

Abstract
CD274 gene encodes programmed death-ligand 1 (PD-L1) protein, also known as B7 homolog 1 (B7-H1), which is a crucial hallmark for highly proliferation cells including cancer cells. PD-1 and PD-L1 interaction is assumed as a negative regulator for immune response which can inhibit the T cell growth and cytokine secretion and supports tumor cells evasion from immune system. therefore, PD-L1 could be assumed as a candidate target for immune-therapy. The predicted structure of PD-L1 indicates (Gly4Ser) 3 linker-based chains links. In that line, different simulation softwares applied to explore the structure of granzyme B (GrB), a serine protease in cytotoxic lymphocytes granules as an apoptosis mediator, was attached to its specific antibody structure (atezolizumab) via an adaptor sequence. Evaluation of accuracy, energy minimization and characterization of biological properties of the final processed structure were performed and our computational outcomes indicated that the employed method for structure prediction has been successfully managed to design the immunotoxin structure. It is necessary to mention that, the precise and accurate design of the immune-therapeutic agents against cancer cells can be confirmed by employment of in-silico approaches. Consequently, based on this approach we could introduce a capable immunotoxin which specifically targeting PD-L1 in an accurate orientation and initiates cancer cell destruction by its toxin domain.
Supplementary Information
The online version contains supplementary material available at 10.1007/s40203-021-00076-z.
Keywords: PD-L1, Immunotoxin, Bioinformatics, Atezolizumab, Granzyme B
Introduction
Programmed death-1 (PD-1) is a protein expressed on the surface of the T and B lymphocytes, monocytes, natural killer T cells as well as dendritic cells. PD-1 has two specific ligands: PD-L1 (B7-H1, CD274) which indicates constant expression on the surface of both hematopoietic and non-hematopoietic cells, and PD-L2 (B7-DC, CD273) which is specifically expressed on the surface of the dendritic cells, macrophages, mast cells and certain B cell populations. The functional interaction between PD-1 and PD-L1 suppresses T cell growth and cytokine release. Therefore, PD-1/PD-L1 interaction can negatively regulate immune responses in the benefit of tumor immune surveillance (Latchman et al. 2001; Rezaeeyan et al. 2017). In the recent years, the PD-1/PD-L1 axis has been considered as a cancer immunotherapy target (Brahmer et al. 2012). An activated T cell can express PD-1 upon antigen recognition which leads to production of interferons such as PD-L1 in multiple tissues. PD-1 binding to its specific ligand (PD-L1) limits T-cell activity and prevents excessive immune stimulation. This immunological mechanism maintains the immune tolerance to self-antigens by negative regulation of the immune responses (Karwacz et al. 2011).
The human protein of PD-L1, also known as B7 homolog 1 (B7-H1), is a 40 kDa type 1 transmembrane protein plays a pivotal role in suppressing of adaptive immune system during certain physiological events such as pregnancy, tissue allografts, autoimmune disease as well as other disease such as hepatitis (Sharpe et al. 2007).
The crystal structure of the human PD-1/PD-L1 complex has already been reported by Zak et al. significant differences have been documented by this study regarding the binding properties of murine and human PD-1/PD-L1. Moreover, the structures of the PD-1/PD-L1 complexes from different species and their individual components have been systematically compared (Zak et al. 2015). Analyses on this information have led to characterization of hot-spot pockets of human PD-1/PD-L1 complex which are crucial to block PD-1/PD-L1interaction. The suggested pharmacophore model for this protein–protein interaction could be employed as a solid starting point for design of chemical probes and potential small-molecule therapeutics to target the PD-1/PD-L1 complex (Zak et al. 2015).
It is reported that PD-L1 upregulation in the tumor microenvironment handicaps immune response in various tumor cells such as lymphoma, melanoma, lung, breast cancer, glioblastoma, ovarian, kidney tumors and bladder cancers (Wang et al. 2016). The PD-1/PD-L1 interaction might induces apoptosis in tumor-specific T cells to inhibit T-lymphocyte proliferation and subsequently pro-inflammatory cytokines release and cytotoxicity. The strategies of protein–protein blockage for manipulation of PD-1/PD-L1 interaction might lead to reversion of exhausted T-cell phenotype to activate antitumor responses. These properties lead to design targeted therapeutic molecules against PD-1/PD-L1 interaction in the context of cancer immunotherapy. It is expected that reversal exhausted T-cell phenotype might provide a therapeutic advantage for treatment of the chronic viral infections (Wang et al. 2016).
The atezolizumab, also known as MPDL3280A, is a fully humanized IgG1 (N298A) monoclonal antibody which targets PD-L1. Atezolizumab disrupts the interaction of the PD-L1/PD-1 and stimulates the reactivation of anti-tumor immune response (Santini and Rudin 2017).
Antibodies engineering can improve the binding affinity therapeutic efficiency of the antibody which are promising factors in human diseases treatment (Payandeh et al. 2018, 2019). Immunotoxins combine the specificity of the antibody-based therapeutics with the cell-killing power of more nature’s toxic proteins. Immunotoxins are chimeric molecules which are consist of a protein toxin fused to a targeting moiety. The targeting domain is the most commonly antigen-binding fragment of a monoclonal antibody. The antibody directs the toxin toward the cancer cell, then the toxin pas through and destroys the cell.
