

Abstract
The formation of secondary organic aerosol (SOA) indoors is one of the many consequences of the rich and complex chemistry that occurs therein. Given particulate matter has well documented health effects, we need to understand the mechanism for SOA formation indoors and its resulting composition. This study evaluates some uncertainties that exist in quantifying gas-to-particle partitioning of SOA-forming compounds using an indoor detailed chemical model. In particular, we investigate the impacts of using different methods to estimate compound vapour pressures as well as simulating the formation of highly oxygenated organic molecules (HOM) via auto-oxidation on SOA formation indoors. Estimation of vapour pressures for 136 α-pinene oxidation species by six investigated methods led to standard deviations of 28–216%. Inclusion of HOM formation improved model performance across three of the six assessed vapour pressure estimation methods when comparing against experimental data, particularly when the NO2 concentration was relatively high. We also explored the predicted SOA composition using two product classification methods, the first assuming the molecule is dominated by one functionality according to its name, and the second accounting for the fractional weighting of each functional group within a molecule. The SOA composition was dominated by the HOM species when the NO2-to-α-terpineol ratio was high for both product classification methods, as these conditions promoted formation of the nitrate radical and hence formation of HOM monomers. As the NO2-to-α-terpineol ratio decreased, peroxides and acids dominated the simple classification, whereas for the fractional classification, carbonyl and alcohol groups became more important.
Keywords: Secondary organic aerosol, Highly oxygenated organic molecules, Vapour pressure, Volatile organic compound, Indoor air chemistry
1. Introduction
In developed countries, we spend approximately 90% of our time indoors (i.e. at home, in the work place, or commuting) and consequently most of our exposure to air pollution occurs indoors. Indoor air pollutants are generated through activities such as cooking (Lee et al., 2002; Farmer et al., 2019), cleaning (Wolkoff et al., 1998; Nazaroff and Weschler, 2004), smoking (Lin et al., 2007) and wood burning (Alberts, 1994), as well as emitted from building materials (Uhde and Salthammer, 2007), whilst some pollutants, such as ozone (O3) or nitrogen oxides (NOx) are transported indoors from outdoors. Consumer products such as cleaning agents, air fresheners and personal care products contain terpene species, such as limonene, α-pinene or α-terpinolene (Nazaroff and Weschler, 2004; Singer et al., 2006). Emissions of such species and subsequent chemical reactions indoors can negatively impact indoor air quality through the formation of numerous multifunctional and sometimes harmful secondary pollutants (Weschler, 2006; Waring and Wells, 2015; Trantallidi et al., 2015).
Some species formed indoors following the oxidation of terpenes and other volatile organic compounds (VOCs) have low enough vapour pressures (Vp) such that they partition to the particle phase to form secondary organic aerosols (SOA) (Sarwar et al., 2004; Waring, 2014). This can happen via nucleation or condensation as shown in Fig. 1.
Fig. 1.
The oxidation of VOCs and subsequent processes to form SOA.
This partitioning process is most likely for multifunctional compounds with high molecular weights of 150–300 g mol−1 and with ambient vapour pressures below 0.1 Pa (Barley and McFiggans, 2010). In order to accurately calculate the rate of partitioning for relevant species from the gas-to aerosol-phase, it is necessary to know their vapour pressures. However, for the vast majority of atmospheric species, measured data do not exist and instead estimation methods must be used to calculate vapour pressure.
There are different methods available to calculate vapour pressure, which have been reviewed by Barley and McFiggans (2010) and O’Meara et al. (2014). Some of the older methods were developed to support the needs of the chemical industry and tended to focus on volatile fluids for which experimental boiling points were readily available (Barley and McFiggans, 2010). However, more recent methods were designed with atmospheric applications as a focus, and for these latter methods, it was also necessary to estimate boiling points (Tb), given the absence of experimental data for most species. For all of the methods, increasing complexity, which generally involves trying to account for increasing multifunctionality, tends to lead to smaller data sets that can become subject to overfitting (Barley and McFiggans, 2010). In other words, if the functionality becomes too defined, there are too few species remaining on which to test whether the estimation method is working.
In terms of estimating Tb, the simplest method is that of Joback and Reid (1987), which includes information for 41 functional groups. It was further developed by Stein and Brown (1994) with 44 additional groups. Finally, the method of Nannoolal et al. (2004) includes both primary and secondary groups along with group interactions and has been suggested as the most accurate method for Tb estimation (Barley and McFiggans, 2010).
Additionally, Barley and McFiggans (2010) suggested that the Nannoolal Vp (NVP) method (Nannoolal et al., 2008) used in conjunction with the estimation method for Tb by Nannoolal et al. (2004) gave the best agreement of modelled-to-measured values in their study. The Myrdal and Yalkowsky (MY) equation (Myrdal and Yalkowsky, 1997) gave the second-best results, but it tended to overestimate Vp. For all tests, errors associated with the determination of Tb tended to dominate the error in the Vp estimate.
O’Meara et al. (2014) carried out an extended analysis with experimentally determined values of the vapour pressure to test against and crucially, with vapour pressures more representative of those compounds likely to exist in the atmosphere. Most of the 90 measured compounds were carboxyl (51), followed by hydroxyl (29), amine (11), ketone (7), aldehyde (5), nitro (9), ester (3), ether (7), methoxy (12), and nitrate (1) species. There were no measurements of peroxyacyl nitrates (PANs), peroxides, or percarboxylic acids available. Note also that many measurements were made at non-ambient temperatures which then had to be extrapolated to atmospheric temperature (i.e. through use of the Antoine equation) leading to further uncertainty (O’Meara et al., 2014). O’Meara et al. (2014) considered 1000 Pa (7.5 Torr) to be the cut-off vapour pressure for significant contribution to the condensed phase.
O’Meara et al. (2014) also evaluated the EVAPORATION (EVAP) model (Compernolle et al., 2011) and the Lee-Kesler (LK-VP) method (Reid et al., 1987). O’Meara et al. (2014) note that some of these tested methods have been produced more recently than others (e.g. EVAP) and have consequently been able to use more measurements for ‘tuning’ when compared with methods developed some years ago, so they should be more accurate.
Some observations made by O’Meara et al. (2014) about limitations of the different methods are that several functional groups present in the test-set compounds are not covered by the EVAP model (including aromatics); the NVP method provides a poor estimate of Vp for compounds with three or more oxygenated groups; the MY equation overestimates Vp as volatility decreases. Overall, the LK-VP or NVP methods were preferred, with the former preferable when many H-bonds were present. Although EVAP was most accurate, it was considered to have more limited use owing to consideration of fewer functional groups than other methods.
The different methods were then tested in a model to estimate SOA loading to compare against measurements. The MY-equation using the Nannoolal et al. (2004) Tb gave the best results (O’Meara et al., 2014), though this was likely due in part to bias (underestimation of Vp as volatility increases) in the calculation of Vp improving the accuracy of the SOA prediction. Most of the methods overestimated SOA concentrations. In the atmosphere, lower volatility compounds are expected to be more oxidized and/or have higher molecular weights. The NVP and LK-VP methods have relatively low accuracy for high SOA loading when volatility is higher, but without the positive bias that some other methods (e.g. MY) have to compensate (O’Meara et al., 2014).
An emerging aspect for SOA prediction is the recent discovery that SOA can be enhanced by rapid formation of low-volatility species, so-called highly oxygenated organic molecules (HOMs) following oxidation of VOCs (Ehn et al., 2014; Bianchi et al., 2019; McFiggans et al., 2019; Pagonis et al., 2019). The HOMs result through auto-oxidation of the organic peroxy radicals (RO2) that are formed following oxidation of the parent VOC (Peräkylä et al., 2020). Indeed, when the RO2 radical lifetime is sufficiently long, auto-oxidation can be repeated several times and HOMs can be formed (Bianchi et al., 2019).
HOMs are highly multifunctional compounds, and their volatility is expected to be very low (Ehn et al., 2014). However, the Vp of HOM species remains uncertain (Kurtén et al., 2016; Kurtén et al., 2018). Pagonis et al. (2019) investigated HOM formation and concentration in a museum following oxidation of limonene. HOM concentrations up to 1.7 ppt (~2.08 × 10−5 μg m−3) were noted while aerosol mass concentration increased up to ~1.6 μg m−3. The measurements could be reproduced by a model assuming a HOM yield of 11% following ozonolysis of limonene.
The aim of this paper is to evaluate some of the uncertainties around SOA prediction indoors, including those associated with estimating vapour pressures and the potential impact of HOM formation on predicted concentrations. Using a detailed chemical model for indoor air, we present a new scheme for gas-to-aerosol partitioning of α-pinene, which we evaluate with measured concentrations of α-pinene oxidation SOA from a chamber study. We then use a modified version of the model to explore 21 α-terpineol oxidation experiments, which were carried out under a range of VOC-to-NO2 ratios. We explore the impact of HOM formation indoors using different Vp methods and investigate how they affect the predicted SOA concentrations and composition.
