

Abstract
Purpose
To describe the molecular epidemiology of nonsyndromic retinitis pigmentosa (RP) and Usher syndrome (US) in Italian patients.
Methods
A total of 591 probands (315 with family history and 276 sporadics) were analyzed. For 155 of them, we performed a family segregation study, considering a total of 382 relatives. Probands were analyzed by a customized multigene panel approach. Sanger sequencing was used to validate all genetic variants and to perform family segregation studies. Copy number variants of selected genes were analyzed by multiplex ligation-dependent probe amplification. Four patients who tested negative to targeted next-generation sequencing analysis underwent clinical exome sequencing.
Results
The mean diagnostic yield of molecular testing among patients with a family history of retinal disorders was 55.2% while the diagnostic yield including sporadic cases was 37.4%. We found 468 potentially pathogenic variants, 147 of which were unpublished, in 308 probands and 66 relatives. Mean ages of onset of the different classes of RP were autosomal dominant RP, 19.3 ± 12.6 years; autosomal recessive RP, 23.2 ± 16.6 years; X-linked RP, 13.9 ± 9.9 years; and Usher syndrome, 18.9 ± 9.5 years. We reported potential new genotype-phenotype correlations in three probands, two revealed by TruSight One testing. All three probands showed isolated RP caused by biallelic variants in genes usually associated with syndromes such as PERCHING and Senior-Loken or with retinal dystrophy, iris coloboma, and comedogenic acne syndrome.
Conclusions
This is the largest molecular study of Italian patients with RP in the literature, thus reflecting the epidemiology of the disease in Italy with reasonable accuracy.
Keywords: retinitis pigmentosa, Usher syndrome, next-generation sequencing
Retinitis pigmentosa (RP; MIM PS268000), also known as rod-cone dystrophy, is the most common hereditary retinal dystrophy, affecting about 1 of 3000 of the general population.1 It is distinguished by primary degeneration of rod photoreceptors, which may combine with cone involvement as the disease progresses. It causes early impairment of peripheral vision and narrowing of the visual field2 and is therefore a leading cause of chronic visual disability in developed countries. Management of RP is usually limited to periodic assessment of retinal function; detection and treatment of complications, including macular edema, secondary choroidal neovascularization, cataract, and glaucoma; and provision of visual aids, but no definitive treatment is yet available. Nevertheless, progress in understanding the pathologic basis of RP and the development of gene-targeted and innovative treatments are creating new prospects for families affected by this condition.3
RP is genetically heterogeneous and may be transmitted by autosomal dominant (AD), autosomal recessive (AR), or X-linked (XL) patterns of inheritance. Hundreds of deleterious variants in approximately 90 genes have so far been associated with RP (https://sph.uth.edu/retnet/sum-dis.htm#A-genes). The clinical implications of achieving molecular confirmation of RP are multifold. As phenotypic differentiation with respect to other partially overlapping hereditary retinal dystrophies can be difficult in specific circumstances, molecular testing may help classification and prognosis in cases with known genotype-phenotype correlations. Molecular confirmation of the inheritance pattern is also crucial for appropriate genetic counseling and identification of carrier or pre/oligosymptomatic relatives. Finally, new therapies are magnifying the potential impact of molecular nosology in the tailored management of RP.
Molecular testing of RP in a clinical context is not easy due to extreme genetic heterogeneity,4,5 which is also complicated by molecular overlap with other hereditary retinal disorders, such as Usher syndrome (US, the most common syndromic presentation of RP), and current incomplete understanding of its molecular basis.
Today, custom-targeted next-generation sequencing gene panels can be applied in hereditary retinal disorders and RP to allow faster, cheaper, and more accurate molecular diagnosis.6,7 These panels can virtually consider the coding sequence of all genes known to be associated with a variable number of hereditary eye disorders in a single run. Given the continuous discovery of new genes associated with RP and other retinal dystrophies, diagnostic laboratories are forced to periodically update their molecular tools and/or bioinformatics pipelines to offer competitive diagnostic accuracy.
Here we describe our findings in a cohort of 591 probands with RP, enrolled in a single specialized service (Retinal Dystrophies Unit, Division of Ophthalmology, ASST Santi Paolo e Carlo Hospital, Milan, Italy), over a period of about 8 years. Molecular testing was carried out by a multigene panel approach, including a progressively increasing number of disease-genes. This is the largest study on the molecular nosology of RP in Italy to date.
Materials and Methods
Selection of Patients
All probands and their affected and unaffected relatives were enrolled at the Retinal Dystrophy Unit of ASST Santi Paolo e Carlo Hospital, University of Milan (Italy), from 2011 to early 2019. They underwent detailed clinical examination, including best-corrected visual acuity, slit-lamp examination, visual field analysis, spectral-domain optical coherence tomography (SD-OCT), fundus autofluorescence (FAF), and dark- and light-adapted electroretinogram (ERG). Diagnosis of RP was based on typical clinical signs (optic disc pallor, bone spicule pigmentation, retinal vessel attenuation), characteristic full-field electoretinographic patterns, varying from reduced A- and B-wave amplitudes in early stages to nondetectable ERG in advanced stages (as established by the International Society for Clinical Electrophysiology of Vision), visual field constriction, and particular SD-OCT and FAF patterns. Patients suspected to have US (retinitis pigmentosa and sensorineural bilateral hearing loss) also underwent complete audiometric evaluation.
Detailed demographic data and family history were recorded by interview. For each proband, the disease (RP or US) and the inheritance model (AD-RP, AR-RP, or XL-RP) were suspected on the basis of clinical data and pedigree, when available. Patients without a family history of eye disorders were indicated as simplex cases (SP-RP). Molecular testing was always preceded by accurate genetic counseling by a doctor specialized in medical genetics. All patients (or their relatives/legal guardians) gave their written informed consent to use their anonymized clinical and genetic data for publication. The study conformed to the Declaration of Helsinki of 1984 and subsequent amendments. Ethics committee approval was obtained.
Molecular and Bioinformatics Analysis
The genetic testing protocols were originally developed by team S6 of the Department of Genetics of the Institut de la Vision (Paris, France), and their technical aspects have been published elsewhere.6,8
In brief, after full clinical assessment and genetic counseling, blood and/or saliva samples were obtained and sent to the MAGI laboratories (MAGI's Lab s.r.l., Rovereto, TN, and MAGI Euregio s.c.s., Bolzano, Italy). DNA was extracted using a commercial kit (E.Z.N.A. Blood DNA kit; Omega Bio-Tek, Norcross, GA, USA) and analyzed by next-generation sequencing (NGS) on an Illumina MiSeq personal sequencer (Illumina, San Diego, CA, USA) using an in-house bioinformatics pipeline as previously described.9,10
The gene panels were periodically updated with new disease-associated genes reported in the literature, so that four different custom NGS panel releases were used in the 8-year period of observation. The list of genes in the latest multigene panel (used to test 267 samples between 2017 and 2019) is shown in Supplementary Table S1; a comparison of genes included in the other three panels can be found in Supplementary Table S2.
Initial variant filtering considered a minor allele frequency (MAF) of 0.05 and discarded all intronic and synonymous variants not known or predicted to affect splicing. Subsequently, individual MAFs were used for selected genes responsible for genetic/clinical subtypes of RP when epidemiologic data were available. All variants were sought in the Human Gene Mutation Database, professional version 2017.2 (https://www.portal.biobase-international.com/hgmd/pro/); dbSNP (https://www.ncbi.nlm.nih.gov/snp/); ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/); and gnomAD (https://gnomad.broadinstitute.org/) databases. The pathogenicity of all variants identified was evaluated according to American College of Medical Genetics and Genomics (ACMG) guidelines11 and sustained by comparison with the VarSome database (https://varsome.com/).12 Only variants of unknown significance (VUS), likely pathogenic (LP) variants, and pathogenic (P) variants were considered in our analysis; likely benign and benign variants were set aside for further study.
