- Huang Y.-L.; Cheng Y.-H.; Hsien K.-C.; Chen Y.-L.; Kao C.-L. Concise Bromodecarboxylation of Cinnamic Acids to B-Bromostyrenes. Tetrahedron Lett. 2009, 50, 1834–1837. 10.1016/j.tetlet.2009.02.003. [DOI] [Google Scholar]
- Kuang C.; Senboku H.; Tokuda M. Stereoselective Synthesis of (E)-β-Arylvinyl Bromides by Microwave-Induced Reaction of Anti-3-Aryl-2,3-Dibromopropanoic Acids Using an AgOAc–AcOH System. Tetrahedron 2005, 61, 637–642. 10.1016/j.tet.2004.10.102. [DOI] [Google Scholar]
- Trumbull E. R.; Finn R. T.; Ibne-Rasa K. M.; Sauers C. K. Kinetics of the Decarboxylative Elimination of Cinnamic Acid Dibromides. J. Org. Chem. 1962, 27, 2339–2344. 10.1021/jo01054a012. [DOI] [Google Scholar]
- Kim S. H.; Wei H.-X.; Willis S.; Li G. A Mild Procedure for the Stereospecific Transformation Oftranscinnamic Acid Derivatives To cis-β-Bromostyrenes. Synth. Commun. 1999, 29, 4179–4185. 10.1080/00397919908085891. [DOI] [Google Scholar]
- Kuang C. X.; Senboku H.; Tokuda M. Convenient and Stereoselective Synthesis of (Z)-1-Bromo-1-Alkenes by Microwave-Induced Reaction. Tetrahedron Lett. 2001, 42, 3893–3896. 10.1016/S0040-4039(01)00595-0. [DOI] [Google Scholar]
- Tang K. G.; Kent G. T.; Erden I.; Wu W. Cis-Beta-Bromostyrene Derivatives from Cinnamic Acids Via a Tandem Substitutive Bromination-Decarboxylation Sequence. Tetrahedron Lett. 2017, 58, 3894–3896. 10.1016/j.tetlet.2017.08.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y. L.; Liu Q.; Sun R.; Zhang Q.; Xu X. X. Iododecarboxylation Reactions of A-Oxo Ketene Dithioacetals: The New Route to A-Iodo Ketene Dithioacetals. Synth. Commun. 2004, 34, 463–469. 10.1081/SCC-120027285. [DOI] [Google Scholar]
- Wang M.; Xu X.-X.; Liu Q.; Xiong L.; Yang B.; Gao L.-X. Iododecarboxylation of A-Carboxylate, A-Cinnamoyl Ketene Cyclic Dithioacetals. Synth. Commun. 2002, 32, 3437–3443. 10.1081/SCC-120014773. [DOI] [Google Scholar]
- Muller T.; Vaccher C.; Vaccher M.-P.; Flouquet N. A New Process for Preparing Dibromides by Decarboxylation of Unsaturated Acids with NBS. Synth. Commun. 1998, 28, 2343–2354. 10.1080/00397919808004287. [DOI] [Google Scholar]
- Chowdhury S.; Roy S. The First Example of a Catalytic Hunsdiecker Reaction: Synthesis of Beta-Halostyrenes. J. Org. Chem. 1997, 62, 199–200. 10.1021/jo951991f. [DOI] [PubMed] [Google Scholar]
- Chowdhury S.; Roy S. Manganese(Ii) Catalysed Hunsdiecker Reaction: A Facile Entry to Alpha-(Dibromomethyl) Benzenemethanol. Tetrahedron Lett. 1996, 37, 2623–2624. 10.1016/0040-4039(96)00343-7. [DOI] [Google Scholar]
- Naskar D.; Roy S. Synthesis of A-Bromo-B-Lactam Via a Novel Catalytic Hunsdiecker Like Protocol. J. Chem. Soc., Perkin Trans. 1 1999, 1, 2435–2436. 10.1039/a904515e. [DOI] [Google Scholar]
- Kuang C. X.; Yang Q.; Senboku H.; Tokuda M. Stereoselective Synthesis of (E)-Beta-Arylvinyl Bromides by Microwave-Induced Hunsdiecker-Type Reaction. Synthesis 2005, 1319–1325. 10.1055/s-2005-865283. [DOI] [Google Scholar]
- Homsi F.; Rousseau G. 4-Endo-Trig Cyclization Processes Using Bis(Collidine)Bromine(I) Hexafluorophosphate as Reagent: Preparation of 2-Oxetanones, 2-Azetidinones, and Oxetanes. J. Org. Chem. 1999, 64, 81–85. 10.1021/jo9810361. [DOI] [PubMed] [Google Scholar]