2. Material and methods
2.1. INDCM model overview
An INdoor Detailed Chemical Model (INDCM) (Carslaw, 2007; Carslaw et al., 2012; Kruza et al., 2017) has been developed to investigate gas-to-particle partitioning for α-pinene oxidation. The INDCM is a near explicit zero-D box model for studying indoor air chemistry. It uses a comprehensive chemical mechanism called the Master Chemical Mechanism, MCM v3.3.1 (Jenkin et al., 1997). The degradation process of VOCs is initiated by reactions with OH, O3, NO3, and photolysis where relevant. Radicals are generated as intermediate products, such as oxy (RO) and peroxy (RO2) radicals, excited and stabilized Criegee (R’R”COO) species, and these radical species can then undergo a number of further reactions. A range of products such as alcohols, carbonyls, and nitrates are formed, until carbon dioxide and water are produced at the end of the oxidation chain. The MCM also includes an inorganic scheme including reactions of ozone, nitrogen oxides (NOx), and carbon monoxide (Jenkin et al., 1997, 2003; Saunders et al., 2003). The modified INDCM includes approximately 5900 species and 20,300 gas-phase reactions (including photolysis), as well as a representation of indoor-outdoor air exchange, internal emissions, and surface deposition of some species (Kruza et al., 2017). The input parameters of the model can be adjusted for different indoor and outdoor settings.
2.2. Model development
2.2.1. Development of α-pinene partitioning reactions
Until now the INDCM only included gas-to-particle partitioning for limonene, where 48 limonene-oxidation species were identified and assumed to partition as described by Carslaw et al. (2012). We have now extended the model adding terms describing the partitioning for α-pinene oxidation products. The α-pinene gas-phase oxidation scheme is available from the MCM web site (http://mcm.leeds.ac.uk/MCM/)This scheme was investigated and all non-radical species with more than five carbon atoms were identified, which provided 136 species for further consideration.
Gas-to-particle partitioning was defined for the 136 species according to the theory of Pankow (1994), where the partitioning phase of the species is defined by the thermodynamic equilibrium between the gas-phase and condensed organic-phase (Jenkin, 2004; Bateman et al., 2009). The partitioning coefficient, Kp (m3 μg−1), is defined as:
| (1) |
where R is the ideal gas constant (8.314 J K−1 mol−1); T is the temperature (K); MWom is the mean molecular weight of the absorbing particulate organic material (g mol−1); γom is the activity coefficient of the species in the condensed organic-phase. We have assumed that the aerosol is well-mixed and that in this instance, the γom value is 1.0. Past studies have shown that SOA prediction is likely to depend more on the Vp method used than the value of the activity coefficient adopted (McFiggans et al., 2010; Barley et al., 2011; Topping et al., 2011, 2013); Vp is the liquid vapour pressure of the species (Torr). The initial value of MWom was assumed to be 120 g mol−1 (Sarwar and Corsi, 2007), but subsequent values are calculated as the model run proceeds, by accounting for the molecular weights and proportions of each individual component of the overall particle mass.
The partitioning process is represented dynamically as a balance between absorption and desorption presented in Equation (2) (Jenkin, 2004; Bateman et al., 2009). A temperature and species independent value of 6.2 × 10−3 m3 μg−1 s−1 was used for kon, which was calculated based on the estimated collision rate of gaseous molecules with a monodisperse aerosol with a diameter of ~50 nm (Bateman et al., 2009; Carslaw et al., 2012). The value of koff was then found assuming equilibrium conditions exist (Jenkin, 2004).
| (2) |
A range of Vp estimation methods were then used as described in section 2.3. For each of them, we used the method presented by Nannoolal et al. (2004) to calculate Tb for each species as recommended by Johnson et al. (2006) and Barley and McFiggans (2010). This equilibrium absorptive partitioning method will overestimate the condensed amount of all species (including HOMs) because there will be a finite kinetic uptake to the aerosol (and to walls in chamber experiments); i.e., it will not be in equilibrium (Garmash et al., 2020).
2.2.2. Incorporating HOM in the INDCM model
HOMs are formed in the atmosphere via different reaction pathways, such as reactions with O3 and the OH radical (Berndt et al., 2016). Ozonolysis is an efficient pathway to form HOMs, but reaction with OH radicals can also contribute, but with a smaller molar yield than the ozonolysis pathway (Bianchi et al., 2019). There is limited information on how much HOM formation occurs via these different oxidation pathways, so our preliminary assumptions were based on the limited information available in the literature (Pagonis et al., 2019; McFiggans et al., 2019) and ongoing (not yet published) experimental work. We assumed that when α-pinene is oxidized by OH and O3, 8 and 10% respectively of the initial oxidation step forms the highly oxidized HOM peroxy radicals (HOMRO2), with the remaining 92 and 90% respectively following the original oxidation route according to the MCM reaction protocol (Jenkin et al., 1997).
It was then assumed that the HOMRO2 underwent reaction with HO2, NO3, NO, other RO2 in the model, and that a HOM monomer was formed as a result. It was also assumed that HOMRO2 could dimerize in the gas phase to form HOM dimers. We made no assumptions about the HOM and HOMRO2 structures for this work. The mechanistic details and rate coefficients adopted from the MCM are provided in the Supplementary Information (see Table S1).
Given that HOMs are found to be mainly of low volatility with a small fraction being semivolatile, we assume (ignoring the semivolatile HOM fraction) that the vapour pressure for HOM is 3.59 × 10−10 Torr, which is equivalent to the lowest Vp already being used in the model (e.g. for one of the α-pinene oxidation products). Given that HOM dimers were assumed to have low volatility, and probably even extremely low volatility (Peräkylä et al., 2020), we assume the Vp for the HOM dimer is 0. The molecular mass was assumed to be 294 g mol−1 and 462 g mol−1 for HOM and the HOM dimer, respectively. Clearly, measurements of these values in the future would aid model development.
2.3. Development of α-terpineol scheme
Given that there is not an α-terpineol scheme in the MCM at present, we have added the initial oxidation steps of α-terpineol explicitly to the INDCM mechanism and then assumed that the products of these oxidation steps are identical to those from α-pinene oxidation, including the subsequent partitioning to SOA. Rate coefficients for reactions of α-terpineol with O3, OH, and NO3 were taken from the literature at 3 × 10−16, 1.9 × 10−10 and 16 × 10−12 cm3 molecule−1 s−1, respectively (Wells, 2005; Jones and Ham, 2008). The resulting RO2 radicals and other intermediate products reacted according to the MCM protocol (Jenkin et al., 1997) with the assumptions about HOM formation described in the previous section also included here.
2.4. Vapour pressure methods
The vapour pressures for the α-pinene oxidation species were calculated using some of the methods discussed in the Introduction section such as the Myrdal and Yalkowsky equation (MY), the EVAPORATION model (EVAP), the Nannoolal (NVP) method, as well as the Antoine method, the Mackay method, and the modified version of the Grain-Watson equation given the latter three are used in the EPI – Suite v4.11 software (Ketenoglu, 2018; EPI-Suite v4.11). For each method, we used the estimation of Tb proposed by Nannoolal et al. (2004) at a temperature of 295 K and relative humidity (RH) of 50%.
2.5. Experimental data
This study uses two experimental data sets to evaluate the model. The experiments were carried out in a 100 L Teflon-film reaction chamber, maintained at ~20 °C and 50% relative humidity. A total volume of 80 L was used for these experiments. Ozone was produced by photolyzing air in separate Teflon-film ozone chambers (150–200 ppm) and NO2 (100 ppm in balance of N2) was received from a cylinder for the second experimental data set. The purity of these species was separately verified by ozone and NOx monitors (Thermo Electron Models 49i and 42i (Waltham, MA), respectively.) The terpene was introduced as an injection into the reaction chamber via a heated syringe injection port with diluent air to achieve a specific concentration defined for each experiment. Ozone was added to the reaction chamber by either gas-tight syringe or a 10 L Teflon chamber connected to the reaction chamber port. For the addition of NO2 to the reaction chamber, NO2 + air was added to the reaction chamber by a gas-tight syringe. Given that reaction mixtures were directly injected into the 80 L reaction chamber, dilution occurred by mixing in the chamber. Particle samples were monitored with a scanning mobility particle sizer (SMPS, TSI 3091), providing detection over the particle size range of 9.82–414.2 nm in 105 size bins, every 2.27 min, at a sample flow rate of 1 L/min. The sample collection tube was 45 in long 3/16 in internal diameter Teflon tubing. Particles were assumed to be spherical with a density of 1.0 g/mL.