All genetic variants were validated by Sanger sequencing using a Beckman Coulter CEQ 8000 sequencer (Beckman Coulter, Milan, Italy). Sanger sequencing was also used for family segregation studies when additional family members were available. Whole-gene or intragenic rearrangements of selected genes (i.e., EYS, RP2, and USH2A) were analyzed by multiplex ligation-dependent probe amplification (MLPA) (www.mrc-holland.com) using the Beckman Coulter CEQ 8000 sequencer. This test was initially performed in probands strongly suspected to have recessive inheritance on the basis of family history, for whom sequencing analysis revealed a single heterozygous variant in a gene known from the literature to be subject to such rearrangements.
Selected patients who tested negative to targeted NGS analysis underwent clinical exome sequencing using the Illumina commercial kit “TruSight One sequencing panel” on the MiSeq platform, a test comprising about 5000 disease-associated genes (http://www.illumina.com/products/trusight-one-sequencing-panel.ilmn).
Probands were considered genetically solved on the basis of the ACMG variant pathogenicity classification, evidence in the literature, family segregation study, homozygosity, or compound heterozygosity for recessive genes. Only data from solved probands were used for the epidemiologic presentation of RP. Genetic variants of unsolved probands were considered putatively associated with the disease.
Statistical Analysis
Student's t-test was used to compare pairs of means (MedCalc Software, Mariakerke, Belgium). Quantitative data are reported as mean ± SD.
Results
A total of 591 probands were enrolled (age range at diagnosis, 9–92 years; mean age at genetic diagnosis, 44.8 ± 15 years; mean age at onset, 22.2 ± 15.3 years; female/male ratio, 0.96). In 155 cases, one or more relatives were available for family segregation studies, making a total of 382 additional patients (66 clinically affected, 316 clinically unaffected). Based on clinical observation and pedigree study, families/probands were assigned to a genetic class (AD-RP, 67, 11.3%; AR-RP, 103, 17.4%; XL-RP, 37, 6.3%; US, 108, 18.3%; SP-RP, 276, 46.7%). Among the 591 probands, 308 (52.1%) carried one or more variants in a causative gene classified as VUS, LP, and/or P (Supplementary Tables S3–S7). A total of 335 different variants were identified (Supplementary Table S7), 147 of which were unpublished (Table 1, Fig. 1).
Table 1.
List of the 147 New Variants Identified in This Study
| Genetic Class | Gene | RefSeq | Exon/Intron | Nucleotide Change | Amino Acid Change | Allele State | dbSNP rs | VARS-OME |
|---|---|---|---|---|---|---|---|---|
| USH | ADGRV1 | NM_032119 | ex20 | c.3746T>G | p.(Leu1249*) | HET | rs775348911 | P |
| USH | ADGRV1 | NM_032119 | ex21 | c.4523delA | p.(His1508Profs*38) | HET | Not available | P |
| USH | ADGRV1 | NM_032119 | ex28 | c.5954del | p.(Asn1985Thrfs*17) | HOM | Not available | P |
| USH | ADGRV1 | NM_032119 | ex37 | c.8401G>T | p.(Gly2801*) | HET | Not available | P |
| USH | ADGRV1 | NM_032119 | int57 | c.11940+2T>A | p.(?) | HET | Not available | P |
| USH | ADGRV1 | NM_032119 | ex80 | c.17406_17412del | p.(Gln5803Glyfs*3) | HET | Not available | P |
| AR-RP | C2orf71 | NM_001029883 | ex1 | c.8G>A | p.(Cys3Tyr) | HOM | rs1420546201 | VUS |
| USH | CDH23 | NM_022124 | ex41 | c.5280del | p.(Arg1762Glufs*32) | HET | Not available | P |
| USH | CDH23 | NM_022124 | ex43 | c.5584G>A | p.(Glu1862Lys) | HET | rs773004408 | LP |
| USH | CDH23 | NM_022124 | ex50 | c.6831del | p.(Lys2278Serfs*2) | HET | rs1200012430 | P |
| AR-RP | CEP290 | NM_025114 | ex4 | c.223A>G | p.(Lys75Glu) | HET | rs779010679 | LP |
| AR-RP | CEP290 | NM_025114 | int23 | c.2368-1G>A | p.(?) | HET | Not available | P |
| AR-RP | CERKL | NM_201548 | ex2 | c.334C>T | p.(Gln112*) | HET | rs772748858 | P |
| AR-RP | CERKL | NM_201548 | ex11 | c.1297_1298del | p.(Ile433Cysfs*29) | HET | Not available | P |
| AR-RP | CNGA1 | NM_000087 | int8 | c.450-2A>G | p.(?) | HOM | Not available | P |
| AR-RP | CNGB1 | NM_001297 | ex7 | c.451G>C | p.(Asp151His) | HET | rs761126116 | VUS |
| AR-RP | CNGB1 | NM_001297 | ex11 | c.827_834del | p.(Ile276Thrfs*4) | HET | Not available | P |
| AR-RP | CNGB1 | NM_001297 | ex16 | c.1333G>T | p.(Glu445*) | HET | Not available | P |
| AR-RP | CNGB1 | NM_001297 | ex20 | c.1949C>T | p.(Pro650Leu) | HOM | rs780961773 | VUS |
| AR-RP | CNGB1 | NM_001297 | int21 | c.1957+2T>G | p.(?) | HET | rs755398007 | P |
| AR-RP | CNGB1 | NM_001297 | ex23 | c.2296T>C | p.(Cys766Arg) | HOM | rs773682702 | VUS |
| AR-RP | CNGB1 | NM_001297 | ex26 | c.2629G>A | p.(Gly877Arg) | HET | rs200963831 | VUS |
| AR-RP | CRB1 | NM_201253 | ex5 | c.1155T>G | p.(Cys385Trp) | HOM | Not available | LP |
| AR-RP | CRB1 | NM_201253 | ex6 | c.1445_1453del | p.(Ile482_Thr484del) | HET | rs780580887 | LP |
| AR-RP | CRB1 | NM_201253 | ex7 | c.2305C>T | p.(Arg769Cys) | HET | rs746307301 | LP |
| AR-RP | CRB1 | NM_201253 | ex7 | c.2405C>T | p.(Pro802Leu) | HET | rs1558132026 | LP |
| AR-RP | CRB1 | NM_201253 | ex7 | c.2676G>C | p.(Lys892Asn) | HET | Not available | LP |
| AR-RP | CRB1 | NM_201253 | ex8 | c.2816_2817delinsAA | p.(Cys939*) | HET | Not available | P |
| AR-RP | CRB1 | NM_201253 | ex9 | c.3548C>A | p.(Ser1183*) | HET | Not available | P |
| AR-RP | CRB1 | NM_201253 | ex11 | c.3961T>G | p.(Cys1321Gly) | HET | rs62635649 | LP |
| AR-RP | EYS | NM_001142800 | ex10 | c.1561_1562insTATA | p.(Asn521Ilefs*10) | HOM | Not available | LP |
| AR-RP | EYS | NM_001142800 | ex11 | c.1688_1689del | p.(Tyr563Serfs*6) | HET | Not available | LP |
| AR-RP | EYS | NM_001142800 | ex20 | c.3131A>G | p.(Asn1044Ser) | HET | rs985352881 | VUS |
| AR-RP | EYS | NM_001142800 | int21 | c.3164+1G>A | p.(?) | HET | rs1029564423 | P |
| AR-RP | EYS | NM_001142800 | int22 | c.3443+5G>A | p.(?) | HOM | Not available | VUS |
