

SUMMARY
GenomePaint (https://genomepaint.stjude.cloud/) is an interactive visualization platform for whole-genome, whole-exome, transcriptome, and epigenomic data of tumor samples. Its design captures the inter-relatedness between DNA variations and RNA expression, supporting in-depth exploration of both individual cancer genomes and full cohorts. Regulatory non-coding variants can be inspected and analyzed along with coding variants, and their functional impact further explored by examining 3D genome data from cancer cell lines. Further, GenomePaint correlates mutation and expression patterns with patient outcomes, and supports custom data upload. We used GenomePaint to unveil aberrant splicing that disrupts the RING domain of CREBBP, discover cis-activation of the MYC oncogene by duplication of the NOTCH1-MYC enhancer in B-lineage acute lymphoblastic leukemia, and explore the inter- and intra-tumor heterogeneity at EGFR in adult glioblastomas. These examples demonstrate that deep multi-omics exploration of individual cancer genomes enabled by GenomePaint can lead to biological insights for follow-up validation.
In Brief:
Zhou et al. develop GenomePaint for visualizing coding and non-coding variants in cancer. With a primary focus on pediatric cancer, GenomePaint enables detection of common and rare driver variants within cancer subtypes and discovery of novel oncogenic events by integrating DNA, RNA and epigenetic data from individual cancer genomes.
INTRODUCTION
Multi-omics next-generation sequencing (NGS) has become mainstream for cancer research in recent years (Hoadley et al. 2018; Ma et al. 2018; Rusch et al. 2018). While whole-genome sequencing (WGS) enables identification of somatically acquired coding and non-coding point mutations and structural variants, the incorporation of tumor transcriptome sequencing (RNA-seq) is essential for assessing the potential impact of DNA alterations on transcription deregulation. Applications of integrative analysis of DNA and RNA data include mutant allele expression status, aberrant RNA splicing caused by splice site mutations, gene fusions, and allele-specific expression (Mansour et al. 2014; Herranz et al. 2014; Zhang et al. 2016; Bahr et al. 2018; Ma et al. 2018). More importantly, in order to evaluate the potential functional effect of non-coding variants, chromosome conformation capture assays such as Hi-C and NGS of chromatin immunoprecipitation of transcription factors and histone marks (ChIP-seq) are needed to unveil perturbations to the 3D structure caused by non-coding regulatory variants (Zimmerman et al. 2018; Hnisz et al. 2016). While the inter-relatedness of these diverse data is critical for investigating gene dysregulation events, existing cancer genome browsers such as IGV (Robinson et al. 2011) and UCSC Genome Browser (Kent et al. 2002) do not address this need as they were primarily designed based on a “one data type, one track” model. Even the recently-published cancer genomic visualization tool Xena (Goldman et al. 2020) primarily focuses on depicting the mutation prevalence or expression profile of a research cohort and has limited features for exploring individual tumor genomes, which are critical for discovery of unique genomic drivers in “N of 1" case studies or rare cancer subtypes.
To meet these challenges, we developed a dynamic web-based visualization tool GenomePaint (https://genomepaint.stjude.cloud/) with a design that captures the inter-relatedness of diverse cancer omics data both at the cohort and individual sample levels, the latter of which enables in-depth exploration of genomes of rare cancer subtypes which account for >50% pediatric cancer. We used GenomePaint to present somatic DNA variation, RNA expression, and clinical outcome information for 3,854 pediatric cancer genomes mostly profiled by the St. Jude/Washington University Pediatric Cancer Genome Project (PCGP) or NCI Therapeutically Applicable Research to Generate Effective Treatment (TARGET). Genomic, transcriptomic and epigenomic data generated from pediatric leukemia and neuroblastoma cancer cell lines were also included to enable the assessment of the functional impact of non-coding variants. We present rich data visualization features along with use cases to demonstrate how GenomePaint visualizations move beyond basic data querying to empower biological discovery.
RESULTS
Pediatric cancer datasets
We assembled somatic variants and RNA-seq gene expression of 3,854 pediatric cancers representing 16 histotypes (Tables S1 and S2). Somatic variants in both coding and non-coding regions included 810,277 single-nucleotide variants (SNVs) and small insertion/deletions (indels), 321,686 copy number variations (CNV), 56,484 loss of heterozygosity events (LOH), 52,142 structural variations (SV), and 65 internal tandem duplications (ITD), and 2,990 fusion genes. Whole-genome (WGS) and whole-exome (WES) sequencing of paired tumor-normal samples were profiled for 40% (1,546) and 48% (1,875) of the cases respectively while 49% (1,890) of the tumors were profiled by RNA-seq (see release notes). Mutational signatures and patient outcome data were available for 915 and 1,102 tumors respectively as part of the TARGET pan-cancer initiative (Ma et al. 2018). To assist interpretation of non-coding regulatory variants in pediatric cancers, epigenetic data including histone modification, transcription factor ChIP-Seq, and Hi-C chromatin interaction were uploaded for 8 pediatric cancer cell lines (Zimmerman et al. 2018; Tian et al. 2019) along with their corresponding WGS and RNA-seq data (Table S3). The dataset is primarily available in GRCh37 (hg19), while the variants and gene expression data can also be explored in GRCh38 (hg38).
Navigation in GenomePaint
To visualize comprehensive pediatric cancer omics data GenomePaint offers three interactive and interconnected views: Cohort View, Matrix View and Sample View. The Cohort View entry point can be accessed by 1) selecting a significantly mutated gene from a landscape map presented on the pediatric cancer (PeCan) knowledgebase portal (https://pecan.stjude.cloud, Figure 1A), 2) directly querying a genomic region or a gene of interest (Figure 1B). The landscape map can be pre-assembled by one of the pediatric pan-cancer studies (Ma et al. 2018; Gröbner et al. 2018) or constructed dynamically by user selection of a cancer subtype. Cohort View displays the omics data for the entire cohort along with the relevant genomic annotation. From the Cohort View, a user can explore further by either selecting a sample of interest to launch the Sample View, or genes of interest to launch the Matrix View (Figure 1B). Key features of each view are described below, and their functionality is fully described in the online user tutorials (STAR Methods).
Figure 1: Navigation in GenomePaint.
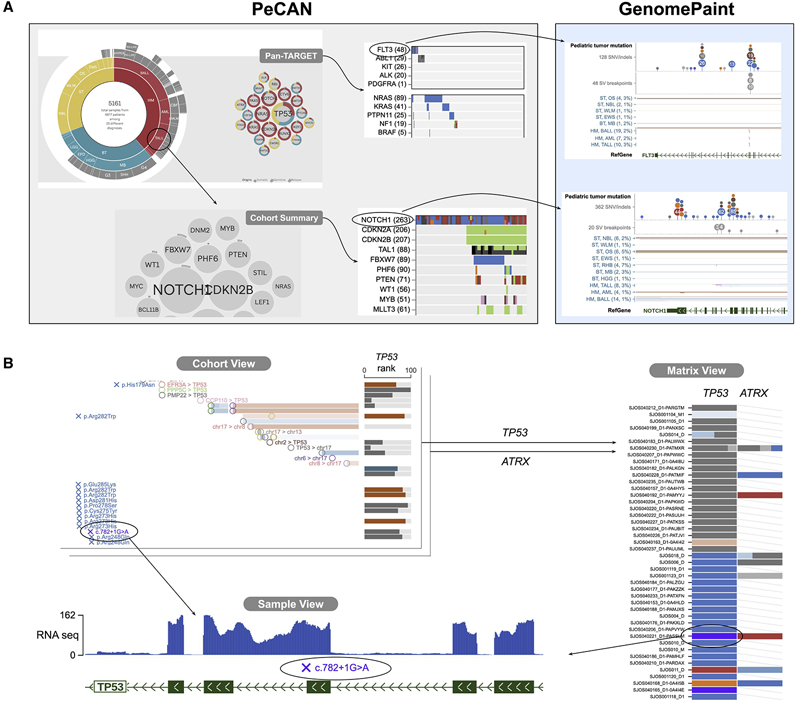
(A) On PeCan (https://pecan.stjude.cloud), users can access a heatmap of significantly mutated genes (left panel) and click on a gene to launch the GenomePaint view (right panel). The heatmap can be assembled by one of those published pan pediatric cancer studies (top panel) or constructed dynamically by a user selected histotype (bottom).
(B) In GenomePaint, Cohort View is the entry point where a user can query a chromosomal location or gene of interest to simultaneously visualize variants and gene expression. Clicking on a genomic variant pops up a navigation panel for accessing multiple views, including the Sample View where details of DNA variants, RNA expression and epigenomic data of a single cancer genome can be examined. Clicking on a histotype launches the Matrix View which shows a data matrix with variant data across multiple loci. Clicking on a data point within the Matrix View launches the Sample View.