- Kuang C. X.; Senboku H.; Tokuda M. Stereoselective Synthesis of (E)-Beta-Arylvinyl Halides by Microwave-Induced Hunsdiecker Reaction. Synlett 2000, 2000, 1439–1442. [Google Scholar]
- Jiang Y. B.; Kuang C. X. A New Convenient Access to Highly Functionalized (E)-2-Arylvinyl Bromides. J. Chem. Sci. 2009, 121, 1035–1040. 10.1007/s12039-009-0116-6. [DOI] [Google Scholar]
- Naskar D.; Chowdhury S.; Roy S. Is Metal Necessary in the Hunsdiecker-Borodin Reaction?. Tetrahedron Lett. 1998, 39, 699–702. 10.1016/S0040-4039(97)10639-6. [DOI] [Google Scholar]
- Naskar D.; Roy S. Catalytic Hunsdiecker Reaction and One-Pot Catalytic Hunsdiecker–Heck Strategy: Synthesis of α,β-Unsaturated Aromatic Halides, A-(Dihalomethyl)Benzenemethanols, 5-Aryl-2,4-Pentadienoic Acids, Dienoates and Dienamides. Tetrahedron 2000, 56, 1369–1377. 10.1016/S0040-4020(99)01035-2. [DOI] [Google Scholar]
- Naskar D.; Das S. K.; Giribabu L.; Maiya B. G.; Roy S. Novel Catalytic Hunsdiecker-Heck (CHH) Strategy toward All-E Stereocontrolled Ferrocene-Capped Conjugated Push-Pull Polyenes. Organometallics 2000, 19, 1464–1469. 10.1021/om000020+. [DOI] [Google Scholar]
- Das J. P.; Roy S. Catalytic Hunsdiecker Reaction of Alpha, Beta-Unsaturated Carboxylic Acids: How Efficient Is the Catalyst?. J. Org. Chem. 2002, 67, 7861–7864. 10.1021/jo025868h. [DOI] [PubMed] [Google Scholar]
- Jones A. S.; Verhelst G.; Walker R. T. The Synthesis of the Potent Anti-Herpes Virus Agent, E-5(2-Bromovinyl)-2′-Deoxyuridine and Related Compounds. Tetrahedron Lett. 1979, 20, 4415–4418. 10.1016/S0040-4039(01)86605-3. [DOI] [Google Scholar]
- Herdewijn P.; De Clercq E.; Balzarini J.; Vanderhaeghe H. Synthesis and Antiviral Activity of the Carbocyclic Analogues of (E)-5-(2-Halovinyl)-2′-Deoxyuridines and (E)-5-(2-Halovinyl)-2′-Deoxycytidines. J. Med. Chem. 1985, 28, 550–555. 10.1021/jm50001a003. [DOI] [PubMed] [Google Scholar]
- Izawa T.; Nishiyama S.; Yamamura S.; Kato K.; Takita T. Synthesis of Carbocyclic Nucleosides: Synthesis of (±)-2,2-Bis(Hydroxymethyl)Cyclopropyl Nucleosides. J. Chem. Soc., Perkin Trans. 1 1992, 1, 2519–2525. 10.1039/P19920002519. [DOI] [Google Scholar]
- Bazin M.-A.; Kihel L. E.; Lancelot J.-C.; Rault S. Original One-Pot Microwave-Promoted Hunsdiecker–Suzuki Strategy: Straightforward Access to Trans-1,2-Diarylethenes from Cinnamic Acids. Tetrahedron Lett. 2007, 48, 4347–4351. 10.1016/j.tetlet.2007.04.114. [DOI] [Google Scholar]
- Rajanna K. C.; Reddy N. M.; Reddy M. R.; Saiprakash P. K. Micellar Mediated Halodecarboxylation of α,β-Unsaturated Aliphatic and Aromatic Carboxylic Acids—a Novel Green Hunsdiecker–Borodin Reaction. J. Dispersion Sci. Technol. 2007, 28, 613–616. 10.1080/01932690701282690. [DOI] [Google Scholar]
- Novaes L. F. T.; Pastre J. C. Formal Total Synthesis of Actinoranone and Asymmetric Synthesis of Labda-7,13-(E)-Dien-15-Ol. Org. Lett. 2017, 19, 3163–3166. 10.1021/acs.orglett.7b01287. [DOI] [PubMed] [Google Scholar]
- Katritzky A. R.; Majumder S.; Jain R. Microwave-Induced Halodecarboxylation of α,β-Unsaturated Carboxylic Acids. J. Indian Chem. Soc. 2003, 80, 1073–1075. [Google Scholar]
- Hazarika D.; Phukan P. TsNBr2 Promoted Decarboxylative Bromination of A, B-Unsaturated Carboxylic Acids. Tetrahedron Lett. 2018, 59, 4593–4596. 10.1016/j.tetlet.2018.11.041. [DOI] [Google Scholar]
