

Abstract
Water is generally considered an enemy of metal halide perovskites, being responsible for their rapid degradation and, consequently, undermining the long-term stability of perovskite-based solar cells. However, beneficial effects of liquid water have been surprisingly observed, and synthetic routes including water treatments have shown to improve the quality of perovskite films. This suggests that the interactions of water with perovskites and their precursors are far from being completely understood, as water appears to play a puzzling dual role in perovskite precursor solutions. In this context, studying the basic interactions between perovskite precursors in the aqueous environment can provide a deeper comprehension of this conundrum. In this context, it is fundamental to understand how water impacts the chemistry of iodoplumbate perovskite precursor species, PbIx2–x. Here, we investigate the chemistry of these complexes using a combined experimental and theoretical strategy to unveil their peculiar structural and optical properties and eventually to assign the species present in the solution. Our study indicates that iodide-rich iodoplumbates, which are generally key to the formation of lead halide perovskites, are not easily formed in aqueous solutions because of the competition between iodide and solvent molecules in coordinating Pb2+ ions, explaining the difficulty of depositing lead iodide perovskites from aqueous solutions. We postulate that the beneficial effect of water when used as an additive is then motivated by its behavior being similar to high coordinative polar aprotic solvents usually employed as additives in one-step perovskite depositions.
1. Introduction
Lead halide perovskites have been subjected to great research from the global scientific community in the last decades in virtue of their outstanding optoelectronic properties.1,2 These allowed their employment as innovative materials for photovoltaics, leading to amazing efficiencies up to 25% reported in state-of-the-art devices.3,4 On the other hand, the stability of these materials can be strongly affected by factors such as UV light, temperature, water, and ambient humidity.5 In particular, the origin of water-induced instability has been investigated from different points of view, ranging from the dissolution mechanism of methylammonium lead iodide (CH3NH3PbI3)6−8 to formation of defects9 and the interaction of water with perovskite surfaces.10 The detrimental action of water on perovskites can be summarized by the degradation pathway reported in eq 1
| 1 |
leading to the intermediated (CH3NH3)4PbI6 hydrated phase and PbI2. Nevertheless, the role of water is not limited to this: interestingly enough, the beneficial effects of water have been recently reported.11,12 In particular, it has been proved that if the degradation pathway of eq 1 is blocked at the first stage, that is, the reversible formation of the monohydrate phase, the crystallization process of the perovskite film is enhanced with increased grain size and nonradiative recombination centers are deactivated.12,13 Moreover, controlled concentrations of water have been proven to passivate defects.13−15 Therefore, the nature of the water–perovskite interaction appears to also depend on the actual amount of water in contact with the material, thus potentially being a game-changing factor in perovskite synthesis. In this regard, however, the wide literature of studies on this subject is far from reaching a unanimous consensus.16−29 Conings et al. reported that the presence of a fraction of water up to 10% volume in the precursor solution does not affect the photovoltaic performance of the prepared perovskite thin film.16 In contrast, Clegg and Hill observed that low concentration of water can be simultaneously detrimental for the photovoltaic performances but beneficial for the long-term stability of the device.17 In contrast to these studies, controlled amounts of water and humidity have improved the performances of perovskite solar cells: small amounts of H2O as an additive in the perovskite precursor solution improves film crystallization and, consequently, photovoltaic performances.18−22 Similar effects are encountered considering ambient moisture23−28 directly interacting in the precursor solution, during the annealing stage or in post-treatment processes. Zhang et al. recently reported a prenucleation method for the fabrication of perovskite solar cells in ambient conditions, in which water promotes the formation of lead complexes.29 In this context, it is evident that the dual nature of water when involved in the interaction with perovskites is a consequence of its rich chemistry, which brings to bear a plethora of possible reaction pathways.
As metal halide perovskites can be easily synthesized in solution, for example, through spin-coating and blade-coating,30−32 the solvent has a crucial role in determining the final perovskite quality. This directly affects the type and amount of precursors in solution, usually in the form of iodoplumbate species, that is, Pb2+ and I– complexes such as PbI+, PbI2, PbI3–, PbI42–, PbI53–, and PbI64–, which are forerunner species to the perovskite formation.33−38 Solvent and processing conditions are in turn linked to the presence of defects incorporated in the perovskite film,33,34,38−41 with possible serious consequences on both material stability and overall optoelectronic quality. Highly coordinating solvents generally employed in synthetic routes, such as dimethyl sulfoxide (DMSO) and N,N-dimethylformamide (DMF), are toxic and harmful for the environment, raising an issue for safe and sustainable development of perovskite manufacturing.42,43 In this sense, water would be an environmentally friendly solution to this problem, but because of the abovementioned stability problems and the general low solubility of precursor lead salts (e.g., PbI2) in this solvent, it has only rarely been employed for perovskite synthesis as a precursor. For example, the use of Pb(NO3)2 as a precursor has been attempted owing to its enhanced water solubility:44−46 Hsieh et al. reported on the formation of CH3NH3PbI3 starting from Pb(NO3)2 in aqueous solution. Through this synthesis method, they were able to achieve materials with cell performances close to those obtained with a standard PbI2/DMF solution.44 Following this work, Sveinbjörnsson et al. produced perovskites with Pb(NO3)2 and CsNO3 obtaining good results,45 while Zhai et al. employed Pb(NO3)2 to manufacture perovskite solar cells on plastic.46 Mixed halide perovskite nanocrystals were also successfully prepared by Geng et al. through synthesis in an aqueous environment in acidic conditions.47 Therefore, it has been proven that it is not impossible to synthesize a perovskite using water as the principal solvent, but the numerous reports on its detrimental effect and the low solubility of some of the common precursors generally employed certainly suggest that it is not a straightforward process.
To gain insights into this peculiar chemistry and to understand how water can impact the synthesis of perovskite through interaction with precursors, we here investigate the nature of iodoplumbates in aqueous solutions. We aim, in particular, to obtain information on how water influences the stability and optical properties of these complexes and on the possible differences with other more common solvent environments such as DMSO, DMF, γ-butyrolactone (GBL), and acetonitrile (ACN). Formation of complex iodoplumbate species, such as PbI3– and PbI42–, has already been tracked in a typical synthetic solution environment by many studies, mainly by means of UV–vis spectroscopy,11,33,35,40,41,48 highlighting how these species are preferentially produced in a low coordinating solvent environment, that is, following the general trend ACN > GBL > DMF > DMSO well described by the increasing Gutmann’s donor number with the corresponding values of 14.1, 18.0, 26.6, and 29.8 kcal/mol, respectively.34,49 In this sense, being transparent to UV and visible radiations because of its large optical energy gap,50−53 water is an ideal solvent to explore the spectroscopic properties of iodoplumbates in a spectral range larger than those accessible with the solvents commonly employed for the synthesis of perovskites. Here, we investigate the nature of iodoplumbate precursors in aqueous solution by studying the PbI2 + CH3NH3I (MAI) solutions by means of UV–vis spectroscopy, ab initio molecular dynamics (MD) simulations, and time-dependent density functional theory (TDDFT) excited-state calculations including relativistic effects. By supporting experiments with high-level quantum mechanical simulations, we achieve a deeper comprehension of the chemistry of iodoplumbates in aqueous solutions in terms of structure, coordination number, and equilibria. We find that formation of iodoplumbates containing a high number of coordinated iodide ions is hindered in aqueous environments and it only takes place when a very high quantity of MAI is added in the solution. This is interesting if we only consider the low donor number of water (18.0 kcal/mol),49 for which a favorable formation of iodoplumbates would be expected, confirming the uncertainties that affect this quantity when it comes to protic solvents and suggesting, at the same time, that other factors such as the high polarity of water molecules and the strong solvation of I– are acting on these complex equilibria.