Immunotoxins, as highly promising therapeutic agents, which are composed of two main parts: a toxic part derived from different kinds of toxins such as fungal, bacterial and human apoptotic agents, and a coupled antibody part with high affinity to a specific receptor on the surface of malignant cells. Immunotoxins, using their toxic parts, tend to knock-down the target cells after its binding (Madhumathi et al. 2016). As an effective molecule, granzyme B (GrB) activates apoptotic pathways in the tumor or virus-infected cells. Regarding enzymatic cytotoxicity, the GrB should be amended to candidate as immunotoxin (Hehmann-Titt et al. 2013). GrB is important molecule because it can active apoptosis pathway by activating caspase. After binding immunotoxin to its target in tumor cell, entrance of GrB to the target cell is started by prf1 releasing and cleaving Glu and Asp residues. Finally DNA is fragmented (Hehmann-Titt et al. 2013; Ganji et al. 2020; Saito et al. 2011).
In this study, we designed an immunotoxin composed of the GrB molecule and atezolizumab (anti-PD-L1 antibody molecule). In this line, we applied an integrated in silico approach and different computational tools/software. Bioinformatics tools/software facilitate understanding of the molecular mechanisms for different macromolecules supporting the rational design of the novel therapeutic agents. The bioinformatics prediction/modeling can reduce failure rate of the experimental methods and also expenses to figure out novel results. Our findings showed that the designed immunotoxin has an effective interaction with the PD-1 causing the GrB part applies its toxic effects on the target cells.
Methods
Sequence availability
To explore the atezolizumab antibody 3D structure, the Protein data bank (PDB) was investigated. 5XXY (Zhang et al. 2017) records were found for atezolizumab antibody 3D structure on PDB which belongs to crystal structure of PD-L1 complexed with atezolizumab fab at 2.9 A.
The acquired structure was in its trimeric form. Accordingly obtained structures were subjected to MOE software to remove extra chains to achieve atezolizumab antibody and PD-L1structure, respectively.
Furthermore, registered patents were explored for registered sequences of the atezolizumab antibody. PD-L1 and GrB sequences were received from UniProtKB.
Linker sequence addition
As regards, the light and heavy chains of the antibody are considered separately without any linking amino acids in 3D structure prediction, for recombinant production of the antibody, a linker was added to join two chains. The (Gly4Ser)n is the most prevalent linker employed in joining of two chains in antibody studies.
Three repeats of the linker sequence, (Gly4Ser) 3, were added to join the heavy and light chains. modeler 9.21 software was applied to predict the antibody 3D structure with linker. The predicted 3D structure of antibody in previous step, was used as the template structure in modeler 9.21 software for homology modelling. To estimate the homology between the final antibody after linker addition and the antibody structure without linker, Discovery Studio software and Dali Server were used to compute the RMSD and Z-score between them, respectively.
Immunotoxin design and modelling
GrB structure, as the toxic domain of the immunotoxin, was obtained from the PDB. The acquired structure was in dimeric form with ligand interactions. TheMOE software was employed to eliminate the ligands and one of the monomers. In this regard, an adaptor sequence was attached beforehand of the interpolation of the antibody and the GrB, to increase the GrB toxicity. In the following, to predict the immunotoxin final structure combined with = GrB and the antibody structures modeler homology modeling software was employed. The homology modeling prediction is based on template structure. In this regard, modeler aligns the immunotoxin sequence with the pre-existing structures. The modeler software was applied to attain the immunotoxin structure based on antibody (containing linker) and preparing GrB as templates.
Loop and structure refinement
Structure refinement was applied on the immunotoxin structure to achieve a more native and more stable molecule. We used the loop model class in MODELLER to refine the conformation of the loops. The loop optimization method relies on a scoring function and optimization schedule adapted for loop modeling.
The ModRefiner is an algorithm for atomic-level and high-resolution protein structure refinement, which can start from the either C-alpha trace, main-chain model or full-atomic model. Both side-chain and backbone atoms are completely flexible during structure refinement simulations, where conformational search is guided by a composite of physics- and knowledge-based force field.
One of the main aims of structure refinement is to draw the initial starting models closer to their native status, in the terms of hydrogen bonds, backbone topology and side-chain positioning. It also generates significant improvement in physical quality of the local structures. The standalone program also supports Ab initio full-atomic relaxation, where the refined model is not restrained by the initial model or the reference model.
Model validation
Estimating the quality of the protein structure models is a vital step in protein structure prediction. QMEAN Qualitative Model Energy Analysis was employed for the quality estimation of immunotoxin structure model. The RamPage (Ramachandran Plot Analysis) available was used to validate the quality of the immunotoxin 3D model before and after the refinement.
One of the ways that can corroborate with the 3D structure accuracy is secondary structure determination of the sequence. in order to predict the immunotoxin, secondary structure following software and services were used: Jpred, PSIPRED and SOPMA.
In the next step, the secondary structure of the modeled molecules was compared with the predicted secondary structure acquired from MOE software to indicate if they are in concordance with each other.
Immunotoxin characterization
Further characterization of the immunotoxin was carried out using following software and services: ProtParam is a tool which allows the computation of various physical and chemical parameters. The computed parameters include the molecular weight, theoretical pI, amino acid composition, atomic composition, extinction coefficient, estimated half-life, instability index, aliphatic index and grand average of hydropathicity (GRAVY).
Periscop is a sequence-based predictor that estimates the expression levels and yield of soluble proteins in the periplasm of E. coli.