- Fursule R. A.; Patil P. O.; Shewale B. D.; Kosalge S. B.; Deshmukh P. K.; Patil D. A. Novel System for Decarboxylative Bromination of Alpha, Beta-Unsaturated Carboxylic Acids with Diacetoxyiodobenzene. Chem. Pharm. Bull. 2009, 57, 1243–1245. 10.1248/cpb.57.1243. [DOI] [PubMed] [Google Scholar]
- Han J. L.; Wei Y. P. Decarboxylative Bromination of Cinnamic Acids by 2-Iodoxybenzoic Acid with Tetrabutylammonium Bromide. J. Chem. Res. 2012, 36, 247–248. 10.3184/174751912X13324352705972. [DOI] [Google Scholar]
- Telvekar V. N.; Arote N. D.; Herlekar O. P. Mild and Efficient Method for Decarboxylative Bromination of A, B-Unsaturated Carboxylic Acids with Dess-Martin Periodinane. Synlett 2005, 2495–2497. 10.1055/s-2005-917077. [DOI] [Google Scholar]
- Telvekar V. N.; Chettiar S. N. A Novel System for Decarboxylative Bromination. Tetrahedron Lett. 2007, 48, 4529–4532. 10.1016/j.tetlet.2007.04.137. [DOI] [Google Scholar]
- Homsi F.; Rousseau G. Halodecarboxylation of α,β-Acetylenic and α,β-Ethylenic Acids. Tetrahedron Lett. 1999, 40, 1495–1498. 10.1016/S0040-4039(99)00021-0. [DOI] [Google Scholar]
- Wang C.; Bao W.; Zhang X. Synthesis of Vinyl Bromides from the Reaction of A, B-Unsaturated Carboxylic Acids with Ionic Liquid [BMIM][Br3]/[BMIM]Br. J. Chem. Res. 2005, 2005, 617–619. 10.3184/030823405774663110. [DOI] [Google Scholar]
- Hosseinzadeh R.; Tajbakhsh M.; Mavvaji M.; Lasemi Z. Synthesis of β-Aryl Vinyl Bromides from α,β-Unsaturated Carboxylic Acids by Use of Ethylenebis(N-Methylimidazolium) Ditribromide. Res. Chem. Intermed. 2015, 41, 2427–2436. 10.1007/s11164-013-1357-z. [DOI] [Google Scholar]
- You H.-W.; Lee K.-J. Halodecarboxylation of A, B-Unsaturated Carboxylic Acids Bearing Aryl and Styrenyl Group at B-Carbon with Oxone® and Sodium Halide. Synlett 2001, 2001, 105–107. 10.1055/s-2001-9734. [DOI] [Google Scholar]
- Krawczyk H.; Albrecht Ł. Bromodecarboxylation of (E)-3-Aryl-2-(Diethoxyphosphoryl)Acrylic Acids: A Facile Route to Diethyl Arylethynylphosphonates. Synthesis 2007, 2007, 1877–1881. 10.1055/s-2007-966057. [DOI] [Google Scholar]
- Kumar M. S.; Rajanna K. C.; Venkanna P.; Venkateswarlu M.; Sudhakar Chary V. Ultrasonically Assisted Decarboxylative Bromination of α,β-Unsaturated Carboxylic Acids under Vilsmeier-Haack Conditions. Synth. React. Inorg., Met.-Org., Nano-Met. Chem. 2016, 46, 642–646. 10.1080/15533174.2014.989573. [DOI] [Google Scholar]
- Zych A. J.; Wang H.-J.; Sakwa S. A. Synthesis and Suzuki–Miyaura Reactions of 5-Halo-3,4-Dihydropyrimidin-2(1H)-ones. Tetrahedron Lett. 2010, 51, 5103–5105. 10.1016/j.tetlet.2010.07.093. [DOI] [Google Scholar]
- Telvekar V. N.; Takale B. S. A Novel Method for Bromodecarboxylation of A, B-Unsaturated Carboxylic Acids Using Catalytic Sodium Nitrite. Tetrahedron Lett. 2011, 52, 2394–2396. 10.1016/j.tetlet.2011.02.101. [DOI] [Google Scholar]
- Hatvate N. T.; Takale B. S.; Ghodse S. M.; Telvekar V. N. Transition Metal Free Large-Scale Synthesis of Aromatic Vinyl Chlorides from Aromatic Vinyl Carboxylic Acids Using Bleach. Tetrahedron Lett. 2018, 59, 3892–3894. 10.1016/j.tetlet.2018.09.034. [DOI] [Google Scholar]
- Ye C.; Shreeve J. M. Structure-Dependent Oxidative Bromination of Unsaturated C-C Bonds Mediated by Selectfluor. J. Org. Chem. 2004, 69, 8561–8563. 10.1021/jo048383x. [DOI] [PubMed] [Google Scholar]
