

Abstract
Purpose
To investigate use of the third-generation sequencing (TGS) Oxford Nanopore system as a new approach for preimplantation genetic testing (PGT).
Methods
Embryos with known structural variations underwent multiple displacement amplification to create fragments of DNA (average ~ 5 kb) suitable for sequencing on a nanopore.
Results
High-depth sequencing identified the deletion interval for the relatively large HBA1/2--SEA alpha thalassemia deletion. In addition, STRs were able to be identified in the primary sequence data for potential use in conventional PGT-M linkage confirmation. Sequencing of amplified embryo DNA carrying a translocation enabled balanced embryos to be identified and gave the precise identification of translocation breakpoints, offering the opportunity to differentiate carriers from non-carrier embryos. Low-pass sequencing gave reproducible profiles suitable for simple identification of whole-chromosome and segmental aneuploidies.
Conclusion
TGS on the Oxford Nanopore is a possible alternative and versatile approach to PGT with potential for performing economical workups where the long read sequencing information can be used for assisting in a traditional PGT workup to design an accurate and reliable test. Additionally, application of TGS has the possibility of providing combined PGT-A/SR or in selected stand-alone PGT-M cases involving pathogenic deletions. Both of these applications offer the opportunity for simultaneous aneuploidy detection to select either balanced embryos for transfer or additional carrier identification. The low cost of the instrument offers new laboratories economical entry into onsite PGT.
Keywords: Third-generation sequencing (TGS), Preimplantation genetic testing (PGT)
Introduction
Sequencing approaches to chromosome analyses in embryo preimplantation assessments have taken over from array CGH [1]. Early arrays used BAC probes that were hybridized with whole-genome amplification (WGA) DNA material from biopsy pieces with the total material costs a combination of the WGA process, the fluorescent labeling, and the array slides and reagents [2]. Major equipment includes hybridization and slide wash systems and moderate- to-high-resolution scanners, depending on the array system chosen. While manageable for many medium to large laboratories, total equipment and maintenance costs were often prohibitive for the smaller analysis groups and so preimplantation genetic testing for aneuploidy (PGT-A) remained a costly and controlled process overall. With the advent of NGS approaches [3, 4], the material costs to analyze an embryo reduced significantly (to under $100USD), bringing PGT-A into wider application in routine IVF screening. While the starting point of PGT-A by either array or NGS still includes some aspect of WGA with its reagent costs, other technical requirements for NGS were reduced compared to array CGH; however, capital item expenditures were increased. While many of the larger groups quickly converted to the sequencing-based approach for its advantages of reduced sample handling, potential for semi-automation and overall reduced reagent costs, smaller groups were again faced with prohibitive capital costs or the only other alternative of outsourcing analyses to commercial service suppliers.
In the last few years, the next era of DNA sequencing technology, termed third-generation sequencing (TGS), has gained a place in many different areas of chromosome analysis [5]. The most common of these technologies are based on nanopore cells that can rapidly sequence long stretches of DNA at moderate cost per run. It is the ability of TGS approaches to sequence long stretches of DNA, typically 1–100 kb fragments that make them especially useful for chromosome assembly but somewhat less considered for situations where the fragment sizes are smaller and fragment numbers required for analysis are high or if sequencing accuracy is important. Several reports have identified TGS as being useful for investigating structural chromosome rearrangements [6, 7], gene fusions [8], and/or in situations where DNA deletions or insertions are involved [9].
Two instruments, PacBio and Oxford Nano, are the market leaders in TGS and have similar material costs per run but a significant difference for instrument CAPEX—the latter aspect often being important for laboratories on a smaller budget. There have been only a few reports on the use of these TGS instruments for PGT applications [10–13]. In one of these studies [13], the testing for embryo aneuploidy was aimed at capitalizing on the real-time data analysis available with TGS but approached the testing in a manner similar to NGS with small fragment WGA products being analyzed. This latter approach did show promise for rapid analysis for situations where fresh embryo transfer was required. The low sequencing depth used to meet time requirements did limit applications to whole-chromosome ploidy analysis with an inability to see segmental changes. In this study, we used DNA from embryos of known chromosome status as DNA models to explore the opportunities for the use of the Oxford Nanopore system to analyze longer genome fragments at higher sequencing depths and to investigate detail of structural rearrangements in couples seeking PGT as well as any potential utility in analyzing their embryos for chromosome balance prior to transfer.
