

Abstract
A catalyst-free, stereoselective visible-light-driven annulation reaction between alkenes and N,N-substituted dialkyl anilines for the synthesis of substituted tetrahydroquinolines is presented. The reaction is driven by the photoexcitation of an electron donor–acceptor (EDA) complex, and the resulting products are obtained in good to high yields with complete diastereoselectivity. Mechanistic rationale and photochemical characterization of the EDA-complex are provided.
Introduction
Electron donor–acceptor (EDA) complexes have in recent years gained considerable attention in the field of organic chemistry due to their photochemical properties which can be used to mediate a number of advanced chemical transformations.1−5 An EDA complex is the result of a weak association between an electron-rich donor and an electron-deficient acceptor and is characterized by a charge-transfer band in the absorption spectrum.4 Due to the bathochromic shift of the charge-transfer band, with respect to the donor and acceptor, visible light can often be employed to induce a single electron transfer from the donor to the acceptor. The resulting radical pair can be used in synthetic chemistry,1−4,6−12 including enantioselective alkylations,13−17 aromatic alkylations,16 and biaryl couplings.18
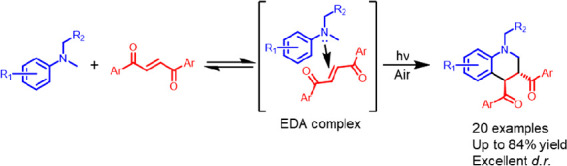
Recently, we reported an EDA-complex-driven protocol for the UV-light-induced generation of α-aminoalkyl radicals and their addition to maleimides to form fused tetrahydroquinolines (THQs) (Scheme 1a).19
Scheme 1. Light-Induced Construction of Tetrahydroquinolines.

The THQ is a highly desirable structural target in synthesis, as it can be found in a variety of biologically active compounds.20,21 Examples include molecules with antiviral,22−24 antibiotic,25,26 and cytotoxic27,28 activity. Thus, a diverse set of methodologies for the synthesis of the THQ-scaffold has been developed, and among the most versatile strategies are the aza-Diels–Alder, the Grieco, and the Povarov reactions.22−24
Additionally, several methods for the visible-light-driven construction of substituted THQ have been reported (Scheme 1b,c).29−34 However, the use of photocatalysts is typically required, and substrates are generally limited to cyclic alkenes (Scheme 1a,c) or alkenes that cannot undergo photoisomerization (Scheme 1b).35−37 To the best of our knowledge, no diastereoselective annulations between tertiary amines and alkenes that can undergo photoisomerization have been reported under photochemical conditions. Clearly, the possibility of alkene E/Z-isomerization under photochemical conditions poses a significant synthetic challenge for selectivity in the desired cyclization. But if successful, it could lead to the diastereoselective functionalization of THQ in the 3 and 4 positions (Scheme 1d). Inspired by these opportunities, we envisioned that the more challenging acyclic internal alkenes could function as acceptors in novel EDA complexes with aromatic amines for the synthesis of substituted THQ (Scheme 1d) and that the cumbersome photoisomerization could be kept to a minimum with a light source operating in the visible region of the light spectrum.
Herein, we present a catalyst-free EDA-mediated diastereoselective synthesis of substituted THQ from tertiary amines and 1,2-dibenzoylethylene (DBE).
Results and Discussion
To initiate our study, the interaction between 4′-N,N-trimethylaniline (1a) and 1,2-DBE (2a) was investigated (Figure 1). UV–vis measurements of 1a, 2a, and a mixture of the two compounds were performed. Upon mixing, a color change from light yellow to orange was observed (Figure 1A), and a new absorption band appeared in the visible region (Figure 1B), indicating an EDA interaction.
Figure 1.

(A) Photos of 1a, 2a, and 1a + 2a in acetonitrile. (B) UV–vis absorption spectra of 1a (0.1 M), 2a (0.1 M), and 1a + 2a in acetonitrile.
Next, the impact of visible light irradiation of a mixture of 1a and 2a was studied. Compact fluorescent light bulbs (CFLs) were used as the irradiation source (SI, Figure S2 for emission spectrum). The desired cyclized product 3a was formed in 25% yield using acetonitrile as solvent (Table 1, entry 1). 1,4-Dioxane proved to be the best solvent for the reaction, providing the desired THQ in 65% yield (Table 1, entry 8). Other solvents, both polar and nonpolar, showed inferior results, correlating with previously published findings.19,38 The impact of irradiation time on the formation of 3a was investigated using gas chromatography, and a reaction time of 4 h was determined to be optimal (SI, Figure S5). Significant degradation of the desired product was observed after prolonged irradiation. A large stoichiometric excess of the amine was proven to be important, as using 4 molar equiv lowered the yield significantly (Table 1, entry 9). This can be rationalized as a result of increased concentration of the photoactive EDA complex with higher loadings of the donor (KEDA = 0.42 M–1, calculated using the Benesi–Hildebrand method, Figure S3). The role of the oxidant, oxygen, was then evaluated. When the reaction mixture was irradiated under inert atmosphere, the reaction was suppressed (Table 1, entry 10). Irradiating under an atmosphere of oxygen gas on the other hand resulted in a low yield and a sluggish reaction (Table 1, entry 11). Changing the oxidant to persulfate resulted in decreased yield (Table 1, entry 13). Addition of acetic acid increased the yield of 3a to 73% (Table 1, entry 16), a result that correlates with the literature.39 Alternative light sources, such as blue LED (450 nm) or UV-CFL (365 nm) performed worse than the regular CFL (Table 1, compare entries 14 and 17 with 8). However, the use of blue LED under the optimized reaction conditions (1,4-dioxane as solvent and acetic acid as additive) and longer reaction time provide the desired product in the yield of 60% (Table 1, entry 19). The exclusion of light resulted in complete suppression of the formation of the desired product, confirming the need for an excitation source (Table 1, entry 18).
Table 1. Screening of Reaction Conditionsa.

| entry | solvent | light source | yield of 3a (%)b | d.r.b |
|---|---|---|---|---|
| 1 | acetonitrile | CFL | 25 | >25:1 |
| 2 | methanol | CFL | 14 | >25:1 |
| 3 | tetrahydrofuran | CFL | 29 | >25:1 |
| 4 | ethyl acetate | CFL | 17 | >25:1 |
| 5 | dichloromethane | CFL | 40 | >25:1 |
| 6 | 1,2-dichloroethane | CFL | 41 | >25:1 |
| 7 | 1,2-dimethoxyethane | CFL | 32 | >25:1 |
| 8 | 1,4-dioxane | CFL | 65 | >25:1 |
| 9 | 1,4-dioxane | CFL | 30c | >25:1 |
| 10 | 1,4-dioxane | CFL | 0d | – |
| 11 | 1,4-dioxane | CFL | 12e | >25:1 |
| 12 | 1,4-dioxane | CFL | 37f | >25:1 |
| 13 | 1,4-dioxane/water (2:1) | CFL | 47f | >25:1 |
| 14 | 1,4-dioxane | blue LED | 23 | >25:1 |
| 15 | 1,4-dioxane | CFL | 68g | >25:1 |
| 16 | 1,4-dioxane | CFL | 73h | >25:1 |
| 17 | 1,4-dioxane | UV-CFL | 30 | >25:1 |
| 18 | 1,4-dioxane | – | 0i | – |
| 19 | 1,4-dioxane | blue LED | 60h,j | >25:1 |
Reaction conditions: 1a (0.7 mmol) and 2a (0.1 mmol) in 3 mL of solvent irradiated with two 15 W compact fluorescent lamps in room temperature for 4 h.
Determined by NMR with 1,2,4,5-tetramethylbenzene as internal standard.
4 equiv of amine.
Under Ar.
Under O2.
K2S2O8 (2 equiv) used as an additive.
Acetic acid (30 equiv) used as additive.
Acetic acid (80 equiv) used as additive.
Reaction performed in absence of light.
Reaction time of 12 h.
With our optimized reaction conditions in hand, the generality of the reaction was examined (Scheme 2). Excellent diastereoselectivity toward the anti-diastereomer (confirmed by X-ray analysis, entry 3q) was obtained for all substrates, and the syn-isomer of 3 was never observed. Symmetric DBEs with simple aliphatic substituents provided the corresponding THQs in moderate yields in combination with 4′-N,N-trimethylaniline (Scheme 2, 3b–e). A clear effect of the electronic properties of substituents in the p-position on the 1,2-DBE can be observed as the yield decreases when more electron-donating substituents are introduced: p-H 73%, p-Me 63%, i-Bu 52%, and tert-Bu 45% (Scheme 2, 3a–3d). The introduction of two methyl groups in m- and p-position likewise decreased the yield (Scheme 2, 3e) as well as p-OMe (Scheme 2, 3f). Mildly σ-withdrawing groups such as p-Br or p-Cl resulted in no change or a slight increase in the yield (Scheme 2, 3h–i). When an unsymmetrical dibenzoyl ethylene was used as the substrate, the product was obtained as an inseparable mixture of the two regioisomers (Scheme 2, 3j).
Scheme 2. Substrate Scope of Anilines and Dibenzoylethylenes,
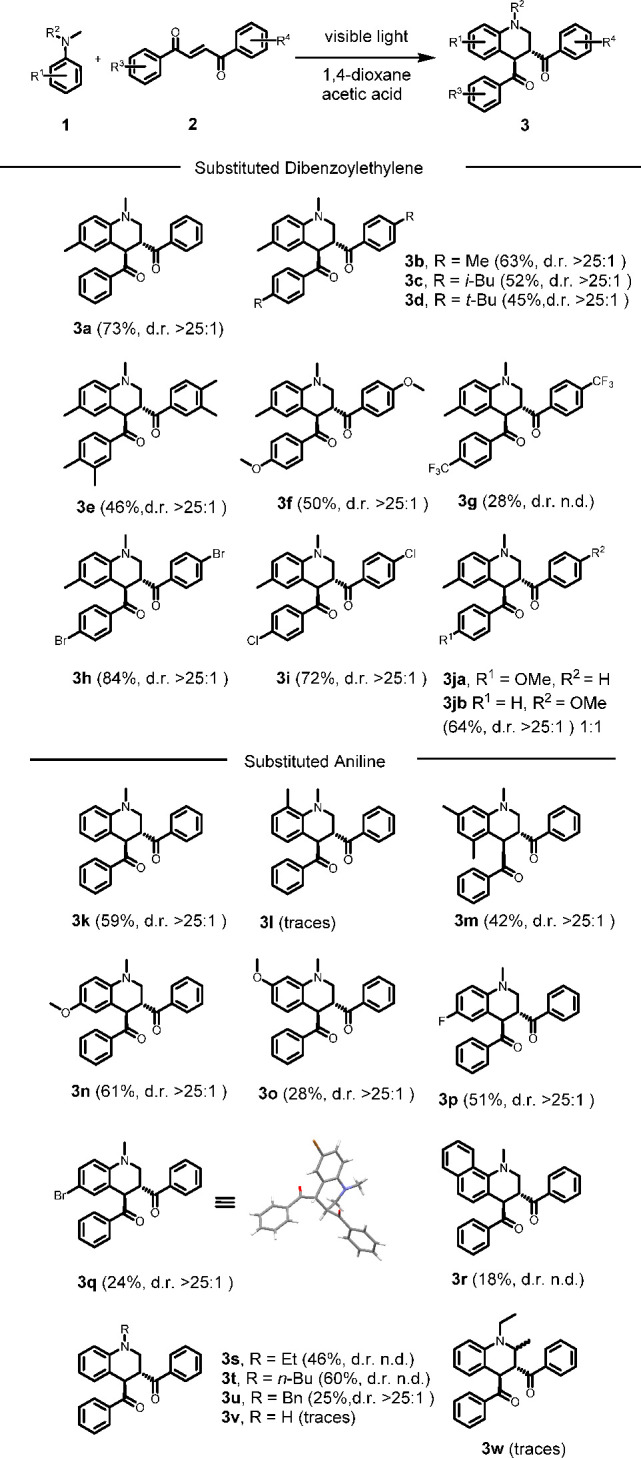
Reaction conditions: Substituted 1,2-DBE (0.25 mmol) and aniline (1.75 mmol) in 1,4-dioxane (6 mL) and acetic acid (1 mL) was irradiated with CFL lamp for 4 h.
n.d. = not determined.