- Li C.-T.; Yuan X.; Tang Z.-Y. Transition Metal Free Decarboxylative Fluorination of Cinnamic Acids with Selectfluor®. Tetrahedron Lett. 2016, 57, 5624–5627. 10.1016/j.tetlet.2016.11.003. [DOI] [Google Scholar]
- Sinha J.; Layek S.; Mandal G. C.; Bhattacharjee M. A Green Hunsdiecker Reaction: Synthesis of Beta-Bromostyrenes from the Reaction of Alpha, Beta-Unsaturated Aromatic Carboxylic Acids with KBr and H2O2 Catalysed by Na2MoO4·2H2O in Aqueous Medium. Chem. Commun. 2001, 1916–1917. 10.1039/b104540g. [DOI] [PubMed] [Google Scholar]
- Roy S.; Bhar S. Sodium Tungstate-Catalyzed “on-Water” Synthesis of B-Arylvinyl Bromides. Green Chem. Lett. Rev. 2010, 3, 341–347. 10.1080/17518253.2010.491095. [DOI] [Google Scholar]
- Choudary B. Molybdate-Exchanged Mg–Al–LDH Catalyst: An Eco-Compatible Route for the Synthesis of B-Bromostyrenes in Aqueous Medium. Catal. Commun. 2004, 5, 215–219. 10.1016/j.catcom.2004.02.003. [DOI] [Google Scholar]
- Roy S. C.; Guin C.; Maiti G. A Mild and Efficient Method for Oxidative Halodecarboxylation of α,β-Unsaturated Aromatic Acids Using Lithium Bromide/Chloride and Ceric Ammonium Nitrate. Tetrahedron Lett. 2001, 42, 9253–9255. 10.1016/S0040-4039(01)01936-0. [DOI] [Google Scholar]
- Nikishin G. I.; Sokova L. L.; Makhaev V. D.; Kapustina N. I. Solid-Phase Oxidative Halodecarboxylation of β-Arylacrylic Acids with the Ceric Ammonium Nitrate—Alkali Halide System. Russ. Chem. Bull. 2008, 57, 118–123. 10.1007/s11172-008-0018-7. [DOI] [Google Scholar]
- Sodre L. R.; Esteves P. M.; de Mattos M. C. S. A Green Hunsdiecker Reaction of Cinnamic Acids. J. Braz. Chem. Soc. 2013, 24, 212–218. 10.5935/0103-5053.20130027. [DOI] [Google Scholar]
- Bi M. X.; Qian P.; Wang Y. K.; Zha Z. G.; Wang Z. Y. Decarboxylative Bromination of Alpha, Beta-Unsaturated Carboxylic Acids Via an Anodic Oxidation. Chin. Chem. Lett. 2017, 28, 1159–1162. 10.1016/j.cclet.2017.04.030. [DOI] [Google Scholar]
- Kee-Jung Lee K. W. L.; Dae Yoon Chi. Bromodecarboxylation of Arylpropiolic Acids with Oxone® and Sodium Bromide. Bull. Korean Chem. Soc. 2001, 22, 549–550. [Google Scholar]
- Cohen M. J.; McNelis E. Oxidative Decarboxylation of Propiolic Acids. J. Org. Chem. 1984, 49, 515–518. 10.1021/jo00177a025. [DOI] [Google Scholar]
- Naskar D.; Roy S. 1-Haloalkynes from Propiolic Acids: A Novel Catalytic Halodecarboxylation Protocol. J. Org. Chem. 1999, 64, 6896–6897. 10.1021/jo990434g. [DOI] [PubMed] [Google Scholar]
- Jayaraman A.; Cho E.; Irudayanathan F. M.; Kim J.; Lee S. Metal-Free Decarboxylative Trichlorination of Alkynyl Carboxylic Acids: Synthesis of Trichloromethyl Ketones. Adv. Synth. Catal. 2018, 360, 130–141. 10.1002/adsc.201701116. [DOI] [Google Scholar]
- Jayaraman A.; Cho E.; Kim J.; Lee S. Decarboxylative Tribromination for the Selective Synthesis of Tribromomethyl Ketone and Tribromovinyl Derivatives. Adv. Synth. Catal. 2018, 360, 3978–3989. 10.1002/adsc.201800851. [DOI] [Google Scholar]
- Niknam K.; Zolfigol M. A.; Madrakian E.; Ghaemi E. Tribromoisocyanuric Acid/NaNO2: A New Reagent for Mononitration of Phenols under Mild and Heterogeneous Conditions. S. Afr. J. Chem. 2007, 60, 109–112. [Google Scholar]