Cohort View
The Cohort View presents DNA variants and RNA expression in two side-by-side panels for user-selected cancer histotypes. In the DNA variation panel, site-specific variants such as SNV/indels and structural variation breakpoints are displayed either in aggregate, using the dense mode, or at the individual sample level using the expanded mode. In the expanded mode, site-specific variants are overlaid on segmental variants such as CNV and LOH enabling the review of co-occurrence of different types of somatic variants from the same tumor sample. This feature, coupled with the aligned RNA expression view shown on a separate panel, integrates different data types generated from the same tumor sample, aiding the assessment of somatic variant pathogenicity. For example, co-occurrence of TP53 point mutation with LOH or copy-number loss is present in the majority (83%, 33 out of 40) of adrenocortical tumors, consistent with the bi-allelic loss of TP53 tumor suppressor function required for cancer development (online example 1). By contrast, the mutual exclusivity of point mutation and copy number loss of IKZF1 in B-cell acute lymphoblastic leukemia suggests IKZF1 haploinsufficiency contributes to leukemogenesis (online example 2).
A unique feature of Cohort View enables exploration of the long-range interaction between a candidate non-coding regulatory variant and its target gene, e.g. a regulatory CNV causing aberrant cis-activation of its target gene located outside the CNV boundary (Liu et al. 2020; Hnisz et al. 2016; Northcott et al. 2017; Yang et al. 2020). Specifically, a user can select one or multiple genes for display on the expression panel, to identify a potential target affected by a cis-regulatory variant under inspection in the DNA variation panel. To illustrate this feature, we show TAL1 and PRLR, two oncogenes aberrantly cis-activated by common and rare regulatory variants respectively in the T-cell acute lymphoblastic leukemia (T-ALL) cohort hosted on GenomePaint (Figure 2).
Figure 2: Visualizing common or rare regulatory variants in T-ALL samples on Cohort View.
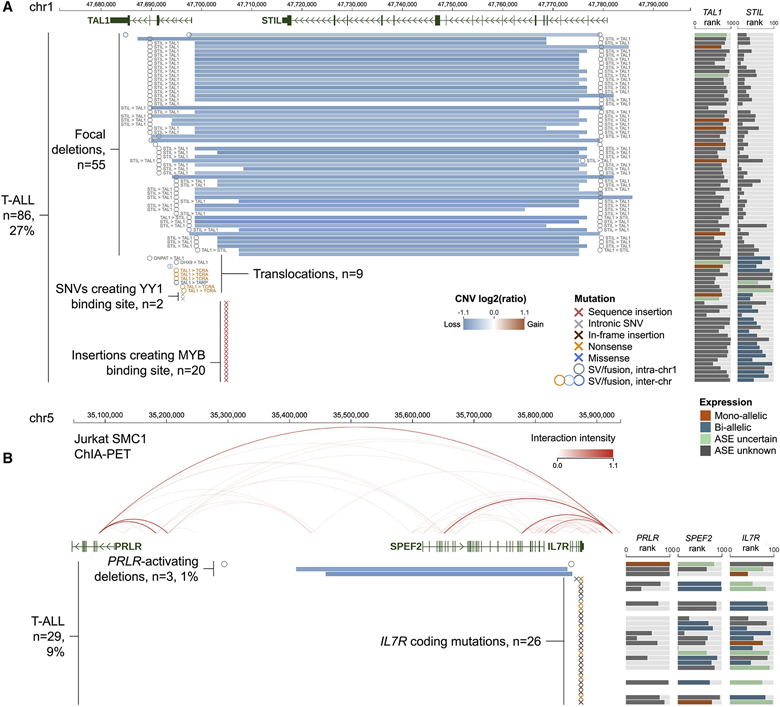
(A) Activation of TAL1 oncogene in 27% of the T-ALL by multiple regulatory variants including STIL-TAL1 deletion, translocation, somatic insertion at upstream intergenic region or somatic SNV in intron 1. Cohort View displays genomic variants (left panel) and expression rank (right panel) for each sample. In the genomic variant panel, the CNV segments are colored as blue (deletion) or red (amplification) lines, SV breakpoints are shown as circles, and SNVs/indels are marked by “x”. The expression panel colors the allele-specific expression (ASE) status for each sample.
(B) A rare focal deletion of SPEF2 and IL7R leading to activation of PRLR in 3 (~1%) T-ALL samples. The deletion of the top sample was detected as a structural variation due to its subclonality. The arc graph displays Jurkat chromatin interaction by SMC1 ChIA-PET imported as a reference track.
See also Figure S1. Live link: https://proteinpaint.stjude.org/gp/tall.tal1.prlr.html
TAL1 is a transcription factor normally expressed only in the early stages of thymocyte development (e.g. hematopoietic stem cells), but activated in a subset of T-ALLs (O’Neil and Look 2007). Figure 2A shows a 130 Kb region surrounding TAL1 with a variety of somatic alterations present in 86 (27%) of the 316 T-ALL samples (Figure S1A). The somatic deletion encompassing STIL, a house-keeping gene, is the most prevalent alteration in this region. Examination of TAL1 and STIL expression side-by-side in the expression panel shows uniform over-expression of TAL1 accompanied by mono-allelic expression suggesting cis-activation in contrast to the highly variable expression of STIL. This pattern recapitulates the known functional impact of the deletion, i.e. despite its genomic localization at STIL, the deletion removes the boundary elements of an insulated neighborhood that suppresses TAL1 expression (Hnisz et al. 2016) causing oncogenic activation of TAL1 in T-ALL. Cohort View also presents alternative mechanisms leading to cis-activation of TAL1, including hijacking of the T-cell receptor regulatory elements by translocation (n=6) and creation of de novo transcription factor binding sites either by somatic indel (MYB binding site) in the upstream intergenic region (n=20) (Mansour et al. 2014) or somatic SNV (YY1 binding site) in intron 1 (n=2) (Liu et al. 2020).
PRLR is a cytokine receptor located >800 Kb upstream of IL7R, a known oncogene in T-ALL (Liu et al. 2017). Cohort View showed that this region contains a rare >400 Kb deletion, detected as a SV in 1 out of 30 WGS samples or as a CNV in 2 out of 265 samples analyzed by SNP array (Figure S1A). The deletion removes exon 1 or an upstream non-coding region of IL7R and the entirety of the intervening SPEF2 gene (Figure 2B). The expression panel showed that PRLR is uniformly over-expressed, in contrast to the varying expression level in IL7R and SPEF2. When examining expression values using the FPKM boxplot, these three samples exhibit the highest PRLR expression in the T-ALL cohort and are outliers compared with the remaining T-ALLs (Figure S1B). Notably, the deletion can cause reduction of IL7R expression, which initially appears paradoxical given the presence of recurrent gain-of-function, oncogenic mutations in IL7R (Zhang et al. 2012) in 26 T-ALL samples in this cohort (shown as colored x in Figure 2B). Importantly, a highly significant cis-activation signature was identified in one of the samples profiled by WGS (Liu et al. 2020), suggesting PRLR is likely the target gene for this deletion.
The effect of the deletion on 3D genome architecture can be examined by importing ChIA-PET chromatin interaction data generated from the T-ALL cell line Jurkat (Hnisz et al. 2016) as an arc graph on Cohort View. This shows that the region contains multiple boundary elements of an insulated neighborhood, implying that the mechanism of PRLR cis-activation is similar to that of TAL1 activation by STIL deletion.
Cohort View allows the user to correlate clinical outcome with DNA genomic alterations or gene expression levels. A “mutated” group of tumor samples harboring diverse types of somatic alterations within a selected genomic region can be defined and compared to non-affected samples, and their event-free and overall survival visualized in a Kaplan-Meier plot. For example, the MYB oncogene can be activated by somatic alterations such as amplifications, hotspot mutations (missense variants or in-frame insertions), and translocations in the T-ALL cohort, and the group of tumors with these alterations have significantly worse outcomes than the non-mutated group (Liu et al. 2017, online example 3).
Differential gene expression across different cancer types can be viewed as boxplots by clicking a sample on the RNA expression panel. Within each cancer type, expression-based survival analysis bins tumor samples into distinct groups by their expression level for a gene of interest. GenomePaint shows that high MDM2 expression is associated with worse outcome in acute myeloid leukemia, replicating a previously published finding (Kojima et al. 2005, online example 4).
Matrix View
The Matrix View allows a user to assemble multiple genes or loci in order to evaluate their mutational patterns across a set of tumors, and associate the patterns with clinical outcome. For example, among the driver genes identified in neuroblastoma, exclusivity of activating mutations in MYCN and loss-of-function mutations in ATRX are readily detectable, while this pattern is absent in the gene pair of MYCN and ALK. By dividing these patients into four groups based on the co-mutation pattern of MYCN and ALK, a Kaplan-Meier plot shows that MYCN and ALK co-mutation is associated with the worst outcome (online example 5).
Sample View
From Cohort View and Matrix View, clicking on a DNA variant will launch the Sample View, which shows detailed “omics” data for the selected tumor, centered on the variant of interest. Sample View supports a dynamic range of genome visualization – users can explore at base-pair resolution and zoom out at various scales up to the entire chromosome.