The ccSol predicts protein solubility using physico-chemical properties. Further researches were performed to investigate the recombinant protein antigenicity and toxicity. VaxiJen is a tool for prediction of protective antigens and subunit vaccines. ToxinPred as an in-silico method is developed to predict and design toxic/non-toxic peptides. AlgPred was used to determine the possible allergic features of the recombinant molecules. To specify and characterize the existing epitopes within the immunotoxin sequence, various softwares were used. Propred and Propred-1 were utilized to specify T-cell epitopes 1 and 2, respectively.
In order to identify the B cell epitopes, Ellipro, Bepipred and SVMtrip were used.
Immunotoxin binding efficiency
The interaction possibility between the immunotoxin and specific receptor can be investigated using 3D structure for molecular docking study. The possible interactions between PD-L1and immunotoxin was predicted by Zdock server. ClusPro server can be used to explore the designed immunotoxin functionality. To calculate the Gibbs free energy of the reaction between these two proteins PRODIGY (PROtein binDIng enerGY prediction) server was applied. Immunotoxin-PDL and Atezolimab-PDL complexes were subjected to this analysis.
Results
Sequence retrieval
The sequence source for atezolizumab antibody was drug bank with Accession Number DB11595. The sequences under the UniProtKB ID of Q9NZQ7 and P10144 were obtained for the PD-L1 and GrB protein (Supplementary Fig. 1).
The 5XXY Protein Data Bank (PDB) record was detected for the crystal structure of PD-L1 complexed with atezolizumab fab at 2.9A. The acquired structure was in its trimeric form. Chain H and L belong to atezolizumab chains (heavy and light) and chain A belongs to PD-L1. All non-protein atoms were eliminated to attain the Antibody structure using MOE software. Figure 1 shows atezolizumab and PD-L1 structure before and after monomerization.
Fig. 1.

a Crystal structure of PD-L1 (blue) complexed with atezolizumab fab (orange and violet). b Atezolizumab antibody. c PD-L1. MOE software was used to generate the molecular graphics
Linker sequence addition
Modeler 9.21 was an effective approach in modeling of the antibody structure with the linker sequences. 5 different structures predicted by Modeler 9.21. Discovery Studio software and Dali Server were applied to determine the homology between the structures after the linker addition and original atezolizumab. The best structure with the lowest RSMD and highest Z-score selected for further analyzes. Table 1 shows the results for the RSMD and Z-score investigations of the predicted structures.
Table 1.
RSMD and Z-score evaluations for the predicted structures
| Z-score | RMSD | |
|---|---|---|
| 1 | 20.0 | 1.0 |
| 2 | 22.0 | 0.8 |
| 3 | 22.2 | 0.6 |
| 4 | 21.6 | 1.0 |
| 5 | 21.0 | 1.1 |
Figure 2 demonstrates the best selected structure in which two chains of the antibody are linked as a single chain. The RMSD was calculated to be 0.6 Å. The results showed that linker sequence addition does not make any significant changes in the original structure of the antibody. Figure 3 shows the superimposed structures of the antibody structure before/after linker addition.
Fig. 2.
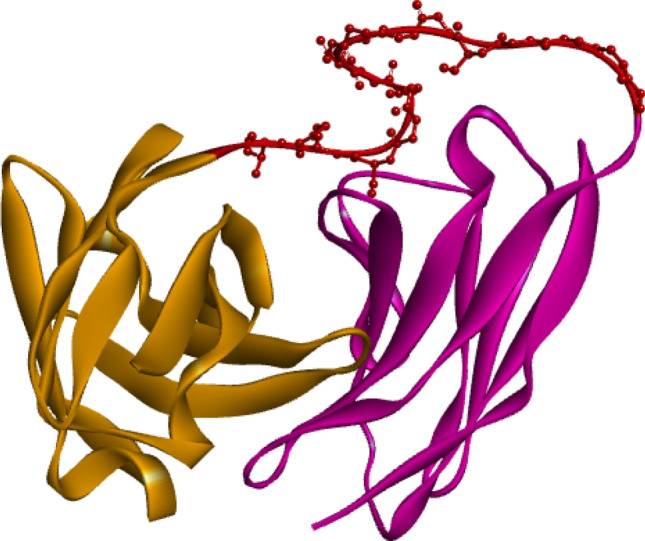
Atezolizumab with linker. Light chain sequence (brown) is followed by the heavy chain sequence (violet) and the linker is shown in ball and stick model (red)
Fig. 3.