- de Nooy A. E. J.; Besemer A. C.; van Bekkum H. Selective Oxidation of Primary Alcohols Mediated by Nitroxyl Radical in Aqueous Solution. Kinetics and Mechanism. Tetrahedron 1995, 51, 8023–8032. 10.1016/0040-4020(95)00417-7. [DOI] [Google Scholar]
- Jayaraman A.; Lee S. Silver-Mediated Decarboxylative Fluorodiiodination of Alkynoic Acids: Synthesis of Regio- and Stereoselective Fluoroalkenes. Org. Lett. 2019, 21, 3485–3489. 10.1021/acs.orglett.9b00597. [DOI] [PubMed] [Google Scholar]
- Reddy V. P.Organofluorine Compounds in Biology and Medicine; Elsevier Science, 2015. [Google Scholar]
- Cady G. H.; Kellogg K. B. Trifluoroacetyl Hypofluorite1. J. Am. Chem. Soc. 1953, 75, 2501–2502. 10.1021/ja01106a504. [DOI] [Google Scholar]
- Menefee A.; Cady G. H. Pentafluoropropionyl Hypofluorite. J. Am. Chem. Soc. 1954, 76, 2020–2021. 10.1021/ja01636a095. [DOI] [Google Scholar]
- Grakauskas V. Aqueous Fluorination of Carboxylic Acid Salts. J. Org. Chem. 1969, 34, 2446–2450. 10.1021/jo01260a040. [DOI] [Google Scholar]
- Habibi M. H.; Mallouk T. E. Photochemical Selective Fluorination of Organic-Molecules Using Mercury (II) Fluoride. J. Fluorine Chem. 1991, 51, 291–294. 10.1016/S0022-1139(00)80299-7. [DOI] [Google Scholar]
- Halpern D. F.; Robin M. L.. Method for Fluorodecarboxylation. USUS4996371, 1990.
- Patrick T. B.; Johri K. K.; White D. H. Fluoro-Decarboxylation with Xenon Difluoride. J. Org. Chem. 1983, 48, 4158–4159. 10.1021/jo00170a072. [DOI] [Google Scholar]
- Patrick T. B.; Johri K. K.; White D. H.; Bertrand W. S.; Mokhtar R.; Kilbourn M. R.; Welch M. J. Replacement of the Carboxylic-Acid Function with Fluorine. Can. J. Chem. 1986, 64, 138–141. 10.1139/v86-024. [DOI] [Google Scholar]
- Patrick T. B.; Khazaeli S.; Nadji S.; Heringsmith K.; Reif D. Mechanistic Studies of Fluorodecarboxylation with Xenon Difluoride. J. Org. Chem. 1993, 58, 705–708. 10.1021/jo00055a026. [DOI] [Google Scholar]
- Adcock W.; Blokhin A. V.; Elsey G. M.; Head N. H.; Krstic A. R.; Levin M. D.; Michl J.; Munton J.; Pinkhassik E.; Robert M.; et al. Manifestations of Bridgehead-Bridgehead Interactions in the Bicyclo[1.1.1]Pentane Ring System. J. Org. Chem. 1999, 64, 2618–2625. 10.1021/jo982124o. [DOI] [PubMed] [Google Scholar]
- Bandak D.; Babii O.; Vasiuta R.; Komarov I. V.; Mykhailiuk P. K. Design and Synthesis of Novel 19f-Amino Acid: A Promising 19F NMR Label for Peptide Studies. Org. Lett. 2015, 17, 226–229. 10.1021/ol503300m. [DOI] [PubMed] [Google Scholar]
- Chatalova-Sazepin C.; Binayeva M.; Epifanov M.; Zhang W.; Foth P.; Amador C.; Jagdeo M.; Boswell B. R.; Sammis G. M. Xenon Difluoride Mediated Fluorodecarboxylations for the Syntheses of Di- and Trifluoromethoxyarenes. Org. Lett. 2016, 18, 4570–4573. 10.1021/acs.orglett.6b02208. [DOI] [PubMed] [Google Scholar]
- Zhang Q. W.; Brusoe A. T.; Mascitti V.; Hesp K. D.; Blakemore D. C.; Kohrt J. T.; Hartwig J. F. Fluorodecarboxylation for the Synthesis of Trifluoromethyl Aryl Ethers. Angew. Chem., Int. Ed. 2016, 55, 9758–9762. 10.1002/anie.201604793. [DOI] [PubMed] [Google Scholar]
- Eisenberg M.; DesMarteau D. D. The Reaction of Xenon Difluoride with Some Strong Oxy-Acids. Inorg. Nucl. Chem. Lett. 1970, 6, 29–34. 10.1016/0020-1650(70)80279-3. [DOI] [Google Scholar]