Sample View presents sequencing coverage for DNA and RNA, enabling manual curation of copy number variation detected in whole genome (WGS) or whole exome (WES) sequencing and aberrant splicing in RNA-seq, respectively. For RNA-seq, Sample View also provides a splice junction graph depicting RNA-seq read count spanning canonical or novel splice junctions. DNA variations can be presented alongside the splice junction graph, enabling users to evaluate its potential effect on RNA transcription. A TP53 splice site variant from an osteosarcoma, NM_000546:c.782-1G>A, is selected for detailed inspection in Sample View (online example 6). The RNA-seq coverage plot shows increased coverage of intron 7, suggesting intron retention, while the splice site junction graph depicts a >90% reduction of canonical junction reads between exons 7 and 8 when compared to the two neighboring junctions (10 junction reads for exon 7-8, >100 reads for exon 6-7 and exon 8-9). The predominance of intron retention over canonical splicing suggests a second DNA hit: this can be evaluated by inspecting the Allelic Imbalance (AI) plot to check the difference in the variant allele fraction of the matched tumor and normal WGS or WES data. In this example, a zoomed-out view of WES AI plot reveals a 39 Mb LOH region encompassing the TP53 loci thereby confirming a second hit resulting in the mono-allelic state of aberrant TP53 splicing in tumor RNA (online example 6).
A zoomed-out chromosomal view of the somatic DNA variant track can also be highly informative for examining global patterns of mutational signatures. Somatic SNVs in this track, color-coded by their most probable mutational signatures based on our previous analysis (Ma et al. 2018), are shown in a “rainfall” plot with the y-axis depicting their inter-mutation distance (online example 7). This enables the inspection of patterns of mutational clusters (i.e. the “rainfall”) that are indicative of potential cancer etiology. For example, co-occurrence of SVs with clusters of SNVs bearing the APOBEC mutational signatures in an osteosarcoma suggests kataegis, a process of mutagenesis generating localized hypermutation associated with APOBEC activity (Nik-Zainal et al. 2012). By contrast, the non-kataegis regions in the same tumor are dominated by SNVs bearing COSMIC signature 3 indicating defects in repairing DNA double-strand breaks by homologous recombination (online example 7).
Where available, Sample View displays epigenetic and 3D genome data; the current release includes ChIP-seq, Hi-C and Capture-C data generated from pediatric cancer cell lines. When exploring structural variants, the re-arranged genome is used as the reference upon which genomic, transcriptomic, and epigenomic data are overlaid. This feature greatly facilitates the discovery of aberrant transcription caused by events such as enhancer hijacking. Using an example from our previous discovery of enhancer hijacking leading to MYC activation in a subset of high-risk neuroblastoma (Zimmerman et al. 2018), we show how this interface enables identification of candidate non-coding regulatory SVs. From the Cohort View, the presence of a t(4:8) SV upstream of MYC, coupled with the outlier MYC expression in the neuroblastoma cell line NB69, suggests that the SV disrupts gene regulation (Figure 3A). Selecting the t(4:8) SV for Sample View reveals the juxtaposition of the HAND2 enhancer on chromosome 4, defined by strong H3K27Ac peaks, to the MYC promoter region on chromosome 8 (Figure 3B). NB69 Hi-C data, shown as a triangular contact map on the axis of the rearranged genome, unveils direct contact between the MYC promoter and the HAND2 enhancer in the 3D genome sphere (Figure 3B), validating the hypothesis that the aberrant MYC activation is likely caused by the (4:8) SV. By examining the MYC expression and the presence of inter-chromosomal translocation in the region, Sample View may reveal that a similar mechanism may cause MYC activation in other pediatric cancer subtypes (e.g. a B-lineage ALL and a high-grade glioma shown in Figure 3A).
Figure 3: Sample View of interchromsomal rearrangement leading to MYC dysregulation by enhancer hijacking.

(A) Inter-chromosomal translocations in various pediatric primary tumors and cell lines in the 1 Mb region spanning MYC (orange vertical highlight) in the Cohort View. Circles of different colors represent different inter-chromosomal translocations including three neuroblastoma (NBL) cell lines previously reported to show MYC overexpression (e.g. t(4;8) in both NB69 and SKNAS, t(7;8) in SH-SY5Y), as well as three previously unpublished cases (e.g. t(8;9) in PASFIC, t(8; 10) in SJHGG001_A, and t(2;8) in SJBALL247_D). Variant frequency is based on 204, 76, 545 samples analyzed by WGS in NBL, HGG, and B-ALL, respectively. The right panel shows MYC expression rank (bar width) and ASE (bar colors) in each respective tumor.
(B) Sample View of in situ Hi-C data of NBL cell line NB69 shows extensive chromatin interaction between MYC and chromosome 4 along the axis of t(4;8) translocation detected by WGS (oval). The chromosome 4 region also shows strong H3K27ac signal (box), indicating MYC-regulating enhancers. Live link: https://proteinpaint.stjude.org/gp/myc.sv.nb69.html
Use case 1: Discovery of disruption of the CREBBP RING domain as a likely pathogenic event
CREBBP encodes the transcriptional coactivator and histone acetyltransferase (HAT) and is known to be involved in relapse of pediatric B-ALL (Mullighan et al. 2011; Li et al. 2020). In the Cohort View we found a recurrent somatic SNV (NM_004380.2:c.3779+1G>A) at the splice donor site of CREBBP exon 20 in four B-ALLs profiled by TARGET or PCGP (Figures 4A and S2A). A tooltip shows that this variant matches a pathogenic germline variant in ClinVar (Figure 4B) for Rubinstein-Taybi Syndrome (Roelfsema et al. 2005) and an in vitro transfection experiment showed that the variant could cause exon skipping (Dauwerse et al. 2016). In our cohort RNA-seq is available for one of these four B-ALLs (PANWHJ). By examining the splice junction graph in Sample View, we confirmed that exon 20 skipping occurred in vivo in leukemia. This result was based on the following data: 1) the presence of a previously unrecognized splice junction between exon 19 and 21 (percent spliced in: 55%); and 2) an approximately 50% reduction of coverage of exon 20. The event is predicted to be in-frame, as shown by the tooltip over the novel junction (Figure 4C). A more compact view of the splice junctions, RNA-seq coverage, and the splice site mutation is shown when the display is switched from genomic coordinates to mRNA coordinates (Figure 4D) where protein domains are shown. This view reveals that exon 20 skipping disrupts the RING domain of CREBBP. While prior studies focused on CREBBP mutations located in the HAT domain, this recurrent splice site variant indicates that disruption of the RING domain, a negative regulator of the HAT domain (Delvecchio et al. 2013), may have a comparably deleterious effect. Indeed, further inspection using the Cohort View revealed 7 additional B-ALLs harboring somatic alterations disrupting the RING domain of CREBBP (Figure S2). These include two cases with missense mutations on exons 20 and 21, three cases with exon 21 splice site/region mutations, and two cases with focal deletion of exon 20 (Figure S2). Collectively these data show that disruption of the CREBBP RING domain is a recurrent pathogenic event caused by multiple types of somatic alterations in B-ALL.
Figure 4: Assessing aberrant splicing in CREBBP caused by somatic splice site variants.

(A) Zoomed-out “dense” Cohort View of somatic SNV/indels across the genomic region encoding CREBBP. Dots represent variants, where color indicates mutation class and size corresponds to the number of cases. Zoomed-in detail shows a recurrent variant (detected in 4 (0.3%) out of 1,201 B-ALL samples analyzed by WGS or WES) disrupting the canonical splice donor site (cyan) in intron 20.
(B) Variant tooltip panel for the variant from (A), showing the four harboring tumors, matching dbSNP ID, and pathogenic ClinVar classification.
(C) Sample View of one of the tumors (PANWHJ) showing RNA-seq splice junction and coverage. In the splice junction track, dot size and Y-axis position indicates junction read count. A novel junction (box arrow) skips exon 20. Diagram of this exon skipping event (box detail) displays junction read counts and translation frame prediction (in-frame).
(D) CREBBP view of NM_004380, with the same set of tracks in (C). The RING domain (blue) spans exons 19 to 21.
See also Figure S2. Live link: https://proteinpaint.stjude.org/gp/crebbp.splicesite.html
Use case 2: Discovery of MYC activation in B-ALL caused by duplication of NOTCH1 MYC enhancer (N-ME)
Somatic alterations in lineage-specific enhancers at the 8q24 region downstream of MYC are an important mechanism for MYC overexpression in cancer (Lancho and Herranz 2018). The NOTCH1 MYC enhancer (N-ME, 1.4 Mb downstream of MYC) and the blood enhancer cluster (BENC, 1.7 Mb downstream of MYC) are recurrently duplicated in T-lineage acute lymphoblastic leukemia (T-ALL) (Herranz et al. 2014; Liu et al. 2017) and acute myeloid leukemia (AML) (Bahr et al. 2018) respectively, causing MYC overexpression in these two types of leukemia. Consistent with these reports, Cohort View shows that duplications of N-ME and BENC are present in 23 T-ALLs and 6 AMLs respectively, and the majority of these events are accompanied by high MYC expression (Figure 5A). Examining focal somatic copy number gain in this region across the entire pediatric cancer cohort, we found duplication of BENC and N-ME in previously unreported cancer subtypes, including recurrent duplication of both enhancers in B-ALLs as well as BENC duplication in one rhabdomyosarcoma (at both diagnosis and relapse) and one high grade glioma (Figure S3A).