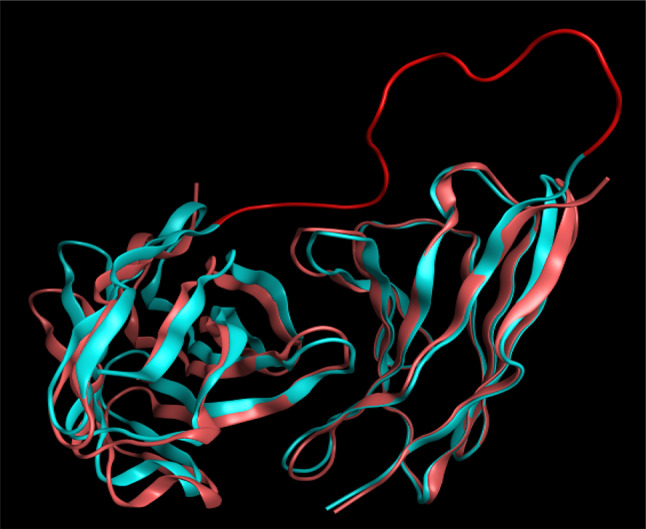
The RMSD between atezolizumab before/after linker addition. The structure with linker is in blue and the one without it is shown in pink. The linker is shown in red
Immunotoxin assembly
The structure under the PDB record of 1FQ3 (Estébanez-Perpiñá et al. 2000) was found for the GrB structure. To obtain a monomeric GrB, all the non-protein atoms and the extra GrB structures were discarded by MOE software. All sequences of the antibody, GrB, adaptor and linker sequences were applied to synthesize the final immunotoxin sequence (Supplementary Fig. 2). Figure 4 shows GrB structure before/after monomerization. Homology modelling successfully determined whole of the immunotoxin structure.
Fig. 4.

Granzyme B 3D structure. a Is the 1FQ3 and b is the GrB without extra non-protein and protein molecules
Loop and structure refinement and model validation
Ramachandran plot indicates that some amino acids in the predicted immunotoxin structure are located at outlier region. Hence, refinement of the model could improve the quality. One aim of ModRefiner is to draw the initial starting models closer to their native state, in terms of hydrogen bonds, backbone topology and side-chain positioning. It also generates significant improvement in the physical quality of the local structures. Ramachandran plot of the refined model was improved. In initial model the percentage of the residues in the favored region was 71.2 while 77.2% in the loop refined and 89.3%in final structure refined model. 11.5%residues in outlier region in the initial model decrease to 9.0% in the loop refined and 0.4% in final structure refined model (Table 2). Figure 5 shows the Ramachandran plot of the immunotoxin before/after refinement. Figure 6 shows the final structure of the immunotoxin coloring based on different domains.
Table 2.
Ramachandran plot result of initial loop refined and final structure refined model
| Favored region (%) | Allowed region (%) | Outlier region (%) | |
|---|---|---|---|
| Initial model | 71.2 | 17.3 | 11.5 |
| Loop refinement by modeller | 77.2 | 13.8 | 9.0 |
| Loop and structure refinement by modeller and modrefiner | 89.3 | 10.3 | 0.4 |
Fig. 5.

Rampage structural validation of the immunotoxin (a initial model, b loop refinement by modeller and c loop and structure refinement by modeller and modrefiner)
Fig. 6.
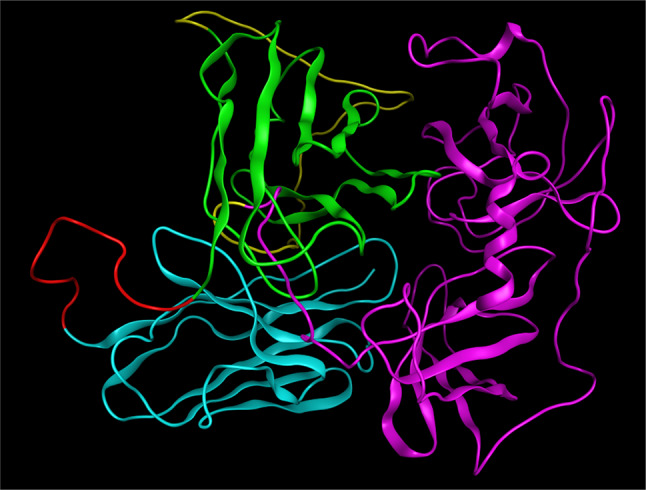
The final structure of the immunotoxin in solid ribbon model. The antibody light chain structure is in blue, antibody heavy chain structure is in green, the linker structure is in red, the adaptor structure is in yellow and the GrB structure is violet
The RMSD between the antibody and the immunotoxin was 1.623. This results showed that there are no significant changes in the native structure of scFv moiety of the immunotoxin. All of the immunotoxin domains can have their own function in the environment without being compromised. Figure 7 shows the alignment of atezolizumab fab and immunotoxin.
Fig. 7.

Structure alignment of atezolizumab fab and immunotoxin. The immunotoxin is shown in green and the antibody is shown in violet. The RMSD between the antibody and the immunotoxin is 1.623
The secondary structures of the modeled structure and the predicted secondary structure of the immunotoxin molecule were compared. The predicted secondary structure is in accordance with the secondary structure of the predicted 3D model which indicates the reliability of the predicted 3D model and employed approach. The accuracy of the secondary structure prediction software, is often better than the 3D protein model predictions. Therefore, the consistency between the secondary structure prediction and the secondary structure is associated with the predicted 3D model which could be a good indication of thehigh-quality 3D structure prediction.
Immunotoxin characterization
Based on the ProtParam results, the molecular weight of the immunotoxin was estimated to be 55,199.40D, the pI was predicted to be 9.51 and it was predicted that the protein half-life in E. coli is 10 h. The protein expression content of the recombinant gene in E. coli was expected to be 9.2622 mg/L by Periscope server. Although, the recombinant protein was estimated to be soluble. ToxinPred predicts no toxic peptide along the immunotoxin sequence. The allergenicity prediction results indicated the designed immunotoxin contains no significant allergen potency (Score = − 1.0586434, Threshold = − 0.4). The results for overall prediction for the protective antigen demonstrated that the designed immunotoxin is a probable ANTIGEN (Score = − 0.6881, Threshold = − 0.4).