- Ramsden C. A.; Shaw M. M. Fluorodecarboxylation, Rearrangement and Cyclisation: The Influence of Structure and Environment on the Reactions of Carboxylic Acids with Xenon Difluoride. Tetrahedron Lett. 2009, 50, 3321–3324. 10.1016/j.tetlet.2009.02.090. [DOI] [Google Scholar]
- Rueda-Becerril M.; Sazepin C. C.; Leung J. C.; Okbinoglu T.; Kennepohl P.; Paquin J. F.; Sammis G. M. Fluorine Transfer to Alkyl Radicals. J. Am. Chem. Soc. 2012, 134, 4026–4029. 10.1021/ja211679v. [DOI] [PubMed] [Google Scholar]
- Meyer D.; Jangra H.; Walther F.; Zipse H.; Renaud P. A Third Generation of Radical Fluorinating Agents Based on N-Fluoro-N-Arylsulfonamides. Nat. Commun. 2018, 9, 4888. 10.1038/s41467-018-07196-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung J. C.; Chatalova-Sazepin C.; West J. G.; Rueda-Becerril M.; Paquin J. F.; Sammis G. M. Photo-Fluorodecarboxylation of 2-Aryloxy and 2-Aryl Carboxylic Acids. Angew. Chem., Int. Ed. 2012, 51, 10804–10807. 10.1002/anie.201206352. [DOI] [PubMed] [Google Scholar]
- Rueda-Becerril M.; Mahe O.; Drouin M.; Majewski M. B.; West J. G.; Wolf M. O.; Sammis G. M.; Paquin J. F. Direct C-F Bond Formation Using Photoredox Catalysis. J. Am. Chem. Soc. 2014, 136, 2637–2641. 10.1021/ja412083f. [DOI] [PubMed] [Google Scholar]
- Leung J. C. T.; Sammis G. M. Radical Decarboxylative Fluorination of Aryloxyacetic Acids Usingn-Fluorobenzenesulfonimide and a Photosensitizer. Eur. J. Org. Chem. 2015, 2015, 2197–2204. 10.1002/ejoc.201500038. [DOI] [Google Scholar]
- Ventre S.; Petronijevic F. R.; MacMillan D. W. Decarboxylative Fluorination of Aliphatic Carboxylic Acids Via Photoredox Catalysis. J. Am. Chem. Soc. 2015, 137, 5654–5657. 10.1021/jacs.5b02244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X.; Meng C.; Yuan X.; Jia X.; Qian X.; Ye J. Transition-Metal-Free Visible-Light Photoredox Catalysis at Room-Temperature for Decarboxylative Fluorination of Aliphatic Carboxylic Acids by Organic Dyes. Chem. Commun. 2015, 51, 11864–11867. 10.1039/C5CC04527D. [DOI] [PubMed] [Google Scholar]
- Pieber B.; Shalom M.; Antonietti M.; Seeberger P. H.; Gilmore K. Continuous Heterogeneous Photocatalysis in Serial Micro-Batch Reactors. Angew. Chem., Int. Ed. 2018, 57, 9976–9979. 10.1002/anie.201712568. [DOI] [PubMed] [Google Scholar]
- Tarantino G.; Hammond C. Catalytic Formation of C(sp3)–F Bonds Via Heterogeneous Photocatalysis. ACS Catal. 2018, 8, 10321–10330. 10.1021/acscatal.8b02844. [DOI] [Google Scholar]
- Yin F.; Wang Z.; Li Z.; Li C. Silver-Catalyzed Decarboxylative Fluorination of Aliphatic Carboxylic Acids in Aqueous Solution. J. Am. Chem. Soc. 2012, 134, 10401–10404. 10.1021/ja3048255. [DOI] [PubMed] [Google Scholar]
- Patel N. R.; Flowers R. A. 2nd. Mechanistic Study of Silver-Catalyzed Decarboxylative Fluorination. J. Org. Chem. 2015, 80, 5834–5841. 10.1021/acs.joc.5b00826. [DOI] [PubMed] [Google Scholar]
- Dos Passos Gomes G.; Wimmer A.; Smith J. M.; Konig B.; Alabugin I. V. CO2 or SO2: Should It Stay, or Should It Go?. J. Org. Chem. 2019, 84, 6232–6243. 10.1021/acs.joc.9b00503. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Guo C. Y.; Yang C.; Chen J. P. Ag-Catalyzed Chemoselective Decarboxylative Mono- and Gem-Difluorination of Malonic Acid Derivatives. J. Am. Chem. Soc. 2019, 141, 5617–5622. 10.1021/jacs.9b00681. [DOI] [PubMed] [Google Scholar]