The first experimental data set had initial conditions of 100 ppb O3, 100 ppb of α-pinene, and RH of 50% in the dark with zero NO2 conditions. The experiment was repeated three times and the formed SOA concentration measured over approximately an hour. A further set of 21 chamber experimental runs explored the impact of NO2 on SOA formation initiated by α-terpineol ozonolysis. The NO2 concentration was varied between 0 and 2000 ppb, whilst α-terpineol ranged between 20 and 200 ppb with different VOC-to-NO2 ratios explored. The ozone concentration was in excess at 25 ppm. Under high ozone conditions, NO is removed by O3 and NO2 is the only nitrogen oxide species present (Waring and Wells, 2015). NO concentrations were expected to be minimal under these experimental conditions. The experimental set up using high concentrations ensured that the terpene was oxidized as quickly as possible and also minimised losses to the chamber walls.
Both sets of experiments were conducted in the absence of seed particles and for the second set of experiments, with the use of very high O3 concentrations, in order to minimize wall losses of SOA. Based on Pathak et al. (2007), an aerosol mass loss rate constant was determined for each experimental condition, using the assumption that the loss rate is first order and independent of particle size. The mass corrections due to wall losses using this method ranged from 3 to 30% and have been applied to our experimental data. Wall losses are however, also likely to be influenced by the surface to volume ratios for the experimental chambers. The Pathak chamber surface to volume ratio was 2.8 m−1 versus 20.1 m−1 for the chamber used for these experiments. This difference in surface to volume ratio may imply that our correction is an underestimate. There is currently a great deal of uncertainty around wall loss corrections for SOA and their gas-phase precursors and how these vary according to factors such as chamber size, material and level of cleanliness. Therefore, rather than attempting to correct our measured SOA concentrations further, we present them as lower limits.
Following the experimental data, the model was set to the starting parameters of humidity, temperature and volume of the chamber, as well as the initial concentrations of SOA (0.1 μg m3), VOCs, O3, NO2 in the experiments and described above. For each model run we assumed that there was no exchange with outdoors, no deposition to the Teflon chamber, and experiments were carried out in the dark. The timeframe for each model run was set to 60 min for the α-pinene model runs and 30 min for the α-terpineol model runs.
3. Results and discussion
3.1. Vapour pressure estimations
The six different methods described in Section 2.3 were used to calculate Vp for the 136 species selected as being potentially condensable from the MCM α-pinene scheme. The results show a wide variation in the values of Vp calculated by the different methods for different functionalities including carbonyl, acid, PAN, alcohol, organic nitrate, or peroxide groups (Fig. 2a). Such a variation could consequently have a large bearing on the accuracy of the predicted SOA concentrations. The MY, NVP and EVAP methods show a tendency to predict a wider range of Vps for different functionalities when compared to the other methods. The biggest discrepancy among methods is shown for peroxide species with the EVAP and NVP methods predicting that these species would be much more condensable than predictions by other methods. Fig. 2b shows the Vp values, for 7 selected species of different molecular mass values representing a range of different functionalities and again dem-onstrates the range of predicted Vp values.
Fig. 2.
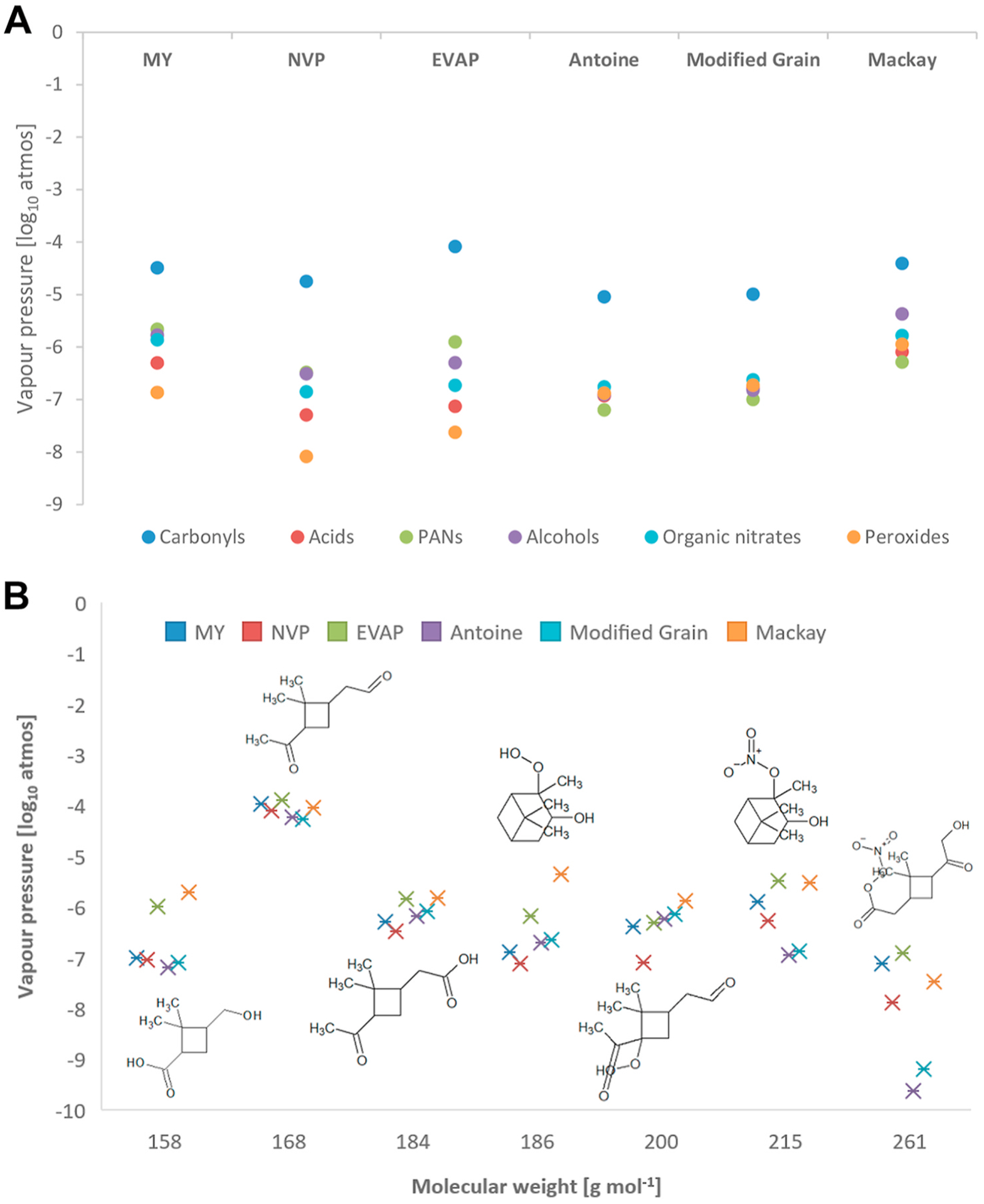
(a) Range of predicted average Log10 Vp (atmospheres) of each class of compounds for each of the 6 methods across the 136 species at 295K. (b) Log10 Vp (atmospheres) for 7 selected species including their chemical structures (reading from the left to the right: dimethylcyclobutanecarboxylic acid, pinonaldehyde, pinonic acid, C10H18O3, C10H16O4, C10H17NO4, C10H15NO7), against molecular weight.
Therefore, based on the inability of some of the older methods to distinguish between different functionalities for reasons outlined in the Introduction, we decided to continue to explore the MY, EVAP and NVP methods, coupled with the Nannoolal et al. (2004) method, for calculating Tb in each case.
3.2. Model sensitivity to HOM formation
We investigated the model sensitivity for one of the selected methods (NVP) to our starting assumption that 8% of the reaction products from the preliminary oxidation of α-pinene by OH and 10% from oxidation by O3 formed HOM material. We varied these percentages from 0 to 10% for the yield of HOM formation from OH and 0–20% for the yield from α-pinene ozonolysis and tried different combinations of these percentages. We then explored the difference between the modelled SOA concentrations and the average from the three α-pinene oxidation experiments over the ~60-min experimental time. The results of the root mean square (rms) difference between the average measured and modelled concentrations of SOA are reported in Table S2.
Table S2 shows that the rms error is reduced slightly as the % of HOMs formed from ozonoylsis or reaction with OH increases, but the extent to which this happens depends on the combination of values assumed. Given the uncertainty around these yields in the absence of sufficient experimental data, we kept our original assumption that the HOM formation yield from OH oxidation was 8% and from ozonolysis was 10%. Note that under these conditions, the HOM monomer and dimer comprised ~56% of the total SOA. Clearly, there is a need for further experimental data to help constrain models further around these assumptions.