| AR-RP | EYS | NM_001142800 | ex29 | c.6047G>A | p.(Gly2016Asp) | HET | rs886061672 | VUS |
| AR-RP | EYS | NM_001142800 | ex30 | c.6124G>T | p.(Glu2042*) | HET | Not available | P |
| AR-RP | EYS | NM_001142800 | ex30 | c.6152del | p.(Pro2051Hisfs*5) | HET | Not available | P |
| AR-RP | EYS | NM_001142800 | ex34-35 | c.(6725+28_6834+2)_(6907_7197)dup | p.(?) | HET | Not available | LP |
| AR-RP | EYS | NM_001142800 | ex36 | c.7138T>C | p.(Cys2380Arg) | HET | rs1344785031 | VUS |
| AR-RP | EYS | NM_001142800 | ex41 | c.7943del | p.(Thr2648Lysfs*34) | HOM | rs1477705832 | P |
| AR-RP | EYS | NM_001142800 | ex42 | c.8159T>C | p.(Ile2720Thr) | HET | rs756985941 | LP |
| AR-RP | EYS | NM_001142800 | ex42 | c.8168del | p.(Gln2723Argfs*18) | HET | rs1168101857 | P |
| AR-RP | EYS | NM_001142800 | ex42 | c.8204A>G | p.(Tyr2735Cys) | HET | Not available | VUS |
| AR-RP | EYS | NM_001142800 | ex43 | c.8545C>T | p.(Arg2849*) | HET | rs1326635278 | P |
| AR-RP | EYS | NM_001142800 | ex43 | c.8569G>T | p.(Glu2857*) | HET | Not available | P |
| AR-RP | EYS | NM_001142800 | ex43 | c.9337_9350del | p.(Lys3113Phefs*8) | HET | Not available | P |
| AR-RP | EYS | NM_001142800 | ex43 | c.9392G>A | p.(Gly3131Glu) | HOM | Not available | VUS |
| AR-RP | HGSNAT | NM_152419 | ex7 | c.743+1del | p.(?) | HET | Not available | P |
| AR-RP | IFT140 | NM_014714 | ex3 | c.142G>A | p.(Glu48Lys) | HOM | rs984306756 | VUS |
| AD-RP | IMPDH1 | NM_001142574 | ex7 | c.674A>C | p.(Asn225Thr) | HET | Not available | VUS |
| AR-RP | IMPG2 | NM_016247 | ex13 | c.2143del | p.(Tyr715Thrfs*10 | HOM | Not available | LP |
| AR-RP | IQCB1 | NM_001023570 | ex8 | c.658_659delinsG | p.(Ser220Glufs*7) | HOM | Not available | P |
| AR-RP | KLHL7 | NM_001031710 | ex1 | c.112C>T | p.(Arg38Trp) | HET | Not available | LP |
| AR-RP | KLHL7 | NM_001031710 | ex5 | c.614A>G | p.(Asp205Gly) | HET | Not available | LP |
| AR-RP | MAK | NM_001242957 | int1 | c.101+1G>T | p.(?) | HET | Not available | P |
| AR-RP | MAK | NM_001242957 | ex11 | c.1519dup | p.(His507Profs*32) | HET | rs748216114 | P |
| AR-RP | MERTK | NM_006343 | ex4 | c.679G>T | p.(Val227Phe) | HET | Not available | VUS |
| AR-RP | MERTK | NM_006343 | ex6 | c.861_866del | p.(Thr288_Glu289del) | HET | Not available | LP |
| AR-RP | MERTK | NM_006343 | ex7 | c.1048C>T | p.(Gln350*) | HOM | Not available | P |
| AR-RP | MERTK | NM_006343 | ex7 | c.1075_1076del | p.(Val359Phefs*5) | HOM | Not available | P |
| USH | MYO7A | NM_000260 | ex13 | c.1448_1451del | p.(Asp483Glyfs*15) | HET | Not available | P |
| USH | MYO7A | NM_000260 | int13 | c.1554+5del | p.(?) | HET | Not available | VUS |
| USH | MYO7A | NM_000260 | ex16 | c.1834_1836del | p.(Ser612del) | HET | Not available | LP |
| USH | MYO7A | NM_000260 | ex24 | c.3070C>T | p.(Gln1024*) | HOM | Not available | P |
| USH | MYO7A | NM_000260 | ex30 | c.3906del | p.(Tyr1302*) | HET | Not available | P |
| USH | MYO7A | NM_000260 | ex37 | c.5168G>A | p.(Arg1723Lys) | HET | Not available | P |
| USH | MYO7A | NM_000260 | int43 | c.5945-4G>T | p.(?) | HET | Not available | VUS |
| XL-RP | OFD1 | NM_003611 | ex8 | c.736C>T | p.(Gln246*) | HEM | Not available | P |
| USH | PCDH15 | NM_033056 | ex4 | c.308T>G | p.(Leu103Arg) | HET | Not available | LP |
| USH | PCDH15 | NM_033056 | ex6 | c.556C>T | p.(Gln186*) | HET | rs1384677442 | P |
| USH | PCDH15 | NM_033056 | ex33 | c.5287_5292del | p.(Ala1763_Pro1764del) | HOM | rs397517465 | VUS |
| AR-RP | PDE6A | NM_000440 | ex17 | c.2100_2122del | p.(Tyr700*) | HET | Not available | P |
| AR-RP | PDE6A | NM_000440 | ex20 | c.2277delC | p.(Met760*) | HET | Not available | P |
| AR-RP* | PDE6A | NM_000440 | int20 | c.2358+1G>A | p.(?) | HET | rs1333137167 | P |
| AR-RP | PDE6B | NM_000283 | ex2 | c.616G>T | p.(Glu206*) | HET | Not available | P |
| AR-RP | PDE6B | NM_000283 | ex4 | c.760G>A | p.(Glu254Lys) | HET | rs146204075 | LP |
| AR-RP | PDE6B | NM_000283 | int11 | c.1467+4del | p.(?) | HET | Not available | VUS |
| AR-RP | PDE6B | NM_000283 | ex12 | c.1532G>T | p.(Cys511Phe) | HET | Not available | VUS |
| AR-RP | PDE6B | NM_000283 | int18 | c.2193+1G>C | p.(?) | HOM | Not available | P |
| AR-RP | POMGNT1 | NM_017739 | ex17 | c.1415T>C | p.(Leu472Pro) | HET | Not available | LP |
| AD-RP | PRPF31 | NM_015629 | ex7 | c.549del | p.(Glu183Aspfs*15) | HET | Not available | P |
| AD-RP | PRPF31 | NM_015629 | ex7 | c.551_552 insCCGGAGCT | p.(Glu185Argfs*16) | HET | Not available | P |
| AD-RP | PRPF31 | NM_015629 | ex10 | c.992G>A | p.(Trp331*) | HET | rs1555794205 | P |
| AD-RP | PRPF31 | NM_015629 | ex12 | c.1205C>A | p.(Ser402*) | HET | Not available | P |
| AD-RP | PRPF31 | NM_015629 | ex13 | c.1307G>A | p.(Gly436Asp) | HET | rs746117107 | VUS |
| AD-RP | PRPF4 | NM_004697 | ex12 | c.1241A>G | p.(Asn414Ser) | HET | rs775720412 | VUS |
| AD-RP | PRPF6 | NM_012469.3 | ex10 | c.1300G>A | p.(Val434Met) | HET | rs753357562 | VUS |
| AD-RP | PRPF8 | NM_006445 | ex15 | c.2111A>C | p.(Asn704Thr) | HET | Not available | VUS |
| AD-RP | PRPF8 | NM_006445 | ex32 | c.5070C>G | p.(Asp1690Glu) | HET | Not available | LP |
| AD-RP | PRPF8 | NM_006445 | ex43 | c.6910T>G | p.(Phe2304Val) | HET | Not available | LP |
| AD-RP | PRPF8 | NM_006445 | ex43 | c.6994G>T | p.(Asp2332Tyr) | HET | Not available | LP |
| AD-RP | PRPF8 | NM_006445 | ex43 | c.6994_7001 delinsTTTACTC | p.(Asp2332Phefs*27) | HET | Not available | P |
| AD-RP | PRPF8 | NM_006445 | ex43 | c.7007G>C | p.(*2336Serext*41) | HET | Not available | LP |
| AD-RP | PRPH2 | NM_000322 | ex1 | c.535T>G | p.(Trp179Gly) | HET | Not available | LP |
| AD-RP | PRPH2 | NM_000322 | ex2 | c.643A>C | p.(Asn215His) | HET | Not available | LP |
| AR-RP | RBP4 | NM_006744 | ex5 | c.457_459del | p.(Phe153del) | HOM | rs1490873010 | LP |
| AR-RP | RDH12 | NM_152443 | ex8 | c.667G>T | p.(Val223Phe) | HOM | rs370015375 | LP |