2. Computational Details
To simulate Pb2+ and the iodoplumbates in aqueous solutions, we first consider a structural configuration of a previously performed MD simulation of liquid water.54 In particular, we consider a periodic cubic cell of length 15.46 Å, containing 128 H2O molecules, Figure 1a, corresponding to the experimental density. Then, we replace a water molecule with a Pb2+ ion and we carry out a DFT-based Born-Oppenheimer MD simulation in the NVT ensemble. A time step of 0.48 fs is employed. A production run of ∼15 ps follows an equilibration run of 5 ps. The target temperature of the simulations, controlled with a Nosé–Hoover thermostat,55,56 is set at 350 K, in order to achieve liquid-like behavior and to approximately take into account nuclear quantum motion.57,58 A similar computational strategy is adopted to study aqueous I–. The simulation of aqueous iodoplumbates (PbI+, PbI2, PbI3–, and PbI42–) is carried out by replacing a water molecule within the first coordination shell of aqueous Pb2+ with a I– ion for each I– of the considered species. The same computational setup used to simulate aqueous Pb2+ is then employed. DFT-based MD simulations are carried out with the rVV10 functional,59,60 which accounts for van der Waals interactions. In particular, the analytic expression of the rVV10 functional depends on an empirical parameter b, which governs the short-range interactions.59 We adopt the value of b = 9.3, as this has been shown to reproduce the experimental mass density in the case of liquid water.61 The freely available CP2K/QUICKSTEP package is used to perform the MD simulations.62 This suite of programs features a combined atomic basis set/plane-wave approach: atom-centered Gaussian-type basis functions are used to describe the orbitals and an auxiliary plane-wave basis set is employed to re-expand the electron density. For all the calculations, we use a triple-ζ correlation-consistent polarized basis set (cc-pVTZ) for O and H atoms63 and double-ζ polarized basis sets for the wave functions for Pb and I. A cutoff of 600 Ry is used for the plane waves.64 Core electrons are described by the analytical Goedecker–Teter–Hutter pseudopotentials.65 The following convergence criteria for the wave function optimization are employed: 10–7 a.u. for the electronic gradient and 10–12 a.u. for the energy difference between the final self-consistent field (SCF) cycles. This computational setup has been found to provide structural and dynamical properties of liquid water and aqueous solutes in good agreement with the experiment.54,66
Figure 1.

(a) Model of PbI2 in aqueous solution employed for the MD calculations. (b) Optimized structures of the complexes formed in aqueous solution by Pb2+, PbI+, PbI2, PbI3–, and PbI42–. O atoms in red, H in white, Pb in brown, and I in pink.
DFT calculations for the optimization of selected iodoplumbate structures are run with the Gaussian 09 software package,67 employing the B3LYP exchange–correlation functional,68,69 Grimme’s dispersion interactions DFT-D3,70 CPM polarizable continuum model for implicit solvation effects, and LANL2DZ basis sets for heavy Pb and I atoms together with the LANL2 pseudopotentials for the core electrons, while for light atoms (C, H, N, O, and S), the 6-31G* basis sets are chosen. Relative energy differences for optimized solvated lead complexes are evaluated following eq 2
| 2 |
where Etot is the total energy of the iodoplumbate with n explicit coordinating solvent molecules and Esolute and Esolv are the total energies of the bare Pb2+/iodoplumbate and the solvent molecule, respectively.
TDDFT calculations are carried out with the ADF 2014.04 program package.71−73 We employ the B3LYP exchange–correlation functional,68,69 a ZORA Hamiltonian to include relativistic spin–orbit coupling (SOC) effects74,75 and the COSMO implicit solvation model.76 A Slater-type TZP basis is used for all the atoms (the cores 1s-2s, 1s-4p, and 1s-4d are kept frozen, respectively, for S, I, and Pb). The absorption spectra are simulated by interpolating the computed transitions by Gaussian functions with a broadening σ = 0.1. To obtain the thermally averaged UV–vis optical absorption spectra of the various investigated species, ∼30 snapshots equally spaced in time from the last 5 ps of each MD simulation of aqueous iodoplumbates were subjected to TDDFT calculations retaining the closest 1–8 water molecules (i.e., the first coordination sphere for each complex) and adding a polarizable continuum solvation model.
Equilibrium chemical composition of solutions are simulated with Spana/Database (version 2020-06-19), a freely available software77 based on the HALTAFALL algorithm78,79 and partially inspired by SOLGASWATER.80
For the theory on calculation of equilibrium dissociation constants of iodoplumbates in aqueous solutions through the thermodynamic integration method, the reader is referred to Section S1, Supporting Information.
3. Experimental Details
PbI2 solutions with concentrations ranging from 2.00 × 10–3 to 6.04 × 10–1 mM in H2O are prepared from a 1.00 mM PbI2 stock solution. A volume of MAI corresponding to a concentration range between 2.00 and 3.76 × 102 mM is added to a 3.00 × 10–2 mM PbI2 solution prepared from the same 1.00 mM stock solution. UV–vis absorption spectra are recorded with a double-beam spectrophotometer Perkin-Elmer Lambda 800 employing quartz cuvettes with two optical walls and an optical path of 0.5 cm.
4. Results and Discussion
To unveil the structural properties of hydrated iodoplumbates, we first carried out MD simulations of the Pb2+, PbI+, PbI2, PbI3–, and PbI42– complexes in an aqueous environment, employing models such as the one reported for PbI2 in Figure 1a. One of the advantages of using MD simulations for water systems over cluster models is that because of the protic nature of water, employing larger solvation shells is a way to take into account the formation of hydrogen bonds between water molecules, thus making it easier to unveil the structure of coordination complexes. Because iodoplumbates with higher iodide content, such as PbI53– and PbI64–, are difficult to find in the diluted solutions generally employed for the investigation of the spectral properties of these systems,33,34,40,41,48 we did not consider these complexes in our analysis.
Pb2+ in aqueous solution is found to occur, on average, as an heptacoordinated complex, Pb(H2O)72+, with the oxygen water atoms pointing toward the metal cation. The complex features a pentagonal-bipyramidal structure, Figure 1b, and is dynamically holodirected with water molecules surrounding the cation from all the directions in line with a previous first-principles simulation.81 The average Pb–O distance of 2.56 Å is consistent with those inferred from X-ray measurements (2.5382 and 2.50 Å83). Consistently, integration of the first peak of the Pb–O radial distribution function (RDF), Figure 2a, provides a Pb–O coordination number nO = 6.91. Further analysis of the distribution reveals not only a majority sevenfold coordination (69% of configurations) but also sixfold coordination (21%) and eightfold coordination (10%) were found in line with the larger coordination numbers usually observed when accounting for van der Waals interactions in the simulations.54,61
Figure 2.
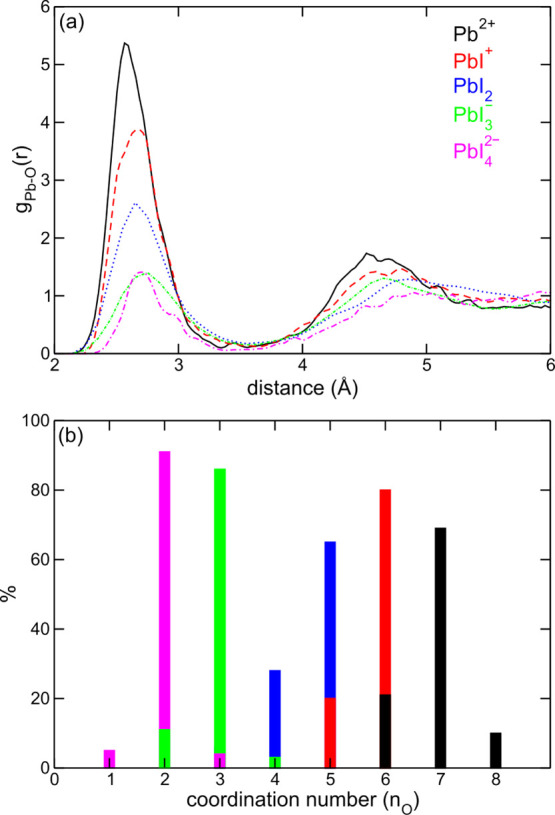
(a) Pb–O RDF for aqueous Pb2+ (black), PbI+ (red), PbI2 (blue), PbI3– (green), and PbI42– (magenta). (b) Average number of water molecules (%) coordinating Pb2+ in the considered aqueous species. The same color code used for panel a) is adopted.
Noteworthy, previous 1H NMR84 and X-ray82 studies report an average sixfold coordination for aqueous Pb2+. However, we note that experimental techniques could be unable to probe loosely coordinated water molecules81 that, instead, we inherently take into account within our approach. For example, the protons belonging to such remote water molecules are likely to not be subject to the influence of the metal cation, thus being not probed by 1H NMR. In this regard, we note that the NMR estimate was questioned because it was found to underestimate the coordination of other metal cations.84 Furthermore, Persson et al.82 predicted the coordination number of aqueous Pb2+ by coupling the experiment with inferred average Pb–O bond lengths (2.5–2.6 Å, in line with this work). Again, such an estimate may not account for all water molecules within the first shell. In fact, nO = 6.00 in our simulation corresponds to integration of the first peak of the RDF up to a distance of 2.9 Å, a value ∼0.4 Å shorter than that corresponding to the first minimum of the RDF.