3.3. Model evaluation with the α-pinene chamber results
The α-pinene experimental results described in section 2.4 have been used as the basis for testing the model’s ability to calculate SOA concentrations using the three different Vp estimation methods identified in the previous section and with and without the inclusion of HOMs. The analysis of the results (Fig. 3) shows that without the inclusion of HOMs, the model under-estimates the measured values, both the steep rise at the start and the final concentrations after ~60 min. Compared to the final measured value of 128 μg m−3 (averaged across three experiments), the model predicts concentrations of 76 (NVP), 21 (MY) and 56 μg m−3 (EVAP) to 2 significant figures.
Fig. 3.
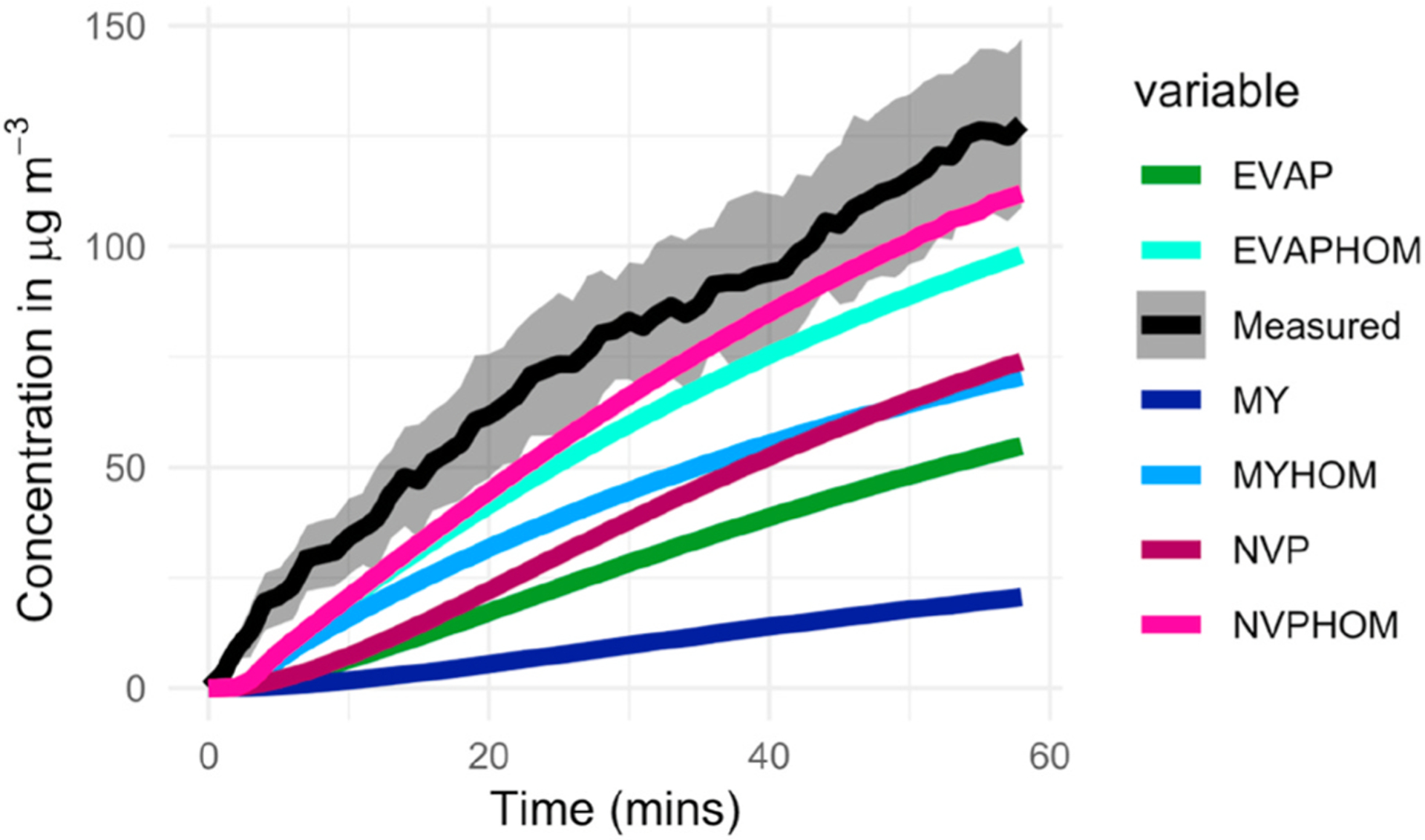
Comparison of measured and modelled SOA concentrations (μg m−3), following the addition of 100 ppb of O3 to 100 ppb of α-pinene in the chamber. The measurements are shown with a shaded grey area that defines the standard deviation for the three experiments. The model runs are shown for the three different vapour pressure methods and with (EVAPHOM, MYHOM, NVPHOM) and without (EVAP, MY, NVP) HOM formation.
Including the formation of HOMs improves the model predictions throughout the 60 min and in particular, better captures the steep rise in particle concentrations at the start of the experiment (Fig. 3). The predicted SOA concentration after ~60 min increased to 115 (NVP), 100 (EVAP) and 72 (MY) μg m−3. In general, adding HOM into the model improves the SOA prediction for all the methods. Table 1 presents the rms difference between modelled and averaged measured concentrations of SOA over the experimental period for the three Vp methods and with and without the inclusion of HOM.
Table 1.
The rms difference between the average measured and the modelled SOA concentrations over the 60-min experiment time with and without HOM.
| EVAP | EVAP HOM | NVP | NVPHOM | MY | MY HOM | |
|---|---|---|---|---|---|---|
| 60 min average | 0.63 | 0.24 | 0.50 | 0.16 | 0.87 | 0.44 |
3.4. α-terpineol experiments
3.4.1. Model SOA concentrations
In order to test the model over more reactant conditions, we used the information from the 21 experimental runs described in Section 2 of this paper, which incorporate a wide range of VOC and NO2 concentrations. The INDCM was modified to include α-terpineol oxidation as described in section 2.4 and used to estimate the SOA concentration for the three selected methods with and without HOMs and then compared with the experimental results. Fig. 4 shows the modelled versus measured SOA for each of the 21 experiments (see Table S3 in the Supplementary Information for further details).
Fig. 4.
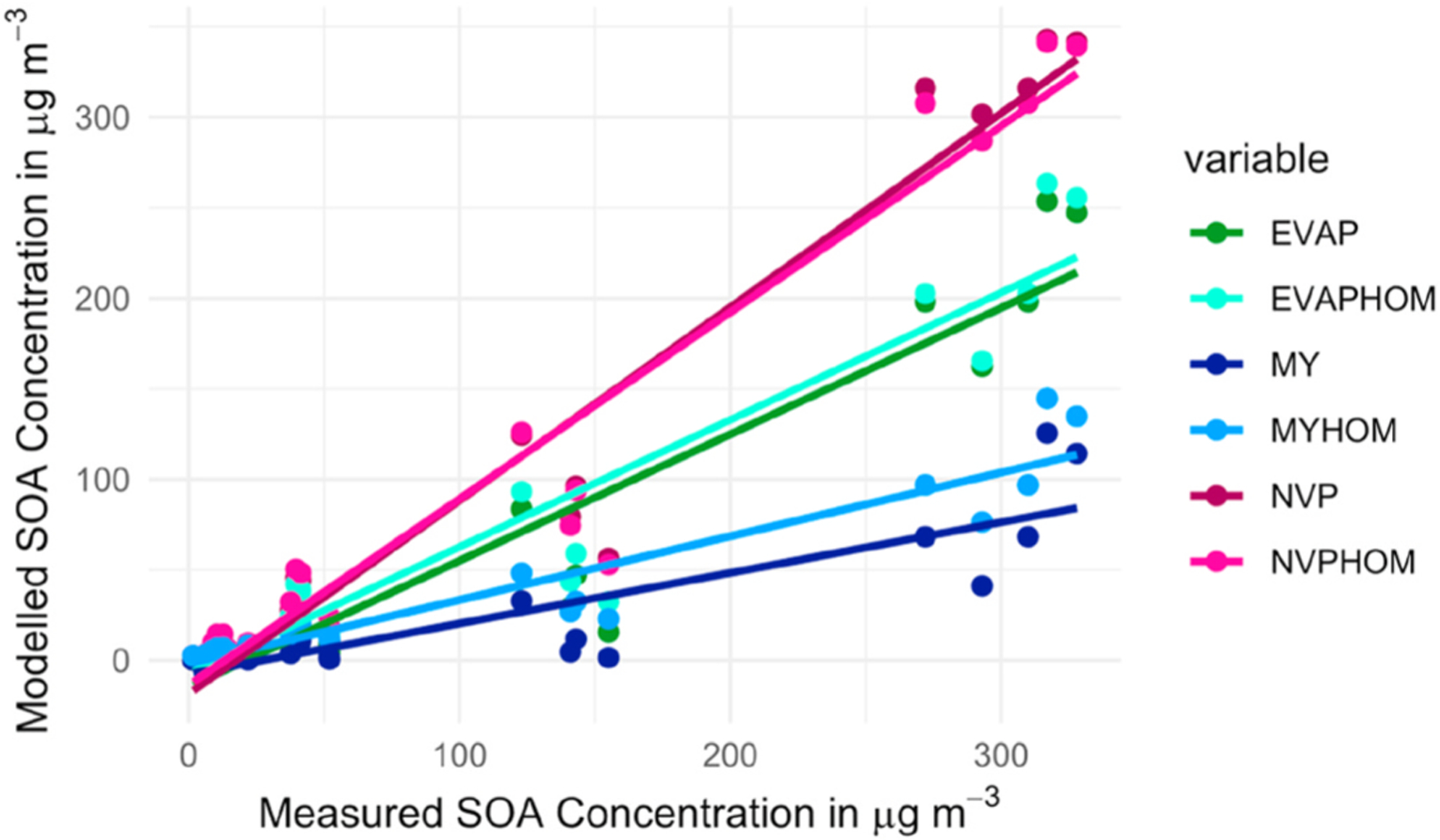
Comparison of SOA (μg m−3) modelled and measured SOA concentrations for 21 experiments for a range of initial α-terpineol and NO2 concentrations. The model results show the EVAP, NVP and MY method with and without the inclusion of HOMs. The line function equations and the coefficient of determination for each method are presented in the Supplementary Information (see Table S4).