| AD-RP | RDH12 | NM_152443 | ex8 | c.680_684delinsT | p.(Ala227Valfs*50) | HET | P | |
| AD-RP | RHO | NM_000539 | ex3 | c.559T>A | p.(Cys187Ser) | HET | Not available | P |
| AD-RP | RHO | NM_000539 | ex4 | c.907_911delinsGC | p.(Pro303_Val304delinsAla) | HET | Not available | LP |
| AD-RP | RHO | NM_000539 | ex5 | c.1032G>C | p.(Gln344His) | HET | rs749753555 | LP |
| AR-RP | RLBP1 | NM_000326 | int3 | c.(13-2)_(13-1)delinsCC | p.(?) | HET | Not available | P |
| AR-RP | RP1 | NM_006269 | Int3 | c.788-1G>C | p.(?) | HOM | Not available | P |
| AR-RP | RP1 | NM_006269 | ex4 | c.1234dup | p.(Met412Asnfs*7) | HET | rs760283610 | P |
| AR-RP | RP1 | NM_006269 | ex4 | c.1720_1721del | p.(Ser574Asnfs*8) | HET | Not available | P |
| AD-RP | RP1 | NM_006269 | ex4 | c.2447dup | p.(Asn816Lysfs*9) | HET | Not available | P |
| XL-RP | RP2 | NM_006915 | int1 | c.103-2del | p.(?) | HEM | Not available | P |
| XL-RP | RP2 | NM_006915 | exon3 | c.(768+?)_(884-?)del | p.(?) | HEM | Not available | LP |
| AD-RP | RP9 | NM_203288 | ex5 | c.436C>T | p.(Arg146*) | HET | rs1426378506 | P |
| AR-RP | RPE65 | NM_000329 | ex10 | c.1112C>T | p.(Pro371Leu) | HET | rs770760551 | LP |
| XL-RP | RPGR | NM_001034853 | ex2 | c.101_102insA | p.(Asn34Lysfs*2) | HET | Not available | P |
| XL-RP | RPGR | NM_001034853 | Int3 | c.248-1G>C | p.(?) | HEM | Not available | P |
| XL-RP | RPGR | NM_001034853 | ex4 | c.299T>C | p.(Leu100Pro) | HEM | rs1064797366 | VUS |
| XL-RP | RPGR | NM_001034853 | ex7 | c.739delinsAA | p.(Gln247Lysfs*36) | HEM | Not available | P |
| XL-RP | RPGR | NM_001034853 | ex7 | c.752G>T | p.(Gly251Val) | HEM | Not available | LP |
| XL-RP | RPGR | NM_001034853 | ex12 | c.1473_1477del | p.(Glu491Aspfs*6) | HEM | Not available | P |
| XL-RP | RPGR | NM_001034853 | ex15 | c.2340_2371dup | p.(Gly791Glufs*35) | HEM | Not available | P |
| AD-RP | SNRNP200 | NM_014014 | ex11 | c.1376A>T | p.(Glu459Val) | HET | Not available | LP |
| AR-RP | TULP1 | NM_003322 | ex5 | c.450_451insCT | p.(Ser151Leufs*6) | HET | Not available | LP |
| AR-RP | TULP1 | NM_003322 | ex11 | c.1063G>A | p.(Asp355Asn) | HET | rs1085307806 | LP |
| USH | USH1C | NM_005709 | ex4 | c.348_373del | p.(His116Glnfs*24) | HOM | Not available | P |
| USH* | USH2A | NM_206933 | ex2 | c.194del | p.(Thr65Ilefs*80) | HET | Not available | P |
| USH | USH2A | NM_206933 | ex5 | c.(785-?)_(1840+?)del | p.(?) | HET | Not available | P |
| USH | USH2A | NM_206933 | ex5-10 | c.(784+?)_(1841-?)del | p.(?) | HOM | Not available | P |
| AR-RP | USH2A | NM_206933 | int8 | c.1551-5T>G | p.(?) | HET | rs770011395 | VUS |
| AR-RP | USH2A | NM_206933 | ex9 | c.1571C>A | p.(Ala524Asp) | HET | rs772624410 | LP |
| AR-RP | USH2A | NM_206933 | ex11 | c.1891G>C | p.(Asp631His) | HET | rs552400144 | LP |
| AR-RP | USH2A | NM_206933 | ex12 | c.1991G>T | p.(Cys664Phe) | HET | Not available | LP |
| USH | USH2A | NM_206933 | ex13 | c.2435C>T | p.(Thr812Ile) | HET | rs768560709 | LP |
| USH | USH2A | NM_206933 | ex14 | c.2953T>C | p.(Cys985Arg) | HET | rs1171264735 | LP |
| AR-RP* | USH2A | NM_206933 | ex17 | c.3332T>G | p.(Leu1111*) | HET | Not available | LP |
| AR-RP | USH2A | NM_206933 | ex23 | c.4862T>A | p.(Ile1621Asn) | HET | Not available | LP |
| USH | USH2A | NM_206933 | ex27 | c.5438_5443del | p.(Ser1813_Ser1 815delinsCys) | HET | rs752992414 | LP |
| AR-RP* | USH2A | NM_206933 | ex27 | c.5330G>A | p.(Arg1777Gln) | HET | rs541275063 | LP |
| USH | USH2A | NM_206933 | int31 | c.6164-3C>G | p.(?) | HET | rs755593389 | VUS |
| USH | USH2A | NM_206933 | ex32 | c.6317_6318del | p.(Ile2106Serfs*51) | HET | Not available | P |
| AR-RP | USH2A | NM_206933 | ex36 | c.6929C>T | p.(Thr2310Met) | HET | rs151057466 | LP |
| USH | USH2A | NM_206933 | ex41 | c.7939C>T | p.(Pro2647Ser)† | HET | Not available | VUS |
| AR-RP | USH2A | NM_206933 | ex42 | c.8395G>C | p.(Gly2799Arg) | HOM | Not available | LP |
| AR-RP | USH2A | NM_206933 | ex48 | c.9433_9437delins AGGAGATCATATCCATTCCATAGGA | p.(Leu3145Argfs*22) | HET | Not available | P |
| AR-RP | USH2A | NM_206933 | ex53 | c.10429T>C | p.(Ser3477Pro) | HET | Not available | LP |
| USH | USH2A | NM_206933 | ex56 | c.10975dup | p.(Thr3659Asnfs*24) | HET | Not available | P |
| AR-RP | USH2A | NM_206933 | ex60 | c.11660G>C | p.(Trp3887Ser) | HET | Not available | LP |
| AR-RP | USH2A | NM_206933 | ex63 | c.12358C>T | p.(Arg4120Cys) | HET | rs727503718 | LP |
| USH | USH2A | NM_206933 | ex68 | c.14815del | p.(Ser4939Argfs*11) | HET | Not available | P |
| USH | USH2A | NM_206933 | ex71 | c.15373C>T | p.(Arg5125Cys) | HET | rs771243585 | VUS |
All variants were classified for pathogenicity according to American College of Medical Genetics and Genomics guidelines. ex, exon; int, intron; HEM, hemizygous; HET, heterozygous; HOM, homozygous; rs, dbSNP accession number.
*Recurrent variant.
†Probably in haplotype with the known LP variant p.(Pro4735Arg).
Figure 1.

Overview of the 147 unpublished variants: (A) distribution in genes, (B) disease distribution, and (C) classification of pathogenicity.
From a total of 468 potentially pathogenic variants, 279 were only found once (i.e., in a single proband), while 56 recurred in two or more unrelated probands. In particular, 51 previously known variants recurred in a total of 178 probands, while 5 previously unpublished variants were found in 2 probands each. After testing, families/probands were reassigned to a specific class based on genetic results, including segregation study if possible (Fig. 2A). Variants classified by type are reported in Table 2. Results by genetic class (i.e., AD-RP, AR-RP, XL-RP, US) after molecular testing are reported in Supplementary Tables S3–S6.
Figure 2.

Prevalence of the four classes of RP based on genetic diagnosis in the whole population of 591 probands. (A) Probands, n = 591. (B) Relatives, n = 382.