- Mizuta S.; Stenhagen I. S.; O’Duill M.; Wolstenhulme J.; Kirjavainen A. K.; Forsback S. J.; Tredwell M.; Sandford G.; Moore P. R.; Huiban M.; et al. Catalytic Decarboxylative Fluorination for the Synthesis of Tri- and Difluoromethyl Arenes. Org. Lett. 2013, 15, 2648–2651. 10.1021/ol4009377. [DOI] [PubMed] [Google Scholar]
- Zhang X. Insights into the Mechanism of Silver-Catalyzed Decarboxylative Fluorination. Comput. Theor. Chem. 2016, 1082, 11–20. 10.1016/j.comptc.2016.02.016. [DOI] [Google Scholar]
- Krishanmoorthy S.; Schnell S. D.; Dang H.; Fu F.; Prakash G. K. S. Fluorodecarboxylation: Synthesis of Aryl Trifluoromethyl Ethers (ArOCF3) and Thioethers (ArSCF3). J. Fluorine Chem. 2017, 203, 130–135. 10.1016/j.jfluchem.2017.07.017. [DOI] [Google Scholar]
- Goh Y. L.; Adsool V. A. Radical Fluorination Powered Expedient Synthesis of 3-Fluorobicyclo[1.1.1]Pentan-1-Amine. Org. Biomol. Chem. 2015, 13, 11597–11601. 10.1039/C5OB02066B. [DOI] [PubMed] [Google Scholar]
- Kokhan S. O.; Tymtsunik A. V.; Grage S. L.; Afonin S.; Babii O.; Berditsch M.; Strizhak A. V.; Bandak D.; Platonov M. O.; Komarov I. V.; et al. Design, Synthesis, and Application of an Optimized Monofluorinated Aliphatic Label for Peptide Studies by Solid-State 19F Nmr Spectroscopy. Angew. Chem., Int. Ed. 2016, 55, 14788–14792. 10.1002/anie.201608116. [DOI] [PubMed] [Google Scholar]
- Phae-Nok S.; Soorukram D.; Kuhakarn C.; Reutrakul V.; Pohmakotr M. Silver-Mediated Decarboxylative Fluorination of Paraconic Acids: A Direct Entry to β-Fluorinated γ-Butyrolactones. Eur. J. Org. Chem. 2015, 2015, 2879–2888. 10.1002/ejoc.201500023. [DOI] [Google Scholar]
- Phae-Nok S.; Pohmakotr M.; Kuhakarn C.; Reutrakul V.; Soorukram D. Site-Specific Synthesis of β-Fluorinated γ-Butyrolactams Via Decarboxylative Fluorination of β-Carboxyl-γ-Butyrolactams. Eur. J. Org. Chem. 2019, 2019, 4710–4720. 10.1002/ejoc.201900685. [DOI] [Google Scholar]
- Dong Y.; Wang Z.; Li C. Controlled Radical Fluorination of Poly(Meth)Acrylic Acids in Aqueous Solution. Nat. Commun. 2017, 8, 277. 10.1038/s41467-017-00376-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing R.; Li Y.; Yu H. Preparation of Fluoro-Functionalized Graphene Oxide Via the Hunsdiecker Reaction. Chem. Commun. 2016, 52, 390–393. 10.1039/C5CC08252H. [DOI] [PubMed] [Google Scholar]
- Tarantino G.; Botti L.; Dimitratos N.; Hammond C. Catalytic Formation of C(sp3)–F Bonds Via Decarboxylative Fluorination with Mechanochemically-Prepared Ag2O/TiO2 Heterogeneous Catalysts. RSC Adv. 2017, 7, 30185–30190. 10.1039/C7RA06180C. [DOI] [Google Scholar]
- Wang C. M.; Mallouk T. E. New Photochemical Method for Selective Fluorination of Organic Molecules. J. Am. Chem. Soc. 1990, 112, 2016–2018. 10.1021/ja00161a066. [DOI] [Google Scholar]
- Lai C.; Kim Y. I.; Wang C. M.; Mallouk T. E. Evidence for Carbocation Intermediates in the Titanium Dioxide-Catalyzed Photochemical Fluorination of Carboxylic Acids. J. Org. Chem. 1993, 58, 1393–1399. 10.1021/jo00058a019. [DOI] [Google Scholar]
- Huang X.; Liu W.; Hooker J. M.; Groves J. T. Targeted Fluorination with the Fluoride Ion by Manganese-Catalyzed Decarboxylation. Angew. Chem., Int. Ed. 2015, 54, 5241–5245. 10.1002/anie.201500399. [DOI] [PubMed] [Google Scholar]