Next, the effect of substituents on the aniline reaction partner was evaluated. Electron-donating groups proved to be well tolerated (Scheme 2, 3n) compared to σ-withdrawing groups such as p-F or p-Br (Scheme 2, 3p–q). Introduction of a methoxy group in m-position resulted in a significantly decreased the yield (Scheme 2, 3o). A significant steric effect was observed when a methyl group was introduced in the o-position on the aniline, as the reaction was completely suppressed (Scheme 2, 3i). This result could be explained by a significantly weaker EDA complex formed between the reaction partners, something also indicated by the lack of color change upon mixing. The steric clash between the o-Me and the N-Me groups leads to a less planar and less conjugated aromatic amine weakening the interaction with the alkene. Introduction of two methyl groups in the less sterically demanding m-positions did not lead to a similar decrease in yield (Scheme 2, 3m). We were also interested in investigating the regioselectivity of the reaction regarding the aniline reactant. Changing one of the N-methyl substituents to aliphatic in benzylic groups resulted in a complete selectivity toward reaction of the N-methyl group (Scheme 2, 3s–u). When N,N-diethylaniline was used, no reaction took place (Scheme 2, 3w). These results highlight the selectivity of the reaction toward N-methyl substituted anilines. However, subjecting monosubstituted N-methyl aniline to the reaction conditions resulted in complete suppression of reactivity (Scheme 2, 3v). The impact of geometrical isomerism of the DBE substrate was then examined. It is reported that E-DBE undergoes isomerization to the Z-isomer under visible light irradiation.40 In order to evaluate the impact of this background reaction, Z-DBE was subjected to the reaction conditions to furnish the desired product 3a, albeit in a lower yield of 55% (Scheme 3). Complete diastereoselectivity toward the anti-diastereomer was observed, suggesting that the cyclization reaction is not of concerted character. Notably, other acyclic activated olefins were not tolerated in the reaction such as benzylideneacetophenone, benzylidenemalonitrile, cinnamaldehyde, nitrostyrene, diethylfumarate, or diethylmaleate. These olefins did not provide the corresponding desired annulation products under the developed conditions.
Scheme 3. Impact of Stereochemistry of the Substrate Dibenzoylethylene.

To further probe the impact of light on the reaction, the illumination was cycled on/off, resulting in suppression of product formation in the absence of light (SI, Figure S6). This affirms the fact that the reaction requires constant illumination. In addition, the quantum yield of the reaction was determined to be 4.5 (SI, Section 8), which suggests that a radical chain process is involved in the mechanism. The role of oxygen was also investigated. The observation that the reaction is suppressed in the absence of oxygen (Scheme 1, entry 10) and the evidence for presence of hydrogen peroxide in the reaction mixture after irradiation (SI, Figure S4), demonstrates the role of ambient oxygen as an external oxidant. Based on these findings and support in the literature,19,41−43 we propose the following mechanism (Scheme 4): The reaction commences with the formation of an EDA complex between reactants 1a and 2a. Upon irradiation with light, an electron transfer occurs, a key step which is supported by calculation of the Gibbs free energy change for the photoinduced electron transfer (SI Section 1.8). Subsequent proton-transfer results in the formation of the α-amino alkyl radical A and enol radical B. The enol radical B reacts with oxygen, regenerating 2a and forming a hydroperoxyl radical and thus starting the self-propagating radical chain mechanism. In the next step, 2a reacts with α-amino alkyl radical A to form C, and after cyclization, the radical intermediate D is formed. In the final step, D is oxidized to the desired product 3a via two possible alternative pathways. The cyclohexadienyl radical intermediate D is oxidized by molecular oxygen41,42 yielding 3a and a second hydroperoxyl radical which in turn can propagate a radical chain reaction by oxidizing 1a to A.43,44 Alternatively, D can be intersected by a hydroperoxyl radical yielding 3a and hydrogen peroxide as a byproduct.
Scheme 4. Proposed Reaction Mechanism.

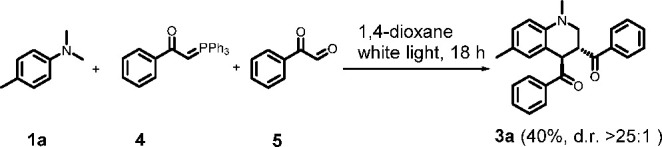
In order to improve the step economy of the reaction, we were interested in the possibility of running the annulation as a multicomponent version with 2a formed in situ. The one-pot version would circumvent a purification step and give potential access to a broader scope of the target compounds (3) while using simpler starting materials. As it turns out, the reaction between 1a, phosphonium ylide 4, and phenylglyoxal 5 works well, and 3a could be obtained in 40% yield (Scheme 5), which corresponds to a yield per bond formed of 80%.
Scheme 5. Multicomponent Synthesis of 3a.
To investigate the synthetic usefulness of the 3,4-dibenzoyl-THQ structure, compound 3a was treated with ammonium acetate in acetic acid45 to yield fused 3H-pyrrole derivative 6 (Scheme 6). Subjecting 3a to acid in acetic anhydride afforded fused furan derivative 7.46 These results highlight the possibility of efficient construction of fused heterocycles from the 3,4-dibenzoyl-THQ.
Scheme 6. Synthetic Applications.

NH4OAc, AcOH, 120 °C, 5 h.
Ac2O, HCl, 80 °C, 18 h.
In summary, a protocol for the visible-light-mediated synthesis of substituted THQs has been developed. The reaction requires no photocatalyst and relies on photoexcitation of an EDA complex formed between N-alkyl-N-methylaniline and a 1,2-dibenzoyl ethylene, where atmospheric oxygen functions as the terminal oxidant. A broad substrate scope is presented, demonstrating the tolerance for common functional groups in the reaction. The resulting 3,4-dibenzoyl-THQ structure is further derivatized and proven to be a useful building block for the construction of fused heterocycles. Synthetic applications of other EDA complexes are currently being explored by our research group.
Experimental Section
General Information
All reagents and solvents were purchased from Sigma-Aldrich and Alfa Aesar and used without any further purification unless specified. Purification of products was performed by an automated column chromatography Biotage Isolera Spektra One with Biotage SNAP-10 g KP-silica column together with a 1 g samplet cartridge using petroleum ether (40–60 °C)/ethyl acetate as the solvent mixture unless otherwise noted. 1H (400 MHz) and 13C (101 MHz) NMR spectra were acquired on an Agilent NMR machine at 25 °C. The chemical shifts for 1H and 13C NMR spectra are reported in parts per million (ppm) relative to the residual peak from solvent CDCl3 as the internal standard: 1H NMR at δ 7.26 ppm and 13C NMR at δ 77.0 ppm for CDCl3. All coupling constants (J) are reported in hertz (Hz) and multiplicities are indicated by s (singlet), d (doublet), dd (doublet of doublet), td (triplet of doublet), ddd (doublet of doublets of doublets), triplet (t), dt (doublet of triplet) and m (multiplet). Fourier-transform infrared (FT-IR) spectra were recorded on a PerkinElmer series FT-IR spectrometer and are reported in wavenumber (cm–1). High-resolution mass spectrometry measurements (HRMS) were performed by CMSI service at Chalmers University of Technology, Gothenburg using an Agilent 6520 equipped with an electrospray interface operated in the positive ionization mode with quadrupole time-of-flight mass analyzer. UV–vis absorption spectra were recorded on a Cary 4000 UV–vis spectrophotometer, using 1 × 1 cm quartz cuvettes. All light promoted reactions were carried out in Biotage microwave vials (10–20 mL) under irradiation with two 20 W white CFL bulbs (Osram, 1200 lm) at a distance of 5 cm. Gas chromatographic studies were carried out using an Agilent 7820A gas chromatograph with an Agilent HP-5 19091J-413 column, and detection was accomplished using a flame ionization detector. Crystals of 3q for single-crystal X-ray diffraction were grown using the layering technique from 3q in dichloromethane and fresh hexane. The colorless yellow prism-shaped crystals appeared after 4 days. A Bruker D8 VENTURE Kappa Duo with a PHOTON III detector was used for the data collection. Collections were carried out at low temperature [120(2) K] using a Cryostream SAINT (version 7.60a; Bruker AXS, 2016) software to perform data reduction and unit cell refinement. The atomic coordinates were located using direct methods employed by SHELXS.47 The successive refinements, once the atoms were placed in their postulated positions, were made using SHELXL.48 X-Seed49 was used for the data refinement. All non-hydrogen atoms were then refined anisotropically. The hydrogen atoms were placed in idealized positions in a riding model, after location on a Fourier difference map. An isotropic refinement was used for all hydrogen atoms, and temperature factors of 1.2 or 1.5 times that of the parent atoms were assigned.
Synthesis of Starting Materials
Amines 1i–k were synthesized following a reported method.50 To a solution of N-methylaniline (1.07 g, 10 mmol, 1 equiv) and potassium carbonate (4.8 g, 35 mmol, 3.5 equiv) in acetonitrile (9 mL) was added appropriate alkyl bromide (12 mmol, 1.2 equiv). The mixture was refluxed for 72 h and then cooled to room temperature. Solvent was removed under reduced pressure, and the residue was partitioned between water and dichloromethane. The aqueous phase was extracted three times with dichloromethane, and the combined organic phases were dried over anhydrous sulfate and concentrated under reduced pressure to yield an oily residue that was purified using column chromatography (SiO2, petroleum spirits/ethyl acetate, 9:1).