The sevenfold coordination and the pentagonal-bipyramidal structure are retained for PbI+ and PbI2 in aqueous solution with iodide ions replacing water molecules in the first coordination shell, Figure 1b, as evidenced by calculated nO = 5.90 and 4.65 for PbI+ and PbI2, respectively. However, solvation of the iodide/water complexes is somehow more rigid than that of the bare Pb2+ ion. In fact, an eightfold coordination (iodide + water) is not observed during MD simulations for PbI+ and PbI2. Furthermore, we notice an average elongation of the Pb–O distance from 2.56 Å of Pb2+ to 2.65 Å and to 2.68 Å of PbI+ and PbI2, respectively, Figure 2a. We address both the difference in the coordination number and the increase in Pb–O bond distances to the reduced positive charge on the lead cation as a consequence of the strong interaction with the iodide anions, which makes Pb2+ less available to bond with other water molecules and partially prevents their interaction because of steric hindrance. This effect becomes even more apparent when considering PbI3– and PbI42–, which instead show a sixfold coordination and an octahedral structure, Figure 1b, with nO = 2.93 and 1.99, respectively, thus indicating that a reduced number of water molecules can interact with the metal cation. For a detailed structural description of these complexes, the reader is referred to Section S2, Supporting Information.
It is also interesting to comment on the coordination of iodide by surrounding water molecules in the aqueous iodoplumbates. Analysis of the I–H RDF indicates that three water molecules coordinate, on average, with I– for each complex, Figure S2, Supporting Information. We also notice that the average distance between iodide and hydrogen atoms is reduced from PbI+ (2.74 Å) to PbI42– (2.61 Å). In fact, as the interaction with the central metal cation involves more iodide anions, their partial negative charge increases, becoming available for a stronger hydrogen bond interaction with water molecules. To sum up, in Table 1, we report the average Pb–O and I–H coordination number, nO and nH, along with the average Pb–O and I–H bond distances. As we go down the table, that is, passing from Pb2+ to PbI42–, the number of coordinating water molecules is decreased from ca. 7 to 2, according to the increasing presence of iodide anions. In contrast with this, the number of water molecules coordinating iodide anions is essentially constant for all the studied iodoplumbates (∼3), for a total fourfold coordination, in line with the solvation of the aqueous iodide, Figure S2, Supporting Information. At the same time, we can see that the Pb–O bonds moves from Pb2+ to PbI42–, while for the I–H bonds, we see an opposite trend, which is motivated by the weakening and the strengthening of Pb–O and I–H bonds, respectively.
Table 1. Calculated Values of the Average Number of Water Molecules Coordinating Pb2+ (nO) and Hydrogen Atoms Coordinating I– (nH) in the Considered Iodoplumbatesa.
| species | nO | nH | r(Pb–O) | r(I–H) |
|---|---|---|---|---|
| Pb2+ | 6.91 | 2.56 | ||
| PbI+ | 5.90 | 3.09 | 2.68 | 2.74 |
| PbI2 | 4.65 | 3.14 | 2.65 | 2.68 |
| PbI3– | 2.93 | 3.00 | 2.75 | 2.65 |
| PbI42– | 1.99 | 3.04 | 2.75 | 2.61 |
Average Pb–O and I–H distances, r(Pb–O) and r(I–H), respectively, are also reported (Å).
We next investigate the optical properties of solvated iodoplumbates by measuring their UV–vis spectra. We start by analyzing the spectra of bare PbI2 at different concentrations. PbI2 is poorly soluble in water (solubility limit around 1.66 × 10–3 mol PbI2/Kg H2O at 298.15 K, corresponding to 0.76 g PbI2/l H2O), with a solubility product, Kps, ranging from 6.3 × 10–9 to 1.2 × 10–8 at room temperature85 (in the following, we are using the default value implemented in Spana, 7.94 × 10–9). We prepared solutions with concentrations ranging from 2.00 × 10–3 to 6.04 × 10–1 mM, by dilution of a 1.00 mM stock solution. At such concentrations, PbI2 is almost completely dissociated and one retrieves the spectrum of the solvated Pb2+ ion overlapping with that of iodide at ∼226 nm, Figure 3a. By increasing the PbI2 concentration, we observe the emergence of a band at 270 nm, which can be assigned to the lowest energy absorption feature of solvated PbI+, Figure 3a. Further increasing the concentration of the solution, one may expect to observe higher coordinated iodoplumbates. At the highest considered concentration of 6.04 × 10–1 mM, PbI2 amounts to ca. 0.2% of the total, Table S1, Supporting Information, thus essentially not contributing to the overall solution absorbance. Formation of PbI3– and PbI42– complexes is negligible in the concentration range here considered.
Figure 3.
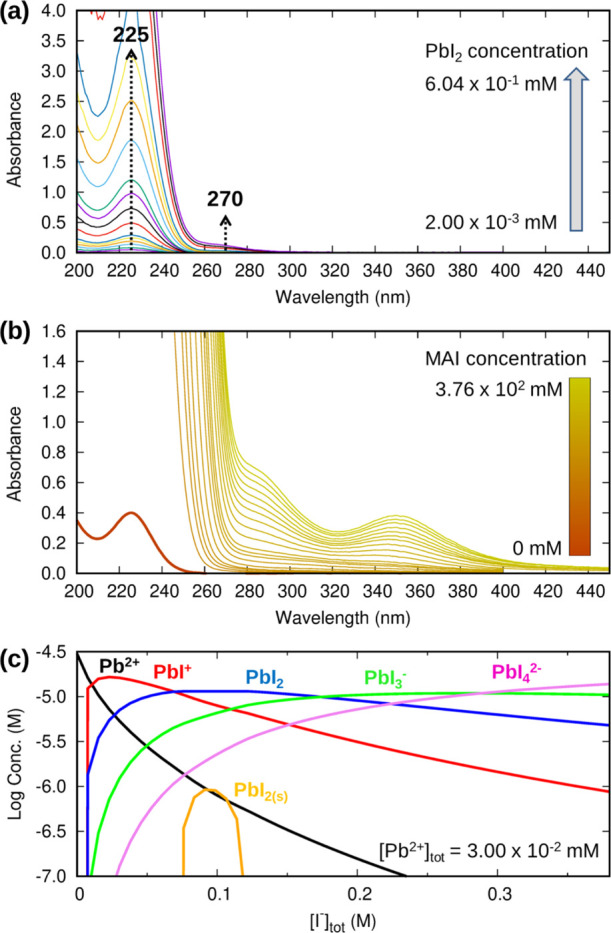
(a) Experimental UV–vis spectra of PbI2 at different concentrations. (b) Experimental UV–vis spectra of PbI2 3.00 × 10–2 mM + MAI (from 2.00 to 3.76 × 102 mM). (c) Estimated concentration diagram of aqueous iodoplumbates for a 3.00 × 10–2 mM PbI2 solution upon addition of MAI with a maximum concentration of 3.76 × 102 mM. The concentration of I– is out of range.
On the basis of this preliminary analysis, we chose to start from a PbI2 solution with a concentration of 3.00 × 10–2 mM, for which we are confidently in a range dominated by fully dissociated PbI2. Upon the first addition of MAI to this solution, we can see an increase of absorbance at 226 nm (with the detector that quickly gets saturated) and in the region between 250 and 280 nm, Figure 3b. The increase in absorption at 226 nm is mainly due to I–.86 Upon addition of ∼40 mM of MAI, corresponding to a 1333 [PbI2]/[MAI] ratio, we see the rise of absorbances at 287 and 345 nm. These signals increase with further addition of MAI, taking a shoulder-like and band-like spectral shape, respectively, starting at an MAI concentration of ∼131 mM. Interestingly, in the case of the 345 nm band, we can see that the absorption maximum is gradually shifted toward longer wavelengths, from 345 to 351 nm. This shift is likely indicative of the formation of PbI2, PbI3–, and PbI42– species. Notably, the absence of scattering signals even at such high concentrations of MAI suggests that no aggregates are present in the solution. We then simulate the concentration profile for the Pb2+, PbI+, PbI2, PbI3–, PbI42–, and I– species, solving the corresponding equilibria with the Spana software by employing the equilibrium formation constants at zero ionic strength reported by Tur’yan.85,87 The results, reported in Figure 3c, confirm that a large quantity of MAI is needed to induce the formation of iodine-rich iodoplumbates, thus implying that it is not easy for the iodide anion to displace water molecules interacting with the Pb2+ center.14 We also note that the amount of solid PbI2 as evaluated by Spana is negligible, thus indicating that metal halide is almost completely dissolved. Although the diluted PbI2 solutions we employ here are not representative of the deposition conditions used for perovskites, they are still useful to probe the relative strength of formation of iodoplumbates. In particular, this means that it is difficult to form iodide-rich iodoplumbates, such as PbI53– and PbI64–, that are considered necessary for the formation of good-quality perovskites.33−35,88
To assign the observed optical features to the specifically investigated complexes, we resort to a direct comparison between experiment and calculated optical spectra. To evaluate the optical properties of aqueous iodoplumbates, the thermally averaged structures of solvated Pb2+, PbI+, PbI2, PbI3–, and PbI42– species extracted from MD simulations were considered. Here, we explicitly include in the excited-state calculations the first solvation sphere considering a cutoff distance for a Pb–O bond length of 3.25 Å, corresponding to the first minimum of the RDF, and we represent the bulk solvent by an implicit solvation model, see Computational Details. Time-averaged spectra obtained for each species are compared with experimental UV–vis spectra of PbI2 with increasing amounts of added MAI. Because of the limited number of simulated optical transitions, we are only able to describe the lowest absorption band for each solvated species. Additional TDDFT simulations including an extended number of excited states are reported in Figure S3, Supporting Information. The results obtained by the thermally averaged TDDFT calculations are reported in Figure 4a. As one may notice, the calculated spectra follow the experimental trend quite accurately. Starting with solvated Pb2+, we calculate an absorption maximum at 218 nm, in good agreement with the experimental maximum at 226 nm, which, as stated before, is also related to the absorption of I–. Absorption maxima at 294 – 314, 336, 349, and 361 nm are respectively calculated for PbI+, PbI2, PbI3–, and PbI42– and reported, together with the experimental maxima as per Figure 3b, in Table 2.