According to the prediction provided by the Propred and Propred-1, the immunotoxin contains 75 and 94 T cell epitopes of type 1 and type 2, respectively. In addition, according to the Ellipro prediction results the immunotoxin contains 19 linear and 4 structural B cell epitopes. Bepipred and SVMtrip identified 24 and 8 linear epitopes in the immunotoxin sequence respectively.
Immunotoxin binding efficiency
The Zdock, Cluspro and Hex8 results demonstrated the possibility of the immunotoxin interaction to the PD-L1. The results demonstrated that PD-L1 loops contribute to reaction with the immunotoxin. The directional binding of the immunotoxin shows that this molecule is able to bind to PD-L1 via integrin binding region. Such interaction enables the immunotoxin to hamper leukocytes from binding to PD-L1 expressing cells in addition to directing the immunotoxin to its desired target. This phenomenon enhances the cytotoxic activity. Figure 8 indicates the interactions of the immunotoxin with PD-L1.
Fig. 8.

Docking results of mmunotoxin with PD-L1 (9 position with the best dock score). The immunotoxin is shown in rainbow and PD-L1 in violet
The best docking result according to the experimental data was selected. The docked pose details of immunotoxin in the integrin-binding region of PD-L1 is shown in Fig. 9. Residues D49, A52, I54, Y56, E58, N63, Q66, V68, M59, V111, R113, M115, G119, A121, D122, Y123 and R125 from PD-L1 interact with residues 153S, 154N, 176Y, 178G, 179S, 181R, 222N and 224D from the heavy chain and 95P, 96R, 153S, 154N, 176Y, 222N and 224D from the light chain of antibody of the immunotoxin.
Fig. 9.
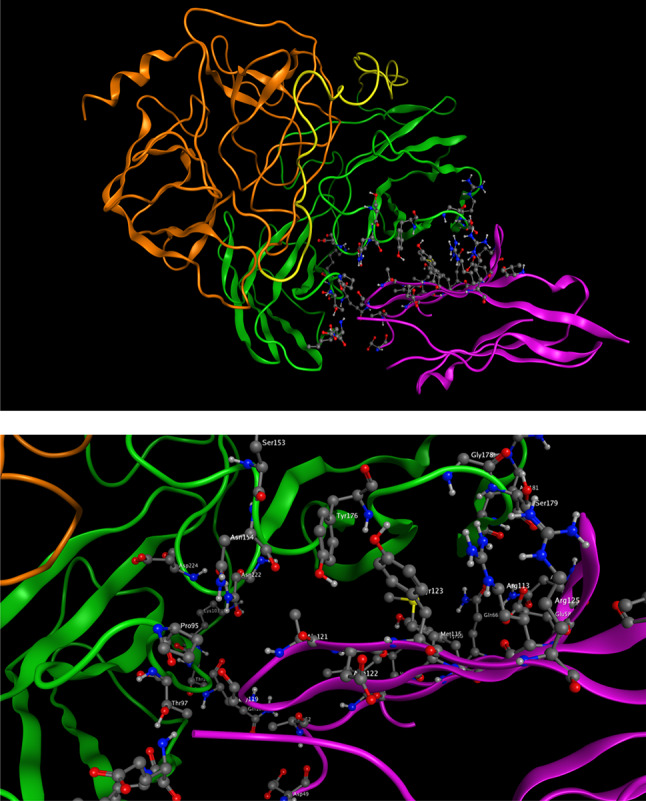
The best docked structure of immunotoxin and PD-L1. The docked pose of immunotoxin in the integrin-binding region of PD-L1 in two formats, top) complete proteins down) close up of interactions between immunotoxin and PD-L1. The antibody structure is in green, the adaptor structure is in yellow and the GrB structure is orange
The results of binding energy calculation indicates that immunotoxin binds to the PD-L1 with high affinity. The Atezolimab-PDL1 complex was checked for its binding affinity as a positive control to be compared with the Immunotoxin-PDL1 complex. Table 3 indicates the binding energy between the PDL-1 and Atezolimab as well as Immunotoxin molecules (Fig. 10).
Table 3.
The binding energy between the PDL-1 and atezolimab and immunotoxin molecules
| Protein–protein complex | ΔG (kcal/mol) | Kd (M) at 25.0 °C |
|---|---|---|
| Immunotoxin-PDL | − 11.1 | 7.8E−09 |
| Atezolimab-PDL | − 10.9 | 9.3E−09 |
Fig. 10.

Immunotoxins function: when designed immunotoxins bind to PD-L1 on tumor cell, it blocks the interaction of PD-1/PD-L1, as a result, apoptosis is not induced in tumor-specific T cells to inhibit T-lymphocyte proliferation and subsequently pro-inflammatory cytokines release and cytotoxicity. On the other hand, when immunotoxins bind to PD-L1 on tumor cell, GrB, leads to apoptosis in cancer cells
Discussion
Analyzing the functional properties of a designed macromolecule is one of the essential requirements in biological modeling studies. Since the structural and functional analyses are costly and time consuming, exploiting the molecular modeling methods and protein 3D structures could circumvent numerous challenges ahead empirical experimentation. Even models with low accuracy could be useful to make assumptions about the fictional model of an unknown antigen (2001, 2003). Using molecular modeling methods would help the researchers to decrease the number of the necessary experimental assessments and to avoid different ethical issues (Mohammadpour et al. 2018; Khalili et al. 2017a, b, 2018). Given these properties of molecular modeling methods, we have launched an in-silico study to model and assess an immunotoxin functionality against PD-L1 molecule. Various analyses were carried out to assess the designed immunotoxin which indicated promising functional properties for the final molecule.