N-Ethyl-N-methylaniline (1i)
Purified using column chromatography (SiO2, petroleum spirits/ethyl acetate, 9:1), yellow oil, 900 mg (67%). Spectroscopic data in accordance with the literature:511H NMR (400 MHz, chloroform-d) δ 7.24 (m, 2H), 6.76–6.66 (m, 3H), 3.41 (qd, J = 7.1, 1.6 Hz, 2H), 2.91 (s, 3H), 1.16–1.10 (m, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d) δ 149.1, 129.2, 116.0, 112.4, 46.8, 37.4, 11.2 ppm.
N-Butyl-N-methylaniline (1j)
Purified using column chromatography (SiO2, petroleum spirits/ethyl acetate, 9:1), yellow oil 1.45 g (89%). Spectroscopic data in accordance with the literature:521H NMR (400 MHz, chloroform-d) δ 7.27–7.21 (m, 2H), 6.75–6.65 (m, 3H), 3.36–3.29 (m, 2H), 2.93 (s, 3H), 1.63–1.51 (m, 2H), 1.43–1.29 (m, 2H), 1.01–0.92 (t, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d) δ 149.5, 129.3, 115.9, 112.2, 52.7, 38.4, 29.0, 20.5, 14.1 ppm.
N-Benzyl-N-methylaniline (1k)
Purified using column chromatography (SiO2, petroleum spirits/ethyl acetate, 9:1), yellow oil, 1.2 g (60%). Spectroscopic data in accordance with the literature:521H NMR (400 MHz, chloroform-d) δ 7.35–7.22 (m, 6H), 7.19 (m, 2H), 6.63–6.56 (m, 2H), 4.51 (s, 2H), 3.01 (s, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d) δ 148.7, 138.5, 131.9, 128.8, 127.2, 126.7, 114.0, 108.5, 56.7, 38.9 ppm.
1,2-Dibenzoyl ethylenes 2b–j were synthesized according to a reported method.40 The appropriate acetophenone (2 mmol, 1 equiv), iodine (4 mmol, 2 equiv), and copper(II)bromide (0.4 mmol, 0.2 equiv) were heated in DMF (2 mL) at 80 °C for 18–24 h. The reaction mixture was then cooled to room temperature, and excess iodine was quenched by addition of sodium thiosulfate solution. The aqueous mixture was extracted three times with ethyl acetate, and the combined organic phases were dried over sodium sulfate. After removal of the solvent, the solid residue was recrystallized from boiling heptane/ethyl acetate to yield the desired substituted Z-1,2-DBEs 2b–j.
(E)-1,4-Di-p-tolylbut-2-ene-1,4-dione (2b)
Yellow solid, 100 mg (38%), spectroscopic data in accordance with the literature:401H NMR (400 MHz, chloroform-d) δ 8.01–7.94 (m, 6H), 7.35–7.30 (m, 4H), 2.44 (s, 6H) ppm; 13C{1H} NMR (101 MHz, chloroform-d) δ 189.5, 145.1, 135.1, 134.6, 129.7, 129.2, 21.9 ppm.
(E)-1,4-Bis(4-isobutylphenyl)but-2-ene-1,4-dione (2c)
Yellow solid, 193 mg (55%), spectroscopic data in accordance with the literature:401H NMR (400 MHz, chloroform-d) δ 8.04–7.96 (m, 6H), 7.30 (d, J = 8.2 Hz, 4H), 2.57 (d, J = 7.2 Hz, 4H), 1.93 (dt, J = 13.6, 6.8 Hz, 2H), 0.92 (d, J = 6.6 Hz, 12H) ppm; 13C{1H} NMR (101 MHz, chloroform-d) δ 189.6, 148.8, 135.1, 134.9, 129.8, 129.1, 45.6, 30.3, 22.5 ppm.
(E)-1,4-Bis(4-(tert-butyl)phenyl)but-2-ene-1,4-dione (2d)
Yellow solid, 180 mg (52%), spectroscopic data in accordance with the literature:401H NMR (400 MHz, chloroform-d) δ 8.05–7.99 (m, 6H), 7.58–7.52 (m, 4H), 1.36 (s, 18H) ppm; 13C{1H} NMR (101 MHz, chloroform-d) δ 189.6, 158.0, 135.1, 134.6, 129.1, 126.0, 35.4, 31.2 ppm.
(E)-1,4-Bis(3,4-dimethylphenyl)but-2-ene-1,4-dione (2e)
Yellow solid, 200 mg (68%); mp 139–141 °C; 1H NMR (400 MHz, chloroform-d) δ 8.00 (d, J = 2.4 Hz, 2H), 7.88–7.78 (m, 4H), 7.31–7.24 (m, 2H), 2.35 (s, 12H) ppm; 13C{1H} NMR (101 MHz, chloroform-d) δ 189.6, 143.7, 137.4, 134.9 (2C), 130.1, 123.0, 126.7, 20.2, 19.8 ppm; ATR-FTIR νmax = 1645, 1601, 1409, 1299, 961, 752, 723 cm–1; HRMS (ESI) m/z calcd C20H21O2 [M + H]+ 293.1536, found 293.1550.
(E)-1,4-Bis(4-methoxyphenyl)but-2-ene-1,4-dione (2f)
Yellow solid, 148 mg (50%), spectroscopic data in accordance with the literature:401H NMR (400 MHz, chloroform-d) δ 8.12–8.04 (m, 4H), 8.02 (s, 2H), 7.04–6.95 (m, 4H), 3.90 (s, 6H) ppm; 13C{1H} NMR (101 MHz, chloroform-d) δ 188.3, 164.3, 134.8, 131.5, 130.3, 114.3, 55.7 ppm.
(E)-1,4-Bis(4-(trifluoromethyl)phenyl)but-2-ene-1,4-dione (2g)
Yellow solid, 86 mg (24%), spectroscopic data in accordance with the literature:401H NMR (400 MHz, chloroform-d) δ 8.20–8.14 (m, 4H), 8.02 (s, 2H), 7.85–7.79 (m, 4H) ppm; 13C{1H} NMR (101 MHz, chloroform-d) δ 188.7, 139.4 (t, C–F, 4JC–F = 1.3 Hz), 135.35 (q, C–F, 2JC–F = 33 Hz), 135.27, 129.3, 126.2 (q, C–F, 3JC–F = 3.7 Hz), 123.6 (q, C–F, 1JC–F = 272.8 Hz) ppm.
(E)-1,4-Bis(4-bromophenyl)but-2-ene-1,4-dione (2h)
Yellow solid, 275 mg (70%); mp 191–192 °C; 1H NMR (400 MHz, chloroform-d) δ 7.97 (s, 2H), 7.91 (m, 4H), 7.72–7.65 (m, 4H) ppm; 13C{1H} NMR (101 MHz, chloroform-d) δ 188.7, 135.6, 135.0, 132.5, 130.5, 129.6 ppm; HRMS (ESI) m/z calcd C16H11Br2O2 [M + H]+ 392.9120, found 392.9124.
(E)-1,4-Bis(4-chlorophenyl)but-2-ene-1,4-dione (2i)
Yellow solid, 100 mg (33%), spectroscopic data in accordance with the literature:401H NMR (400 MHz, chloroform-d) δ 8.02–7.98 (m, 4H), 7.97 (s, 2H), 7.56–7.44 (m, 4H) ppm; 13C{1H} NMR (101 MHz, chloroform-d) δ 188.4, 140.7, 135.2, 135.0, 130.4, 129.4 ppm.
(E)-1-(4-Methoxyphenyl)-4-phenylbut-2-ene-1,4-dione (2j)
Isolated using column chromatography (SiO2, 5% ethyl acetate in petroleum ether), yellow solid, 50 mg (19%), spectroscopic data in accordance with the literature:401H NMR (400 MHz, chloroform-d) δ 8.12–8.03 (m, 4H), 8.00 (dd, J = 2.1, 0.5 Hz, 2H), 7.66–7.59 (m, 1H), 7.56–7.48 (m, 2H), 7.03–6.94 (m, 2H), 3.89 (s, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d) δ 190.06, 188.05, 164.34, 137.08, 135.41, 134.52, 133.90, 131.46, 130.11, 128.98, 114.26, 55.70 ppm.
General Procedure for the Oxidative Annulation Reaction
To a 10 mL microwave vial were added substituted 1,2-DBE 2 (0.25 mmol), appropriate aniline derivative 1 (1.75 mmol), 1,4-dioxane (6 mL), and glacial acetic acid (1 mL). The mixture was irradiated using two 20 W white light CLF lamps, with a distance from the vial of 5 cm, for 4 h. To allow sufficient atmospheric oxygen into the reaction mixture, the vial was kept open during the course of the reaction. Solvent was then removed under reduced pressure, and the residue was purified using column chromatography (SiO2, petroleum spirits/ethyl acetate) to yield the THQ 3.
(1,6-Dimethyl-1,2,3,4-tetrahydroquinoline-3,4-diyl)bis(phenylmethanone) (3a)
Purified using column chromatography (SiO2, 5% ethyl acetate in hexane), yellow solid, 67 mg, (73%); mp 123–124 °C; 1H NMR (400 MHz, chloroform-d) δ 8.11 (dd, J = 8.4, 1.4 Hz, 2H), 7.99–7.94 (m, 2H), 7.64–7.55 (m, 2H), 7.54–7.44 (m, 4H), 7.01–6.95 (m, 1H), 6.70–6.59 (m, 2H), 5.39 (dt, J = 9.2, 0.9 Hz, 1H), 4.49 (td, J = 9.6, 4.8 Hz, 1H), 3.49 (dd, J = 11.3, 4.9 Hz, 1H), 3.30 (dd, J = 11.3, 9.8 Hz, 1H), 2.90 (s, 3H), 2.11 (s, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d) δ 202.5, 200.5, 144.0, 138.0, 136.1, 133.53, 133.48, 129.1 (2C), 129.0 (2C), 128.9 (2C), 128.65 (2C), 128.6, 128.2, 126.9, 122.5, 112.2, 53.2, 46.2, 45.5, 39.5, 20.5 ppm; ATR-FTIR νmax = 1687, 1669, 1511, 1213, 967, 774, 690 cm–1; HRMS (ESI) m/z calcd C25H24NO2 [M + H]+ 370.1807, found 370.1825.
(1,6-Dimethyl-1,2,3,4-tetrahydroquinoline-3,4-diyl)bis(p-tolylmethanone) (3b)
Purified using column chromatography (SiO2, 5% ethyl acetate in hexane), yellow solid, 63 mg (63%); mp 150–153 °C; 1H NMR (400 MHz, chloroform-d) δ 8.03–7.96 (m, 2H), 7.88–7.81 (m, 2H), 7.32–7.21 (m, 4H), 6.95 (ddd, J = 8.3, 1.4, 0.8 Hz, 1H), 6.67–6.58 (m, 2H), 5.34 (d, J = 9.3 Hz, 1H), 4.44 (td, J = 9.7, 4.8 Hz, 1H), 3.45 (dd, J = 11.2, 4.8 Hz, 1H), 3.27 (dd, J = 11.3, 9.9 Hz, 1H), 2.89 (s, 3H), 2.43 (s, 3H), 2.40 (s, 3H), 2.09 (s, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d) δ 202.1, 200.1, 144.44, 144.35, 144.1, 135.6, 133.6, 129.7 (2C), 129.6 (2C), 129.3 (2C), 128.9 (2C), 128.6, 128.5, 126.8, 122.9, 112.1, 53.4, 46.0, 45.36, 39.60, 21.9, 21.8, 20.5 ppm; ATR-FTIR νmax = 1665, 1604, 1515, 1271, 1182, 828, 789 cm–1; HRMS (ESI) m/z calcd C27H28NO2 [M + H]+ 398.2120, found 398.2132.