Figure 4.
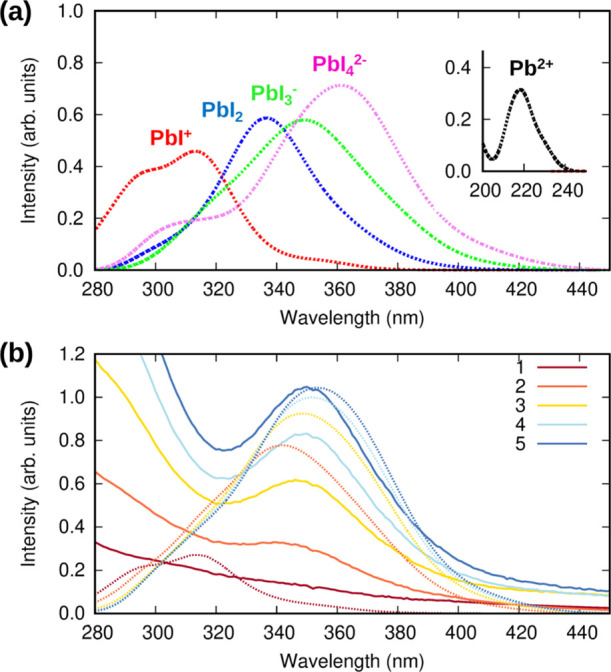
(a) Time-averaged spectra of solvated Pb2+ (black), PbI+ (red), PbI2 (blue), PbI3– (green), and PbI42– (magenta) complexes. The inset shows the spectrum of the solvated Pb2+ complex. (b) Selected UV–vis spectra of PbI2 3.00 × 10–2 mM + MAI (continuous lines) compared with time-averaged spectra of simulated iodoplumbates weighted for their concentrations as evaluated by Spana software (dotted lines) in the 280–450 nm region. Each color corresponds to a specific MAI concentration: 1–57 mM, 2–131 mM, 3–231 mM, 4–311 mM, and 5–376 mM. Experimental intensities were scaled to compare with the calculated ones.
Table 2. Theoretical Absorption Maxima for the Investigated Iodoplumbates Compared with the Experimental Absorption Peaks from PbI2 + MAI Solutions at Different Concentrations of MAI (CMAI).
| species (theo.) | abs. max (nm) | solutions (exp.) | abs. max (nm) |
|---|---|---|---|
| Pb2+ | 218 | PbI2 | 226 |
| PbI+ | 294 | PbI2 + MAI (CMAI < 40 mM) | ∼270 |
| 314 | PbI2 + MAI (40 mM < CMAI < 131 mM) | 287, 345 | |
| PbI2 | 336 | PbI2 + MAI (131 mM < CMAI < 167 mM) | 345–351 |
| PbI32– | 349 | ||
| PbI4– | 361 |
Considering the complexity of the involved equilibria with multiple overlapping absorptions from different species, a global approach for comparing theory and experiment is required. To this end, we analyze the absorption profile obtained by multiplying the concentrations determined through the Spana software, Figure 3c, by the calculated thermally averaged absorption profiles of the various species. Results, reported in Figure 4b, show a good agreement between the variation of the experimental absorbance as a function of MAI additions and the calculated absorption profile, especially for high added MAI amounts. Because of the limited number of excitation energies calculated here, we are only able to reproduce the longest wavelength absorption band centered at ∼350 nm and the emergence of a shoulder at ∼315 nm, which, however, trustfully reproduce the experimental spectra. The main feature at ∼350 nm is dominated by the absorption of PbI3– and PbI42– with a residual contribution from PbI2, these being the only species which absorb at such long wavelength.
Interestingly, these absorptions are quite different from those found for iodoplumbates in organic solvents, such as GBL, for which the absorption maxima lie around 330, 370, and 423 nm for PbI2, PbI3–, and PbI42–, respectively.33,34,41,48 This behavior is unexpected if one only considers Gutmann’s donor number (D.N.), or donicity, as a metric of solvent coordination strength to Pb2+: with a value of 18.0 kcal/mol,49 the same as GBL, one would expect a similar coordination to Pb2+ by water. In fact, the experimental D.N. trend is in agreement with the relative formation energies of the Pb2+(solv)n complexes, evaluated for n = 1–7 and solv = DMSO, DMF, GBL, ACN, and H2O as per eq 2, see Table 3. Following the work of Hamill et al.,89 we also consider the relative formation energy for the PbI2(solv)n adducts with n = 1 as a supplementary indicator, c.f. Table 3, the observed trend being consistent with that provided by the energetics of Pb2+–solvent interactions. Notably, GBL and H2O shares the same D.N. but the calculations show that hydrated Pb2+ complexes with low number of coordinated solvent molecules are less stable compared to GBL ones, while the energetics tends to equalize for highly solvated complexes, that is, for n > 3. From this analysis, we can conclude that, while D.N. has been proved to correctly describe the coordination to Pb2+ of commonly employed organic solvents, this seems not to be the case for water, which again has to be treated as a special case. In this regard, we pinpoint that the D.N. is determined by the heat of the 1:1 reaction between SbCl5 and the compound of interest dissolved in dichloroethane. However, because of the interactions with SbCl5, D.N. has been determined by indirect methods for protic solvents.90,91
Table 3. Relative Formation Energies (in eV) of Solvated Pb2+(solv)n, with n = 1–7, and PbI2(solv) Complexes, for solv (D.N., kcal/mol) = DMSO (29.8), DMF (26.6), GBL (18.0), H2O (18.0), and ACN (14.1).
| DMSO | DMF | GBL | H2O | ACN | |
|---|---|---|---|---|---|
| Pb2+(solv) | –2.412 | –2.166 | –1.781 | –1.494 | –1.400 |
| Pb2+(solv)2 | –2.108 | –1.904 | –1.599 | –1.409 | –1.278 |
| Pb2+(solv)3 | –1.883 | –1.712 | –1.488 | –1.343 | –1.185 |
| Pb2+(solv)4 | –1.680 | –1.502 | –1.305 | –1.198 | –1.031 |
| Pb2+(solv)5 | –1.483 | –1.340 | –1.177 | –1.083 | –0.912 |
| Pb2+(solv)6 | –1.366 | –1.221 | –1.091 | –1.021 | –0.818 |
| Pb2+(solv)7 | –1.295 | –1.108 | –1.027 | –0.953 | –0.738 |
| PbI2(solv) | –1.273 | –1.084 | –0.863 | –0.827 | –0.648 |
The difference between the optical properties of iodoplumbates in water and organic solvents is indicative that such properties are highly dependent on both the coordination number and donor strength of the solvent. Increasing the coordination number generally leads to an absorption blue shift, see the calculated absorption spectra of Pb2+(H2O)n and PbI2(H2O)n complexes at varying n, Figure S4, Supporting Information. Increasing the solvent donor strength at a fixed number of coordinated solvent molecules (n = 2, n = 3) in Pb2+(solv)n complexes leads, on the other hand, to an absorption red shift, see Figure S5, Supporting Information.