Material and methods
DNA source
The research study was approved by the Ethics Committee of the First Peoples’ Hospital of Kunming (Approval number YLS2019-27). DNA sources were embryos previously analyzed by PGT-SR, PGT-A, and PGT-M and considered unsuitable for transfer (Table 1). It was decided that whole embryos of known chromosome status which still required genome amplification and had no indications of mosaicism provided a good model for our approach to conduct detailed structural and ploidy analyses. The embryos originally came from the PGT cycles of two couples: one couple with known carrier status for alpha thalassemia having PGT-M and the other couple having PGT-SR because one was a reciprocal translocation carrier. The embryos from the translocation couple were identified with segmental chromosome aneuploidies arising from meiotic non-disjunction of the reciprocal translocation and included a whole-chromosome aneuploidy coincidental to the translocation analysis by NGS [4, 14]. Embryos from the second couple contained a microdeletion associated with a common alpha thalassemia deletion which was detected during PGT-M using GAP-PCR [15].
Table 1.
Study embryos
| Couple | Carrier status | PGT method | Embryo | PGT results (biopsy) | TGS results (biopsied embryo) | Concordance between PGT and TGS |
|---|---|---|---|---|---|---|
| 1 | Paternal and maternal carrier of HBA1/2 SEA deletion | GAP-PCR + NGS | B2 | --SEA/--SEA deletion, 45, XN, −22 |
Homozygous 14-kb HBA deletion 45, XN, −22 |
Concordant |
| B3 | --SEA/--SEA deletion, 46, XN |
Homozygous 14-kb HBA deletion 46, XN |
Concordant | |||
| 2 | Paternal carrier t(1;18)(q42;q12.3) | NGS | L3 |
45, XN, −4 −18q12.3qter +1q42qter |
45, XN, −4 −18q12.3qter +1q42qter |
Concordant |
| L4 |
46, XN, +18q12.3qter −1q42qter |
46, XN, +18q12.3qter −1q42qter |
Concordant | |||
| L5 |
46, XN, +18q12.3q23 −1q42qter |
46, XN, +18q12.3q23 −1q42qter |
Concordant | |||
| L10 | 47, XN, +4 | 47, XN, +4 | Concordant |
Generation of embryo DNA libraries
Multiple displacement amplification (MDA) is a preferred method used by many different groups for its ability to reliably and uniformly amplify small picogram amounts of DNA to microgram levels. The final product runs on an agarose gel at an apparent size of 30–50 Kb. Whole embryos (estimated at 80–150 cells each) of known chromosome profile were chosen rather than smaller biopsy fragments in order to avoid possible issues of limited mosaicism often observed in other PGT biopsy studies while still utilizing a genome amplification process. Several studies have suggested that a biopsy and the whole embryo are concordant in major chromosomes whereas they may vary for mosaic levels, segmental changes, and low to medium level mosaic whole chromosomes [16]. Each embryo had the zona removed and was washed in PBS prior to tubing and sending out for amplification and commercial TGS analysis (GrandOmics, Wuhan, China). Embryos underwent a Repli-G Single Cell MDA WGA using a phi29 polymerase process (QIAGEN) according to the manufacturer protocol. After isothermal amplification, 1.5 μg of the amplified DNA was prepared by digestion of single-stranded regions using T7 endonuclease (NEB) to reduce or remove single-strand displacement loops and branched structures which are a feature of this amplification mode. DNA was then blunt end repaired and addition of Oxford Nanopore–specific sequencing adapters was performed according to manufacturer recommendations (1D Genomic DNA by Ligation; Oxford Nanopore Technologies).