(1,6-Dimethyl-1,2,3,4-tetrahydroquinoline-3,4-diyl)bis((4-isobutylphenyl)methanone) (3c)
Purified using column chromatography (SiO2, 5% ethyl acetate in hexane), yellow solid, 61.9 mg (52%); mp 113–114 °C; 1H NMR (400 MHz, chloroform-d) δ 8.00 (d, J = 8.3 Hz, 2H), 7.87 (d, J = 8.3 Hz, 2H), 7.29–7.19 (m, 4H), 6.95 (ddt, J = 8.3, 2.1, 0.8 Hz, 1H), 6.66–6.60 (m, 2H), 5.35 (d, J = 9.4 Hz, 1H), 4.46 (td, J = 9.7, 4.8 Hz, 1H), 3.47 (dd, J = 11.2, 4.8 Hz, 1H), 3.29 (dd, J = 11.2, 9.9 Hz, 1H), 2.90 (s, 3H), 2.54 (dd, J = 11.1, 7.2 Hz, 4H), 2.10 (s, 3H), 1.99–1.82 (m, 2H), 0.96–0.87 (m, 12H) ppm; 13C{1H} NMR (101 MHz, chloroform-d) δ 202.0, 200.1, 148.0, 147.9, 143.9, 135.8, 133.8, 129.5 (2C), 129.4 (2C), 128.9 (2C), 128.6 (2C), 128.50, 128.3, 126.6, 122.8, 112.0, 53.3, 46.1, 45.5, 45.4, 45.2, 39.5, 30.1, 22.4, 22.4, 22.3, 20.4 ppm; ATR-FTIR νmax = 1668, 1603, 1514, 1270, 1181, 860, 799 cm–1; HRMS (ESI) m/z calcd C33H40NO2 [M + H]+ 482.3059, found 482.3070.
(1,6-Dimethyl-1,2,3,4-tetrahydroquinoline-3,4-diyl)bis((4-(tert-butyl)phenyl)methanone) (3d)
Purified using column chromatography (SiO2, 5% ethyl acetate in hexane), yellow solid, 54.5 mg (45%); mp 179–181 °C; 1H NMR (400 MHz, chloroform-d) δ 8.02 (d, J = 8.6 Hz, 2H), 7.89 (d, J = 8.6 Hz, 2H), 7.47 (dd, J = 15.8, 8.6 Hz, 3H), 6.99–6.92 (m, 1H), 6.67–6.59 (m, 2H), 5.35 (d, J = 9.4 Hz, 1H), 4.46 (td, J = 9.7, 4.8 Hz, 1H), 3.47 (dd, J = 11.3, 4.8 Hz, 1H), 3.28 (dd, J = 11.3, 9.9 Hz, 1H), 2.90 (s, 3H), 2.10 (s, 3H), 1.36 (s, 9H), 1.33 (s, 9H) ppm; 13C{1H} NMR (101 MHz, chloroform-d) δ 202.0, 199.9, 157.2, 157.0, 143.9, 135.4, 133.4, 128.9 (2C), 128.6 (2C), 128.5, 128.3, 126.7, 125.8 (2C), 125.7 (2C), 122.8, 111.9, 53.2, 45.9, 45.2, 39.5, 35.1, 35.1, 31.1, 31.0, 20.4 ppm; ATR-FTIR νmax = 1668, 1604, 1514, 1270, 1188, 803 cm–1; HRMS (ESI) m/z calcd C33H40NO2 [M + H]+ 482.3059, found 482.3069.
(1,6-Dimethyl-1,2,3,4-tetrahydroquinoline-3,4-diyl)bis((3,4-dimethylphenyl)methanone) (3e)
Purified using column chromatography (SiO2, 5% ethyl acetate in hexane), yellow solid, 43.4 mg (46%); mp 129–130 °C; 1H NMR (400 MHz, chloroform-d) δ 7.85 (dd, J = 10.2, 2.4 Hz, 2H), 7.70 (dd, J = 10.2, 2.5 Hz, 2H), 7.25–7.18 (m, 2H), 6.95 (dd, J = 8.3, 2.2 Hz, 1H), 6.67–6.59 (m, 2H), 5.34 (d, J = 9.4 Hz, 1H), 4.45 (td, J = 9.7, 4.7 Hz, 1H), 3.49–3.41 (m, 1H), 3.28 (dd, J = 11.2, 10.0 Hz, 1H), 2.90 (d, J = 1.1 Hz, 3H), 2.34 (s, 3H), 2.32 (s, 3H), 2.31 (s, 3H), 2.29 (s, 3H), 2.09 (s, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d) δ 202.4, 200.4, 144.0, 143.2, 143.1, 137.23, 137.20, 136.1, 134.1, 130.2, 130.14, 130.08, 129.9, 128.6, 128.5, 127.0, 126.8, 126.5, 123.1, 112.0, 53.5, 46.0, 45.3, 39.6, 20.5, 20.22, 20.18, 20.0, 19.9 ppm; ATR-FTIR νmax = 1662, 1603, 1518, 1267, 1208, 794 cm–1; HRMS (ESI) m/z calcd C29H32NO2 [M + H]+ 426.2433, found 426.2436.
(1,6-Dimethyl-1,2,3,4-tetrahydroquinoline-3,4-diyl)bis((4-methoxyphenyl)methanone) (3f)
Purified using column chromatography (SiO2, 5% ethyl acetate in hexane), off-white solid, 53.3 mg (50%); mp 139–140 °C; 1H NMR (400 MHz, chloroform-d) δ 8.08 (d, J = 9.0 Hz, 2H), 7.95 (d, J = 8.9 Hz, 2H), 7.08–6.86 (m, 5H), 6.70–6.56 (m, 2H), 5.32 (dt, J = 9.7, 1.0 Hz, 1H), 4.44 (td, J = 10.0, 4.8 Hz, 1H), 3.88 (s, 3H), 3.86 (s, 3H), 3.44 (dd, J = 11.2, 4.8 Hz, 1H), 3.28 (dd, J = 11.2, 10.2 Hz, 1H), 2.90 (s, 3H), 2.09 (s, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d) δ 200.9, 199.0, 163.9, 163.8, 144.0, 131.5 (2C), 131.2, 131.0 (2C), 129.1, 128.5, 128.4, 126.8, 123.1, 114.1 (2C), 114.0 (2C), 112.2, 55.59, 55.58, 53.6, 46.0, 45.0, 39.6, 20.5 ppm; ATR-FTIR νmax = 1661, 1598, 1572, 1509, 1316, 1255, 1170, 845 cm–1; HRMS (ESI) m/z calcd C27H28NO4 [M + H]+ 430.2018, found 430.2024.
(1,6-Dimethyl-1,2,3,4-tetrahydroquinoline-3,4-diyl)bis((4-(trifluoromethyl)phenyl)methanone) (3g)
Purified using column chromatography (SiO2, 5% ethyl acetate in hexane), thick yellow-orange oil, 35 mg (28%); 1H NMR (400 MHz, chloroform-d) δ 8.25–8.13 (m, 2H), 8.08–8.01 (m, 2H), 7.83–7.71 (m, 4H), 6.99 (dq, J = 8.2, 0.9 Hz, 1H), 6.66 (d, J = 8.4 Hz, 1H), 6.54–6.48 (m, 1H), 5.32 (d, J = 9.5 Hz, 1H), 4.46 (td, J = 9.7, 5.1 Hz, 1H), 3.55–3.44 (m, 1H), 3.29 (dd, J = 11.2, 10.1 Hz, 1H), 2.90 (d, J = 0.7 Hz, 3H), 2.10 (s, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d) δ 201.6, 199.8, 143.8, 140.5, 138.7, 134.8 (q, C–F, 2JC–F = 33 Hz), 134.7 (q, C–F, 2JC–F = 33 Hz), 129.4 (2C), 129.0 (2C), 128.2, 127.3, 126.4–125.8 (m, C–F, 4C), 123.7 (q, C–F, 1JC–F = 272.6 Hz), 123.6 (q, C–F, 1JC–F = 272.6 Hz), 121.9, 121.2, 112.4, 52.9, 46.6, 46.2, 39.5, 20.46 ppm; 19F NMR (470 MHz, chloroform-d) δ −63.16(s, 3F), −63.23(s, 3F) ppm; ATR-FTIR νmax = 1683, 1512, 1322, 1170, 1128, 1067 cm–1; HRMS (ESI) m/z calcd C27H22F6NO2 [M + H]+ 506.1555, found 506.1548.
(1,6-Dimethyl-1,2,3,4-tetrahydroquinoline-3,4-diyl)bis((4-bromophenyl)methanone) (3h)
Purified using column chromatography (SiO2, 5% ethyl acetate in hexane), yellow solid, 113.4 mg (84%); mp 128–130 °C; 1H NMR (400 MHz, chloroform-d) δ 8.00–7.88 (m, 2H), 7.85–7.77 (m, 2H), 7.71–7.56 (m, 4H), 6.97 (ddt, J = 8.3, 2.2, 0.8 Hz, 1H), 6.65 (d, J = 8.3 Hz, 1H), 6.52 (dt, J = 2.0, 0.9 Hz, 1H), 5.30–5.16 (m, 1H), 4.39 (td, J = 9.9, 5.0 Hz, 1H), 3.44 (dd, J = 11.3, 5.0 Hz, 1H), 3.26 (dd, J = 11.3, 10.1 Hz, 1H), 2.90 (s, 3H), 2.09 (s, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d) δ 201.4, 199.6, 143.8, 136.67, 134.7, 132.34 (2C), 132.28 (2C), 130.6 (2C), 130.2 (2C), 128.99, 128.98, 128.8, 128.3, 127.1, 122.3, 112.3, 53.2, 46.3, 45.6, 39.5, 20.5 ppm; ATR-FTIR νmax = 2856, 1670, 1654, 1582, 1507, 1067, 1005, 816 cm–1; HRMS (ESI) m/z calcd C25H22Br2NO2 [M + H]+ 526.0017, found 526.0025.