Figure 5: MYC dysregulation by enhancer duplications in pediatric cancers.
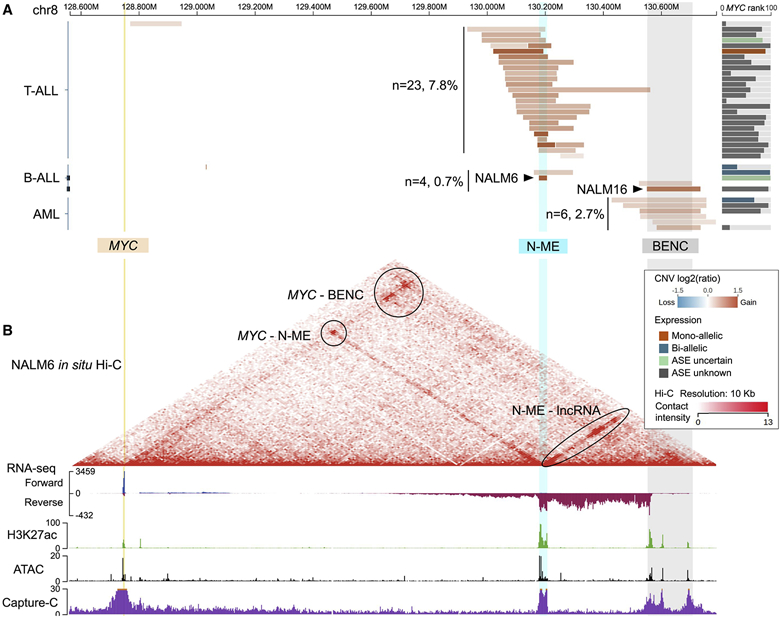
(A) Copy number gains from B-ALL, T-ALL, and AML over the 2 Mb region spanning MYC (yellow, left), and two enhancers (N-ME: cyan, BENC: gray). The number of samples with focal CNV segments (<2 Mb) overlapping with N-ME (cyan) or BENC (gray) is labeled for each cancer type; this count excludes two T-ALL samples with focal amplifications outside the N-ME and BENC enhancer regions. Arrows point to amplifications in NALM6 and NALM16 that overlap with N-ME and BENC respectively. The right panel shows MYC expression rank (bar width) and ASE (bar colors) in each respective tumor. Variant frequency is based on 298 T-ALLs, 545 B-ALLs and 219 AMLs analyzed by WGS or SNP array.
(B) Detailed assay results of NALM6, including in situ Hi-C (heatmap), stranded total RNA-seq (blue for forward strand, red for reverse strand), H3K27ac ChIP-seq (green), ATAC-seq (black), and Capture-C using MYC promoter as bait (purple).
See also Figure S3. Live link: https://proteinpaint.stjude.org/gp/myc.enhancer.nalm6.html
To further study this observation, we reviewed the details of two B-ALL cell lines, NALM6 and NALM16, in the Sample View. NALM6 belongs to the B-ALL subtype defined by IGH-DUX4 translocation which activates DUX4, a transcription factor that contains 2 homeobox domains; it has a five-copy N-ME amplification. NALM16 is a hypodiploid B-ALL and has a three-copy BENC amplification. Both cell lines had high MYC expression (tooltip shows FPKM of 194 and 110 respectively) which are ranked at >95 percentile of the entire B-ALL cohort (Figure 5A expression panel). Genome-wide 3-D conformation data generated by in situ Hi-C from our previous study (Tian et al. 2019) were uploaded to GenomePaint (Figure S3B). In NALM6, the MYC promoter interacts with the amplified N-ME enhancer—an interaction previously only reported in T-ALL. On the other hand, MYC-BENC interaction was found in both NALM6 and NALM16, even though BENC amplification was present only in NALM16. Further investigation on NALM6 was possible as we uploaded RNA-seq, H3K27ac ChIP-seq, and ATAC-seq data generated from our prior study (Tian et al. 2019), and we were able to detect transcription of long non-coding RNAs (likely enhancer RNAs), enhancer activity and open chromatin structure at both BENC and N-ME regions in Sample View (Figure 5B). Subsequently, we performed a Capture-C experiment in NALM6 using the MYC promoter as the bait, which verified MYC promoter interaction with both N-ME and BENC (Figure 5B, Capture-C signal track; STAR Methods).
To characterize the cis-regulatory effect of N-ME amplification in NALM6, we phased the heterozygous SNPs on the NALM6 genome into two haplotypes using digital long-read Chromium WGS data ((Tian et al. 2019), Supplementary Notes) to enable haplotype phasing of RNA-seq, ChIP-seq, ATAC-seq, and Capture-C. These phased tracks can all be reviewed in Sample View (Figure S4A). As expected, WGS showed allelic imbalance (AI) of heterozygous SNPs with a median fraction of 0.82 of the amplified haplotype at the N-ME region with 5 copy-gain, but not at the diploid flanking regions (60 Kb left flanking with WGS median fraction 0.55, 350 Kb right flanking with WGS median fraction 0.53, Figure 8A). Notably, AI was detected in RNA-seq at all three loci. At N-ME, RNA-seq AI is more pronounced with a median fraction of 0.89 and extended into the 350 Kb downstream diploid flanking region with a median fraction of 0.74 (Figure 6A). The downstream region contains long non-coding RNA genes LINC00977 and CCDC26, and it is evident that the elevated transcription is driven by the amplified N-ME in cis as the Hi-C data shows extensive physical contact between N-ME and this lncRNA-containing region (Figure 5B, oval)—a contact present exclusively in NALM6 (Figure S3B). We verified the N-ME-driven allele-specific expression (ASE) in NALM6 cells by performing sequential RNA-DNA fluorescence in situ hybridization (Figure 6B). Specifically, mono-allelic expression was detected in 58% of 55 analyzed cells, and allelic imbalance was detected in 30% of the cells, confirming the allelic imbalance observed in phased RNA-seq. The sequential FISH image data also show that the MYC locus is surrounded by an RNA cloud that is transcribed from the enhancer region, further validating the MYC/N-ME interaction projected from Hi-C and Capture-C data.
Figure 6: N-ME amplification drives imbalanced enhancer RNA transcription.

(A) Top: total sequencing coverage of NALM6 WGS and RNA-seq in a region surrounding N-ME. Elevated enhancer RNA transcription is observed at both N-ME and the 350 Kb flanking region on right. Bottom: NALM6 phased heterozygous SNPs are used to generate boxplots comparing N-ME-amplified allele fractions between WGS and RNA-seq, for the three regions (N-ME and two flanking regions). Only SNPs with total WGS coverage >10 are included. In the boxplots, boxes represent the IQR bisected by the median, and whiskers represent the maximum and minimum range of the data that do not exceed 1.5 times the IQR.
(B) Sequential RNA-DNA FISH (fluorescence in situ hybridization) of NALM6 cells. The cells are first hybridized with DNA probe without denature to detect enhancer RNA at the N-ME locus, and then are denatured and rehybridized with DNA probes of both N-ME and MYC promoter loci. 55 cells were assessed to derive the percentage of four categories of allelic expression status. Scale bars: 10μm. N-ME probe is based on BAC clone RP11-282K23; MYC probe is based on BAC clone RP11-237F24.
See also Figure S4.
N-ME interaction with MYC is expected to be NOTCH1-dependent (Herranz et al. 2014). However, NOTCH1 expression is low in NALM6 when compared to the T-ALLs (Figure S4B). Western blot confirmed the low levels of both the full length NOTCH1 protein and the NOTCH Transmembrane (NTM) subunit in NALM6 compared to T-ALL cell lines (Figure S4C). This suggests that alternative transcription factors may bind to the N-ME enhancer leading to MYC activation. Since DUX4 was aberrantly activated in NALM6, we uploaded NALM6 DUX4 ChIP-seq data to evaluate whether DUX4 could be a potential candidate. Using Sample View, we found dual DUX4 peaks close to the 100 bp N-ME core region where NOTCH1 binding in a T-ALL cell line was shown to drive the enhancer activity (Figure S4D) (Herranz et al. 2014). The location of the dual DUX4 peaks correspond to the two DUX4 binding sites predicted by FIMO, and match the location of the NOTCH1 peak detected in T-ALL cell line Jurkat (Figure S4D). These data suggest that DUX4 may contribute to N-ME enhancer activation in NALM6. Interestingly, the sole patient B-ALL sample (SJERG002) in our cohort with N-ME duplication is also of the IGH-DUX4 subtype. Similar to NALM6, this primary B-ALL patient sample has high MYC expression (FPKM=156) which is ranked at the 98th percentile of the B-ALL cohort.