Table 2.
Types of Putative Pathogenic Variants
| Variant Type | Hemizygous, No. | Heterozygous, No. | Homozygous, No. | Total, No. |
|---|---|---|---|---|
| Large deletions/insertions | 1 | 8 | 4 | 13 |
| Missense | 4 | 178 | 23 | 205 |
| Nonsense | 4 | 71 | 12 | 87 |
| Frameshift | 10 | 65 | 18 | 93 |
| In frame | 11 | 2 | 13 | |
| Splice site | 3 | 43 | 11 | 57 |
| Total | 22 | 376 | 70 | 468 |
Segregation study confirmed disease transmission and provided evidence of genotype-phenotype association in 51 of the 66 affected relatives and their families. In the remaining 15 affected relatives, molecular testing suggested that the candidate variant did not segregate in the family and was unlikely to cause the disease (Fig. 2B). A total of 170 relatives were classified as carriers.
MLPA analysis was performed on the USH2A, EYS, and RP2 genes in 33 patients (17 probands and 16 relatives) and proved positive in 9 (52.9%) probands and 1 affected relative (6.25%), while 10 healthy relatives (62.5%) were found to be healthy (heterozygous) carriers.
The diagnostic yields of molecular testing are reported in Table 3. Diagnostic yield in patients with family history was 73.1%, 62.1%, 56.8%, and 37% for AD-RP, AR-RP, XL-RP, and US patients, respectively, and 55.2% when considering the four classes together.
Table 3.
Inheritance Based on Family History and Genetic Testing
| Presumed Inheritance Based on Observation | Diagnostic Yield of Cases With Family History: Solved Cases/Total No. (% Solved) | Prevalence of RP Inheritance Patterns of Solved Cases/Total No. (% Solved) | |||
|---|---|---|---|---|---|
| AD-RP | 67/591 (11.3) | 49/67 (73.1) | AD-RP | 59/591 (10) | |
| AR-RP | 98/591 (16.6) | ||||
| XL-RP | 24/591 (4.1) | ||||
| US | 40/591 (6.8) | ||||
| AR-RP | 103/591 (17.4) | 64/103 (62.1) | |||
| XL-RP | 37/591 (6.3) | 21/37 (56.8) | |||
| US | 108/591 (18.3) | 40/108 (37) | |||
| Total | 315/591 (53.3) | Mean diagnostic yield 174/315 (55.2) | |||
| SP-RP | 276/591 (46.7) | Diagnostic yield of cases without family history: solved cases/total No. (% solved) | |||
| AD-RP | 10/276 (3.6) | ||||
| AR-RP | 30/276 (10.9) | Overall diagnostic yield 221/591 (37.4) | |||
| XL-RP | 2/276 (0.7) | ||||
| Total | 42/276 (15.2) | ||||
Diagnostic yield of cases with family history of retinal disorders is the fraction (percentage) of cases for which the inheritance based on genetic testing matched the inheritance presumed from observation. The overall diagnostic yield is the rate of solved cases in the whole population of 591 probands, therefore including sporadic cases.
Among 276 sporadic cases, 71 had potentially pathogenic variants. For 63 of them, family segregation analysis was performed and made it possible to solve 30 cases (2 AD, 27 AR, 1 XL); 12 were solved without segregation analysis (8 AD, 3 AR, 1 XL). Molecular testing was therefore useful in establishing an inheritance pattern in 42 sporadic cases (15.2%; 10 tested positive for AD-RP, 30 for AR-RP, and 2 for XL-RP). Taken together, on 591 probands, our results demonstrated a prevalence of 10% (59 probands) AD-RP, 16.6% (98 probands) AR-RP, 4.1% (24 probands) XL-RP, and 6.8% US (40 probands). Considering the whole population of 591 probands, 221 (37.4%) were solved, while the other 370 (62.6%) remained unsolved.
Mean age at onset by genetic class after molecular testing is reported in Figure 3. The mean age at onset of XL-RP (13.9 ± 9.9 years) was significantly lower (P = 0.0072) than that of the three other genetic classes taken together (21.2 ± 14.4 years).
Figure 3.
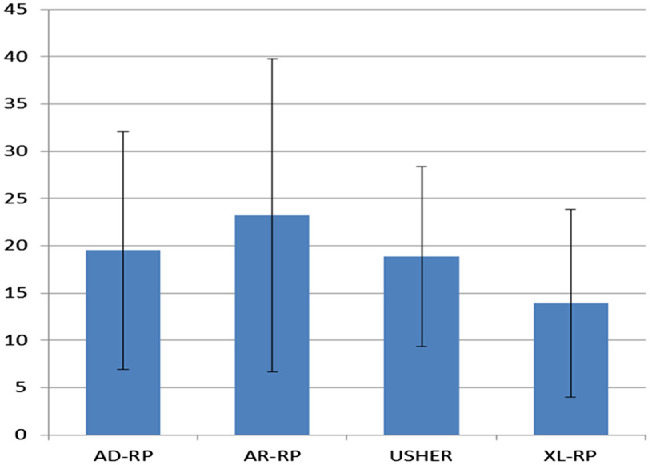
Mean age of onset of the different classes of RP calculated on 214 genetically solved patients (probands and affected relatives). AD-RP, 19.5 ± 12.6 years; AR-RP, 23.2 ± 16.6 years; XL-RP, 13.9 ± 9.9 years; US, 18.9 ± 9.5 years. Bars indicate ± SD.
Only 19 of a total of 70 homozygous variants (27.1%) were caused by established consanguinity of the proband's parents. Ten of 13 large deletions/insertions (76.9%) were in EYS, 2 (15.4%) in USH2A, and 1 (7.7%) in RP2.
We report two de novo variants, one in RHO causing AD-RP in a 35-year-old woman (AD31) (Supplementary Table S3) and one in CRB1 causing AR-RP in a 49-year-old woman (AR18) (Supplementary Table S4).
For one patient, it was not possible to establish the genetic cause of RP due to multiple pathogenic variants. Indeed, this patient (AR12) carried CNGB1 and EYS variants, indicating AR-RP, but lack of a segregation study prevented us from determining whether the variants were in cis or trans configuration (Supplementary Table S4).
The new USH2A p.(Pro2647Ser) variant listed in Table 1 is probably in haplotype with the likely pathogenic variant p.(Pro4735Arg) as it was found in two unrelated patients, probands AR128 (Supplementary Table S4) and US63 (Supplementary Table S5). The segregation study performed in the family of AR128 confirmed that the variants are in cis configuration.
We also found the USH2A p.(Leu1572Phe) variant, previously described as pathogenic,13–15 in haplotype with the variant p.(Glu767Serfs*21)16–18 in six unrelated patients (five US patients and one AR-RP patient; data not shown). Caution is needed in basing a diagnosis on these two variants, since they were in cis configuration; identification of another variant on the second allele would be necessary to solve this case.
The following genes did not harbor any disease-causing variant in our proband cohort: ABCA4, AGBL5, ARHGEF18, ARL2BP, ARL6, BBS2, BEST1, C8orf37, CA4, CIB2, CLRN1, CRX, DHDDS, FSCN2, GUCA1B, GUCY2D, HARS, IDH3B, IFT172, KIZ, LRAT, NEK2, NRL, PDE6G, PDZD7, PRCD, PROM1, RBP3, REEP6, RGR, ROM1, SAG, SEMA4A, SLC7A14, TTC8, USH1G, ZNF408, and ZNF513.
Sixty-eight patients who tested negative at the initial analysis underwent a second analysis with one of the later panel releases (64 probands) or clinical exome analysis by TruSight One (4 probands). This second analysis was decisive for 26 of them (22 cases with a subsequent panel release and 4 with TruSight One).