(1,6-Dimethyl-1,2,3,4-tetrahydroquinoline-3,4-diyl)bis((4-chlorophenyl)methanone) (3i)
Purified using column chromatography (SiO2, 5% ethyl acetate in hexane), yellow solid, 83.1 mg (72%); mp 149–150 °C; 1H NMR (400 MHz, chloroform-d) δ 8.03 (d, J = 8.6 Hz, 2H), 7.89 (d, J = 8.7 Hz, 2H), 7.46 (dd, J = 16.1, 8.6 Hz, 4H), 6.97 (ddt, J = 8.3, 2.1, 0.8 Hz, 1H), 6.65 (d, J = 8.4 Hz, 1H), 6.54 (dt, J = 2.0, 0.9 Hz, 1H), 5.28 (dd, J = 9.6, 1.1 Hz, 1H), 4.41 (td, J = 9.9, 5.0 Hz, 1H), 3.45 (dd, J = 11.3, 5.0 Hz, 1H), 3.27 (dd, J = 11.2, 10.1 Hz, 1H), 2.90 (s, 3H), 2.10 (s, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d) δ 201.2, 199.4, 143.8, 140.2, 140.2, 136.3, 134.3, 130.5 (2C), 130.1 (2C), 129.34 (2C), 129.28 (2C), 128.8, 128.3, 127.0, 122.3, 112.2, 53.2, 46.3, 45.6, 39.5, 20.5 ppm; ATR-FTIR νmax = 2859, 1673, 1587, 1512, 1399, 1204, 1090, 818 cm–1; HRMS (ESI) m/z calcd C25H22Cl2NO2 [M + H]+ 438.1028, found 438.1028.
(3-Benzoyl-1,6-dimethyl-1,2,3,4-tetrahydroquinolin-4-yl)(4-methoxyphenyl)methanone (3ja) and (4-benzoyl-1,6-Dimethyl-1,2,3,4-tetrahydroquinolin-3-yl)(4-methoxyphenyl)methanone (3jb)
Purified using column chromatography (SiO2, 1% ethyl acetate in hexane), thick yellow oil, 66 mg (64%); 1H NMR (400 MHz, chloroform-d) δ 8.11–8.06 (m, overlapping 3ja and 3jb, 4H), 7.98–7.92 (m, overlapping 3ja and 3jb, 4H), 7.62–7.53 (m, overlapping 3ja and 3jb, 2H), 7.52–7.42 (m, overlapping 3ja and 3jb, 4H), 7.00–6.90 (m, overlapping 3ja and 3jb, 6H), 6.67–6.57 (m, overlapping 3ja and 3jb, 4H), 5.38–5.31 (m, overlapping 3ja and 3jb, 2H), 4.52–4.40 (m, overlapping 3ja and 3ja, 2H), 3.89 (s, OMe 3ja, 3H), 3.86 (s, OMe 3jb, 3H), 3.50–3.42 (m, overlapping 3ja and 3jb, 2H), 3.34–3.25 (m, overlapping 3ja and 3jb, 2H), 2.90 (s, NMe, 3H), 2.89 (s, NMe, 3H), 2.10 (s, ArMe 3ja, 3H), 2.09 (s, ArMe 3jb, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d) δ 202.7, 200.8, 200.7, 198.8, 163.93, 163.89, 144.0, 138.1, 136.1, 133.5, 133.4, 131.5 (2C), 131.12, 131.05 (2C), 130.5, 129.1 (2C), 128.92 (2C), 128.85 (2C), 128.7 (2C), 128.6, 128.52, 128.49 (2C), 128.2, 126.81, 126.76, 122.8, 114.1, 114.0, 112.11, 112.09, 55.6 (2C), 53.5, 53.3, 46.3, 45.8, 45.4, 45.1, 39.57, 39.55, 20.49, 20.47 ppm; ATR-FTIR νmax = 1660, 1598, 1572, 1509, 1317, 1257, 1170 cm–1; HRMS (ESI) m/z calcd C26H26NO3 [M + H]+ 400.1911, found 400.1913.
(1-Methyl-1,2,3,4-tetrahydroquinoline-3,4-diyl)bis(phenylmethanone) (3k)
Purified using column chromatography (SiO2, 5% ethyl acetate in hexane), yellow oil, 44.3 mg (59%); mp 104–105 °C; 1H NMR (400 MHz, chloroform-d) δ 8.09 (d, J = 8.4 Hz, 2H), 8.00–7.94 (m, 2H), 7.59 (dtd, J = 8.5, 7.3, 1.3 Hz, 2H), 7.53–7.43 (m, 4H), 7.14 (tt, J = 7.2, 0.9 Hz, 1H), 6.78 (dd, J = 7.6, 1.3 Hz, 1H), 6.71 (dt, J = 8.4, 1.2 Hz, 1H), 6.60–6.54 (m, 1H), 5.38 (dd, J = 9.4, 1.2 Hz, 1H), 4.55–4.46 (m, 1H), 3.56–3.46 (m, 1H), 3.35 (ddd, J = 11.2, 10.1, 1.1 Hz, 1H), 2.93 (d, J = 1.1 Hz, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d) δ 202.1, 200.5, 145.8, 137.9, 136.0, 133.5, 133.4, 129.0 (2C), 128.84 (2C), 128.77 (2C), 128.6 (2C), 127.9, 127.8, 122.1, 117.3, 111.7, 52.9, 46.1, 45.0, 39.2 ppm; ATR-FTIR νmax = 1669, 1598, 1578,1506, 1448, 1228, 1202, 753, 703 cm–1; HRMS (ESI) m/z calcd C24H22NO2 [M + H]+ 356.1651, found 356.1661.
(1,5,7-Trimethyl-1,2,3,4-tetrahydroquinoline-3,4-diyl)bis(phenylmethanone) (3m)
Purified using column chromatography (SiO2, 5% ethyl acetate in hexane), white solid, 39.9 mg (42%); mp 198–199 °C; 1H NMR (400 MHz, chloroform-d) δ 8.10–8.02 (m, 2H), 7.78 (dd, J = 7.4, 1.2 Hz, 2H), 7.63–7.52 (m, 2H), 7.52–7.40 (m, 4H), 6.52–6.38 (m, 2H), 5.46–5.33 (m, 1H), 3.83 (q, J = 1.5 Hz, 1H), 3.46 (dd, J = 11.9, 3.4 Hz, 1H), 3.41–3.31 (m, 1H), 2.71 (s, 3H), 2.26 (s, 3H), 2.06 (s, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d) δ 201.8, 198.9, 146.9, 137.0, 136.3, 136.1, 135.6, 133.5, 133.0, 129.1 (2C), 128.8 (2C), 128.6 (2C), 128.3 (2C), 121.6, 116.0, 111.0, 50.4, 44.3, 43.2, 40.1, 21.8, 20.6 ppm; ATR-FTIR νmax = 1680, 1577, 1447, 1331, 1268, 1214, 970, 702 cm–1; HRMS (ESI) m/z calcd C26H26NO2 [M + H]+ 384.1964, found 384.1991.
(6-Methoxy-1-methyl-1,2,3,4-tetrahydroquinoline-3,4-diyl)bis(phenylmethanone) (3n)
Purified using column chromatography (SiO2, 5% ethyl acetate in hexane), yellow solid, 58.6 mg (61%); 129–130 °C; 1H NMR (400 MHz, chloroform-d) δ 8.14–8.04 (m, 2H), 7.96 (dt, J = 7.0, 1.5 Hz, 2H), 7.58 (dddd, J = 9.0, 7.0, 5.2, 1.4 Hz, 2H), 7.53–7.39 (m, 4H), 6.81–6.61 (m, 2H), 6.43 (dd, J = 2.9, 1.3 Hz, 1H), 5.41 (d, J = 9.2 Hz, 1H), 4.51 (tdd, J = 9.5, 4.9, 1.3 Hz, 1H), 3.57 (s, 3H), 3.46 (ddd, J = 11.2, 5.0, 1.3 Hz, 1H), 3.29–3.19 (m, 1H), 2.87 (s, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d) δ 201.9, 200.4, 152.0, 140.9, 138.0, 136.1, 133.57, 133.56, 129.1 (2C), 129.0 (2C), 128.9 (2C), 128.7 (2C), 123.9, 114.6, 113.3, 113.2, 55.7, 53.5, 46.2, 45.4, 40.0 ppm; ATR-FTIR νmax = 1663, 1594, 1579, 1510, 1447, 1209, 803, 689 cm–1; HRMS (ESI) m/z calcd C25H24NO3 [M + H]+ 386.1756, found 386.1753.
(7-Methoxy-1-methyl-1,2,3,4-tetrahydroquinoline-3,4-diyl)bis(phenylmethanone) (3o)
Purified using column chromatography (SiO2, 5% ethyl acetate in hexane), yellow solid, 26.9 mg (28%); mp 152–153 °C; 1H NMR (400 MHz, chloroform-d) δ 8.07–8.00 (m, 2H), 7.85–7.77 (m, 2H), 7.57–7.49 (m, 2H), 7.46–7.38 (m, 4H), 7.13 (td, J = 8.3, 0.6 Hz, 1H), 6.42 (dd, J = 8.4, 0.8 Hz, 1H), 6.30 (dd, J = 8.2, 0.9 Hz, 1H), 5.46 (d, J = 5.5 Hz, 1H), 4.00 (ddd, J = 6.5, 5.5, 3.4 Hz, 1H), 3.50 (s, 3H), 3.44 (ddd, J = 11.7, 3.4, 0.6 Hz, 1H), 3.36–3.28 (m, 1H), 2.83 (s, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d) δ 203.3, 199.6, 157.0, 148.0, 137.0, 136.1, 133.2, 132.7, 128.8 (2C), 128.57 (2C), 128.55 (2C), 128.4 (2C), 128.2, 110.5, 105.8, 100.6, 55.1, 51.6, 44.6, 41.5, 40.0 ppm; ATR-FTIR νmax = 1677, 1579, 1447, 1211, 967, 700 cm–1; HRMS (ESI) m/z calcd C25H24NO3 [M + H]+ 386.1756, found 386.1751.
(6-Fluoro-1-methyl-1,2,3,4-tetrahydroquinoline-3,4-diyl)bis(phenylmethanone) (3p)
Purified using column chromatography (SiO2, 5% ethyl acetate in hexane), yellow solid, 49.9 mg (51%); mp 132–133 °C; 1H NMR (400 MHz, chloroform-d) δ 8.13–8.07 (m, 2H), 8.00–7.90 (m, 2H), 7.65–7.55 (m, 2H), 7.54–7.43 (m, 4H), 6.85 (dddd, J = 9.0, 8.1, 2.9, 0.8 Hz, 1H), 6.63 (dd, J = 9.0, 4.8 Hz, 1H), 6.54 (ddd, J = 9.4, 2.9, 1.1 Hz, 1H), 5.38 (dd, J = 8.9, 1.0 Hz, 1H), 4.46 (td, J = 9.2, 4.9 Hz, 1H), 3.50 (dd, J = 11.4, 4.9 Hz, 1H), 3.29 (dd, J = 11.3, 9.5 Hz, 1H), 2.88 (s, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d) δ 201.5, 200.1, 155.5(d, C–F, 1JC–F = 236.2 Hz), 142.8(d, C–F, 4JC–F = 1.9 Hz), 137.5, 135.9, 133.8, 133.7, 129.10 (2C), 129.06 (2C), 128.9 (2C), 128.6 (2C), 123.4 (d, C–F, 3JC–F = 6.9 Hz), 114.82 (d, C–F, 2JC–F = 23.3 Hz), 114.31 (d, C–F, 2JC–F = 21.8 Hz), 112.8 (d, C–F, 3JC–F = 7.7 Hz), 53.00, 45.86, 45.07, 39.77 ppm; 19F NMR (470 MHz, chloroform-d) δ −127.15 to −127.23(m) ppm; ATR-FTIR νmax = 1670, 1591, 1577, 1505, 1448, 1211, 906, 729, 685 cm–1; HRMS (ESI) m/z calcd C24H21FNO2 [M + H]+ 374.1556, found 374.1572.