The best overall agreement between the measured and modelled SOA values is for the NVP method. The agreement is worse for the MY method compared to the other two, although the addition of HOMs improves the predictions. The inclusion of HOMs reduced the root mean square difference between the modelled and measured values by 29% for EVAP, 19% for NVP and 27% for MY (Table S3, Supplementary Information). The improvement was particularly marked for the experiments with higher NO2 concentrations. Adding HOMs into the model improved the agreement between the measured and modelled results for our experimental conditions and for all Vp methods we tested.
We further explored the relationship between model and measured agreement for the three methods with the α-terpineol and NO2 concentrations (Fig. 5a) and with the measured SOA concentration (Fig. 5b).
Fig. 5a.
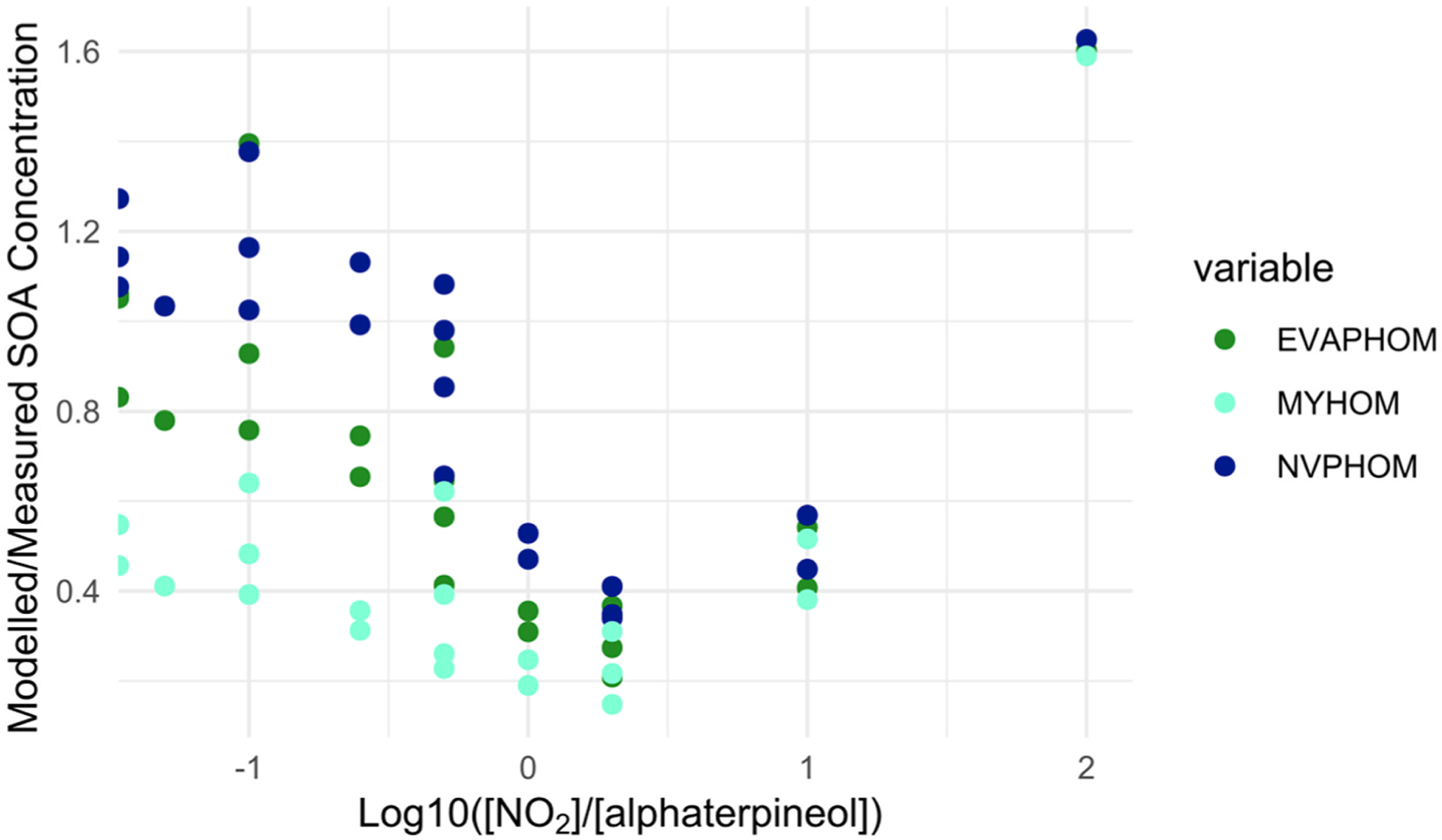
Modelled/measured SOA concentrations at 30 min output for the 21 NO2 and α-terpineol (AT) experiments for the EVAP, NVP and MY method with HOMs included against log10 [NO2]/[α-terpineol]. Note that the x-axis is shown using a log10 scale and that the points furthest to the left are those for which NO2 is zero.
Fig. 5b.
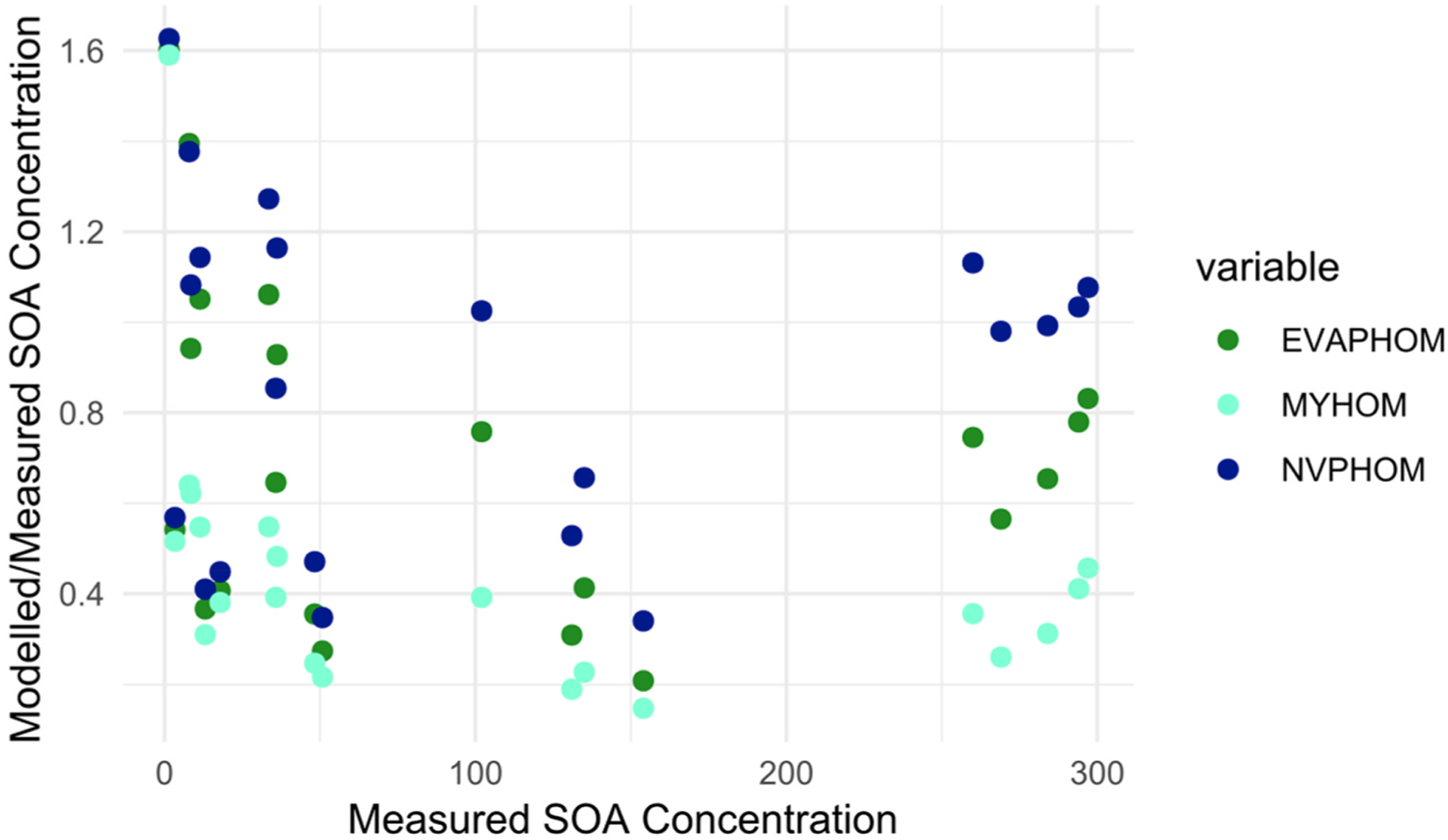
Modelled/measured SOA concentrations at 30 min output for the 21 NO2 and α-terpineol experiments for the EVAP, NVP and MY method with HOMs included against measured SOA.