The designed immunotoxin is composed of the atezolizumab antibody sequence as the targeting moiety and the GrB as the toxic moiety. The atezolizumab monoclonal antibody as a fully humanized IgG1 would bring about proper targeting of the PD-L1/PD-1 interaction on the cancer cells. This would prompt the successful delivery of the immunotoxin to the cancer cell. Moreover, being fully humanized would reduce the possible adverse effects and undesired immune activation against the immunotoxin. These humanized antibodies would compensate for reduced efficacy of the mouse-derived antibody sequences owing to their higher immunogenicity (Nelson et al. 2010). An approved sequence of the atezolizumab was used to predict its 3D structure with different modeling approaches. Using more than one approach to model the 3D structures would lead to higher accuracy of the final model and the chance to find the best approach to obtain a native like structure for the protein. It has already been shown that changing the length of the linker sequence to join the variable domains of the light and heavy chains of an antibody (to form single-chain antibody fragment (scFv) would have a drastic effect on the orientation of their assembly and the valence of the formed complex. Different applications of single-chain variable fragment (scFv) for therapeutic and imaging purposes are directly affected by the valency, flexibility and the size. Inclusion of linkers with at least 12 amino acids in thelength predominantly leads to formation of the monomers with varying amounts of dimer and higher molecular mass oligomers in equilibrium (Kortt et al. 2001). An ideal linker sequence is expected to effectively reduce the interference amongst domains and indicates the lowest effect on the folding properties of the joined domains. Our results indicated that linking two variable domains of the atezolizumab light and heavy chains using a 12 amino acids glycine and serine reach sequence did not change the overall structure of the linked domains. The obtained RMSD values confirmed the lowest effect of the added linker on the joined domains. Using scFv as the targeting moiety instead of whole antibody molecule (which were employed in early generations of immunotoxins) effectively reduces the final size of the immunotoxin. Smaller antigens are endowed with better penetration capability into the tissues and slower renal plasma clearance (Kreitman 2003; Akbari et al. 2017).
The GrB moiety was used to confer the toxicity effect of the immunotoxin. GrB is a serine protease which induces apoptosis via activation of several pathways within the cytoplasm and nucleus of target cells. Proteolytic activation of the caspase 3 is one of the major pathways of GrB mediated apoptosis activation (Chowdhury and Lieberman 2008; Waugh et al. 2000). Various substrates of apoptotic induction, like the inhibitor of caspase activated deoxyribonuclease and Lamin B, could be produced following the direct proteolysis of caspase 3 via GrB (Boivin et al. 2009). GrB mediated cleavage of Bcl-2 and Bid protein could also initiate the caspase 3 pathway which is associated with caspase-8 activation (Waugh et al. 2000; Hengartner 2000). Bid cleavage ultimately leads to permeabilization of the mitochondrial outer membrane and release of pro-apoptotic factors such as Smac/DIABLO, Omi/HtrA2, cytochrome c and apoptosis inducing factor (Boivin et al. 2009). The formation of the apoptosome complex is induced by release of cytochrome c which in turn leads to downstream activation of Caspase 3. Permeabilization of the mitochondrial outer membrane could be further promoted by GrB mediated cleavage of the anti-apoptotic Mcl-1 and following release of Bim protein as a member of the pro-apoptotic Bcl-2 family (Kägi et al. 1994). The substrates of the GrB activated effector caspases including NuMa, ICAD, Tubulin, PARP-1and Lamin B are also among the substrates of the GrB (Chowdhury and Lieberman 2008; Boivin et al. 2009; Hiebert and Granville 2012). Alternatively, apoptosis activation could occur via other immune cell/GrB-mediated roots. Perforin multimerization is needed in this root to build pores in the target membrane through which the GrB trafficking into the cell would be possible (Boivin et al. 2009). Given these facts, targeting of the GrB against cancer cells could eventually lead to activation of apoptosis in these cells and their elimination.
The 3D modeling of the final immunotoxin indicated that each component of the immunotoxin does not experience any drastic changes. The calculated RMSD values confirmed the retained structural properties of each component of the immunotoxin. The predicted high chance of the immunotoxin to have soluble expression in the periplasm of E. coli is accounted as a desirable property. This property is important due to presence of 3 disulfide bonds of the GrB molecule; the periplasmic space is an oxidizing compartment in genetically un-modified E. coli cells which is equipped with various chaperones and foldases (Boivin et al. 2009). Given these properties this compartment could bring about the suitable environment to form these bonds. Forming these bonds is imperative to restore the original biological functions of the designed immunotoxin. It has also been shown that the designed immunotoxin could be expressed as a soluble protein. of note, soluble proteins are associated with much more simple downstream purification processes and they are more likely to be biologically functional with proper folding. Therefore, the designed immunotoxin could have a appropriate therapeutic function upon expression and easier purification processes. The interaction between the immunotoxin and the integrin binding region of the PD-L1 molecule indicates that the designed antigen could properly target the cancer sites and hamper the interaction between leukocytes and PD-1expressing cells. Low binding energy between the immunotoxin and PD-1expressing cells was calculated by docking analyses. It implies that the interaction between these two molecules would be strong enough to exert the immunotoxin functions. High antigenicity and lack of toxicity and allergenecity are also the desirable properties that confirmed the suitable design of the final immunotoxin.