AD-RP
AD-RP accounted for 10% of our RP cohort (Fig. 2A). It is usually described as the mildest form of the disease due to a later onset.4 In our cohort, the average age of onset in AD-RP was not statistically different from that of AR-RP (P = 0.13) and US (P = 0.8) but was significantly higher than that of XL-RP (P = 0.04) (Fig. 3). The most frequent disease-causing genes in our AD-RP probands were RHO (29%) and RP1 (22%). In line with the literature, 16 of 17 RHO variants (94%) were missense, which probably modify protein function (gain of function) or interfere with wild-type protein function (dominant-negative variants). All RP1 variants were nonsense or frameshift variants (small deletions) that cause haploinsufficiency (loss of function).19,20
For PRPF31, we found clinically relevant variants in 13 patients, including four probands and nine relatives. Segregation analysis detected the familial variant in four clinically unaffected relatives from three families. This is in line with the phenotypic nonpenetrance of PRPF31-associated retinitis pigmentosa and the wide range of age of onset described by other authors; both are probably caused by many factors such as allelic heterogeneity and genetic modifiers.21,22 Figure 4 illustrates genetic variant percentages in AD-RP probands and mean age at onset per gene.
Figure 4.
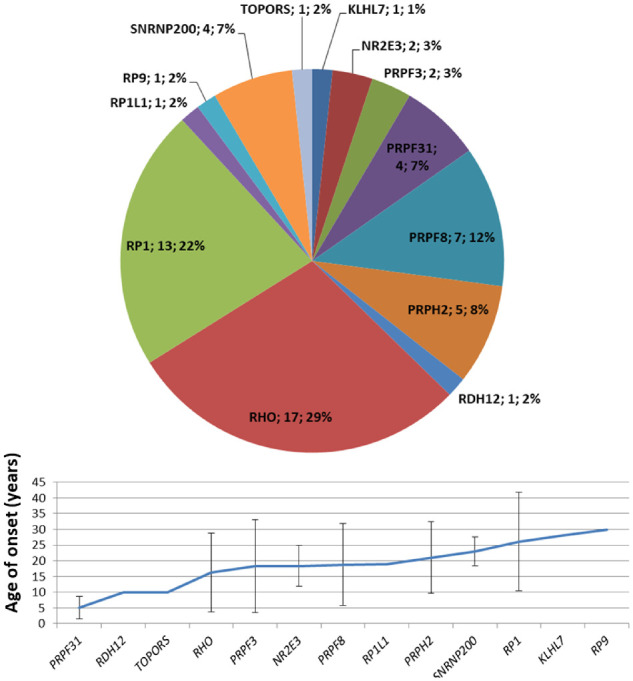
Genetic variants identified in AD-RP probands. Mean age of onset for each gene was calculated on 56 genetically solved patients (probands and affected relatives). Bars indicate ± SD.
AR-RP
AR-RP accounted for 16% of our RP cohort (Fig. 2A). According to the literature, AR-RP is generally more severe and has earlier onset than AD-RP, typically in the first decade,4 but our data were unable to confirm this in our cohort. In fact, mean age at diagnosis of AR-RP was not significantly different from that of AD-RP and US, whereas it was significantly higher than that of XL-RP (P = 0.0039) (Fig. 3).
Regarding USH2A variants, 66.3% (67 of 101) were missense and the other 33.7% were nonsense, small deletions/insertions, and intronic splice site variants. Of the 30 solved USH2A patients, 50% carried biallelic missense variants. Conversely, only 23.4% (11 of 47) of EYS variants were missense, with small deletions/insertions being the most common (12 of 47, 25.5%) followed by large deletions/insertions (9 of 47, 19.1%). Seventeen of 20 solved patients (89.6%) with EYS-associated RP had nonsense, small deletions/insertions and intronic splice site variants in one of the alleles. In addition, 12 solved patients (60%) had homozygous pathogenic variants.
In our cohort, all genes associated with AR-RP proved to be almost fully penetrant. Variants in USH2A and EYS occurred in more than half (51%) of all AR-RP cases (Fig. 5).
Figure 5.

Genetic variants identified in AR-RP probands. Mean age of onset for each gene was calculated on 93 genetically solved patients (probands and affected relatives). Bars indicate ± SD.
XL-RP
XL-RP accounted for 4% of our RP cohort (Fig. 2A) and showed earlier onset than the other forms (Fig. 3), in line with previous reports.4,23 Variants in RPGR accounted for most cases (75%), followed by variants in RP2 (20%) and OFD1 (4%) (Fig. 6). Only five probands (21%) showed missense hemizygous variants, and four were classified as pathogenic and likely pathogenic. Most probands had small deletions/insertions and nonsense variants (64%).
Figure 6.

Genetic variants identified in XL-RP probands. Mean age of onset for each gene was calculated on 30 genetically solved patients (probands and affected relatives). Bars indicate ± SD.
Family segregation study was only possible in 10 of 25 probands, all of whom carried pathogenic variants in RPGR. Among heterozygous females with the RPGR variant (3 probands and 16 relatives), 52.6% (10/19) were affected: tapetal reflex alone, as seen in XL carriers, was not considered a sign of disease.
Usher Syndrome
US patients comprised 7% of our cohort. The syndrome is characterized by concomitant RP, sensorineural deafness, and, in some types, vestibular dysfunction. Three clinical types are distinguished on the basis of clinical severity, but it is not always easy to predict the underlying genes due to genetic and phenotypic heterogeneity.24 It is the most frequent syndromic form of RP and is caused by variants in several genes, the most frequent in our cohort affecting USH2A (65%) and MYO7A (17%) (Fig. 7).
Figure 7.

Genetic variants identified in probands with US. Mean age of onset for each gene was calculated on 35 genetically solved patients (probands and affected relatives). Bars indicate ± SD.
The highest percentage of USH2A variants found in US probands included mainly small deletions/insertions (33.3%). Of the 26 solved probands, only 2 (7.7%) had missense biallelic variants, all in homozygous state, while 14 (53.8%) had a biallelic combination of nonsense, frameshift, or splicing variants, and 10 (38.5%) had a missense variant in one allele and another variant in the second allele. Of the MYO7A variants, 68.2% were loss of function with a large proportion of splice site variants (31.8%). Inversely, pathogenic CDH23 variants were mostly missense (60%). All the genotypes identified were compatible with autosomal recessive inheritance. No case of digenic/triallelic inheritance was identified in our cohort.
All US genes proved to be fully penetrant. The mean age at onset of disease caused by these genes (18.9 ± 9.5 years) was only slightly lower than that of nonsyndromic AD-RP and AR-RP cases taken together (21.8 ± 15.3 years), and the difference was not statistically significant (P = 0.28) (Fig. 3).
Potential New Genotype-Phenotype Correlations
Proband AR63 with RP and his affected brother carried two KLHL7 VUS variants in a compound heterozygous state (Supplementary Table S4). Their parents and a sister proved to be healthy carriers. This gene is known to be associated with AD-RP 42 (MIM 612943) and the autosomal recessive PERCHING syndrome (MIM 617055), which is characterized by global developmental delay, dysmorphic facial features, feeding and respiratory difficulties with poor overall growth, axial hypotonia, and joint contractures. The two affected siblings were screened for the specific PERCHING syndrome features and turned out to have nonsyndromic RP. Our data therefore suggest the possibility of autosomal recessive inheritance for variants in KLHL7, also in the case of isolated RP. Interestingly, PERCHING syndrome is currently only associated with homozygous variants,25,26 while affected individuals in this family are compound heterozygous.
In two patients with isolated RP, TruSight One revealed correlations with genes usually associated with other hereditary eye disorders. Patient AR81 with nonsyndromic RP harbored the homozygous variant p.(Phe153del) in RBP4 (MIM 180250), a gene specifically associated with an autosomal recessive syndrome featuring retinal dystrophy, iris coloboma, and comedogenic acne syndrome (MIM 615147) (Supplementary Table S4). The family segregation study confirmed that the variant was found in the homozygous state in the proband's brother, also affected by isolated RP, whereas the healthy mother and sister were heterozygous. Dermatologic and cardiologic examinations excluded the concomitance of other systemic features in the two affected siblings. Although there is very little literature on variants in this gene, patient AR81 seems to be the first with biallelic variants in RBP4 and nonsyndromic RP.