There is a strong dependency of the degree of model agreement with the measurements on the NO2 and α-terpineol concentrations (Fig. 5a). It has been observed that SOA formation is suppressed in the presence of NOX, although this suppression is due to NO reacting with RO2 (Chan et al., 2010) and we do not expect high NO concentrations under these experimental conditions. However, even for the same x-axis value in Fig. 5a, there are sometimes large differences between measured: modelled agreement (e.g. at −1), depending on the concentration of SOA. Agreement generally improves as SOA concentration increases, although Fig. 5b shows that there is a wide degree of scatter.
However, for the majority of runs, the model performs reasonably well. We now move on to discuss differences in predicted SOA compositions for these runs and given the better performance of the EVAP and NVP methods, focus only on these for the remainder of the paper.
3.4.2. Model predicted composition
One of the benefits of the INDCM is its inherent chemical detail, which facilitates detailed investigation of the chemical composition of the SOA. For the 21 α-terpineol/NO2 chamber experiments, the composition as well as the resulting concentration will vary quite significantly. The resulting SOA composition can be explored in two ways. First, a relatively simple species-level classification can be used. So if a species ends with the OH (hydroxyl) name (e.g. CH3OH), it can be grouped as an alcohol or a PAN-type species if it ends in PAN (e.g. MPAN) based on the MCM protocol (Jenkin et al., 1997). However, this method of assigning functionality does not account for the fact that many oxidation species have multifunctionality. For instance, it is not uncommon for α-pinene oxidation species to have three carbonyl groups, an alcohol group, and an acid group. Thus, the species-level classification could overlook the more detailed compositional information available.
We therefore compared the ‘simple’ species-level classification discussed above with a method where we counted functional groups and then assigned the parent molecule according to the proportion of each functional group (‘complex’). For example, the species LIMALNO3 from the MCM (see Fig. S1 Supplementary Information) would from our simple classification method be defined as an organic nitrate. However, it contains a hydroxyl group and 2 carbonyl groups as well as the nitrate group. For our more detailed classification system, it would therefore contain 25% organic nitrate, 50% carbonyl and 25% hydroxyl character.
We have run the comparison for the EVAP method for the 21 experiments as an example (Fig. 6). The results from using the NVP method were very similar to these. At the end of each model run, the concentration of each of the 136 species is assigned functionality using either the ‘simple’ or ‘complex’ classifications. For both classifications, the total carbonyl (etc.) contributions from each species are added to find the percentage of the total SOA made up by carbonyl (etc.) functionality for each run.
Fig. 6.
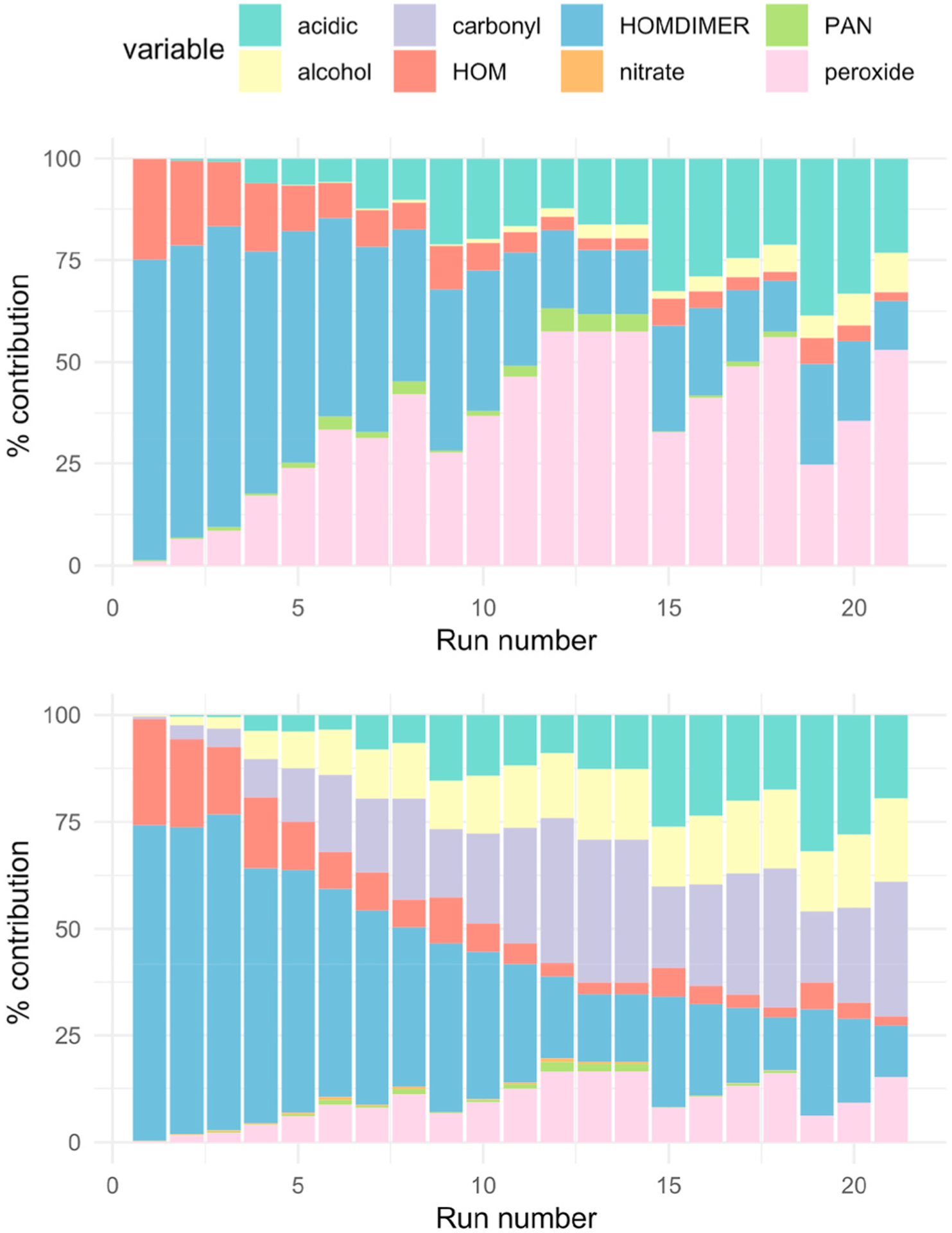
The ‘simple’ (upper) and ‘complex’ (lower) classification of the SOA composition for the EVAP method for the 21 sets of experimental conditions.
Fig. 6 shows that the simpler classification identifies more of the material as peroxide in nature, although the complex classification highlights the carbonyl and hydroxyl groups on the multifunctional compounds. The contribution of the carbonyls is minor for the simple classification. The more complex classification gives a better indication of the functionality of the species, particularly given that the functional groups will affect volatility.