Use case 3: Exploring intra-tumor heterogeneity in adult glioblastoma (GBM).
To test broad applicability of GenomePaint on adult cancer, we re-analyzed 38 TCGA GBM samples that have both paired tumor-normal WGS and tumor RNA-seq (STAR Methods). The resulting data, including high-quality, expert-curated somatic variants (i.e. SNV/indel, CNV, SV, and fusion) as well as output from our analysis pipeline (i.e. sequencing coverage, gene FPKM, and RNA splice junctions), were uploaded to GenomePaint for visual exploration. Our investigation focused on EGFR, the most frequently altered oncogene affected by amplification or point mutations in GBM (Brennan et al. 2013). The Cohort View depicts the overall prevalence of somatic alterations, with 21 samples (55%) exhibiting focal amplifications at EGFR locus accompanied by elevated EGFR expression (Figure 7A). Notably, amplified samples all contain additional somatic SVs and/or SNVs in EGFR. SVs are dispersed across the entire EGFR locus, with two hotspots (marked by boxes in Figure 7A) representing the EGFRvIII isoforms with defective extracellular domain (n=8) or truncation of the C-terminal autophosphorylation domain encoded by exons 25–28 (n=5), both of which are oncogenic (Brennan et al. 2013). Variant allele fraction (VAF) can be ascertained by mouse-over an SNV or SV. Amongst these 21 amplified samples, point mutation can be the sole variant type with VAF ranging 0.80-0.96 clone (n=5, 24%, Figure S5A), or accompanied by one or multiple SVs within EGFR (n=7, 33%, Figure S5B), or absent in samples that harbor multiple SVs (n=9, 43%), suggesting intra-tumor heterogeneity at EGFR locus in the majority of the tumor samples.
Figure 7: Visualizing intratumor heterogeneity of TCGA GBM at EGFR locus.
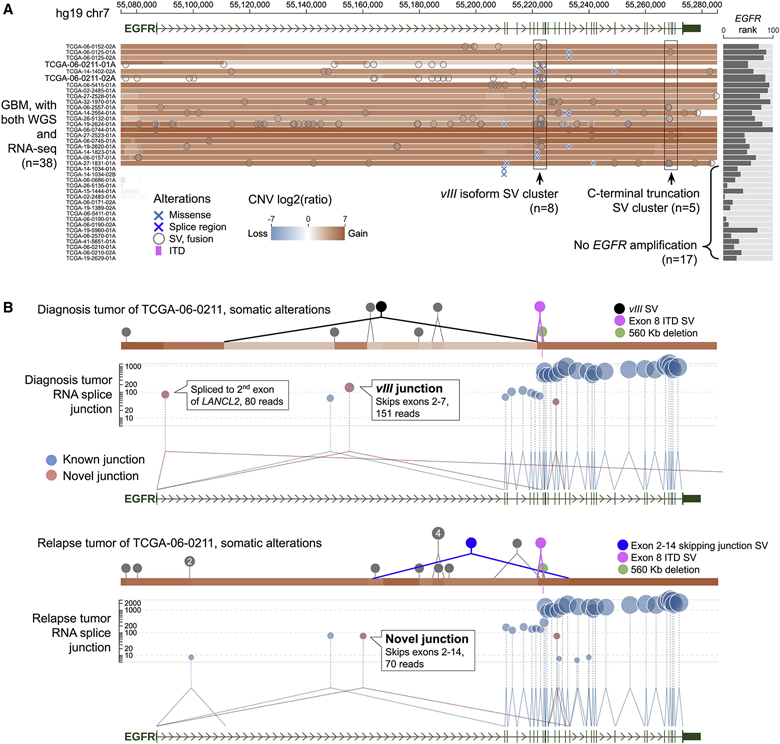
(A) Cohort View of somatic alterations and gene expression of 38 TCGA GBM samples with both WGS and RNA-seq data. Across the cohort, 21 tumors with amplifications at EGFR locus exhibit high EGFR expression. In each sample, structural variation breakpoints (circles) and coding mutations (X marks) are shown on top of CNV segments. Rectangular regions shown SV clusters for EGFRvIII (n=8, left) and EGFR C-terminal truncation (n=5, right).
(B) Sample View of TCGA-06-0211-01A, a diagnosis tumor (top), and its matching relapsed sample TCGA-06-0211-02A (bottom). SVs are shown as solid circles, highlighting representative SVs that are diagnosis-specific (EGFRvIII), relapse-specific (intron 1-14 deletion), or shared (exon 8 ITD and 560K deletion) by color. In RNA-seq track, novel junctions corresponding to diagnostic-specific EGFRvIII and relapse-specific exons 2-14 skipping are labeled.
See also Figures S5 and S6. Live link: https://proteinpaint.stjude.org/gp/tcga.gbm.html
By exploring complex SV patterns along with aberrant transcription profiles of EGFR in individual tumors using Sample View, we found that clonal composition and diversity of a GBM tumor can change dramatically from diagnosis to relapse. For example, TCGA-06-0211, a case with samples collected at diagnosis (01A) and relapse (02A), harbors multiple SVs that can be diagnosis-specific, relapse-specific or shared. Specifically, somatic deletion of exons 2-7 was detected in ~95% of the WGS reads at diagnosis, making EGFRvIII isoform the predominant transcript in RNA-seq (Figure 7B, top). By contrast, EGFRvIII was absent at relapse, with no DNA SV in WGS nor vIII junction reads in RNA-seq. Similar pattern was found previously in a diagnosis-relapsed pair of pediatric high grade glioma SJHGG019 (Xu et al. 2019). In the relapsed tumor of TCGA-06-0211, aberrant splicing at exon 1 was dominated by an abnormal isoform that skips exons 2-14 due to the presence of a relapse-specific DNA SV with breakpoints in intron 1 and 14 (Figure 7B, bottom). Notably, two SVs, both involving exon 8, persisted from diagnosis to relapse: one is a 2.3 Kb internal tandem duplication (ITD) encompassing partial exon 7 and the full exon 8, and the other is a 560 Kb large deletion starting from the upstream intergenic region. The ITD SV resulted in the expression of an in-frame exon 8 ITD transcript at diagnosis and relapse (Figure S6) while the large 560 Kb deletion, with a 10-fold increase of its mutant allele fraction at relapse, may account for the elevated expression of exons 8-28 at relapse despite the absence of EGFRvIII. These data indicate selection for mutant EGFR isoforms from diagnosis to relapse (deCarvalho et al. 2018).
DISCUSSION
The visualization of cancer genomic data improves our understanding of the molecular mechanisms in the initiation and progression of both pediatric and adult cancers (Robinson et al. 2011; Gao et al. 2013; Zhou et al. 2016). Current efforts primarily focus on visualizing variants that alter the function or dosage (i.e. copy number) of protein-coding genes, while visualization of non-coding variants is poorly supported. GenomePaint offers a suite of features designed for direct inspection of aberrant transcription that may be caused by regulatory non-coding variants in a cancer cohort (Cohort View) and for in-depth exploration of genomic alterations of individual cancer genomes via integration of multi-omics data (Sample View). Here, each cancer genome can be visualized as a N-of-one case study in addition to being one-of-N cases in a research cohort. This dual-focus functionality is particularly important for pediatric cancer, a disease with many rare subtypes which requires learning the unique genomic profile of every patient. Similar usage can be found in adult cancer studies where both WGS and RNA-seq were performed to closely examine cancer genomes that may exhibit unusual clinical courses (i.e. exceptional responders), cancer recurrence or metastases (Iyer et al. 2012; Al-Ahmadie et al. 2014). Our use case for adult GBM samples illustrates how deep exploration of the complex SV patterns and aberrant isoforms on GenomePaint enables analysis of intra-tumor heterogeneity and clonal evolution from diagnosis to relapse at the EGFR locus. By contrast, other cancer genome browsers such as UCSC Xena focus primarily on depicting mutational prevalence in a cancer cohort, which lacks the capability for in-depth visualization at the individual sample level.
Genomic profiling has become an integral part of clinical testing, and genomic data sharing is critical to the advancement of cancer research and clinical care (Rusch et al. 2018; Zehir et al. 2017). Currently, GenomePaint hosts somatic alterations from 3,854 pediatric cancers representing 16 histotypes, which serve as an extremely valuable resource to the biomedical research community for investigating the pathogenesis of pediatric cancer, a rare disease with devastating effect. The number of cases and cancer histotypes on GenomePaint will continue to expand as we will regularly update genomics data content and present release notes for data sets generated from published research studies as well as from our prospective clinical sequencing program, which performs 3-platform sequencing of whole-genome, whole-exome and RNA-seq for every eligible childhood cancer patient (Rusch et al. 2018). The highly interactive visualization interface enables researchers with no computing background to carry out in-depth online analysis. The ease of visualizing data compiled from multiple studies highlights patterns of recurrence that can be easily overlooked in a single cohort or cancer subtype, while the detailed presentation of Sample View enables users to perform fully integrated analysis. Our first two use cases illustrate the potential for discovery using these features.