(6-Bromo-1-methyl-1,2,3,4-tetrahydroquinoline-3,4-diyl)bis(phenylmethanone) (3q)
Purified using column chromatography (SiO2, 5% ethyl acetate in hexane), off-white solid, 26 mg (24%); mp 192–194 °C; 1H NMR (400 MHz, chloroform-d) δ 8.10–8.03 (m, 2H), 7.97–7.88 (m, 2H), 7.67–7.55 (m, 2H), 7.54–7.42 (m, 4H), 7.22 (ddt, J = 8.7, 1.6, 0.7 Hz, 1H), 6.88–6.84 (m, 1H), 6.55 (dd, J = 8.9, 1.2 Hz, 1H), 5.36–5.30 (m, 1H), 4.44–4.33 (m, 1H), 3.53 (ddd, J = 11.5, 4.7, 1.3 Hz, 1H), 3.34 (ddd, J = 11.8, 9.3, 1.2 Hz, 1H), 2.88 (d, J = 1.3 Hz, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d) δ 201.6, 200.0, 145.1, 137.5, 135.9, 133.9, 133.7, 130.8, 130.6, 129.2 (2C), 129.1 (2C), 129.0 (2C), 128.7 (2C), 123.8, 113.3, 109.3, 52.5, 45.8, 44.8, 39.4 ppm; ATR-FTIR νmax = 1686, 1668, 1591, 1499, 1447, 1209, 699, 691 cm–1; HRMS (ESI) m/z calcd C24H21BrNO2 [M + H]+ 434.0756, found 434.0752.
(1-Methyl-1,2,3,4-tetrahydrobenzo[h]quinoline-3,4-diyl)bis(phenylmethanone) (3r)
Purified using column chromatography (SiO2, 5% ethyl acetate in hexane), white solid, 19.2 mg (18%); mp 197–200 °C; 1H NMR (400 MHz, chloroform-d) δ 8.26–8.15 (m, 2H), 8.04–7.99 (m, 2H), 7.77–7.72 (m, 1H), 7.67–7.42 (m, 9H), 7.38 (d, J = 8.6 Hz, 1H), 6.99 (d, J = 8.5 Hz, 1H), 5.85 (d, J = 9.8 Hz, 1H), 4.82 (ddd, J = 11.3, 9.8, 3.3 Hz, 1H), 3.66 (dd, J = 13.5, 3.3 Hz, 1H), 3.39–3.28 (m, 1H), 3.28 (s, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d) δ 203.0, 200.4, 138.3, 136.1, 133.7, 133.7, 133.6, 129.4 (2C), 129.09 (2C), 129.05 (2C), 128.99, 128.68, 128.6 (2C), 128.5, 126.0, 125.9, 124.0 (2C), 123.96, 123.89, 54.7, 46.0, 44.7, 40.3 ppm; ATR-FTIR νmax = 1667, 1596, 1446, 1266, 1216, 1001, 986, 797, 783 cm–1; HRMS (ESI) m/z calcd C28H24NO2 [M + H]+ 406.1807, found 406.1826.
(1-Ethyl-1,2,3,4-tetrahydroquinoline-3,4-diyl)bis(phenylmethanone) (3s)
Purified using column chromatography (SiO2, 1% ethyl acetate in hexane), yellow oil, 42.4 mg (46%); 1H NMR (400 MHz, chloroform-d) δ 8.13–8.06 (m, 2H), 8.03–7.91 (m, 2H), 7.64–7.55 (m, 2H), 7.54–7.42 (m, 4H), 7.11 (t, J = 7.8 Hz, 1H), 6.82–6.69 (m, 2H), 6.53 (dd, J = 8.3, 6.7 Hz, 1H), 5.38 (d, J = 9.4 Hz, 1H), 4.59–4.40 (m, 1H), 3.64–3.18 (m, 4H), 1.16 (t, J = 6.88 Hz, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d) δ 202.3, 200.9, 144.3, 138.2, 136.2, 133.6, 133.5, 129.1 (2C), 129.0 (2C), 128.9 (2C), 128.7 (2C), 128.4, 128.1, 121.6, 116.7, 111.6, 50.3, 46.6, 45.7, 44.6, 11.1 ppm; ATR-FTIR νmax = 1675, 1597, 1495, 1449, 1346, 743, 691 cm–1; HRMS (ESI) m/z calcd C25H24NO2 [M + H]+ 370.1807, found 370.1817.
(1-Butyl-1,2,3,4-tetrahydroquinoline-3,4-diyl)bis(phenylmethanone) (3t)
Purified using column chromatography (SiO2, 1% ethyl acetate in hexane), yellow oil, 57.4 mg (60%); 1H NMR (400 MHz, chloroform-d) δ8.12–8.07 (m, 2H), 8.00–7.93 (m, 2H), 7.64–7.53 (m, 2H), 7.54–7.42 (m, 4H), 7.10 (dddd, J = 8.2, 7.3, 1.6, 0.8 Hz, 1H), 6.76 (dt, J = 7.6, 1.4 Hz, 1H), 6.69 (dd, J = 8.4, 1.1 Hz, 1H), 6.51 (td, J = 7.4, 1.2 Hz, 1H), 5.37 (d, J = 9.5 Hz, 1H), 4.47 (ddd, J = 10.1, 9.4, 4.2 Hz, 1H), 3.57 (dd, J = 11.6, 4.3 Hz, 1H), 3.47–3.31 (m, 2H), 3.28–3.15 (m, 1H), 1.64–1.52 (m, 2H), 1.42–1.30 (m, 2H), 0.94 (t, J = 7.3 Hz, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d) δ 202.3, 200.8, 144.5, 138.2, 136.1, 133.63, 133.56, 129.1 (2C), 129.0 (2C), 128.9 (2C), 128.7 (2C), 128.4, 128.1, 121.5, 116.8, 111.8, 51.7, 51.3, 46.6, 44.5, 28.5, 20.5, 14.1 ppm; ATR-FTIR νmax = 1678, 1599, 1502, 1457, 1449, 1218, 744 cm–1; HRMS (ESI) m/z calcd C27H28NO2 [M + H]+ 398.2120, found 398.2110.
(1-Benzyl-1,2,3,4-tetrahydroquinoline-3,4-diyl)bis(phenylmethanone) (3u)
Purified using column chromatography (SiO2, 1% ethyl acetate in hexane), yellow oil, 26.1 mg (25%); 1H NMR (400 MHz, chloroform-d) δ 8.16–8.10 (m, 2H), 7.89–7.82 (m, 2H), 7.65–7.59 (m, 1H), 7.57–7.50 (m, 3H), 7.43–7.37 (m, 2H), 7.37–7.26 (m, 5H), 7.11–7.01 (m, 1H), 6.83 (d, J = 7.4 Hz, 1H), 6.74 (d, J = 8.3 Hz, 1H), 6.59 (t, J = 7.5 Hz, 1H), 5.48 (d, J = 9.2 Hz, 1H), 4.64–4.47 (m, 3H), 3.67 (dd, J = 11.8, 4.3 Hz, 1H), 3.52–3.41 (m, 1H) ppm; 13C{1H} NMR (101 MHz, chloroform-d) δ 202.3, 200.5, 145.0, 138.2, 138.0, 135.9, 133.6, 129.1 (2C), 129.0 (2C), 128.89 (2C), 128.87 (2C), 128.6 (2C), 128.4, 128.2, 127.9, 127.0 (2C), 125.9, 121.4, 117.4, 112.3, 55.4, 51.2, 46.4, 44.7 ppm; ATR-FTIR νmax = 1677, 1599, 1497, 1449, 1220, 749, 696 cm–1; HRMS (ESI) m/z calcd C30H26NO2 [M + H]+ 432.1958, found 432.1971.
Multicomponent Synthesis of 3a
For the multicomponent reaction, Wittig reagent 4 (74.4 mg, 0.2 mmol, 1 equiv), phenylglyoxal (35.5 mg, 0.26 mmol, 1.4 equiv), and amine 1a (164 mg, 1.2 mmol, 6.2 equiv) were mixed in 1,4-dioxane (6 mL), and the reaction mixture was irradiated according to the general procedure for 18 h. Solvent was then removed under reduced pressure, and the product was isolated using flash chromatography (silica gel, 5% ethyl acetate in petroleum ethers) to yield the desired product 3a (29 mg, 40%).
Synthesis of 3a on 1 mmol Scale
To a round-bottom flask were added 1,2-DBE 2a (245.4 mg, 1 mmol), 4′,N,N-trimethylaniline 1a (947 mg, 7 mmol), 1,4-dioxane (25 mL), and glacial acetic acid (4 mL). The mixture was irradiated using two 20 W white light CLF lamps, with a distance from the flask of 5 cm, for 18 h. To allow sufficient atmospheric oxygen into the reaction mixture, the flask was kept open during the course of the reaction. Solvent was then removed under reduced pressure, and the residue was purified using column chromatography (SiO2, 5% ethyl acetate in petroleum spirits) to yield the THQ 3a (204.1 mg, 53%).
Condensation Reaction of 3a with Ammonium Acetate to Pyrrole Derivative 6
Following a modified published procedure,45 diketo compound 3a (106.3 mg, 0.29 mmol, 1 equiv) was dissolved in glacial acetic acid (1.8 mL). Ammonium acetate (189 mg, 2.4 mmol, 8.2 equiv) was then added, and the mixture was heated to 120 °C using a heating block in a sealed vial for 4 h. The resulting deep red solution was concentrated in vacuo, and the product was isolated using column chromatography (SiO2, 5% methanol in dichloromethane) as dark red crystals, 43 mg (43%); mp 223–225 °C; 1H NMR (400 MHz, chloroform-d) δ 8.50 (s, 1H), 8.09 (dd, J = 2.1, 1.0 Hz, 1H), 7.81 (ddd, J = 8.2, 7.2, 1.3 Hz, 4H), 7.47 (t, J = 7.5 Hz, 2H), 7.43–7.29 (m, 4H), 7.22–7.14 (m, 2H), 3.99 (s, 3H), 2.30 (s, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d) δ 145.1, 142.6, 137.5, 137.0, 136.6, 134.9, 130.7, 129.7 (2C), 128.8 (2C), 128.4 (2C), 128.0 (2C), 127.6, 127.4, 127.3, 124.2, 124.0, 117.4, 116.3, 115.4, 43.4, 21.5 ppm; ATR-FTIR νmax = 1591, 1454, 1356, 1225, 807, 765, 694 cm–1; HRMS (ESI) m/z calcd C25H21N2 [M + H]+ 349.1704, found 349.1717.