Both methods show that as the NO2-to-α-terpineol ratio decreases (from run 1 to run 21, Table S3), the proportion of HOM and HOM-DIMER is lower. HOM monomers and dimers dominate composition for the lower runs (albeit at low overall concentrations, see Table S3). This is because for the lower run numbers, NO3 concentrations can be high given the high concentrations of NO2 and O3 and absence of NO. Arata et al. (2018) confirmed that indoors, when ozone is in excess, NO is depleted and converted into NO2, and NO3 production is enhanced. For instance, in Run 1, NO3 concentrations reach 73 ppb after 30 min. The NO3 can react with the HOMRO2 radical to form the HOM monomer (Supplementary Information, Table S1), which can then go on to make the HOM dimers.
Hall and Johnston (2011) and Kourtchev et al. (2016) conclude that SOA composition is dominated by oligomers. The model presented here does not include any particle-phase reactions that could enhance SOA formation via oligomer formation, only the HOM reactions as presented in Table S1, which lead to the formation of the HOM dimers. Inclusion of particle-phase reactions is not possible at the moment with existing knowledge, but could potentially alter our findings.
4. Conclusions
The results of this modelling study show that two Vp estimation methods, NVP and EVAP, have the best agreement with measurement results. The important difference between the NVP and EVAP methods is that carbonyls, peroxides, PANs, and organic nitrates are enhanced using the NVP method, but acid and alcohol formation is enriched with the use of the EVAP method. Adding HOMs into the model improved the agreement between measurement and modelling results for all Vp methods. In general, including HOMs increases the SOA mass but also impacts the resulting SOA composition.
There is still a need to understand more about gas-to-particle partitioning indoors, HOM formation and aging of SOA (Cummings and Waring, 2019). Most of the focus to date has been on relatively few terpene species and their propensity to form SOA indoors (e.g. α-pinene) and less is known about the SOA potential of aromatics and longer-chain alkanes for instance. HOM formation seems to be significant in terms of SOA formation indoors, however there are still many model assumptions that would benefit from further research, e.g. vapour pressure and the partitioning coefficients. Therefore, future studies should aim to quantify these processes in order to improve indoor air models. More measurements of SOA composition indoors would also be beneficial.
Supplementary Material
HIGHLIGHTS.
SOA concentration predictions are highly dependent on selected vapour pressure method used.
Highly oxygenated organic molecules form an important component of indoor SOA.
Composition of indoor SOA is highly complex and depends on indoor conditions.
Acknowledgement
This research is part of MOCCIE (MOdelling Consortium for Chemistry of Indoor Environments), which has received funding from an Alfred P. Sloan Foundation program to study Chemistry of Indoor Environments (CIE) (grant agreement no G-2017-9796).
Footnotes
Publisher's Disclaimer: Disclaimer
Publisher's Disclaimer: The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention/the Agency for Toxic Substances and Disease Registry. Mention of any commercial product or trade name does not constitute endorsement by the Centers for Disease Control and Prevention/NIOSH.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.atmosenv.2020.117784.
References
- Alberts WM, 1994. Indoor air pollution: NO, NO2, CO, and CO2. J. Allergy Clin. Immun 94 (2), 289–295. [PubMed] [Google Scholar]
- Arata C, Zarzana KJ, Misztal PK, Liu Y, Brown SS, Nazaroff WW, Goldstein AH, 2018. Measurement of NO3 and N2O5 in a residential kitchen. Environ. Sci. Technol. Lett 5 (10), 595–599. [Google Scholar]
- Barley MH, McFiggans G, 2010. The critical assessment of vapour pressure estimation methods for use in modelling the formation of atmospheric organic aerosol. Atmos. Chem. Phys 10 (2), 749–767. [Google Scholar]
- Barley MH, Topping D, Lowe D, Utembe S, McFiggans G, 2011. The sensitivity of secondary organic aerosol (SOA) component partitioning to the predictions of component properties-Part 3: investigation of condensed compounds generated by a near-explicit model of VOC oxidation. Atmos. Chem. Phys 11 (24), 13145. [Google Scholar]
- Bateman AP, Nizkorodov SA, Laskin J, Laskin A, 2009. Time resolved molecular characterization of limonene/ozone aerosol using high-resolution electrospray ionization mass spectrometry. Phys. Chem. Chem. Phys 11 (36), 7931–7942. [DOI] [PubMed] [Google Scholar]
- Bianchi F, Kurtén T, Riva M, Mohr C, Rissanen MP, Roldin P, Berndt T, Crounse JD, Wennberg PO, Mentel TF, Wildt J, 2019. Highly Oxygenated Organic Molecules (HOM) from Gas-Phase Autoxidation Involving Peroxy Radicals: A Key Contributor to Atmospheric Aerosol. Chem. Rev 119 (6), 3472–3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berndt T, Richters S, Jokinen T, Hyttinen N, Kurtén T, Otkjær RV, Kjaergaard HG, Stratmann F, Herrmann H, Sipilä M, Kulmala M, 2016. Hydroxyl radical-induced formation of highly oxidized organic compounds. Nat. Commun 7 (1), 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carslaw N, 2007. A new detailed chemical model for indoor air pollution. Atmos. Environ 41 (6), 1164–1179. [Google Scholar]
- Carslaw N, Mota T, Jenkin ME, Barley MH, McFiggans G, 2012. A significant role for nitrate and peroxide groups on indoor secondary organic aerosol. Environ. Sci. Technol 46 (17), 9290–9298. [DOI] [PubMed] [Google Scholar]
- Chan AWH, Chan MN, Surratt JD, Chhabra PS, Loza CL, Crounse JD, Seinfeld JH, 2010. Role of aldehyde chemistry and NOx concentrations in secondary organic aerosol formation. Atmos. Chem. Phys 10 (15), 7169–7188. [Google Scholar]
- Compernolle S, Ceulemans K, Müller JF, 2011. EVAPORATION: a new vapour pressure estimation method for organic molecules including non-additivity and intramolecular interactions. Atmos. Chem. Phys 11 (18), 9431–9450. [Google Scholar]
- Cummings BE, Waring MS, 2019. Predicting the importance of oxidative aging on indoor organic aerosol concentrations using the two-dimensional volatility basis set (2D-VBS). Indoor Air 29 (4), 616–629. [DOI] [PubMed] [Google Scholar]
- Ehn M, Thornton JA, Kleist E, Sipilä M, Junninen H, Pullinen I, Springer M, Rubach F, Tillmann R, Lee B, Lopez-Hilfiker F, 2014. A large source of low-volatility secondary organic aerosol. Nature 506 (7489), 476. [DOI] [PubMed] [Google Scholar]
- The EPI-Suite v4.11 software. https://www.epa.gov/tsca-screening-tools/epi-suitetm-estimation-program-interface. Accessed July 19, 2019. [Google Scholar]
- Farmer DK, Vance ME, Abbatt JP, Abeleira A, Alves MR, Arata C, Boedicker E, Bourne S, Cardoso-Saldaña F, Corsi R, DeCarlo PF, 2019. Overview of HOMEChem: house observations of microbial and environmental chemistry. Environ. Sci. Process. Impacts 21 (8), 1280–1300. [DOI] [PubMed] [Google Scholar]
- Garmash O, Rissanen MP, Pullinen I, Schmitt S, Kausiala O, Tillmann R, Percival C, Bannan TJ, Priestley M, Hallquist ÅM, Kleist E, 2020. Multi-generation OH oxidation as a source for highly oxygenated organic molecules from aromatics. Atmos. Chem. Phys 20, 515–537. [Google Scholar]
- Hall IV WA, Johnston MV, 2011. Oligomer content of α-pinene secondary organic aerosol. Aerosol Sci. Technol 45 (1), 37–45. [Google Scholar]
- Jenkin M, 2004. Modelling the formation and composition of secondary organic aerosol from α-and β-pinene ozonolysis using MCM v3. Atmos. Chem. Phys 4, 1741–1757. [Google Scholar]
- Jenkin ME, Saunders SM, Pilling MJ, 1997. The tropospheric degradation of volatile organic compounds: a protocol for mechanism development. Atmos. Environ 31 (1), 81–104. [Google Scholar]
- Jenkin ME, Saunders SM, Wagner V, Pilling MJ, 2003. Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part B): tropospheric degradation of aromatic volatile organic compounds. Atmos. Chem. Phys 3 (1), 181–193. [Google Scholar]
- Joback KG, Reid RC, 1987. Estimation of pure-component properties from group-contributions. Chem. Eng. Commun 57, 233–243. [Google Scholar]
- Johnson D, Utembe SR, Jenkin ME, Derwent RG, Hayman GD, Alfarra MR, Coe H, McFiggans G, 2006. Simulating regional scale secondary organic aerosol formation during the TORCH 2003 campaign in the southern UK. Atmos. Chem. Phys 6 (2), 403–418. [Google Scholar]
- Jones BT, Ham JE, 2008. Ɑ-terpineol reaction with nitrate radical: rate constant and gas-phase products. Atmos. Environ 42 (27), 6689–6698. [Google Scholar]
- Ketenoglu O, 2018. Determination of antoine constants from estimated vapor pressures of selected food contaminants. Curr. J. Appl. Sci. Technol 1–11. [Google Scholar]
- Kourtchev I, Giorio C, Manninen A, Wilson E, Mahon B, Aalto J, Kajos M, Venables D, Ruuskanen T, Levula J, Loponen M, 2016. Enhanced volatile organic compounds emissions and organic aerosol mass increase the oligomer content of atmospheric aerosols. Sci. Rep 6 (1), 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruza M, Lewis AC, Morrison GC, Carslaw N, 2017. Impact of surface ozone interactions on indoor air chemistry: a modeling study. Indoor Air 27 (5), 1001–1011. [DOI] [PubMed] [Google Scholar]
- Kurtén T, Tiusanen K, Roldin P, Rissanen M, Luy JN, Boy M, Ehn M, Donahue N, 2016. α-Pinene autoxidation products may not have extremely low saturation vapor pressures despite high O: C ratios. J. Phys. Chem. A 120 (16), 2569–2582. [DOI] [PubMed] [Google Scholar]
- Kurtén T, Hyttinen N, D’Ambro EL, Thornton J, Prisle NL, 2018. Estimating the saturation vapor pressures of isoprene oxidation products C 5 H 12 O 6 and C 5 H 10 O 6 using COSMO-RS. Atmos. Chem. Phys 18 (23), 17589–17600. [Google Scholar]
- Lee SC, Li WM, Ao CH, 2002. Investigation of indoor air quality at residential homes in Hong Kong - case study. Atmos. Environ 36 (2), 225–237. [Google Scholar]
- Lin HH, Ezzati M, Murray M, 2007. Tobacco smoke, indoor air pollution and tuberculosis: a systematic review and meta-analysis. PLoS Med. 4 (1), 173–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The master chemical mechanism (MCM) V.3.3.1 http://mcm.leeds.ac.uk/MCM. Accessed August 19, 2019.