In conclusion, our results indicated that the designed immunotoxin possess desirable structural, physicochemical and immunological properties to exert its functions at target sites. The interaction between the immunotoxin and the integrin binding region of the PD-L1 molecule indicates its proper orientation, while the low binding energy confirms the stability of this interaction. These properties confirm the appropriateness of this immunotoxin to be employed in fighting against cancer cells. However, performing in vitro analyses would be required for further analyses of the immunotoxin.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
The authors wish to thank Immunology Research Center, Tabriz-Iran and Shahid Sadoughi University of Medical Science, Yazd, Iran for their support by unrestricted free access to the web site for data collection.
Abbreviations
- PD-L1
Programmed death-1
- GrB
Granzyme B
- PDB
Protein data bank
- OPIG
Oxford Protein Informatics Group
- RMSD
Root-mean-square deviation
Author contributions
All authors participated in writing. Writing-review and language correction.
Funding
This work was financially supported by grants from the Iran National Science Foundation (INSF), Iran (project no. 97021150) and Tabriz University of Medical Sciences, Tabriz, Iran (project no. 61892). We gratefully acknowledge them for their contribution to this study.
Availability of data and materials
Data presented in this manuscript is available upon request.
Compliance with ethical standards
Conflict of interest
All authors declare no potential conflicts of interest.
Ethics approval and consent to participate
This article does not contain any studies with human participants or animals performed by any of the authors.
Consent for publication
All authors have read and approved the final version of the manuscript.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Fateme Sefid, Email: sefid.fateme@yahoo.com.
Zahra Payandeh, Email: zpayandeh58@yahoo.com.
Ghasem Azamirad, Email: Azamirad@yazd.ac.ir.
Behzad Baradaran, Email: baradaranb@tbzmed.ac.ir.
Mohsen Nabi Afjadi, Email: mohsennabi66@gmail.com.
Maryam Islami, Email: mahtabi2234@gmail.com.
Maryam Darvish, Email: maryam_darvish@yahoo.com.
Seyed Mehdi Kalantar, Email: kalantarsm@ystp.ac.ir.
References
- Akbari B, Farajnia S, Ahdi Khosroshahi S, Safari F, Yousefi M, Dariushnejad H, et al. Immunotoxins in cancer therapy: review and update. Int Rev Immunol. 2017;36(4):207–219. doi: 10.1080/08830185.2017.1284211. [DOI] [PubMed] [Google Scholar]
- Becker OM, MacKerell AD, Roux B, Watanabe M. Computational biochemistry and biophysics. New York: Marcel Dekker; 2001. [Google Scholar]
- Boivin WA, Cooper DM, Hiebert PR, Granville DJ. Intracellular versus extracellular granzyme B in immunity and disease: challenging the dogma. Lab Invest. 2009;89(11):1195. doi: 10.1038/labinvest.2009.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahmer JR, Tykodi SS, Chow LQ, Hwu W-J, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455–2465. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury D, Lieberman J. Death by a thousand cuts: granzyme pathways of programmed cell death. Annu Rev Immunol. 2008;26:389–420. doi: 10.1146/annurev.immunol.26.021607.090404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estébanez-Perpiñá E, Fuentes-Prior P, Belorgey D, Braun M, Kiefersauer R, Maskos K, et al. Crystal structure of the caspase activator human granzyme B, a proteinase highly specific for an Asp-P1 residue. Biol Chem. 2000;381(12):1203–1214. doi: 10.1515/BC.2000.148. [DOI] [PubMed] [Google Scholar]
- Fiser A, Šali A. Modeller: generation and refinement of homology-based protein structure models. Methods in enzymology. Amsterdam: Elsevier; 2003. pp. 461–491. [DOI] [PubMed] [Google Scholar]
- Ganji M, Khalili S, Mard-Soltani M, Khalesi B, Karkhah A, Amani J. A precisely designed immunotoxin against VCAM1 consisting of a humanized antibody variable domain fused to granzyme: an in silico approach. Int J Pept Res Ther. 2020;26(1):129–137. doi: 10.1007/s10989-019-09822-6. [DOI] [Google Scholar]
- Hehmann-Titt G, Schiffer S, Berges N, Melmer G, Barth S. Improving the therapeutic potential of human granzyme B for targeted cancer therapy. Antibodies. 2013;2(1):19–49. doi: 10.3390/antib2010019. [DOI] [Google Scholar]
- Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407(6805):770. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- Hiebert PR, Granville DJ. Granzyme B in injury, inflammation, and repair. Trends Mol Med. 2012;18(12):732–741. doi: 10.1016/j.molmed.2012.09.009. [DOI] [PubMed] [Google Scholar]