GenomePaint can also serve as a powerful tool for other genetic-based diseases such as adult cancer, as demonstrated by our third use case. Support for custom tracks also furthers this work; for example, somatic SNV/indels, CNVs, and expression data can be downloaded from the NCI Genomics Data Commons (https://gdc.cancer.gov/) and processed into GenomePaint custom track files using scripts provided on our website. As examples, we use the custom track feature to show recurrent deletion of CDKN2A/2B in TCGA diffuse large B-cell lymphoma (online example 8), and the TERT amplification in TCGA melanoma (online example 9). A user can also download the raw genomic data from the public repository, perform a re-analysis and upload the resulting data to GenomePaint as demonstrated in our use case of TCGA GBM samples (Figure 7). Custom track files can be securely hosted on DNANexus and visualized in GenomePaint through the FileViewer mechanism. We plan to incorporate data types including proteomics, methylome, and common germline variants, as well as comprehensive clinical information, to continue enhancing the utility of GenomePaint for cancer genomic research. GenomePaint is freely accessible at https://genomepaint.stjude.cloud/.
STAR★Methods
Resource Availability
Lead Contact
Requests for resources should be directed to and will be fulfilled by the Lead Contact, Xin Zhou (xin.zhou@stjude.org).
Data and Code Availability
NALM16 WGS, RNAseq, Hi-C is available from European Genome-Phenome Archive under accession EGAS00001004669. Additional published data including pediatric cancer and cell line DNA variations, transcriptomes, and epigenomes are listed in Tables S1-S3. The pediatric cancer dataset is freely accessible through GenomePaint at https://genomepaint.stjude.cloud/. Data processing scripts are available on https://github.com/josephpowi/gpdatascript2020. GenomePaint software license is available upon request.
Materials Availability
This study did not generate any unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Culture and Reagents
NALM6 and NALM16 cells were ordered from DSMZ (https://www.dsmz.de/) and cultured in RPMI+ 10% FBS, at 37°C with 5% CO2 according to DSMZ recommendations. The cells were not authenticated.
Method Details
Capture-C validation of MYC-enhancer interactions in NALM6
Capture-C was designed to identify genomic regions interacting with the MYC promoter. Two DpnII cutting sites are present in the MYC promoter region, and two oligos (listed below) were designed near them to capture the MYC promoter. NALM6 was cultured at about 80% confluence and resuspended 107 cells in 10 ml of medium. Cells were crosslinked with 1-2% formaldehyde and quenched by 0.125M glycine. After lysis cells stayed on ice for 20 min, were digested with 500 units of DpnII, then proximity ligation was performed. After reverse crosslinking, ligated DNA was purified, then sheared to 200 bp and prepared as general indexed Illumina libraries. 1-2 μg of 3 different indexed NALM6 samples were captured twice by two biotinylated oligos in the MYC promoter region, then PCR amplified streptavidin beads pulled down libraries. The NG capture-C libraries were sequenced on Miseq V2 paired-end 151 cycles. The sequencing result was analyzed using the pipeline captureC(Davies et al. 2016). In order to avoid high read coverage at the oligo region when visualizing the signals, the reads at the extended 1 kb region of the fragment were filtered. In accordance with Hi-C results, Capture-C showed strong interaction of MYC promoter with both N-Me and BENC (Fig. 2b), despite their great linear distances to MYC promoter (1.4 Mb from N-Me, 1.7 Mb from BENC).
Sequential RNA DNA FISH
Unfixed cells are applied to slides by cytocentrifugation to an appropriate cell density. Cells are then fixed for 5 minutes in 4% PFA followed by permeabilization in 0.05% Tween 20 and NP40 for 5 minutes. Slides are then dehydrated in a graded ethanol series. Probes are prepared by nick translation of genomic DNA clones using fluorophore labeled dUTPs. Labeled DNA is suspended in a hybridization buffer composed of 50% formamide, 10% dextran, 2X SSC and human Cot DNA as competitor to suppress repetitive sequences. Probe mixture is denatured by incubation at 70°C for 5 minutes and is then applied to dehydrated slides and incubated at 37°C overnight. Post hybridization washing is done in 50% formamide, 2X SSC at 37°C for 5 minutes followed by mounting slides in Vectashield mounting medium containing DAPI. Slides are then imaged to identify RNA signals and slide coordinates of imaged fields are recorded. Coverslips are then removed from slides followed by brief rinsing in PBS to remove mounting medium. Slides are then fixed again in 4% PFA followed by a 10 minute treatment in 0.2N HCl containing 0.5% Triton X 100 and then denatured in 70% formamide, 2X SSC at 80°C for 10 minutes. Slides are then dehydrated in a graded ethanol series and a second hybridization is done to detect the location of DNA. Post hybridization washing is repeated as before and slides are then mounted in Vectashield mounting medium. Slides are then reimaged using the slide coordinates established after RNA hybridization to visualize both RNA and DNA together.
Phasing the NALM6 genomic profiling results
NALM6 genomic profiling results, including WGS, stranded total RNA-seq, Capture-C, H3K27ac, and ATAC, are prepared into separate coverage tracks for each haplotype of NALM6 autosomes. NALM6 Chromium WGS (Tian et al. 2019) have identified “phase sets”, that are chromosome segments containing phased heterozygous SNPs. For every such phase set, we name the two haplotypes as “C1” and “C2”; and for each heterozygous SNP within, the two different alleles can be assigned to either C1 or C2. With such phased SNPs, we run bcftools (http://www.htslib.org/) to count allelic sequencing coverage from a BAM file, thus obtaining the haplotype-specific coverage for WGS, RNA-seq, Capture-C, H3K27ac, and ATAC from NALM6 (Figure S14). We found that MYC and the 2.2 Mb surrounding region with the N-Me and BENC enhancers (chr8:128562195-130790266, hg19) are within one phase set of 4.5 Mb in length, providing the basis of enhancer analysis below.
Pediatric cancer data collection
The source data of GenomePaint Data Release V1 is summarized in Table S1 while the sample-level information is recorded in Table S2.
We also collected data for several pediatric tumor-derived cell lines. The data includes WGS, RNA-seq, H3K27ac ChIP-seq, CTCF ChIP-seq, DUX4 ChIP-seq, ATAC-seq, and in-situ Hi-C. Datasets and repository accessions are listed in Table S3.
Pediatric cancer dataset production on hg19/GRCh37 reference genome
Most of the pediatric cancer genomic data collected from publications are hg19-based. In a few cases, hg18-based data is lifted over to hg19. In addition, we conducted the following analysis to obtain additional useful information from the tumor sequencing data and made them available on the hg19 Pediatric Cancer GenomePaint.
We ran CONSERTING (Chen et al. 2015) on the tumor-normal paired whole-exome sequencing (WES) of all pediatric patients to call CNV and LOH, which was not available from the original publications. Resulting CNV and LOH calls are available in GenomePaint and are labeled as “WES” for its DNA sequencing type.
We ran Cis-X (Liu et al. 2020) for allele-specific expression (ASE) analysis on the RNA-seq of all pediatric tumors with both RNA-seq and DNA sequencing (WGS, WES, or CGI). Resulting ASE status is shown alongside gene expression rank.
We use VEP (McLaren et al. 2016) to consistently annotate the SNV/indel variants. The resulting predicted consequences (https://useast.ensembl.org/info/genome/variation/predicted_data.html) are shown as functional classes with a different color for each class; the HGVS protein nomenclatures are used to label the variants.
We run RNApeg (Edmonson et al., unpublished) to call the splice junctions from RNA-seq results.
Pediatric cancer dataset production on hg38/GRCh38 reference genome
GRCh37 (hg19) is the default assembly version for the pediatric cancer genomic dataset. To produce the GRCh38 version, we ran UCSC liftOver for somatic variants and gene internals (for FPKM). Assay tracks of sequencing coverage and splice junctions require remapping of WGS and RNA-seq data to GRCh38, which will be incorporated in upcoming data release.