We found the homozygous IQCB1 p.(Ser220Glufs*7) variant in patient AR62 (Supplementary Table S4) with isolated RP. IQCB1 is usually associated with Senior-Loken syndrome 5 (MIM 609254), characterized by Leber congenital amaurosis and nephronophthisis. Only a few reports have associated IQCB1 (MIM 609237) with isolated RP,27–29 specifically four loss-of-function variants in homozygous or compound heterozygous state. Our case (AR62) confirms the association and expands the corresponding phenotype. In fact, age of onset in this case was 38 years, significantly older than that of previously published cases with isolated RP and variants in IQCB1.
Discussion
Here we reported our 8-year experience in molecular diagnostics of 591 patients with RP, clinically assessed in a single institution and analyzed in a centralized medical genetics service. Molecular testing was first performed with a customized multigene panel approach, integrated with MLPA for genomic rearrangements in selected cases for a restricted number of genes and clinical exomes. During the 8-year observation period, different releases of the diagnostic multigene panels were used, and selected unsolved cases underwent multiple analyses when a new panel was available. All probands and available relatives were evaluated, using the same clinical assessment protocol, by expert ophthalmologists working in a team dedicated to hereditary retinal dystrophies. Genetic counseling always accompanied clinical assessment, before and after molecular testing. To our knowledge, this is the largest molecular study of RP in Italy.
Our study revealed a diagnostic yield of 55.2% among RP probands with a family history. The genetic analysis solved 15.2% of sporadic cases, showing a prevalence in the whole population of 10% for AD-RP, 4.1% for XL-RP, and 6.8% for US. Overall, our study confirmed a genetic basis (and therefore demonstrated a specific Mendelian inheritance pattern) in 37.4% of cases. For forms of RP demonstrated by molecular genetics, autosomal recessive inheritance was therefore the most common transmission pattern in our cohort (16.6%). The remaining 62.4% of unsolved cases can be ascribed to regulatory variants in the genes investigated; genomic rearrangements in genes not tested by MLPA; variants in other, perhaps still unknown causative genes; or genocopies (especially in sporadic cases). We were very strict, considering many parameters like the ACMG variant pathogenicity classification, evidence in the literature, family segregation study, and homozygosity or compound heterozygosity for recessive genes before declaring a case solved. Concerning age at onset, while our data were compatible with an earlier age at onset for XL-RP in males, we were not able to confirm differences between AR-RP compared to AD-RP, as previously demonstrated in other studies. A different genetic background, patient selection bias (our patient cohort was selectively enrolled in a tertiary center, which is assumed to attract severe cases), and heterogeneity in clinical information gathering between different studies may also explain (or contribute to) this divergence in data.
Pathogenicity evaluation of genetic variants detected in patients with RP requires accurate assessment of phenotype, variant features, and family segregation. A good result can only be achieved by close collaboration between geneticists and ophthalmologists. Family segregation study is the best approach for evaluation of VUS, although it may be complicated by incomplete penetrance of some genes.
TruSight One analysis showed that syndromic genes such as RBP4 and IQCB1 may be associated with nonsyndromic RP and may therefore be considered in routine genetic testing for RP and added to custom NGS panels. However, caution is needed in managing cases with an apparently isolated form of RP and causative variants in IQCB1 because the corresponding systemic features may occur later in life.30 The impact of molecular tests on the follow-up of these patients should therefore be considered.
The diagnostic yield by family history of 55.2% achieved by our study is in line with the literature.31 The distribution of RP inheritance classes revealed by our study was similar to data published for other European series32–35 and is in line with ranges reported in the literature.36,37 When we included solved sporadic cases, our diagnostic yield fell to 37.4%, although the following aspects should be considered: (a) as previously mentioned, recessive forms were considered unsolved even if they had two pathogenic variants but lacked a family segregation study to determine whether the alleles were in cis or trans configuration, and (b) among sporadic cases, we also included cases in which the genetic test was conducted for differential diagnosis with respect to other forms of pigmented retinopathies. Although it is difficult to compare the prevalence of the genes involved in our cohort of patients with RP with those of previous European epidemiologic studies, mainly due to the heterogeneity of clinical conditions included in other reports, our results in the US population are in line with those of Carrigan et al.38 in Irish patients. Noteworthy differences can be observed in the group of nonsyndromic patients with RP: in our cohort, the most frequent genes were recessive, such as USH2A (16.6% of all probands) and EYS (11%), while in the Irish population, X-linked (RPGR, 10.5%) and AD genes (RHO, 8.6%) were the most frequent.38
There are also some differences with respect to the Spanish population in the distribution of inheritance patterns of the mutated gene of sporadic cases confirmed by molecular diagnosis and the family segregation study: in the Spanish population, 84.5% of sporadic cases were caused by AR genes, 7.6% by AD genes, and 7.9 by XL genes, while in our population, we found 71.4%, 23.2%, and 4.8% of AR, AD, and XL genes, respectively.39
Although our data are incomplete, we showed that a gene can influence age of onset of the disease. A larger data set is needed to obtain more convincing results.
In our experience, whatever the age of onset, the impact of diagnosis of a disease that can lead progressively to complete blindness and loss of independence is always devastating for a patient. Patients require genetic counseling before and after genetic testing, as well as lifelong psychological support.
Genetic definition of RP is fundamental for predicting familial risk and disease progression and for participation in clinical trials. Gene therapy has recently begun to yield promising and exciting results in the treatment of inherited retinal disorders. A large number of potential therapeutic approaches, such as gene delivery systems, gene replacement, gene silencing, antisense oligonucleotides, and genome editing, are now available and were recently demonstrated to be effective and safe. Preclinical and clinical studies have been conducted for PDE6B-, RPGR-, MERTK-, RPE65-, RDS-, RHO-, ABCA4-, CEP290-, and MYO7A-related inherited retinal dystrophies. The study of RPE65-related retinal dystrophy is the most advanced, and thanks to LUXTURNA (Spark Therapeutics, Philadelphia, PA, USA), the first gene therapy approved by the US Food and Drug Administration for a genetic disease, RPE65-related retinal dystrophy is the only retinal pathology for which a cure can be said to exist.3
It is only a matter of time before new gene therapies become available. Genetically solved patients will have the advantage of being quickly eligible for clinical trials or access to pharmacologic treatment.
Supplementary Material
Acknowledgments
Supported by the Autonomous Province of Trento in the framework of LP 6/99 (dgp 1045/2017), LABEX LIFESENSES (reference ANR-10-LABX-65) with French state funds managed by the Agence Nationale de la Recherche under the Investissements d'Avenir program (ANR-11-IDEX-0004-0), IHU FOReSIGHT (ANR-18-IAHU-0001) with French state funds managed by the Agence Nationale de la Recherche under the Investissements d'Avenir program, and Foundation Fighting Blindness center grant (C-CMM-0907-0428-INSERM04).