- Che C. M.; Lo V. K.; Zhou C. Y.; Huang J. S. Selective Functionalisation of Saturated C-H Bonds with Metalloporphyrin Catalysts. Chem. Soc. Rev. 2011, 40, 1950–1975. 10.1039/c0cs00142b. [DOI] [PubMed] [Google Scholar]
- Xiang J.; Shang M.; Kawamata Y.; Lundberg H.; Reisberg S. H.; Chen M.; Mykhailiuk P.; Beutner G.; Collins M. R.; Davies A.; et al. Hindered Dialkyl Ether Synthesis with Electrogenerated Carbocations. Nature 2019, 573, 398–402. 10.1038/s41586-019-1539-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger M.; Herszman J. D.; Kurimoto Y.; de Kruijff G. H. M.; Schüll A.; Ruf S.; Waldvogel S. R. Metal-Free Electrochemical Fluorodecarboxylation of Aryloxyacetic Acids to Fluoromethyl Aryl Ethers. Chem. Sci. 2020, 11, 6053–6057. 10.1039/D0SC02417A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb E. W.; Park J. B.; Cole E. L.; Donnelly D. J.; Bonacorsi S. J.; Ewing W. R.; Doyle A. G. Nucleophilic (Radio)Fluorination of Redox-Active Esters Via Radical-Polar Crossover Enabled by Photoredox Catalysis. J. Am. Chem. Soc. 2020, 142, 9493–9500. 10.1021/jacs.0c03125. [DOI] [PubMed] [Google Scholar]
- Li J.; Li Y.-L.; Jin N.; Ma A.-L.; Huang Y.-N.; Deng J. A Practical Synthesis of A-Fluoroketones in Aqueous Media by Decarboxylative Fluorination of B-Ketoacids. Adv. Synth. Catal. 2015, 357, 2474–2478. 10.1002/adsc.201500282. [DOI] [Google Scholar]
- Zhang R.; Ni C.; He Z.; Hu J. Decarboxylative Fluorination of β-Ketoacids with N-Fluorobenzenesulfonimide (NFSI) for the Synthesis of A-Fluoroketones: Substrate Scope and Mechanistic Investigation. J. Fluorine Chem. 2017, 203, 166–172. 10.1016/j.jfluchem.2017.08.010. [DOI] [Google Scholar]
- Li Y.-L.; Li J.; Deng J. Practical Access to Difluoromethyl Ketones Via Straightforward Decarboxylative Difluorination of β-Ketoacids. Adv. Synth. Catal. 2017, 359, 1407–1412. 10.1002/adsc.201601315. [DOI] [Google Scholar]
- Shibatomi K.; Katada M.; Kitahara K.; Iwasa S. Catalyst-Free Decarboxylative Fluorination of Tertiary β-Keto Carboxylic Acids. Synlett 2018, 29, 2408–2411. 10.1055/s-0037-1611019. [DOI] [Google Scholar]
- Kawanishi R.; Phongphane L.; Iwasa S.; Shibatomi K. Decarboxylative Fluorination of 2-Pyridylacetates. Chem. - Eur. J. 2019, 25, 7453–7456. 10.1002/chem.201900565. [DOI] [PubMed] [Google Scholar]
- Forrest A. K.; O'Hanlon P. J. The Preparation and Lithiation of Bromofluorofurans Via a Novel Fluorodecarboxylation. Tetrahedron Lett. 1995, 36, 2117–2118. 10.1016/0040-4039(95)00170-H. [DOI] [Google Scholar]
- Wang J.; Scott A. I. Fluoro-Decarboxylation of Pyrrolecarboxylic Acids by F–TEDA–BF4– a Convenient General Synthesis of Fluoropyrroles. J. Chem. Soc., Chem. Commun. 1995, 2399–2400. 10.1039/C39950002399. [DOI] [Google Scholar]
- Yuan X.; Yao J. F.; Tang Z. Y. Decarboxylative Fluorination of Electron-Rich Heteroaromatic Carboxylic Acids with Selectfluor. Org. Lett. 2017, 19, 1410–1413. 10.1021/acs.orglett.7b00335. [DOI] [PubMed] [Google Scholar]
- Wang D. H.; Yuan Z. L.; Liu Q. L.; Chen P. H.; Liu G. S. Decarboxylative Fluorination of Arylcarboxylic Acids Promoted by Ortho-Hydroxy and Amino Groups. Chin. J. Chem. 2018, 36, 507–514. 10.1002/cjoc.201800016. [DOI] [Google Scholar]