TCGA GBM dataset reanalysis
Tumor/germline WGS and tumor RNA-seq BAM files for 38 GBM tumors with both WGS and RNA-seq were downloaded from NCI GDC Legacy Archive. The data were re-analyzed using the hg19 analysis pipeline described in the section “Pediatric cancer dataset production on hg19/GRCh37 reference genome”. For genes recurrently mutated in GBM (Brennan et al. 2013), expert manual curation was performed for their CNV, SV, and coding mutation data. For the rest of the genomic regions, we included variation data from the pipeline output. Upon examining the EGFR locus, we added following intra-chr7 SV events with evidence from WGS reads but were missed by SV calling.
| Sample | Intra-chr7 breakpoints | Reason |
|---|---|---|
| TCGA-06-0211-01A | 55110908-55222135 | Exons 2-7 deletion leading to vIII isoform. |
| 55163884-55232907 | Exons 2-14 deletion with only 1 softclip read support. | |
| TCGA-06-0211-02A | 55163884-55232907 | Exons 2-14 deletion leading to the aberrant splice junction. |
| Both samples | 54663335-55223635 | 560 Kb deletion. |
We noticed the EGFR nonsense mutation Q276* in the diagnosis and relapse tumors of the patient TCGA-06-0211 (Figure S6A). The same variant is present in COSMIC v92 (https://cancer.sanger.ac.uk/cosmic/mutation/overview?id=95519938). In both diagnosis and relapse, the mutant allele is present at subclonal level in DNA, and does not exist in tumor RNA. The mutation is also close to one of the breakpoints underlying the exon 8 ITD detected from RNA-seq by CICERO (Figure S6B). By inspecting the WGS read alignment (Figure S6C), we found the “nonsense” SNV mutant allele is 36 bp away from one SV breakpoint, both of which exist on the same set of reads. Though the SNV appears to be on exon 7, it is actually part of the rearranged intron 8, which is spliced out making the SNV not present in EGFR mRNA. We thus considered the “nonsense” call as a false annotation of this misleading and ultimately inconsequential SNV, and excluded it from our curated set of GBM variants.
Online user tutorials
A user tutorial and instructions for NCI GDC custom track can be found at https://genomepaint.stjude.cloud/#tutorial.
Online examples
Along with the Figure Live Links, we provide the following URLs to showcase various features of GenomePaint. On each URL, users can check the page source to see the detailed customization parameters. For additional details, please refer to Online User Tutorial (https://genomepaint.stjude.cloud/#tutorial) on GenomePaint’s website.
- Example 1: TP53 bi-allelic loss in adrenocortical tumors.
- Example 2: IKZF1 haploinsufficiency in B-ALL.
- Example 3: MYB alteration status in T-ALL and the association with patient outcome.
- Example 4: Association of MDM2 expression in AML with patient outcome.
- Example 5: Matrix View of mutual-exclusive or co-mutation patterns and association with patient outcome.
- Example 6: A TP53 splice site mutation causing intron retention inspected via Sample View.
- Example 7: APOBEC signature is associated with kataegis as revealed by rainfall plot in Sample View.
- Example 8: TCGA DLBC cohort mutation profile over CDKN2A/B locus.
- Example 9: TCGA SKCM cohort mutation profile over TERT.
Quantification and Statistical Analysis
Wilcoxon test is used to compare mean variant allele fraction between WGS and RNA-seq for a group of SNVs. Log-rank test is used to compute p values between pairs of Kaplan-Meier curves. All statistical computing is done in R software.
Supplementary Material
Table S1. List of publications as the sources of the pediatric cancer mutation and gene expression dataset, Related to Figure 1 and STAR Methods.
Table S2. Demographics information and DNA/RNA assay availability for every pediatric tumor used in the pediatric cancer dataset, Related to Figure 1 and STAR Methods.
Key Resource Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Human NOTCH1 | Abcam | ab27526 |
| Deposited Data | ||
| NALM16 WGS, RNAseq, Hi-C | EGA | EGAS00001004669 |
| Table S1. List of publications as the sources of the pediatric cancer mutation and gene expression dataset | ||
| Table S2. Demographics information and DNA/RNA assay availability for every pediatric tumor used in the pediatric cancer dataset. | ||
| Table S3. List of pediatric cancer cell line sequencing datasets available in GenomePaint. | ||
| Experimental Models: Cell Lines | ||
| NALM6 | DSMZ | ACC128 |
| NALM16 | DSMZ | ACC680 |
| Oligonucleotides | ||
| Oligo sequences at MYC promoter for Capture-C: GATCAGAATCGATGCATTTTTTGTGCATGACCGCATTTCCAATAATAAAAGGGGAAAGAGGACCTGGAAAGGAATTAAACGTCCGGTTTGTCCGGGGAGGAAAGAGTTAACGGTTTTTTTC and CTAGGCATCGTTTTCCTCCTTATGCCTCTATCATTCCTCCCTATCTACACTAACATCCCACGCTCTGAACGCGCGCCCATTAATACCCTTCTTTCCTCCACTCTCCCTGGGACTCTTGATC | This paper | N/A |
| Software and Algorithms | ||
| GenomePaint | This paper | https://genomepaint.stjude.cloud// |
| CONSERTING | Chen et al. 2015 | http://ftp.stjude.org/pub/software/conserting/CONSERTING_code.tgz |
| Cis-X | Liu et al. 2020 | https://github.com/stjude/cis-x |
| VEP | McLaren et al. 2016 | https://uswest.ensembl.org/info/docs/tools/vep/index.html |
Highlights:
Access multi-omics data from pediatric cancers, PDX and cell lines.
Evaluate effects of splice site mutations with RNA splicing data.
Evaluate regulatory roles of non-coding alterations with tumor expression data.
Support for adult cancer and user-supplied data.
ACKNOWLEDGEMENTS
We thank Drs. Alexander Gout, Brian Abraham, and David Finkelstein for helpful comments on the manuscript, and Dr. Roel Verhaak for his comments on the GBM data. This research was supported by the National Cancer Institute of the National Institutes of Health under Award Number R01CA216391 to JZ. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. This research was also supported by Cancer Center Support Grant P30CA021765 from the National Institutes of Health and in part by the American Lebanese Syrian Associated Charities (ALSAC).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Al-Ahmadie Hikmat, Iyer Gopa, Hohl Marcel, Asthana Saurabh, Inagaki Akiko, Schultz Nikolaus, Hanrahan Aphrothiti J., et al. 2014. “Synthetic Lethality in ATM-Deficient RAD50-Mutant Tumors Underlies Outlier Response to Cancer Therapy.” Cancer Discovery. 10.1158/2159-8290.cd-14-0380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahr Carsten, Lisa von Paleske, Uslu Veli V., Remeseiro Silvia, Takayama Naoya, Ng Stanley W., Murison Alex, et al. 2018. “A Myc Enhancer Cluster Regulates Normal and Leukaemic Haematopoietic Stem Cell Hierarchies.” Nature 553 (7689): 515–20. [DOI] [PubMed] [Google Scholar]
- Brennan Cameron W., Verhaak Roel G. W., McKenna Aaron, Campos Benito, Noushmehr Houtan, Salama Sofie R., Zheng Siyuan, et al. 2013. “The Somatic Genomic Landscape of Glioblastoma.” Cell 155 (2): 462–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Xiang, Gupta Pankaj, Wang Jianmin, Nakitandwe Joy, Roberts Kathryn, Dalton James D., Parker Matthew, et al. 2015. “CONSERTING: Integrating Copy-Number Analysis with Structural-Variation Detection.” Nature Methods 12 (6): 527–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauwerse Johannes G., van Belzen Martine, van Haeringen Arie, van Santen Gijs, van de Lans Christian, Rahikkala Elisa, Garavelli Livia, Breuning Martijn, Raoul Hennekam, and Peters Dorien. 2016. “Analysis of Mutations within the intron20 Splice Donor Site of CREBBP in Patients with and without Classical RSTS.” European Journal of Human Genetics: EJHG 24 (11): 1639–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies James O. J., Telenius Jelena M., McGowan Simon J., Roberts Nigel A., Taylor Stephen, Higgs Douglas R., and Hughes Jim R.. 2016. “Multiplexed Analysis of Chromosome Conformation at Vastly Improved Sensitivity.” Nature Methods 13 (1): 74–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- deCarvalho Ana C., Kim Hoon, Poisson Laila M., Winn Mary E., Mueller Claudius, Cherba David, Koeman Julie, et al. 2018. “Discordant Inheritance of Chromosomal and Extrachromosomal DNA Elements Contributes to Dynamic Disease Evolution in Glioblastoma.” Nature Genetics 50 (5): 708–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delvecchio Manuela, Gaucher Jonathan, Aguilar-Gurrieri Carmen, Ortega Esther, and Panne Daniel. 2013. “Structure of the p300 Catalytic Core and Implications for Chromatin Targeting and HAT Regulation.” Nature Structural & Molecular Biology 20 (9): 1040–46. [DOI] [PubMed] [Google Scholar]