Disclosure: L. Colombo, None; P.E. Maltese, None; M. Castori, None; S. El Shamieh, None; C. Zeitz, None; I. Audo, None; A. Zulian, None; C. Marinelli, None; S. Benedetti, None; A. Costantini, None; S. Bressan, None; M. Percio, None; P. Ferri, None; A. Abeshi, None; M. Bertelli, None; L. Rossetti, None
References
- 1. Berger W, Kloeckener-Gruissem B, Neidhardt J. The molecular basis of human retinal and vitreoretinal diseases. Prog Retin Eye Res. 2010; 29(5): 335–375. [DOI] [PubMed] [Google Scholar]
- 2. Mrejen S, Audo I, Bonnel S, Sahel JA.. Retinitis pigmentosa and other dystrophies. Dev Ophthalmol. 2017; 58: 191–201. [DOI] [PubMed] [Google Scholar]
- 3. Ziccardi L, Cordeddu V, Gaddini L, et al.. Gene therapy in retinal dystrophies. Int J Mol Sci. 2019; 20(22): 5722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hamel C Retinitis pigmentosa. Orphanet J Rare Dis. 2006; 1: 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Colombo L, Montesano G, Sala B, et al.. Comparison of 5-year progression of retinitis pigmentosa involving the posterior pole among siblings by means of SD-OCT: a retrospective study. BMC Ophthalmol. 2018; 18(1): 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Audo I, Bujakowska KM, Léveillard T, et al.. Development and application of a next-generation-sequencing (NGS) approach to detect known and novel gene defects underlying retinal diseases. Orphanet J Rare Dis. 2012; 7: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Neveling K, Collin RW, Gilissen C, et al.. Next-generation genetic testing for retinitis pigmentosa [published correction appears in Hum Mutat. 2013;34(8):1181]. Hum Mutat. 2012; 33(6): 963–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Boulanger-Scemama E, El Shamieh S, Démontant V, et al.. Next-generation sequencing applied to a large French cone and cone-rod dystrophy cohort: mutation spectrum and new genotype-phenotype correlation. Orphanet J Rare Dis. 2015; 10: 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Maltese PE, Orlova N, Krasikova E, et al.. Gene-targeted analysis of clinically diagnosed long QT Russian families. Int Heart J. 2017; 58(1): 81–87. [DOI] [PubMed] [Google Scholar]
- 10. Marceddu G, Dallavilla T, Guerri G, Manara E, Chiurazzi P, Bertelli M.. PipeMAGI: an integrated and validated workflow for analysis of NGS data for clinical diagnostics. Eur Rev Med Pharmacol Sci. 2019; 23(15): 6753–6765. [DOI] [PubMed] [Google Scholar]
- 11. Richards S, Aziz N, Bale S, et al.. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015; 17(5): 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kopanos C, Tsiolkas V, Kouris A, et al.. VarSome: the human genomic variant search engine. Bioinformatics. 2019; 35(11): 1978–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Song J, Smaoui N, Ayyagari R, et al.. High-throughput retina-array for screening 93 genes involved in inherited retinal dystrophy. Invest Ophthalmol Vis Sci. 2011; 52(12): 9053–9060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhao L, Wang F, Wang H, et al.. Next-generation sequencing-based molecular diagnosis of 82 retinitis pigmentosa probands from Northern Ireland. Hum Genet. 2015; 134(2): 217–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ge Z, Bowles K, Goetz K, et al.. NGS-based molecular diagnosis of 105 eyeGENE probands with retinitis pigmentosa. Sci Rep. 2015; 5: 18287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Eudy JD, Weston MD, Yao S, et al.. Mutation of a gene encoding a protein with extracellular matrix motifs in Usher syndrome type IIa. Science. 1998; 280(5370): 1753–1757. [DOI] [PubMed] [Google Scholar]
- 17. O'Sullivan J, Mullaney BG, Bhaskar SS, et al.. A paradigm shift in the delivery of services for diagnosis of inherited retinal disease. J Med Genet. 2012; 49(5): 322–326. [DOI] [PubMed] [Google Scholar]
- 18. Lenassi E, Saihan Z, Bitner-Glindzicz M, Webster AR.. The effect of the common c.2299delG mutation in USH2A on RNA splicing. Exp Eye Res. 2014; 122: 9–12. [DOI] [PubMed] [Google Scholar]
- 19. Athanasiou D, Aguila M, Bellingham J, et al.. The molecular and cellular basis of rhodopsin retinitis pigmentosa reveals potential strategies for therapy. Prog Retin Eye Res. 2018; 62: 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Diakatou M, Manes G, Bocquet B, Meunier I, Kalatzis V.. Genome editing as a treatment for the most prevalent causative genes of autosomal dominant retinitis pigmentosa. Int J Mol Sci. 2019; 20(10): 2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rose AM, Bhattacharya SS.. Variant haploinsufficiency and phenotypic non-penetrance in PRPF31-associated retinitis pigmentosa. Clin Genet. 2016; 90(2): 118–126. [DOI] [PubMed] [Google Scholar]
- 22. Kiser K, Webb-Jones KD, Bowne SJ, Sullivan LS, Daiger SP, Birch DG.. Time course of disease progression of PRPF31-mediated retinitis pigmentosa. Am J Ophthalmol. 2019; 200: 76–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jauregui R, Takahashi VKL, Park KS, et al.. Multimodal structural disease progression of retinitis pigmentosa according to mode of inheritance. Sci Rep. 2019; 9(1): 10712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. D'Esposito F, Randazzo V, Cennamo G, et al.. Novel USH1G homozygous variant underlying USH2-like phenotype of Usher syndrome [published online ahead of print September 30, 2019]. Eur J Ophthalmol, doi: 10.1177/1120672119879392. [DOI] [PubMed] [Google Scholar]
- 25. Angius A, Uva P, Buers I, et al.. Bi-allelic mutations in KLHL7 cause a Crisponi/CISS1-like phenotype associated with early-onset retinitis pigmentosa [published correction appears in Am J Hum Genet. 2018;102(4):713]. Am J Hum Genet. 2016; 99(1): 236–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Heng LZ, Kennedy J, Smithson S, Newbury-Ecob R, Churchill A.. New macular findings in individuals with biallelic KLHL7 gene mutation. BMJ Open Ophthalmol. 2019; 4(1): e000234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Weisschuh N, Mayer AK, Strom TM, et al.. Mutation detection in patients with retinal dystrophies using targeted next generation sequencing. PLoS One. 2016; 11(1): e0145951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Di Iorio V, Karali M, Brunetti-Pierri R, et al.. Clinical and genetic evaluation of a cohort of pediatric patients with severe inherited retinal dystrophies. Genes (Basel). 2017; 8(10): 280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Stone EM, Andorf JL, Whitmore SS, et al.. Clinically focused molecular investigation of 1000 consecutive families with inherited retinal disease. Ophthalmology. 2017; 124(9): 1314–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Georges B, Cosyns JP, Dahan K, et al.. Late-onset renal failure in Senior-Loken syndrome. Am J Kidney Dis. 2000; 36(6): 1271–1275. [DOI] [PubMed] [Google Scholar]
- 31. Bernardis I, Chiesi L, Tenedini E, et al.. Unravelling the complexity of inherited retinal dystrophies molecular testing: added value of targeted next-generation sequencing. Biomed Res Int. 2016; 2016: 6341870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. van den Born LI, Bergen AA, Bleeker-Wagemakers EM.. A retrospective study of registered retinitis pigmentosa patients in The Netherlands. Ophthalmic Paediatr Genet. 1992; 13(4): 227–236. [DOI] [PubMed] [Google Scholar]
- 33. Gerding H, Busse H.. Häufigkeitsverteilung der verschiedenen Mendel-Erbgänge bei Familien mit Retinopathia pigmentosa. Ergebnisse einer Auswertung des RP-Registers der Universitäts-Augenklinik Münster [Distribution of the frequency of various Mendelian modes of inheritance in families with retinopathia pigmentosa. Results of an evaluation of the RP register of the Munster University Ophthalmology Clinic]. Ophthalmologe. 1994; 91(3): 322–328. [PubMed] [Google Scholar]
- 34. Ayuso C, Garcia-Sandoval B, Najera C, Valverde D, Carballo M, Antiñolo G.. Retinitis pigmentosa in Spain. The Spanish Multicentric and Multidisciplinary Group for Research into Retinitis Pigmentosa. Clin Genet. 1995; 48(3): 120–122. [PubMed] [Google Scholar]
- 35. Bocquet B, Lacroux A, Surget MO, et al.. Relative frequencies of inherited retinal dystrophies and optic neuropathies in southern France: assessment of 21-year data management. Ophthalmic Epidemiol. 2013; 20(1): 13–25. [DOI] [PubMed] [Google Scholar]
- 36. Ayuso C, Millan JM.. Retinitis pigmentosa and allied conditions today: a paradigm of translational research. Genome Med. 2010; 2(5): 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tsang SH, Sharma T.. Autosomal dominant retinitis pigmentosa. Adv Exp Med Biol. 2018; 1085: 69–77. [DOI] [PubMed] [Google Scholar]
- 38. Carrigan M, Duignan E, Malone CP, et al.. Panel-based population next-generation sequencing for inherited retinal degenerations. Sci Rep. 2016; 6: 33248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Martin-Merida I, Avila-Fernandez A, Del Pozo-Valero M, et al.. Genomic landscape of sporadic retinitis pigmentosa: findings from 877 Spanish cases. Ophthalmology. 2019; 126(8): 1181–1188. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.