TGS: mapping and junction fragment identification
In this pilot study, an attempt to see the greatest detail available from any single sample was considered important and so each DNA sample was analyzed on a single flow cell (PromethION, R9.4.1) to yield maximum sequencing data and achieve the highest resolution per test. Sequence runs were performed as per manufacturers’ protocols (SQK-LSK109). The Oxford Nanopore system sequences DNA in real time with fragments analyzed as they elute through each pore. The final sequence files collected continuously for the run period are therefore a primary sequence as well as a reflection of random fragment sequence sampling through the TGS cell. Since, for any discreet fragment, elution through a pore occurs at a random time point during the run, sequence data is essentially a randomized collection of reads for each sample over time. These files were aligned to the hg19 reference genome and standard genome analyses using manufacturer Oxford Nanopore software were performed. Sequence files were then transferred to a second commercial genomics group (Yearth, Changsha, China) for more detailed analyses. Nanopore reads were again aligned to hg19 reference genome using MiniMap2 and NGMLR. PycoQC was used to determine quality of reads and length for each sample. Structural variations were detected using sniffles on the MiniMap2 and NGMLR files. Sniffles did not detect the HBA structural variation and examination of the region was performed manually on alignment files.
Chromosome ploidy analysis was performed by bin plots of defined sizes in a similar manner to most other NGS approaches. Essentially, fragments were mapped and accorded a bin on each chromosome. Bins were plotted against chromosomes on a chromosome position versus log2 scale [4]. In a first elaboration, bins were collated only according to chromosome location. In a refinement, bins of different sizes were created and analyzed as sliding sets with step changes of 500 kb. A simple examination of reproducibility of an aneuploid profile result was achieved by bioinformatically taking sets of fragments, (typically 250,000 or 500,000) by using sequential sequence file numbers grouped into the requisite sets and comparing profiles to other fragment sets from the same run. All of these manipulations, post bin assignment, were similar to those used in many other NGS PGT-A approaches, and technically, these alternatives could also be used for chromosome analysis.
Results
TGS embryo results
Each sequencing flow cell yielded a total 20–50 Gb of embryo DNA sequence data equating to an average depth of coverage of 5–15. This equated to 5,000,000–8,000,000 fragments with an average size of 5 kb (1–30 Kb) reduced from the original 30–50 kb apparent size estimated by agarose gel electrophoresis. This size reduction is a result of the exonuclease removal of single-stranded regions and reflected the complex single-strand/double-strand nature of the original MDA amplification products. The estimated base call error rate for a single pass in TGS is approximately 10% but this did not prevent unique mapping of the majority (80–85%) of fragments using manufacturer software. Unmapped fragments were likely a mix of amplification artifacts and repetitive DNA sequences that cannot be uniquely located. The TGS chromosome analyses for each of the whole biopsied embryos showed complete concordance with previous PGT-M- and NGS-based PGT-SR results obtained from biopsied samples (Table 1).
SEA deletion analysis
The TGS read density plots across the HBA locus for all embryos are shown in Fig. 1a. The --SEA deletion diagnosed by GAP-PCR for PGT-M case 1 embryos B2 and B3 was visually confirmed by the absence of reads across an ~ 14 kb region which overlaps with the known HBA–SEA deletion interval. In contrast, for unrelated case 2 (embryos L3, L4, L5, and L10), there was a relatively consistent read density across this deletion interval, confirming the presence of expected homozygous normal HBA sequences. Sequence examination 1 Mb upstream and downstream of the HBA deletion interval identified a nearby polymorphic STR sequence D16S3400 (Fig. 1b). Based on allelic reads coverage, heterozygous status for the CA repeat was able to be ascertained for embryos L2, L3, and L5.
Fig. 1.