Synthesis of Furan Derivative 7
Following a modified published procedure,46 diketo compound 3a (40.7 mg, 0.11 mmol) was dissolved in acetic anhydride (1.0 mL). Concentrated hydrochloric acid (0.3 mL) was then added at 0 °C. The mixture was heated to 80 °C using a heating block in a sealed vial for 18 h. The resulting orange solution was concentrated in vacuo, and the product was isolated using column chromatography (SiO2, 2% ethyl acetate in hexane) as an air sensitive yellow-orange oil, 21.8 mg (56%); 1H NMR (400 MHz, chloroform-d) δ 7.88–7.81 (m, 2H), 7.61–7.55 (m, 2H), 7.51 (d, J = 2.1 Hz, 1H), 7.49–7.40 (m, 4H), 7.40–7.33 (m, 1H), 7.30 (ddt, J = 7.5, 5.8, 1.0 Hz, 1H), 7.05–6.98 (m, 1H), 6.74 (d, J = 8.3 Hz, 1H), 4.30 (s, 2H), 2.97 (s, 3H), 2.19 (s, 3H) ppm; 13C{1H} NMR (101 MHz, chloroform-d) δ 146.4, 145.1, 144.0, 131.9, 130.9, 129.0 (2C), 128.9 (2C), 128.7, 128.3, 127.9, 127.4 (2C), 127.3, 125.19 (2C), 125.15, 119.24, 119.16, 117.5, 113.2, 49.4, 39.5, 20.6 ppm; ATR-FTIR νmax = 2924, 1598, 1493, 1448, 1242, 1126 cm–1; HRMS (ESI) m/z calcd C25H22NO [M + H]+ 352.1701, found 352.1697.
Acknowledgments
This work was supported by grants from the Swedish research council FORMAS (2019-00699) and from Olle Engkvists Stiftelse. The X-ray crystallographic data used in this work was collected by Ivana Císařová, Department of Inorganic Chemistry, Faculty of Science, Charles University, Hlavova 2030/8, 128 43 Prague 2, Czech Republic.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.0c02819.
Photophysical measurements, synthesis procedures, NMR characterization data, and X-ray crystallographic data for 3q (CCDC 2008622) (PDF)
Accession Codes
CCDC 2008622 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The authors declare no competing financial interest.
Supplementary Material
References
- Yuan Y.; Majumder S.; Yang M.; Guo S. Recent Advances in Catalyst-Free Photochemical Reactions via Electron-Donor-Acceptor (EDA) Complex Process. Tetrahedron Lett. 2020, 61 (8), 151506. 10.1016/j.tetlet.2019.151506. [DOI] [Google Scholar]
- Wei Y.; Zhou Q.-Q.; Tan F.; Lu L.-Q.; Xiao W.-J. Visible-Light-Driven Organic Photochemical Reactions in the Absence of External Photocatalysts. Synthesis 2019, 51 (16), 3021–3054. 10.1055/s-0037-1611812. [DOI] [Google Scholar]
- Crisenza G. E. M.; Mazzarella D.; Melchiorre P. Synthetic Methods Driven by the Photoactivity of Electron Donor-Acceptor Complexes. J. Am. Chem. Soc. 2020, 142 (12), 5461–5476. 10.1021/jacs.0c01416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima C. G. S.; de M. Lima T.; Duarte M.; Jurberg I. D.; Paixão M. W. Organic Synthesis Enabled by Light-Irradiation of EDA Complexes: Theoretical Background and Synthetic Applications. ACS Catal. 2016, 6 (3), 1389–1407. 10.1021/acscatal.5b02386. [DOI] [Google Scholar]
- Postigo A. Electron Donor-Acceptor Complexes in Perfluoroalkylation Reactions. Eur. J. Org. Chem. 2018, 2018 (46), 6391–6404. 10.1002/ejoc.201801079. [DOI] [Google Scholar]
- Guo W.; Tao K.; Xie Z.; Cai L.; Zhao M.; Tan W.; Liu G.; Mei W.; Deng L.; Fan X.; Zheng L. Photodriven Photocatalyst/Metal-Free Direct C-C/C-N Bond Formation: Synthesis of Indoles via EDA Complexes. J. Org. Chem. 2019, 84 (21), 14168–14178. 10.1021/acs.joc.9b01689. [DOI] [PubMed] [Google Scholar]
- Li Z.; Ma P.; Tan Y.; Liu Y.; Gao M.; Zhang Y.; Yang B.; Huang X.; Gao Y.; Zhang J. Photocatalyst- And Transition-Metal-Free α-Allylation of: N -Aryl Tetrahydroisoquinolines Mediated by Visible Light. Green Chem. 2020, 22 (3), 646–650. 10.1039/C9GC04191E. [DOI] [Google Scholar]
- Yang X.; Zhu Y.; Xie Z.; Li Y.; Zhang Y. Visible-Light-Induced Charge Transfer Enables Csp3-H Functionalization of Glycine Derivatives: Access to 1,3-Oxazolidines. Org. Lett. 2020, 22 (4), 1638–1643. 10.1021/acs.orglett.0c00234. [DOI] [PubMed] [Google Scholar]
- Saritha R.; Annes S. B.; Saravanan S.; Ramesh S. Carbazole Based Electron Donor Acceptor (EDA) Catalysis for the Synthesis of Biaryl and Aryl-Heteroaryl Compounds. Org. Biomol. Chem. 2020, 18 (13), 2510–2515. 10.1039/D0OB00282H. [DOI] [PubMed] [Google Scholar]
- Chen L.; Liang J.; Chen Z.; Chen J.; Yan M.; Zhang X. A Convenient Synthesis of Sulfones via Light Promoted Coupling of Sodium Sulfinates and Aryl Halides. Adv. Synth. Catal. 2018, 361 (5), 956–960. 10.1002/adsc.201801390. [DOI] [Google Scholar]
- Morack T.; Mück-Lichtenfeld C.; Gilmour R. Bioinspired Radical Stetter Reaction: Radical Umpolung Enabled by Ion-Pair Photocatalysis. Angew. Chem., Int. Ed. 2019, 58 (4), 1208–1212. 10.1002/anie.201809601. [DOI] [PubMed] [Google Scholar]
- Bartolomeu A. de A.; Silva R. C.; Brocksom T. J.; Noël T.; de Oliveira K. T. Photoarylation of Pyridines Using Aryldiazonium Salts and Visible Light: An EDA Approach. J. Org. Chem. 2019, 84 (16), 10459–10471. 10.1021/acs.joc.9b01879. [DOI] [PubMed] [Google Scholar]
- Arceo E.; Bahamonde A.; Bergonzini G.; Melchiorre P. Enantioselective Direct α-Alkylation of Cyclic Ketones by Means of Photo-Organocatalysis. Chem. Sci. 2014, 5 (6), 2438–2442. 10.1039/c4sc00315b. [DOI] [Google Scholar]
- Cao Z.-Y.; Ghosh T.; Melchiorre P. Enantioselective Radical Conjugate Additions Driven by a Photoactive Intramolecular Iminium-Ion-Based EDA Complex. Nat. Commun. 2018, 9 (1), 3274. 10.1038/s41467-018-05375-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woźniak Ł.; Murphy J. J.; Melchiorre P. Photo-Organocatalytic Enantioselective Perfluoroalkylation of β-Ketoesters. J. Am. Chem. Soc. 2015, 137 (17), 5678–5681. 10.1021/jacs.5b03243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandukuri S. R.; Bahamonde A.; Chatterjee I.; Jurberg I. D.; Escudero-Adán E. C.; Melchiorre P. X-Ray Characterization of an Electron Donor-Acceptor Complex That Drives the Photochemical Alkylation of Indoles. Angew. Chem., Int. Ed. 2015, 54 (5), 1485–1489. 10.1002/anie.201409529. [DOI] [PubMed] [Google Scholar]
- Arceo E.; Jurberg I. D.; Álvarez-Fernández A.; Melchiorre P. Photochemical Activity of a Key Donor-Acceptor Complex Can Drive Stereoselective Catalytic α-Alkylation of Aldehydes. Nat. Chem. 2013, 5 (9), 750–756. 10.1038/nchem.1727. [DOI] [PubMed] [Google Scholar]
- Tobisu M.; Furukawa T.; Chatani N. Visible Light-Mediated Direct Arylation of Arenes and Heteroarenes Using Diaryliodonium Salts in the Presence and Absence of a Photocatalyst. Chem. Lett. 2013, 42 (10), 1203–1205. 10.1246/cl.130547. [DOI] [Google Scholar]
- Hsu C.-W.; Sundén H. α-Aminoalkyl Radical Addition to Maleimides via Electron Donor-Acceptor Complexes. Org. Lett. 2018, 20 (7), 2051–2054. 10.1021/acs.orglett.8b00597. [DOI] [PubMed] [Google Scholar]
- Sridharan V.; Suryavanshi P. A.; Menéndez J. C. Advances in the Chemistry of Tetrahydroquinolines. Chem. Rev. 2011, 111 (11), 7157–7259. 10.1021/cr100307m. [DOI] [PubMed] [Google Scholar]
- Muthukrishnan I.; Sridharan V.; Menéndez J. C. Progress in the Chemistry of Tetrahydroquinolines. Chem. Rev. 2019, 119 (8), 5057–5191. 10.1021/acs.chemrev.8b00567. [DOI] [PubMed] [Google Scholar]
- Su D.-S.; Lim J. J.; Tinney E.; Wan B.-L.; Young M. B.; Anderson K. D.; Rudd D.; Munshi V.; Bahnck C.; Felock P. J.; Lu M.; Lai M.-T.; Touch S.; Moyer G.; DiStefano D. J.; Flynn J. A.; Liang Y.; Sanchez R.; Prasad S.; Yan Y.; Perlow-Poehnelt R.; Torrent M.; Miller M.; Vacca J. P.; Williams T. M.; Anthony N. J. Substituted Tetrahydroquinolines as Potent Allosteric Inhibitors of Reverse Transcriptase and Its Key Mutants. Bioorg. Med. Chem. Lett. 2009, 19 (17), 5119–5123. 10.1016/j.bmcl.2009.07.031. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Zhan P.; Wu J.; Li Z.; Jiang Y.; Ge W.; Pannecouque C.; De Clercq E.; Liu X. Synthesis and Biological Evaluation of Novel 5-Alkyl-2-Arylthio-6-((3,4-Dihydroquinolin-1(2H)-Yl)Methyl)Pyrimidin-4(3H)-Ones as Potent Non-Nucleoside HIV-1 Reverse Transcriptase Inhibitors. Bioorg. Med. Chem. 2011, 19 (14), 4366–4376. 10.1016/j.bmc.2011.05.024. [DOI] [PubMed] [Google Scholar]