Because of the small size of the water molecule and its high polarity and dielectric constant, lead complexes are surrounded by large water coordination spheres, which are difficult to move. This means that the D.N. could not be sufficient to describe the coordination strength of solvents to lead ions in some particular cases. Furthermore, the more pronounced tail in the distribution of Pb–I bonds in all iodoplumbates, Figure S1, Supporting Information, clearly indicates the weakening of Pb–I interactions at increasing contents of iodide in the iodoplumbate complex. This translates in lower energies for iodide removal from the complex and higher dissociation constants. In this regard, starting from MD simulations, we estimate the dissociation constants for each considered iodoplumbate through a grand-canonical formulation of solutes in aqueous solution and the thermodynamic integration method,54,92 see Section S1 in the Supporting Information. The calculated values of dissociation constants for the aqueous iodoplumbates are consistent with the physical picture arising from the structural analysis of the MD simulations, that is, larger iodoplumbates show a stronger tendency to dissociate, in agreement with the trend experimentally observed, cf. Section S1 in the Supporting Information.
Finally, we discuss the solvation of the I– species, which is another aspect worth considering. Integration of the first peak of the I–H RDF indicates that five water molecules coordinate, on average, with the solvated iodide, Figure S6 and Figure S7, Supporting Information. As a polar protic solvent, water better solvates anions than polar aprotic solvents through hydrogen bonding.93,94 This can also be seen from the Mayer’s acceptor number (A.N.), a measure of the strength of a molecule as a Lewis acid. Organic solvents commonly employed in the synthesis of perovskites show an A.N. around 20 ppm (18.9, 17.3, 16.0, and 19.3 ppm for ACN, GBL, DMF, and DMSO, respectively),90,95 while water, with a value of 54.80 ppm,90 is likely to better coordinate with I– (see Section S6 of the Supporting Information for a detailed analysis of iodide solvation), which then becomes less available to interact with Pb2+. We note that A.N. is based on the 31P NMR shift that triethylphosphine oxide shows when dissolved in the compound of interest. In this case, measurements on protic compounds do not suffer from the same issues that are affecting D.N. measurements.96,97
Therefore, because the formation of iodide-rich iodoplumbates is an important step to obtain perovskites, the low propensity to produce these complexes in aqueous environment is indeed a symptom of the possible problems related to using water as a unique solvent for perovskites. Nevertheless, this can partially explain why water can be successfully implemented as an additive to enhance the crystallization of perovskites, as it shows a behavior similar to that typical of high coordinative polar aprotic solvents that are generally employed as an additive in one-step perovskite depositions.
5. Conclusions
We have reported on the study of hydrated perovskite precursors, unveiling their peculiar structural and optical properties by means of a combined theoretical and experimental strategy. The structure and average coordination number of different iodoplumbates have been revealed thanks to ab initio MD simulations. Then, combining the information extracted from excited-state TDDFT calculations and UV–vis spectroscopy has allowed us to characterize the optical properties of iodoplumbates and, consequently, their equilibrium behavior. Compared to other common solvents for perovskite precursors as DMSO, DMF, GBL and ACN, water turns out to be an interesting exception: because of its poor ability to solvate PbI2 and its modest donor number, it cannot be classified as a strongly coordinating solvent to lead ions; nevertheless, it is also difficult to produce iodide-rich iodoplumbates because of the high polarity of water molecules and to the preferential solvation of I– compared to other solvents. With this analysis, we propose an explanation on why it is difficult to deposit perovskites from aqueous solutions and at the same time why water is sometimes employed as an additive for the deposition of perovskites because iodoplumbates species, necessary precursors for the synthesis of perovskite, are hardly formed in this environment. Water is then a very peculiar solvent for the PbI2 + MAI system and an exception to the low donor number/low coordination correlation usually found for organic solvents commonly employed for the synthesis of perovskites.
Acknowledgments
The authors acknowledge the support from the European Union’s Horizon 2020 research and innovation programme under grant agreement no. 764047 of the Espresso project. E.R., F.A., E.M., and F.D.A. thank the Ministero Istruzione dell’Università e della Ricerca (MIUR) and the University of Perugia through the program “Dipartimenti di Eccellenza 2018-2022” (grant AMIS); F.D.A. thanks the Distinguished Scientist Fellowship Program (DSFP) of King Saud University, Riyadh, Saudi Arabia.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpcb.0c08624.
Equilibrium dissociation constants of aqueous iodoplumbates from a grand-canonical formulation of solutes in aqueous solution, structural analysis of aqueous iodoplumbates, concentration of species in PbI2 solutions evaluated with the Spana software, extended spectra of aqueous iodoplumbates, absorption spectra versus number of water molecules and comparison with other solvents, and structural analysis of aqueous iodide (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Burschka J.; Pellet N.; Moon S.-J.; Humphry-Baker R.; Gao P.; Nazeeruddin M. K.; Grätzel M. Sequential Deposition as a Route to High-Performance Perovskite-Sensitized Solar Cells. Nature 2013, 499, 316–319. 10.1038/nature12340. [DOI] [PubMed] [Google Scholar]
- Brenner T. M.; Egger D. A.; Kronik L.; Hodes G.; Cahen D. Hybrid Organic—Inorganic Perovskites: Low-Cost Semiconductors with Intriguing Charge-Transport Properties. Nat. Rev. Mater. 2016, 1, 15007. 10.1038/natrevmats.2015.7. [DOI] [Google Scholar]
- NREL . Photovoltaic Research; NREL. https://www.nrel.gov/pv/ (accessed Aug 1, 2020). [Google Scholar]
- Green M. A.; Ho-Baillie A.; Snaith H. J. The Emergence of Perovskite Solar Cells. Nat. Photonics 2014, 8, 506–514. 10.1038/nphoton.2014.134. [DOI] [Google Scholar]
- Bisquert J.; Juarez-Perez E. J. The Causes of Degradation of Perovskite Solar Cells. J. Phys. Chem. Lett. 2019, 10, 5889–5891. 10.1021/acs.jpclett.9b00613. [DOI] [PubMed] [Google Scholar]
- Khlyabich P. P.; Hamill J. C.; Loo Y.-L. Precursor Solution Annealing Forms Cubic-Phase Perovskite and Improves Humidity Resistance of Solar Cells. Adv. Funct. Mater. 2018, 28, 1801508. 10.1002/adfm.201801508. [DOI] [Google Scholar]
- Zheng C.; Rubel O. Unraveling the Water Degradation Mechanism of CH3NH3PbI3. J. Phys. Chem. C 2019, 123, 19385–19394. 10.1021/acs.jpcc.9b05516. [DOI] [Google Scholar]