Deletion and STR mapping of the alpha thalassemia HBA loci. a --SEA deletion mapping. The positions of reference genes HBZ, HMB, HBA2, and HBA1 are shown at the top of the IGV plots. From read density mapping and position, a homozygous deletion of ~ 14 kb is evident in embryos B2 and B3 (PGT-M case 1). In unrelated embryos L3, L4, L5, and L10 (PGT-SR, case 2), sequences across the deletion are present, confirming homozygous normal sequences. b Identification of a linked polymorphic STR marker D16S3400. Embryos L3, L4, and L5 show evidence of heterozygosity. No reads encompassing this region were found for embryos B2, B3, and L10
Translocation junction breakpoint analysis
Based on deep sequencing (5–15 times depth) of unbalanced embryo L2 from case 2 (Table 1), reads encompassing the actual chromosome 1 and 18 junction breakpoint regions were identified (Fig. 2). Finer sequence review then identified the precise breakpoint coordinates on chromosomes 1 and 18 (Table 1). The breakpoint regions correlated with the approximations identified in the cytogenetic karyotype of the parental translocation carrier.
Fig. 2.
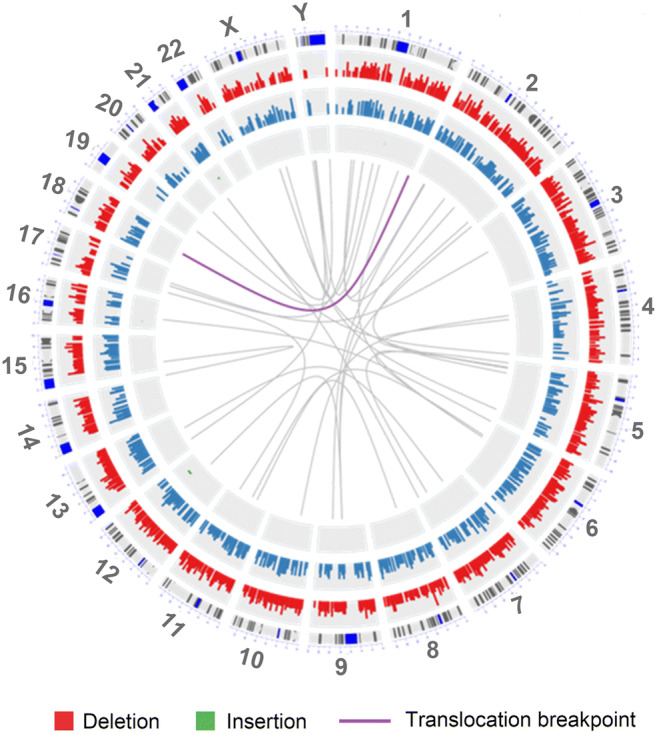
Breakpoint junction circle plots for reciprocal translocation t(1;18)(q42;q12.3). The TGS data from the unbalanced embryo L4 was analyzed for fragments with joined chromosome 1 and 18 sequences. The crossover positions for chromosomes 1 and 18 are indicated by the purple line. Based on the hg19 reference genome, translocation breakpoint coordinates determined by TGS were 244737778 (chromosome 1) and 40471960 (chromosome 18)
Chromosome profiling with low pass TGS
Each sequencing cell output was bioinformatically sampled to investigate the number of DNA sequence reads necessary to achieve a readable ploidy profile. The arbitrary nature of the molecules initially entering the nanopore made such acquisition of random sets of reads straightforward with the final sequence file number being related to the original time of pore capture. Sets of random fragment sequences were achieved by ordered analysis of sequential files of the required fragment numbers. With as few as 250,000 mapped reads (5% of total flow cell reads), simple chromosome ploidy analysis was possible. However, more stable and reproducible profiles were achieved with 500,000 mapped reads (10% of total flow cell reads), where reads were allocated to 5-Mb chromosome bins and ploidy determined using a 0.5-Mb sliding bin analysis (Fig. 3, selected examples embryos B2 and L4). Read numbers of this order are similar to those of commercial processes such as ReproSeq (Thermofisher Scientific). Expected autosome copy number for each embryo showed a high degree of stability for all fragment sub sample sets analyzed. In terms of resolution, both monosomy 22 and segmental aneuploidy for 1q42qter (4.5 Mb) and 18q12.3qter (32.5 Mb) imbalances were clearly visible and reproducibly identified in three replicates.