- Chander S.; Wang P.; Ashok P.; Yang L.-M.; Zheng Y.-T.; Murugesan S. Rational Design, Synthesis, Anti-HIV-1 RT and Antimicrobial Activity of Novel 3-(6-Methoxy-3,4-Dihydroquinolin-1(2H)-Yl)-1-(Piperazin-1-Yl)Propan-1-One Derivatives. Bioorg. Chem. 2016, 67, 75–83. 10.1016/j.bioorg.2016.05.009. [DOI] [PubMed] [Google Scholar]
- Ramesh E.; Manian R. D. R. S.; Raghunathan R.; Sainath S.; Raghunathan M. Synthesis and Antibacterial Property of Quinolines with Potent DNA Gyrase Activity. Bioorg. Med. Chem. 2009, 17 (2), 660–666. 10.1016/j.bmc.2008.11.058. [DOI] [PubMed] [Google Scholar]
- Jarvest R. L.; Berge J. M.; Berry V.; Boyd H. F.; Brown M. J.; Elder J. S.; Forrest A. K.; Fosberry A. P.; Gentry D. R.; Hibbs M. J.; Jaworski D. D.; O’Hanlon P. J.; Pope A. J.; Rittenhouse S.; Sheppard R. J.; Slater-Radosti C.; Worby A. Nanomolar Inhibitors of Staphylococcus Aureus Methionyl TRNA Synthetase with Potent Antibacterial Activity against Gram-Positive Pathogens. J. Med. Chem. 2002, 45 (10), 1959–1962. 10.1021/jm025502x. [DOI] [PubMed] [Google Scholar]
- Muñoz A.; Sojo F.; Arenas D. R. M.; Kouznetsov V. V.; Arvelo F. Cytotoxic Effects of New Trans-2,4-Diaryl-r-3-Methyl-1,2,3,4-Tetrahydroquinolines and Their Interaction with Antitumoral Drugs Gemcitabine and Paclitaxel on Cellular Lines of Human Breast Cancer. Chem.-Biol. Interact. 2011, 189 (3), 215–221. 10.1016/j.cbi.2010.11.010. [DOI] [PubMed] [Google Scholar]
- Kouznetsov V. V.; Merchan-Arenas D. R.; Tangarife-Castaño V.; Correa-Royero J.; Betancur-Galvis L. Synthesis and Cytotoxic Evaluation of Novel 2-Aryl-4-(4-Hydroxy-3-Methoxyphenyl)-3-Methyl-6,7-Methylendioxy-1,2,3,4-Tetrahydroquinolines, Podophyllotoxin-like Molecules. Med. Chem. Res. 2016, 25 (3), 429–437. 10.1007/s00044-015-1486-6. [DOI] [Google Scholar]
- Zhu S.; Das A.; Bui L.; Zhou H.; Curran D. P.; Rueping M. Oxygen Switch in Visible-Light Photoredox Catalysis: Radical Additions and Cyclizations and Unexpected C-C-Bond Cleavage Reactions. J. Am. Chem. Soc. 2013, 135 (5), 1823–1829. 10.1021/ja309580a. [DOI] [PubMed] [Google Scholar]
- Yadav A. K.; Yadav L. D. S. Visible Light Photoredox Catalysis with N-Hydroxyphthalimide for [4 + 2] Cyclization between N-Methylanilines and Maleimides. Tetrahedron Lett. 2017, 58 (6), 552–555. 10.1016/j.tetlet.2016.12.077. [DOI] [Google Scholar]
- Guo J.-T.; Yang D.-C.; Guan Z.; He Y.-H. Chlorophyll-Catalyzed Visible-Light-Mediated Synthesis of Tetrahydroquinolines from N,N-Dimethylanilines and Maleimides. J. Org. Chem. 2017, 82 (4), 1888–1894. 10.1021/acs.joc.6b03034. [DOI] [PubMed] [Google Scholar]
- Liang Z.; Xu S.; Tian W.; Zhang R. Eosin Y-Catalyzed Visible-Light-Mediated Aerobic Oxidative Cyclization of N,N-Dimethylanilines with Maleimides. Beilstein J. Org. Chem. 2015, 11, 425–430. 10.3762/bjoc.11.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls T. P.; Constable G. E.; Robertson J. C.; Gardiner M. G.; Bissember A. C. Brønsted Acid Cocatalysis in Copper(I)-Photocatalyzed α-Amino C-H Bond Functionalization. ACS Catal. 2016, 6 (1), 451–457. 10.1021/acscatal.5b02014. [DOI] [Google Scholar]
- Chen L.; Chao C. S.; Pan Y.; Dong S.; Teo Y. C.; Wang J.; Tan C.-H. Amphiphilic Methyleneamino Synthon through Organic Dye Catalyzed-Decarboxylative Aminoalkylation. Org. Biomol. Chem. 2013, 11 (35), 5922–5925. 10.1039/c3ob41091a. [DOI] [PubMed] [Google Scholar]
- Xin J.-R.; Guo J.-T.; Vigliaturo D.; He Y.-H.; Guan Z. Metal-Free Visible Light Driven Synthesis of Tetrahydroquinoline Derivatives Utilizing Rose Bengal. Tetrahedron 2017, 73 (31), 4627–4633. 10.1016/j.tet.2017.06.030. [DOI] [Google Scholar]
- Nicholls T. P.; Constable G. E.; Robertson J. C.; Gardiner M. G.; Bissember A. C. Bronsted Acid Cocatalysis in Copper(I)-Photocatalyzed α-Amino C-H Bond Functionalization. ACS Catal. 2016, 6 (1), 451–457. 10.1021/acscatal.5b02014. [DOI] [Google Scholar]
- Xu G.-Q.; Li C.-G.; Liu M.-Q.; Cao J.; Luo Y.-C.; Xu P.-F. Dual C-H Functionalization of N-Aryl Tetrahydroisoquinolines: A Highly Diastereoselective Synthesis of Dibenzo[a, f]Quinolizines via Visible-Light Induced Oxidation and Inverse Electron-Demand Aza-Diels-Alder Reaction. Chem. Commun. 2016, 52 (6), 1190–1193. 10.1039/C5CC08833J. [DOI] [PubMed] [Google Scholar]
- Sun D.; Hubig S. M.; Kochi J. K. Oxetanes from [2 + 2] Cycloaddition of Stilbenes to Quinone via Photoinduced Electron Transfer. J. Org. Chem. 1999, 64 (7), 2250–2258. 10.1021/jo981754n. [DOI] [Google Scholar]
- Wu G.; Li Y.; Yu X.; Gao Y.; Chen H. Acetic Acid Accelerated Visible-Light Photoredox Catalyzed N-Demethylation of N,N-Dimethylaminophenyl Derivatives. Adv. Synth. Catal. 2017, 359 (4), 687–692. 10.1002/adsc.201601108. [DOI] [Google Scholar]
- Xu K.; Fang Y.; Yan Z.; Zha Z.; Wang Z. A Highly Tunable Stereoselective Dimerization of Methyl Ketone: Efficient Synthesis of E- and Z-1,4-Enediones. Org. Lett. 2013, 15 (9), 2148–2151. 10.1021/ol4006344. [DOI] [PubMed] [Google Scholar]
- Curran D. P.; Keller A. I. Radical Additions of Aryl Iodides to Arenes Are Facilitated by Oxidative Rearomatization with Dioxygen. J. Am. Chem. Soc. 2006, 128 (42), 13706–13707. 10.1021/ja066077q. [DOI] [PubMed] [Google Scholar]
- Li J.; Bao W.; Zhang Y.; Rao Y. Cercosporin-Photocatalyzed Sp3 (C-H) Activation for the Synthesis of Pyrrolo[3{,}4-c]Quinolones. Org. Biomol. Chem. 2019, 17 (40), 8958–8962. 10.1039/C9OB01946D. [DOI] [PubMed] [Google Scholar]
- Ju X.; Li D.; Li W.; Yu W.; Bian F. The Reaction of Tertiary Anilines with Maleimides under Visible Light Redox Catalysis. Adv. Synth. Catal. 2012, 354 (18), 3561–3567. 10.1002/adsc.201200608. [DOI] [Google Scholar]
- Liu Q.; Li Y.-N.; Zhang H.-H.; Chen B.; Tung C.-H.; Wu L.-Z. Reactivity and Mechanistic Insight into Visible-Light-Induced Aerobic Cross-Dehydrogenative Coupling Reaction by Organophotocatalysts. Chem. - Eur. J. 2012, 18 (2), 620–627. 10.1002/chem.201102299. [DOI] [PubMed] [Google Scholar]
- Yamaguchi M.; Fujiwara S.; Manabe K. Synthesis of 2,2,5-Trisubstituted 2H-Pyrroles and 2,3,5-Trisubstituted 1H-Pyrroles by Ligand-Controlled Site-Selective Dearomative C2-Arylation and Direct C3-Arylation. Org. Lett. 2019, 21 (17), 6972–6977. 10.1021/acs.orglett.9b02559. [DOI] [PubMed] [Google Scholar]
- Xuan J.; Feng Z.-J.; Chen J.-R.; Lu L.-Q.; Xiao W.-J. Visible-Light-Induced C-S Bond Activation: Facile Access to 1,4-Diketones from β-Ketosulfones. Chem. - Eur. J. 2014, 20 (11), 3045–3049. 10.1002/chem.201304898. [DOI] [PubMed] [Google Scholar]
- Sheldrick G. M. A Short History of SHELX. Acta Crystallogr., Sect. A: Found. Crystallogr. 2008, 64 (1), 112–122. 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- Sheldrick G. M. Crystal Structure Refinement with SHELXL. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71 (1), 3–8. 10.1107/S2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbour L. J. X-Seed — A Software Tool for Supramolecular Crystallography. J. Supramol. Chem. 2001, 1 (4), 189–191. 10.1016/S1472-7862(02)00030-8. [DOI] [Google Scholar]
- Mandal A. K.; Sreejith S.; He T.; Maji S. K.; Wang X.-J.; Ong S. L.; Joseph J.; Sun H.; Zhao Y. Three-Photon-Excited Luminescence from Unsymmetrical Cyanostilbene Aggregates: Morphology Tuning and Targeted Bioimaging. ACS Nano 2015, 9 (5), 4796–4805. 10.1021/nn507072r. [DOI] [PubMed] [Google Scholar]
- Katritzky A. R.; Yao G.; Lan X.; Zhao X. The Conversion of Secondary into Tertiary Amides Using Benzotriazole Methodology. J. Org. Chem. 1993, 58 (8), 2086–2093. 10.1021/jo00060a025. [DOI] [Google Scholar]
- Barker T. J.; Jarvo E. R. Umpolung Amination: Nickel-Catalyzed Coupling Reactions of N,N-Dialkyl-N-Chloroamines with Diorganozinc Reagents. J. Am. Chem. Soc. 2009, 131 (43), 15598–15599. 10.1021/ja907038b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.