- Leguy A. M. A.; Hu Y.; Campoy-Quiles M.; Alonso M. I.; Weber O. J.; Azarhoosh P.; van Schilfgaarde M.; Weller M. T.; Bein T.; Nelson J.; et al. Reversible Hydration of CH3NH3PbI3in Films, Single Crystals, and Solar Cells. Chem. Mater. 2015, 27, 3397–3407. 10.1021/acs.chemmater.5b00660. [DOI] [Google Scholar]
- Kye Y.-H.; Yu C.-J.; Jong U.-G.; Chen Y.; Walsh A. Critical Role of Water in Defect Aggregation and Chemical Degradation of Perovskite Solar Cells. J. Phys. Chem. Lett. 2018, 9, 2196–2201. 10.1021/acs.jpclett.8b00406. [DOI] [PubMed] [Google Scholar]
- Mosconi E.; Azpiroz J. M.; De Angelis F. Ab Initio Molecular Dynamics Simulations of Methylammonium Lead Iodide Perovskite Degradation by Water. Chem. Mater. 2015, 27, 4885–4892. 10.1021/acs.chemmater.5b01991. [DOI] [Google Scholar]
- Manser J. S.; Saidaminov M. I.; Christians J. A.; Bakr O. M.; Kamat P. V. Making and Breaking of Lead Halide Perovskites. Acc. Chem. Res. 2016, 49, 330–338. 10.1021/acs.accounts.5b00455. [DOI] [PubMed] [Google Scholar]
- Aranda C.; Guerrero A.; Bisquert J. Crystalline Clear or Not: Beneficial and Harmful Effects of Water in Perovskite Solar Cells. ChemPhysChem 2019, 20, 2587–2599. 10.1002/cphc.201900393. [DOI] [PubMed] [Google Scholar]
- Zhou W.; Zhao Y.; Shi C.; Huang H.; Wei J.; Fu R.; Liu K.; Yu D.; Zhao Q. Reversible Healing Effect of Water Molecules on Fully Crystallized Metal–Halide Perovskite Film. J. Phys. Chem. C 2016, 120, 4759–4765. 10.1021/acs.jpcc.5b11465. [DOI] [Google Scholar]
- Aranda C.; Cristobal C.; Shooshtari L.; Li C.; Huettner S.; Guerrero A. Formation Criteria of High Efficiency Perovskite Solar Cells Under Ambient Conditions. Sustainable Energy Fuels 2017, 1, 540–547. 10.1039/c6se00077k. [DOI] [Google Scholar]
- Solanki A.; Lim S. S.; Mhaisalkar S.; Sum T. C. Role of Water in Suppressing Recombination Pathways in CH3NH3PbI3 Perovskite Solar Cells. ACS Appl. Mater. Interfaces 2019, 11, 25474–25482. 10.1021/acsami.9b00793. [DOI] [PubMed] [Google Scholar]
- Conings B.; Babayigit A.; Vangerven T.; D’Haen J.; Manca J.; Boyen H.-G. The Impact of Precursor Water Content on Solution-Processed Organometal Halide Perovskite Films and Solar Cells. J. Mater. Chem. 2015, 3, 19123–19128. 10.1039/c5ta06199g. [DOI] [Google Scholar]
- Clegg C.; Hill I. G. Systematic Study on the Impact of Water on the Performance and Stability of Perovskite Solar Cells. RSC Adv. 2016, 6, 52448–52458. 10.1039/c6ra11379f. [DOI] [Google Scholar]
- Heo J. H.; Song D. H.; Im S. H. Planar CH3NH3PbBr3 Hybrid Solar Cells with 10.4% Power Conversion Efficiency, Fabricated by Controlled Crystallization in the Spin-Coating Process. Adv. Mater. 2014, 26, 8179–8183. 10.1002/adma.201403140. [DOI] [PubMed] [Google Scholar]
- Gong X.; Li M.; Shi X.-B.; Ma H.; Wang Z.-K.; Liao L.-S. Controllable Perovskite Crystallization by Water Additive for High-Performance Solar Cells. Adv. Funct. Mater. 2015, 25, 6671–6678. 10.1002/adfm.201503559. [DOI] [Google Scholar]
- Wu C.-G.; Chiang C.-H.; Tseng Z.-L.; Nazeeruddin M. K.; Hagfeldt A.; Grätzel M. High Efficiency Stable Inverted Perovskite Solar Cells Without Current Hysteresis. Energy Environ. Sci. 2015, 8, 2725–2733. 10.1039/c5ee00645g. [DOI] [Google Scholar]
- Adhikari N.; Dubey A.; Gaml E. A.; Vaagensmith B.; Reza K. M.; Mabrouk S. A. A.; Gu S.; Zai J.; Qian X.; Qiao Q. Crystallization of a Perovskite Film for Higher Performance Solar Cells by Controlling Water Concentration in Methyl Ammonium Iodide Precursor Solution. Nanoscale 2016, 8, 2693–2703. 10.1039/c5nr06687e. [DOI] [PubMed] [Google Scholar]
- Chiang C.-H.; Nazeeruddin M. K.; Grätzel M.; Wu C.-G. The synergistic effect of H2O and DMF towards stable and 20% efficiency inverted perovskite solar cells. Energy Environ. Sci. 2017, 10, 808–817. 10.1039/c6ee03586h. [DOI] [Google Scholar]
- Petrus M. L.; Hu Y.; Moia D.; Calado P.; Leguy A. M. A.; Barnes P. R. F.; Docampo P. The Influence of Water Vapor on the Stability and Processing of Hybrid Perovskite Solar Cells Made from Non-Stoichiometric Precursor Mixtures. ChemSusChem 2016, 9, 2699–2707. 10.1002/cssc.201600999. [DOI] [PubMed] [Google Scholar]
- You J.; Yang Y.; Hong Z.; Song T.-B.; Meng L.; Liu Y.; Jiang C.; Zhou H.; Chang W.-H.; Li G.; et al. Moisture Assisted Perovskite Film Growth for High Performance Solar Cells. Appl. Phys. Lett. 2014, 105, 183902. 10.1063/1.4901510. [DOI] [Google Scholar]
- Bass K. K.; McAnally R. E.; Zhou S.; Djurovich P. I.; Thompson M. E.; Melot B. C. Influence of Moisture on the Preparation, Crystal Structure, and Photophysical Properties of Organohalide Perovskites. Chem. Commun. 2014, 50, 15819–15822. 10.1039/c4cc05231e. [DOI] [PubMed] [Google Scholar]
- Dubey A.; Kantack N.; Adhikari N.; Reza K. M.; Venkatesan S.; Kumar M.; Khatiwada D.; Darling S.; Qiao Q. Room Temperature, Air Crystallized Perovskite Film for High Performance Solar Cells. J. Mater. Chem. 2016, 4, 10231–10240. 10.1039/c6ta02918c. [DOI] [Google Scholar]
- Eperon G. E.; Habisreutinger S. N.; Leijtens T.; Bruijnaers B. J.; van Franeker J. J.; deQuilettes D. W.; Pathak S.; Sutton R. J.; Grancini G.; Ginger D. S.; et al. The Importance of Moisture in Hybrid Lead Halide Perovskite Thin Film Fabrication. ACS Nano 2015, 9, 9380–9393. 10.1021/acsnano.5b03626. [DOI] [PubMed] [Google Scholar]
- Gangishetty M. K.; Scott R. W. J.; Kelly T. L. Effect of Relative Humidity on Crystal Growth, Device Performance and Hysteresis in Planar Heterojunction Perovskite Solar Cells. Nanoscale 2016, 8, 6300–6307. 10.1039/c5nr04179a. [DOI] [PubMed] [Google Scholar]
- Zhang K.; Wang Z.; Wang G.; Wang J.; Li Y.; Qian W.; Zheng S.; Xiao S.; Yang S. A Prenucleation Strategy for Ambient Fabrication of Perovskite Solar Cells with High Device Performance Uniformity. Nat. Commun. 2020, 11, 1006. 10.1038/s41467-020-14715-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Q.; Armin A.; Burn P. L.; Meredith P. Organohalide Perovskites for Solar Energy Conversion. Acc. Chem. Res. 2016, 49, 545–553. 10.1021/acs.accounts.5b00483. [DOI] [PubMed] [Google Scholar]
- Habibi M.; Zabihi F.; Ahmadian-Yazdi M. R.; Eslamian M. Progress in Emerging Solution-Processed Thin Film Solar Cells – Part II: Perovskite Solar Cells. Renewable Sustainable Energy Rev. 2016, 62, 1012–1031. 10.1016/j.rser.2016.05.042. [DOI] [Google Scholar]
- Jung M.; Ji S.-G.; Kim G.; Seok S. I. Perovskite Precursor Solution Chemistry: From Fundamentals to Photovoltaic Applications. Chem. Soc. Rev. 2019, 48, 2011–2038. 10.1039/c8cs00656c. [DOI] [PubMed] [Google Scholar]
- Rahimnejad S.; Kovalenko A.; Forés S. M.; Aranda C.; Guerrero A. Coordination Chemistry Dictates the Structural Defects in Lead Halide Perovskites. ChemPhysChem 2016, 17, 2795–2798. 10.1002/cphc.201600575. [DOI] [PubMed] [Google Scholar]
- Hamill J. C.; Schwartz J.; Loo Y.-L. Influence of Solvent Coordination on Hybrid Organic–Inorganic Perovskite Formation. ACS Energy Lett. 2018, 3, 92–97. 10.1021/acsenergylett.7b01057. [DOI] [Google Scholar]
- Yan K.; Long M.; Zhang T.; Wei Z.; Chen H.; Yang S.; Xu J. Hybrid Halide Perovskite Solar Cell Precursors: Colloidal Chemistry and Coordination Engineering behind Device Processing for High Efficiency. J. Am. Chem. Soc. 2015, 137, 4460–4468. 10.1021/jacs.5b00321. [DOI] [PubMed] [Google Scholar]