Fig. 3.

Chromosome plots from TGS data. Y-axis copy number. X-axis autosome number. a Embryo B2. b Embryo L4. Three replicates are shown and aneuploidy is marked by red boxes
Discussion
In this study, embryos with known chromosome profiles were used as a model for potential test workup for PGT cases and for demonstrating potential selective PGT-M and PGT-SR applications. These embryos were previously assessed for chromosome profile and alpha thalassemia gene region through trophectoderm biopsy and detailed NGS-based CNV copy number analysis [4, 14]. The embryos were from PGT cases from two different couples—one couple with a deletion of the alpha thalassemia region and in the other couple one partner with a reciprocal translocation. In the former couple, both were carriers of the common alpha thalassemia gene deletion --SEA and had undergone PGT-M to identify any embryos that were homozygous deleted which would result in alpha zero thalassemia and hence were not suitable for transfer. For the second couple, one partner carried a known chromosome rearrangement—a reciprocal translocation t(1,18)(q42,q12.3). In both cases, all embryos were inappropriate for transfer since they were previously assessed as abnormal or affected by a lethal single-gene disorder.
An opportunity for nanopore sequencing is in defining the target regions for relatively simple or straightforward development of patient-specific PCR-based tests for structural rearrangements (PGT-SR). With the knowledge gained from deep sequencing of key participants, it would now be simpler to identify the exact chromosome location and surrounding DNA sequence of any structural rearrangement. This information could then be used to design a less-expensive patient-specific test, such as a PCR junction fragment test, for use during that couple’s actual PGT cycle. A greater time and resource consuming approach using NGS for junction fragment localization through mate pair sequencing analysis followed by an iterative PCR primer design was previously described [12]. The third-generation sequencing approach described herein identifies the junction fragment in the first step, permitting standard PCR primer design, simplifying, and expediting the whole workup phase. In the event that the nature of the rearrangement is not compatible with a PCR test, then biopsy piece(s) can be amplified and analyzed individually on a single cell to reveal the relative chromosome balances, the specific non-disjunction fragment if present, and hence the carrier status of each individual embryo. Such a direct test and carrier analysis would come at elevated cost though similar to the direct analysis of the previous alpha thalassemia deletion assay.
For the two embryos with a purported homozygous HBA gene deletion, deep sequencing (5–15 depth) with the Oxford Nanopore was able to visualize an absence of DNA sequence fragments in ~ 14 kb of the HBA gene region indicating a homozygous deletion. As an example of a non-deleted region, the other embryos from the unrelated couple showed sequence fragments spanning throughout the entire HBA loci, indicating an intact gene region. For the other autosomes, bin regions were in the same ratio confirming that the nanopore could deliver a simple but effective aneuploid screening test. Since the nanopore sequencing was able to identify homozygous deletions of this size in the amplified DNA, it could be considered for direct use in this PGT-M application or potentially in similar cases involving a sufficiently large pathogenic microdeletion or microduplication. For example, in addition to the --SEA deletion analyzed herein, TGS may also be similarly suitable for PGT-M of other large HBA deletions such as the --THAI, −-FIL and --MED deletions [17] that are common in alpha thalassemia major. Such deep sequencing would however again come at a high cost since in order to get the required resolution, only one sample could be run on a single cell.
For routine PGT-A applications, a simple low pass analysis revealed that both balanced and unbalanced chromosome profiles were reproducibly identifiable with 500,000 fragment reads—offering an opportunity for a relatively inexpensive approach when multiplexed samples are run in a single flow cell. For some translocation couples, a balanced profile is sufficient for their requirements, whereas for others, the identification of balanced non-carriers is also desired in order to avoid the same fertility predicament in future generations [12]. The embryos from the translocation couple also simply demonstrated that with deep sequencing, data was able to directly identify the actual chromosome breakpoints as well as the surrounding DNA sequences, and hence, technically identify balanced carriers from non-carriers. Such deep sequencing for every embryo would however result in relatively high cost per embryo tested.