- Kägi D, Ledermann B, Bürki K, Seiler P, Odermatt B, Olsen KJ, et al. Cytotoxicity mediated by T cells and natural killer cells is greatly impaired in perforin-deficient mice. Nature. 1994;369(6475):31. doi: 10.1038/369031a0. [DOI] [PubMed] [Google Scholar]
- Karwacz K, Bricogne C, MacDonald D, Arce F, Bennett CL, Collins M, et al. PD-L1 co-stimulation contributes to ligand-induced T cell receptor down-modulation on CD8+ T cells. EMBO Mol Med. 2011;3(10):581–592. doi: 10.1002/emmm.201100165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalili S, Rasaee MJ, Mousavi SL, Amani J, Jahangiri A, Borna H. In silico prediction and in vitro verification of a novel multi-epitope antigen for HBV detection. Mol Genet Microbiol Virol. 2017;32(4):230–240. doi: 10.3103/S0891416817040097. [DOI] [Google Scholar]
- Khalili S, Zakeri A, Hashemi ZS, Masoumikarimi M, Manesh MRR, Shariatifar N, et al. Structural analyses of the interactions between the thyme active ingredients and human serum albumin. Turk J Biochem. 2017;42(4):459–467. [Google Scholar]
- Khalili S, Rasaee MJ, Bamdad T, Mard-Soltani M, Ghalehni MA, Jahangiri A, et al. A novel molecular design for a hybrid phage-DNA construct against DKK1. Mol Biotechnol. 2018;60(11):833–842. doi: 10.1007/s12033-018-0115-2. [DOI] [PubMed] [Google Scholar]
- Kortt AA, Dolezal O, Power BE, Hudson PJ. Dimeric and trimeric antibodies: high avidity scFvs for cancer targeting. Biomol Eng. 2001;18(3):95–108. doi: 10.1016/S1389-0344(01)00090-9. [DOI] [PubMed] [Google Scholar]
- Kreitman RJ. Immunotoxins for targeted cancer therapy. Chimeric toxins. Boca Raton: CRC Press; 2003. pp. 219–237. [Google Scholar]
- Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. 2001;2(3):261–268. doi: 10.1038/85330. [DOI] [PubMed] [Google Scholar]
- Madhumathi J, Devilakshmi S, Sridevi S, Verma RS. Immunotoxin therapy for hematologic malignancies: where are we heading? Drug Discov Today. 2016;21(2):325–332. doi: 10.1016/j.drudis.2015.05.002. [DOI] [PubMed] [Google Scholar]
- Mohammadpour H, Du W, O'Neill R, Khalili S, Qiu J, Repasky EA, et al. Host-derived serine protease inhibitor 6 provides granzyme B-independent protection of intestinal epithelial cells in murine graft-versus-host disease. Biol Blood Marrow Transplant. 2018;24(12):2397–2408. doi: 10.1016/j.bbmt.2018.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson AL, Dhimolea E, Reichert JM. Development trends for human monoclonal antibody therapeutics. Nat Rev Drug Discov. 2010;9(10):767. doi: 10.1038/nrd3229. [DOI] [PubMed] [Google Scholar]
- Payandeh Z, Rajabibazl M, Mortazavi Y, Rahimpour A, Taromchi AH. Ofatumumab monoclonal antibody affinity maturation through in silico modeling. Iran Biomed J. 2018;22(3):180. doi: 10.22034/ibj.22.3.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payandeh Z, Rajabibazl M, Mortazavi Y, Rahimpour A, Taromchi AH, Dastmalchi S. Affinity maturation and characterization of the ofatumumab monoclonal antibody. J Cell Biochem. 2019;120(1):940–950. doi: 10.1002/jcb.27457. [DOI] [PubMed] [Google Scholar]
- Rezaeeyan H, Hassani SN, Barati M, Shahjahani M, Saki N. PD-1/PD-L1 as a prognostic factor in leukemia. J Hematopathol. 2017;10(1):17–24. doi: 10.1007/s12308-017-0293-z. [DOI] [Google Scholar]
- Saito Y, Kondo H, Hojo Y. Granzyme B as a novel factor involved in cardiovascular diseases. J Cardiol. 2011;57(2):141–147. doi: 10.1016/j.jjcc.2010.10.001. [DOI] [PubMed] [Google Scholar]
- Santini FC, Rudin CM. Atezolizumab for the treatment of non-small cell lung cancer. Expert Rev Clin Pharmacol. 2017;10(9):935–945. doi: 10.1080/17512433.2017.1356717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpe AH, Wherry EJ, Ahmed R, Freeman GJ. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat Immunol. 2007;8(3):239. doi: 10.1038/ni1443. [DOI] [PubMed] [Google Scholar]
- Wang X, Teng F, Kong L, Yu J. PD-L1 expression in human cancers and its association with clinical outcomes. OncoTargets Ther. 2016;9:5023. doi: 10.2147/OTT.S105862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waugh SM, Harris JL, Fletterick R, Craik CS. The structure of the pro-apoptotic protease granzyme B reveals the molecular determinants of its specificity. Nat Struct Mol Biol. 2000;7(9):762. doi: 10.1038/78992. [DOI] [PubMed] [Google Scholar]
- Zak KM, Kitel R, Przetocka S, Golik P, Guzik K, Musielak B, et al. Structure of the complex of human programmed death 1, PD-1, and its ligand PD-L1. Structure. 2015;23(12):2341–2348. doi: 10.1016/j.str.2015.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Qi X, Wang X, Wei D, Wu J, Feng L, et al. Structural basis of the therapeutic anti-PD-L1 antibody atezolizumab. Oncotarget. 2017;8(52):90215. doi: 10.18632/oncotarget.21652. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data presented in this manuscript is available upon request.