- Sharenko A.; Mackeen C.; Jewell L.; Bridges F.; Toney M. F. Evolution of Iodoplumbate Complexes in Methylammonium Lead Iodide Perovskite Precursor Solutions. Chem. Mater. 2017, 29, 1315–1320. 10.1021/acs.chemmater.6b04917. [DOI] [Google Scholar]
- Guo Y.; Shoyama K.; Sato W.; Matsuo Y.; Inoue K.; Harano K.; Liu C.; Tanaka H.; Nakamura E. Chemical Pathways Connecting Lead(II) Iodide and Perovskite via Polymeric Plumbate(II) Fiber. J. Am. Chem. Soc. 2015, 137, 15907–15914. 10.1021/jacs.5b10599. [DOI] [PubMed] [Google Scholar]
- Manser J. S.; Reid B.; Kamat P. V. Evolution of Organic–Inorganic Lead Halide Perovskite from Solid-State Iodoplumbate Complexes. J. Phys. Chem. C 2015, 119, 17065–17073. 10.1021/acs.jpcc.5b05898. [DOI] [Google Scholar]
- Moore D. T.; Sai H.; Tan K. W.; Smilgies D.-M.; Zhang W.; Snaith H. J.; Wiesner U.; Estroff L. A. Crystallization Kinetics of Organic–Inorganic Trihalide Perovskites and the Role of the Lead Anion in Crystal Growth. J. Am. Chem. Soc. 2015, 137, 2350–2358. 10.1021/ja512117e. [DOI] [PubMed] [Google Scholar]
- Stewart R. J.; Grieco C.; Larsen A. V.; Doucette G. S.; Asbury J. B. Molecular Origins of Defects in Organohalide Perovskites and Their Influence on Charge Carrier Dynamics. J. Phys. Chem. C 2016, 120, 12392–12402. 10.1021/acs.jpcc.6b03472. [DOI] [Google Scholar]
- Radicchi E.; Mosconi E.; Elisei F.; Nunzi F.; De Angelis F. Understanding the Solution Chemistry of Lead Halide Perovskites Precursors. ACS Appl. Energy Mater. 2019, 2, 3400–3409. 10.1021/acsaem.9b00206. [DOI] [Google Scholar]
- Park S. Y.; Park J.-S.; Kim B. J.; Lee H.; Walsh A.; Zhu K.; Kim D. H.; Jung H. S. Sustainable Lead Management in Halide Perovskite Solar Cells. Nat. Sustain. 2020, 1–8. [Google Scholar]
- Gardner K. L.; Tait J. G.; Merckx T.; Qiu W.; Paetzold U. W.; Kootstra L.; Jaysankar M.; Gehlhaar R.; Cheyns D.; Heremans P.; et al. Nonhazardous Solvent Systems for Processing Perovskite Photovoltaics. Adv. Energy Mater. 2016, 6, 1600386. 10.1002/aenm.201600386. [DOI] [Google Scholar]
- Hsieh T.-Y.; Wei T.-C.; Wu K.-L.; Ikegami M.; Miyasaka T. Efficient Perovskite Solar Cells Fabricated Using an Aqueous Lead Nitrate Precursor. Chem. Commun. 2015, 51, 13294–13297. 10.1039/c5cc05298j. [DOI] [PubMed] [Google Scholar]
- Sveinbjörnsson K.; Kyi Thein N. K.; Saki Z.; Svanström S.; Yang W.; Cappel U. B.; Rensmo H.; Boschloo G.; Aitola K.; Johansson E. M. J. Preparation of Mixed-Ion and Inorganic Perovskite Films Using Water and Isopropanol as Solvents for Solar Cell Applications. Sustainable Energy Fuels 2018, 2, 606–615. 10.1039/c7se00538e. [DOI] [Google Scholar]
- Zhai P.; Su T.-S.; Hsieh T.-Y.; Wang W.-Y.; Ren L.; Guo J.; Wei T.-C. Toward Clean Production of Plastic Perovskite Solar Cell: Composition-Tailored Perovskite Absorber Made From Aqueous Lead Nitrate Precursor. Nano Energy 2019, 65, 104036. 10.1016/j.nanoen.2019.104036. [DOI] [Google Scholar]
- Geng C.; Xu S.; Zhong H.; Rogach A. L.; Bi W. Aqueous Synthesis of Methylammonium Lead Halide Perovskite Nanocrystals. Angew. Chem., Int. Ed. 2018, 57, 9650–9654. 10.1002/anie.201802670. [DOI] [PubMed] [Google Scholar]
- Stamplecoskie K. G.; Manser J. S.; Kamat P. V. Dual Nature of the Excited State in Organic–Inorganic Lead Halide Perovskites. Energy Environ. Sci. 2015, 8, 208–215. 10.1039/c4ee02988g. [DOI] [Google Scholar]
- Cataldo F. A Revision of the Gutmann Donor Numbers of a Series of Phosphoramides Including TEPA. Eur. Chem. Bull. 2015, 4, 92–97. 10.17628/ecb.2015.4.92-97. [DOI] [Google Scholar]
- Ambrosio F.; Guo Z.; Pasquarello A. Absolute Energy Levels of Liquid Water. J. Phys. Chem. Lett. 2018, 9, 3212–3216. 10.1021/acs.jpclett.8b00891. [DOI] [PubMed] [Google Scholar]
- Heller J. M.; Hamm R. N.; Birkhoff R. D.; Painter L. R. Collective Oscillation in Liquid Water. J. Chem. Phys. 1974, 60, 3483–3486. 10.1063/1.1681563. [DOI] [Google Scholar]
- Shibaguchi T.; Onuki H.; Onaka R. Electronic Structures of Water and Ice. J. Phys. Soc. Jpn. 1977, 42, 152–158. 10.1143/jpsj.42.152. [DOI] [Google Scholar]
- Garbuio V.; Cascella M.; Kupchak I.; Pulci O.; Seitsonen A. P. Proton Disorder in Cubic Ice: Effect on the Electronic and Optical Properties. J. Chem. Phys. 2015, 143, 084507. 10.1063/1.4929468. [DOI] [PubMed] [Google Scholar]
- Ambrosio F.; Miceli G.; Pasquarello A. Redox Levels in Aqueous Solution: Effect of van der Waals Interactions and Hybrid Functionals. J. Chem. Phys. 2015, 143, 244508. 10.1063/1.4938189. [DOI] [PubMed] [Google Scholar]
- Nosé S. A Unified Formulation of the Constant Temperature Molecular Dynamics Methods. J. Chem. Phys. 1984, 81, 511–519. 10.1063/1.447334. [DOI] [Google Scholar]
- Hoover W. G. Canonical Dynamics: Equilibrium Phase-Space Distributions. Phys. Rev. A 1985, 31, 1695–1697. 10.1103/physreva.31.1695. [DOI] [PubMed] [Google Scholar]
- DiStasio R. A.; Santra B.; Li Z.; Wu X.; Car R. The Individual and Collective Effects of Exact Exchange and Dispersion Interactions on the Ab Initio Structure of Liquid Water. J. Chem. Phys. 2014, 141, 084502. 10.1063/1.4893377. [DOI] [PubMed] [Google Scholar]
- VandeVondele J.; Mohamed F.; Krack M.; Hutter J.; Sprik M.; Parrinello M. The Influence of Temperature and Density Functional Models in Ab Initio Molecular Dynamics Simulation of Liquid Water. J. Chem. Phys. 2004, 122, 014515. 10.1063/1.1828433. [DOI] [PubMed] [Google Scholar]
- Vydrov O. A.; Van Voorhis T. Nonlocal van der Waals Density Functional: The Simpler the Better. J. Chem. Phys. 2010, 133, 244103. 10.1063/1.3521275. [DOI] [PubMed] [Google Scholar]
- Sabatini R.; Gorni T.; de Gironcoli S. Nonlocal van der Waals Density Functional Made Simple and Efficient. Phys. Rev. B: Condens. Matter Mater. Phys. 2013, 87, 041108. 10.1103/physrevb.87.041108. [DOI] [Google Scholar]
- Miceli G.; de Gironcoli S.; Pasquarello A. Isobaric First-Principles Molecular Dynamics of Liquid Water with Nonlocal van der Waals Interactions. J. Chem. Phys. 2015, 142, 034501. 10.1063/1.4905333. [DOI] [PubMed] [Google Scholar]
- VandeVondele J.; Krack M.; Mohamed F.; Parrinello M.; Chassaing T.; Hutter J. Quickstep: Fast and Accurate Density Functional Calculations Using a Mixed Gaussian and Plane Waves Approach. Comput. Phys. Commun. 2005, 167, 103–128. 10.1016/j.cpc.2004.12.014. [DOI] [Google Scholar]
- Dunning T. H. Gaussian Basis Sets for Use in Correlated Molecular Calculations. I. The Atoms Boron through Neon and Hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. 10.1063/1.456153. [DOI] [Google Scholar]
- VandeVondele J.; Hutter J. Gaussian Basis Sets for Accurate Calculations on Molecular Systems in Gas and Condensed Phases. J. Chem. Phys. 2007, 127, 114105. 10.1063/1.2770708. [DOI] [PubMed] [Google Scholar]
- Goedecker S.; Teter M.; Hutter J. Separable Dual-Space Gaussian Pseudopotentials. Phys. Rev. B: Condens. Matter Mater. Phys. 1996, 54, 1703–1710. 10.1103/physrevb.54.1703. [DOI] [PubMed] [Google Scholar]
- Miceli G.; Hutter J.; Pasquarello A. Liquid Water through Density-Functional Molecular Dynamics: Plane-Wave vs Atomic-Orbital Basis Sets. J. Chem. Theory Comput. 2016, 12, 3456–3462. 10.1021/acs.jctc.6b00271. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Petersson G. A.; Nakatsuji H., Gaussian 09, Revision D. 01.; Gaussian, Inc.: Wallingford CT, 2016.