In typical workup situations, such junction information for any known structural variant may be acquired from a carrier parent prior to any PGT as part of the workup and could be used for designing a specific, less-expensive PCR-based junction fragment test, similar to that described here for a deletion in the HBA gene region. As with alpha thalassemia PGT-M, such an approach to a detailed PGT-SR could reduce the costs per embryo tested. A PCR junction fragment approach was in fact used for this couple for their actual PGT cycle, but it was not a straight forward nor expedient test development since a mate paired-end NGS approach was used. This required randomly fragmented genomic DNA from the carrier parent, ~ 3 kb length, to initially identify the approximate translocation breakpoint [18]. This information was then used in a step-wise PCR test development program to finally arrive at a PCR junction fragment suitable for testing embryos. The long read sequence approach offered by the nanopore facilitates rapid identification of the breakpoint and so simplifies the final development of any specific PCR-based junction fragment test if requested.
In addition to junction fragment analysis, potential linked polymorphic markers near a gene of interest were able to be determined and interrogated. We showed that analysis of reads near to the HBA region was able to identify a polymorphic STR marker, namely, D16S3400 that has been previously used to assist diagnosis of embryos at risk for alpha thalassemia [19]. Thus, TGS would make the selection of appropriate linked polymorphic markers and design of PCR primers more straightforward for many of the difficult PGT-M cases performed in the past [20]. While SNP analysis might also be considered for linkage confirmations, the underlying base call error rates of 5–15% intrinsic in all current TGS systems does make interpretation not as straightforward. Such screening during workup could substantially reduce the time needed for screening of potential informative markers and thus further simplify composite PGT-M test establishment.
It was demonstrated that the Oxford Nanopore system was capable of both detailed embryo analyses and a more straightforward PGT-A screen. By using different subsets of fragment reads, we could show that as few as 250,000 reads would be necessary for any routine simple euploid test [21]. Since this represented only 5% of cell capacity, it opens the opportunity for barcoded multiplexed samples per cell offering a relatively low-cost approach for routine embryo PGT-A. In situations where high resolution is required, then fewer samples could be accommodated. This versatile approach to analysis means that even small clinics can potentially do effective workups for specific cases by deep sequencing to get the required information and then setting up specific tests for PGT or alternatively just use deep sequencing of embryos as the test itself. In addition, the clinic would have ample capacity to perform their own inhouse PGT-A for any patient at a reasonable cost per embryo tested.
Due to the low cost per sample sequenced, quality of results, and relatively simple workflows, NGS essentially replaced FISH and array CGH for PGT-A applications in most parts of the world [1]. A big impediment though, to wider uptake, especially for small-to-medium-sized clinics, is the initial cost and ongoing maintenance of NGS equipment needed for the sequencing. In groups with only low to medium PGT-A requirements, the economics of the NGS approach make inhouse analyses expensive—often prohibitively so. A versatile platform that can assist in test workup as well as perform basic PGT-A and some more advanced analysis at a low entry cost would enable more groups to participate in both PGT-M test development as well as advanced embryo selection for improved IVF outcomes. We assessed the Oxford Nanopore system to investigate whether it was a potential alternative to NGS systems. Unlike the PacBio TGS system where capital equipment costs are similar to NGS and very high, the Oxford system has, at least for entry level, capital costs that are essentially zero since discovery kits include the basic module needed to run the nanopore. The materials costs for running are similar for both Oxford and PacBio platforms and currently is ~ < $1000 USD per run. The simplicity of this instrument means it can be run from the average laptop, further reducing capital expenditure. Scaling up to very large scale does involve capital expenditure but it is still only a fraction of the price of the NGS basic instrument. Laboratory technical skill requirements are similar in all PGT applications and are no different for the approach described in this study.