- Gao Jianjiong, Aksoy Bülent Arman, Dogrusoz Ugur, Dresdner Gideon, Gross Benjamin, Sumer S. Onur, Sun Yichao, et al. 2013. “Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal.” Science Signaling 6 (269): l1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman Mary J., Craft Brian, Hastie Mim, Repečka Kristupas, Fran McDade Akhil Kamath, Banerjee Ayan, et al. 2020. “Visualizing and Interpreting Cancer Genomics Data via the Xena Platform.” Nature Biotechnology 38 (6): 675–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gröbner Susanne N., Worst Barbara C., Weischenfeldt Joachim, Buchhalter Ivo, Kleinheinz Kortine, Rudneva Vasilisa A., Johann Pascal D., et al. 2018. “The Landscape of Genomic Alterations across Childhood Cancers.” Nature 555 (7696): 321–27. [DOI] [PubMed] [Google Scholar]
- Herranz Daniel, Ambesi-Impiombato Alberto, Palomero Teresa, Schnell Stephanie A., Belver Laura, Wendorff Agnieszka A., Xu Luyao, et al. 2014. “A NOTCH1-Driven MYC Enhancer Promotes T Cell Development, Transformation and Acute Lymphoblastic Leukemia.” Nature Medicine 20 (10): 1130–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz Denes, Weintraub Abraham S., Day Daniel S., Valton Anne-Laure, Bak Rasmus O., Li Charles H., Goldmann Johanna, et al. 2016. “Activation of Proto-Oncogenes by Disruption of Chromosome Neighborhoods.” Science 351 (6280): 1454–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoadley Katherine A., Yau Christina, Hinoue Toshinori, Wolf Denise M., Lazar Alexander J., Drill Esther, Shen Ronglai, et al. 2018. “Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer.” Cell 173 (2): 291–304.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer Gopa, Hanrahan Aphrothiti J., Milowsky Matthew I., Al-Ahmadie Hikmat, Scott Sasinya N., Janakiraman Manickam, Pirun Mono, et al. 2012. “Genome Sequencing Identifies a Basis for Everolimus Sensitivity.” Science 338 (6104): 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent W. James, Sugnet Charles W., Furey Terrence S., Roskin Krishna M., Pringle Tom H., Zahler Alan M., and Haussler David. 2002. “The Human Genome Browser at UCSC.” Genome Research 12 (6): 996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima Kensuke, Konopleva Marina, Samudio Ismael J., Shikami Masato, Cabreira-Hansen Maria, McQueen Teresa, Ruvolo Vivian, et al. 2005. “MDM2 Antagonists Induce p53-Dependent Apoptosis in AML: Implications for Leukemia Therapy.” Blood 106 (9): 3150–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancho Olga, and Herranz Daniel. 2018. “The MYC Enhancer-Ome: Long-Range Transcriptional Regulation of MYC in Cancer.” Trends in Cancer Research 4 (12): 810–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Benshang, Brady Samuel W., Ma Xiaotu, Shen Shuhong, Zhang Yingchi, Li Yongjin, Szlachta Karol, et al. 2020. “Therapy-Induced Mutations Drive the Genomic Landscape of Relapsed Acute Lymphoblastic Leukemia.” Blood 135 (1): 41–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Yu, Easton John, Shao Ying, Maciaszek Jamie, Wang Zhaoming, Wilkinson Mark R., McCastlain Kelly, et al. 2017. “The Genomic Landscape of Pediatric and Young Adult T-Lineage Acute Lymphoblastic Leukemia.” Nature Genetics 49 (8): 1211–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Yu, Li Chunliang, Shen Shuhong, Chen Xiaolong, Szlachta Karol, Edmonson Michael N., Shao Ying, et al. 2020. “Discovery of Regulatory Noncoding Variants in Individual Cancer Genomes by Using Cis-X.” Nature Genetics 52 (8): 811–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour Marc R., Abraham Brian J., Anders Lars, Berezovskaya Alla, Gutierrez Alejandro, Durbin Adam D., Etchin Julia, et al. 2014. “Oncogene Regulation. An Oncogenic Super-Enhancer Formed through Somatic Mutation of a Noncoding Intergenic Element.” Science 346 (6215): 1373–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Xiaotu, Liu Yu, Liu Yanling, Alexandrov Ludmil B., Edmonson Michael N., Gawad Charles, Zhou Xin, et al. 2018. “Pan-Cancer Genome and Transcriptome Analyses of 1,699 Paediatric Leukaemias and Solid Tumours.” Nature 555 (7696): 371–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren William, Gil Laurent, Hunt Sarah E., Riat Harpreet Singh, Ritchie Graham R. S., Thormann Anja, Flicek Paul, and Cunningham Fiona. 2016. “The Ensembl Variant Effect Predictor.” Genome Biology 17 (1): 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullighan Charles G., Zhang Jinghui, Kasper Lawryn H., Lerach Stephanie, Payne-Turner Debbie, Phillips Letha A., Heatley Sue L., et al. 2011. “CREBBP Mutations in Relapsed Acute Lymphoblastic Leukaemia.” Nature 471 (7337): 235–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nik-Zainal Serena, Alexandrov Ludmil B., Wedge David C., Van Loo Peter, Greenman Christopher D., Raine Keiran, Jones David, et al. 2012. “Mutational Processes Molding the Genomes of 21 Breast Cancers.” Cell 149 (5): 979–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northcott Paul A., Buchhalter Ivo, Morrissy A. Sorana, Hovestadt Volker, Weischenfeldt Joachim, Ehrenberger Tobias, Gröbner Susanne, et al. 2017. “The Whole-Genome Landscape of Medulloblastoma Subtypes.” Nature 547 (7663): 311–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neil J, and Look AT. 2007. “Mechanisms of Transcription Factor Deregulation in Lymphoid Cell Transformation.” Oncogene 26 (47): 6838–49. [DOI] [PubMed] [Google Scholar]
- Robinson James T., Thorvaldsdóttir Helga, Winckler Wendy, Guttman Mitchell, Lander Eric S., Getz Gad, and Mesirov Jill P.. 2011. “Integrative Genomics Viewer.” Nature Biotechnology 29 (January): 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roelfsema Jeroen H., White Stefan J., Ariyürek Yavuz, Bartholdi Deborah, Niedrist Dunja, Papadia Francesco, Bacino Carlos A., et al. 2005. “Genetic Heterogeneity in Rubinstein-Taybi Syndrome: Mutations in Both the CBP and EP300 Genes Cause Disease.” American Journal of Human Genetics 76 (4): 572–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusch Michael, Nakitandwe Joy, Shurtleff Sheila, Newman Scott, Zhang Zhaojie, Edmonson Michael N., Parker Matthew, et al. 2018. “Clinical Cancer Genomic Profiling by Three-Platform Sequencing of Whole Genome, Whole Exome and Transcriptome.” Nature Communications 9 (1): 3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Liqing, Shao Ying, Nance Stephanie, Dang Jinjun, Xu Beisi, Ma Xiaotu, Li Yongjin, et al. 2019. “Long-Read Sequencing Unveils IGH-DUX4 Translocation into the Silenced IGH Allele in B-Cell Acute Lymphoblastic Leukemia.” Nature Communications 10 (1): 2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Ke, Ding Liang, Chang Ti-Cheng, Shao Ying, Chiang Jason, Mulder Heather, Wang Shuoguo, et al. 2019. “Structure and Evolution of Double Minutes in Diagnosis and Relapse Brain Tumors.” Acta Neuropathologica 137 (1): 123–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Minjun, Safavi Setareh, Woodward Eleanor L., Duployez Nicolas, Olsson-Arvidsson Linda, Ungerbäck Jonas, Sigvardsson Mikael, et al. 2020. “13q12.2 Deletions in Acute Lymphoblastic Leukemia Lead to Upregulation of FLT3 through Enhancer Hijacking.” Blood 136 (8): 946–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zehir Ahmet, Benayed Ryma, Shah Ronak H., Syed Aijazuddin, Middha Sumit, Kim Hyunjae R., Srinivasan Preethi, et al. 2017. “Mutational Landscape of Metastatic Cancer Revealed from Prospective Clinical Sequencing of 10,000 Patients.” Nature Medicine 23 (6): 703–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Xiaoyang, Choi Peter S., Francis Joshua M., Imielinski Marcin, Watanabe Hideo, Cherniack Andrew D., and Meyerson Matthew. 2016. “Identification of Focally Amplified Lineage-Specific Super-Enhancers in Human Epithelial Cancers.” Nature Genetics 48 (2): 176–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Xin, Edmonson Michael N., Wilkinson Mark R., Patel Aman, Wu Gang, Liu Yu, Li Yongjin, et al. 2016. “Exploring Genomic Alteration in Pediatric Cancer Using ProteinPaint.” Nature Genetics 48 (1): 4–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman Mark W., Liu Yu, He Shuning, Durbin Adam D., Abraham Brian J., Easton John, Shao Ying, et al. 2018. “MYC Drives a Subset of High-Risk Pediatric Neuroblastomas and Is Activated through Mechanisms Including Enhancer Hijacking and Focal Enhancer Amplification.” Cancer Discovery 8 (3): 320–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. List of publications as the sources of the pediatric cancer mutation and gene expression dataset, Related to Figure 1 and STAR Methods.
Table S2. Demographics information and DNA/RNA assay availability for every pediatric tumor used in the pediatric cancer dataset, Related to Figure 1 and STAR Methods.
Data Availability Statement
NALM16 WGS, RNAseq, Hi-C is available from European Genome-Phenome Archive under accession EGAS00001004669. Additional published data including pediatric cancer and cell line DNA variations, transcriptomes, and epigenomes are listed in Tables S1-S3. The pediatric cancer dataset is freely accessible through GenomePaint at https://genomepaint.stjude.cloud/. Data processing scripts are available on https://github.com/josephpowi/gpdatascript2020. GenomePaint software license is available upon request.