- Becke A. D. Density Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]
- Stephens P. J.; Devlin F. J.; Chabalowski C. F.; Frisch M. J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. 10.1021/j100096a001. [DOI] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. A Consistent and Accurate ab initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- ADF 2014, SCM . Theoretical Chemistry; Vrije Universiteit: Amsterdam, The Netherlands, 2014. https://www.scm.com/ (accessed Aug 18, 2018). [Google Scholar]
- Fonseca Guerra C.; Snijders J. G.; te Velde G.; Baerends E. J. Towards An Order-N DFT Method. Theor. Chem. Acc. 1998, 99, 391–403. 10.1007/s002140050021. [DOI] [Google Scholar]
- te Velde G.; Bickelhaupt F. M.; Baerends E. J.; Fonseca Guerra C.; van Gisbergen S. J. A.; Snijders J. G.; Ziegler T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. 10.1002/jcc.1056. [DOI] [Google Scholar]
- van Lenthe E.; Baerends E. J.; Snijders J. G. Relativistic Regular Two-Component Hamiltonians. J. Chem. Phys. 1993, 99, 4597–4610. 10.1063/1.466059. [DOI] [Google Scholar]
- Wang F.; Ziegler T.; van Lenthe E.; van Gisbergen S.; Baerends E. J. The Calculation of Excitation Energies Based on the Relativistic Two-Component Zeroth-Order Regular Approximation and Time-Dependent Density-Functional with Full Use of Symmetry. J. Chem. Phys. 2005, 122, 204103. 10.1063/1.1899143. [DOI] [PubMed] [Google Scholar]
- Pye C. C.; Ziegler T. An Implementation of the Conductor-Like Screening Model of Solvation Within the Amsterdam Density Functional Package. Theor. Chem. Acc. 1999, 101, 396–408. 10.1007/s002140050457. [DOI] [Google Scholar]
- GitHub . ignasi-p/eq-diagr: Chemical Equilibrium Diagrams for aqueous systems. https://github.com/ignasi-p/eq-diagr (accessed Aug 18, 2020).
- Ingri N.; Kakolowicz W.; Sillén L. G.; Warnqvist B. High-Speed Computers as a Supplement to Graphical Methods—V: Haltafall, a general Program for Calculating the Composition of Equilibrium Mixtures. Talanta 1967, 14, 1261–1286. 10.1016/0039-9140(67)80203-0. [DOI] [PubMed] [Google Scholar]
- Warnqvist B.; Ingri N. The HALTAFALL Program-Some Corrections, and Comments on Recent Experience. Talanta 1971, 18, 457–458. 10.1016/0039-9140(71)80069-3. [DOI] [PubMed] [Google Scholar]
- Eriksson G. An Algorithm for The Computation of Aqueous multi-Component, Multiphase Equilibria. Anal. Chim. Acta 1979, 112, 375–383. 10.1016/s0003-2670(01)85035-2. [DOI] [Google Scholar]
- Gourlaouen C.; Gérard H.; Parisel O. Exploring the Hydration of Pb2+: Ab Initio Studies and First-Principles Molecular Dynamics. Chem.—Eur. J. 2006, 12, 5024–5032. 10.1002/chem.200600045. [DOI] [PubMed] [Google Scholar]
- Persson I.; Lyczko K.; Lundberg D.; Eriksson L.; Płaczek A. Coordination Chemistry Study of Hydrated and Solvated Lead(II) Ions in Solution and Solid State. Inorg. Chem. 2011, 50, 1058–1072. 10.1021/ic1017714. [DOI] [PubMed] [Google Scholar]
- Bargar J. R.; Brown G. E.; Parks G. A. Surface complexation of Pb(II) at oxide-water interfaces: II. XAFS and bond-valence determination of mononuclear Pb(II) sorption products and surface functional groups on iron oxides. Geochim. Cosmochim. Acta 1997, 61, 2639–2652. 10.1016/s0016-7037(97)00125-7. [DOI] [Google Scholar]
- Swift T. J.; Sayre W. G. Determination of Hydration Numbers of Cations in Aqueous Solution by Means of Proton NMR. J. Chem. Phys. 1966, 44, 3567–3574. 10.1063/1.1727266. [DOI] [Google Scholar]
- Clever H. L.; Johnston F. J. The Solubility of Some Sparingly Soluble Lead Salts: An Evaluation of the Solubility in Water and Aqueous Electrolyte Solution. J. Phys. Chem. Ref. Data 1980, 9, 751–784. 10.1063/1.555628. [DOI] [Google Scholar]
- Awtrey A. D.; Connick R. E. The Absorption Spectra of I2, I3-, I-, IO3-, S4O6= and S2O3=. Heat of the Reaction I3- = I2 + I-. J. Am. Chem. Soc. 1951, 73, 1842–1843. 10.1021/ja01148a504. [DOI] [Google Scholar]
- Tur’yan Y. I. Zh. Neorg. Khim. 1961, 6, 162–164. [Google Scholar]
- Kim J.; Park B.-w.; Baek J.; Yun J. S.; Kwon H.-W.; Seidel J.; Min H.; Coelho S.; Lim S.; Huang S.; et al. Unveiling the Relationship between the Perovskite Precursor Solution and the Resulting Device Performance. J. Am. Chem. Soc. 2020, 142, 6251–6260. 10.1021/jacs.0c00411. [DOI] [PubMed] [Google Scholar]
- Hamill J. C.; Romiluyi O.; Thomas S. A.; Cetola J.; Schwartz J.; Toney M. F.; Clancy P.; Loo Y.-L. Sulfur-Donor Solvents Strongly Coordinate Pb2+ in Hybrid Organic–Inorganic Perovskite Precursor Solutions. J. Phys. Chem. C 2020, 124, 14496–14502. 10.1021/acs.jpcc.0c03465. [DOI] [Google Scholar]
- Fawcett W. R. Acidity and Basicity Scales for Polar Solvents. J. Phys. Chem. 1993, 97, 9540–9546. 10.1021/j100139a045. [DOI] [Google Scholar]
- Adams D. J.; Dyson P. J.; Tavener S. J., Chemistry in Alternative Reaction Media. Chemistry in Alternative Reaction Media; John Wiley & Sons, Ltd, 2005; pp 1–31. [Google Scholar]
- Cheng J.; Sprik M. Alignment of Electronic Energy Levels at Electrochemical Interfaces. Phys. Chem. Chem. Phys. 2012, 14, 11245–11267. 10.1039/c2cp41652b. [DOI] [PubMed] [Google Scholar]
- Parker A. J. The Effects of Solvation on the Properties of Anions in Dipolar Aprotic Solvents. Q. Rev., Chem. Soc. 1962, 16, 163–187. 10.1039/qr9621600163. [DOI] [Google Scholar]
- Parfenyuk V. I.; Chankina T. I. Studying the Iodide Ion Solvation in Water–Dimethyl Sulfoxide Mixtures by the Method of Volta Potential Differences. Russ. J. Electrochem. 2002, 38, 431–434. 10.1023/a:1015300307626. [DOI] [Google Scholar]
- Wypych A.; Wypych G., 3 - Solvents. In Databook of Solvents, 2nd ed.; Wypych A., Wypych G., Eds.; ChemTec Publishing, 2019; pp 17–798. [Google Scholar]
- Mayer U.; Gutmann V.; Gerger W. The Acceptor Number — a Quantitative Empirical Parameter for the Electrophilic Properties of Solvents. Monatsh. Chem. 1975, 106, 1235–1257. 10.1007/bf00913599. [DOI] [Google Scholar]
- Symons M. C. R.; Eaton G. Triethylphosphine Oxide as an Infrared Probe of Solvent Structure. the Significance of Acceptor Numbers. J. Chem. Soc., Faraday Trans. 1 1982, 78, 3033–3044. 10.1039/f19827803033. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.