- McFiggans G, Topping DO, Barley MH, 2010. The sensitivity of secondary organic aerosol component partitioning to the predictions of component properties-Part 1: a systematic evaluation of some available estimation techniques. Atmos. Chem. Phys 10 (21), 10255. [Google Scholar]
- McFiggans G, Mentel TF, Wildt J, Pullinen I, Kang S, Kleist E, Schmitt S, Springer M, Tillmann R, Wu C, Zhao D, 2019. Secondary organic aerosol reduced by mixture of atmospheric vapours. Nature 565 (7741), 587. [DOI] [PubMed] [Google Scholar]
- Myrdal PB, Yalkowsky SH, 1997. Estimating pure component vapor pressures of complex organic molecules. Ind. Eng. Chem. Res 36 (6), 2494–2499. [Google Scholar]
- Nannoolal Y, Rarey J, Ramjugernath D, Cordes W, 2004. Estimation of pure component properties: Part 1. Estimation of the normal boiling point of non-electrolyte organic compounds via group contributions and group interactions. Fluid Phase Equil. 226, 45–63. [Google Scholar]
- Nannoolal Y, Rarey J, Ramjugernath D, 2008. Estimation of pure component properties: Part 3. Estimation of the vapor pressure of non-electrolyte organic compounds via group contributions and group interactions. Fluid Phase Equil. 269 (1–2), 117–133. [Google Scholar]
- Nazaroff WW, Weschler CJ, 2004. Cleaning products and air fresheners: exposure to primary and secondary air pollutants. Atmos. Environ 38 (18), 2841–2865. [Google Scholar]
- O’Meara S, Booth AM, Barley MH, Topping D, McFiggans G, 2014. An assessment of vapour pressure estimation methods. Phys. Chem. Chem. Phys 16 (36), 19453–19469. [DOI] [PubMed] [Google Scholar]
- Pagonis D, Algrim LB, Price DJ, Day DA, Handschy AV, Stark H, Miller SL, de Gouw JA, Jimenez JL, Ziemann PJ, 2019. Autoxidation of limonene emitted in a university art museum. Environ. Sci. Technol. Lett 6 (9), 520–524. [DOI] [PubMed] [Google Scholar]
- Pankow JF, 1994. An absorption model of the gas/aerosol partitioning involved in the formation of secondary organic aerosol. Atmos. Environ 28 (2), 189–193. [Google Scholar]
- Pathak RK, Stanier CO, Donahue NM, Pandis SN, 2007. Ozonolysis of α-pinene at atmospherically relevant concentrations: temperature dependence of aerosol mass fractions (yields). J. Geophys. Res. Atmos 112 (D3). [Google Scholar]
- Peräkylä O, Riva M, Heikkinen L, Qúeĺever L, Roldin P, Ehn M, 2020. Experimental investigation into the volatilities of highly oxygenated organic molecules (HOM). Atmos. Chem. Phys 20, 649–669. [Google Scholar]
- Reid RC, Prausnitz JM, Poling BE, 1987. Properties of Gases and Liquids. McGraw-Hill, New York, NY, USA. [Google Scholar]
- Sarwar G, Olson DA, Corsi RL, Weschler CJ, 2004. Indoor fine particles: the role of terpene emissions from consumer products. J. Air Waste Manag. Assoc 54 (3), 367–377. [DOI] [PubMed] [Google Scholar]
- Sarwar G, Corsi R, 2007. The effects of ozone/limonene reactions on indoor secondary organic aerosols. Atmos. Environ 41 (5), 959–973. [Google Scholar]
- Saunders SM, Jenkin ME, Derwent RG, Pilling MJ, 2003. Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part A): tropospheric degradation of non-aromatic volatile organic compounds. Atmos. Chem. Phys 3 (1), 161–180. [Google Scholar]
- Singer BC, Destaillats H, Hodgson AT, Nazaroff WW, 2006. Cleaning products and air fresheners: emissions and resulting concentrations of glycol esthersand terpenoids. Indoor Air 16 (3), 179–191. [DOI] [PubMed] [Google Scholar]
- Stein SE, Brown RL, 1994. Estimation of normal boiling points from group contributions. J. Chem. Inf. Comput. Sci 34 (3), 581–587. 10.1021/ci00019a016. [DOI] [PubMed] [Google Scholar]
- Topping DO, Barley MH, McFiggans G, 2011. The sensitivity of Secondary Organic Aerosol component partitioning to the predictions of component properties–Part 2: determination of particle hygroscopicity and its dependence on” apparent” volatility. Atmos. Chem. Phys 11 (15), 7767–7779. [Google Scholar]
- Topping D, Barley M, McFiggans G, 2013. Including phase separation in a unified model to calculate partitioning of vapours to mixed inorganic–organic aerosol particles, 165, 273–288. Faraday discuss. [DOI] [PubMed] [Google Scholar]
- Trantallidi M, Dimitroulopoulou C, Wolkoff P, Kephalopoulos S, Carrer P, 2015. EPHECT III: health risk assessment of exposure to household consumer products. Sci. Total Environ 536, 903–913. [DOI] [PubMed] [Google Scholar]
- Uhde E, Salthammer T, 2007. Impact of reaction products from building materials and furnishings on indoor air quality—a review of recent advances in indoor chemistry. Atmos. Environ 41 (15), 3111–3128. [Google Scholar]
- Waring MS, 2014. Secondary organic aerosol in residences: predicting its fraction of fine particle mass and determinants of formation strength. Indoor Air 24 (4), 376–389. [DOI] [PubMed] [Google Scholar]
- Waring MS, Wells JR, 2015. Volatile organic compound conversion by ozone, hydroxyl radicals, and nitrate radicals in residential indoor air: magnitudes and impacts of oxidant sources. Atmos. Environ 106, 382–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells JR, 2005. Gas-phase chemistry of α-terpineol with ozone and OH radical: rate constants and products. Environ. Sci. Technol 39 (18), 6937–6943. [DOI] [PubMed] [Google Scholar]
- Weschler CJ, 2006. Ozone’s impact on public health: contributions from indoor exposures to ozone and products of ozone-initiated chemistry. Environ. Health Perspect 114 (10), 1489–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolkoff P, Schneider T, Kildesø J, Degerth R, Jaroszewski M, Schunk H, 1998. Risk in cleaning: chemical and physical exposure. Sci. Total Environ 215 (1), 135–156. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.