Conclusions
The Oxford Nanopore offers a cost attractive solution for laboratories of small to medium throughput to enter PGT services and provide inhouse solutions. The flexibility of the platform also enables some detailed investigations of patients for directed test design or altered analysis of embryos to improve resolution of copy number variations of both large and even small insertions/deletions. The variety of available flow cells enable users to adjust scale of analysis or depth of testing/resolution and in combination with newly released barcodes offers the opportunity to run multiple samples on one cell, reducing cost per analysis. The intrinsically higher error rates for TGS compared to NGS mean that identifying point mutations in PGT-M will need some further development. We are currently investigating approaches to direct utilization of the nanopore system for effective use in analyzing DNA fragments holding any specified mutation in PGT-M applications. We are also looking at different ways of data analysis that overcome the reduced accuracy of base calls for SNPs so that TGS can develop further and potentially be considered as a universal approach to all types of PGT applications.
Acknowledgments
We thank all the staff participating in the study.
Funding
The study was supported by grant awarded to Yuanqing Yao by the National Key Research and Development Program of China (2018YFC1003100) and a grant awarded to Don Leigh by the Yunnan Provincial High-end Foreign Experts Project.
Data availability
Not applicable.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
The research study was approved by the Ethics Committee of the First People’s Hospital of Kunming (Approval number YLS2019-27).
Consent to participate
Informed consent was obtained from all individual participants included in the study.
Consent for publication
All authors provided consent for publication.
Code availability
Not applicable.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Li Wang, Email: pkulwang@126.com.
Yuanqing Yao, Email: yqyao_ghpla@163.com.
References
- 1.Viotti M. Preimplantation genetic testing for chromosomal abnormalities: aneuploidy, mosaicism, and structural rearrangements. Genes (Basel) 2020;11:602. doi: 10.3390/genes11060602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gutiérrez-Mateo C, Colls P, Sánchez-García J, Escudero T, Prates R, Ketterson K, Wells D, Munné S. Validation of microarray comparative genomic hybridization for comprehensive chromosome analysis of embryos. Fertil Steril. 2011;95:953–958. doi: 10.1016/j.fertnstert.2010.09.010. [DOI] [PubMed] [Google Scholar]
- 3.Fiorentino F, Biricik A, Bono S, Spizzichino L, Cotroneo E, Cottone G, Kokocinski F, Michel CE. Development and validation of a next-generation sequencing-based protocol for 24-chromosome aneuploidy screening of embryos. Fertil Steril. 2014;101:1375–1382. doi: 10.1016/j.fertnstert.2014.01.051. [DOI] [PubMed] [Google Scholar]
- 4.Wang L, Cram DS, Shen J, Wang X, Zhang J, Song Z, et al. Validation of copy number variation sequencing for detecting chromosome imbalances in human preimplantation embryos. Biol Reprod. 2014;91:37. doi: 10.1095/biolreprod.114.120576. [DOI] [PubMed] [Google Scholar]
- 5.Jain M, Koren S, Miga KH, Quick J, Rand AC, Sasani TA, Tyson JR, Beggs AD, Dilthey AT, Fiddes IT, Malla S, Marriott H, Nieto T, O’Grady J, Olsen HE, Pedersen BS, Rhie A, Richardson H, Quinlan AR, Snutch TP, Tee L, Paten B, Phillippy AM, Simpson JT, Loman NJ, Loose M. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat Biotechnol. 2018;36:338–345. doi: 10.1038/nbt.4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Au CH, Ho DN, Ip BBK, Wan TSK, Ng MHL, Chiu EKW, Chan TL, Ma ESK. Rapid detection of chromosomal translocation and precise breakpoint characterization in acute myeloid leukemia by nanopore long-read sequencing. Cancer Genet. 2019;239:22–25. doi: 10.1016/j.cancergen.2019.08.005. [DOI] [PubMed] [Google Scholar]
- 7.Chow JFC, Cheng HHY, Lau EYL, Yeung WSB, Ng EHY. High-resolution mapping of reciprocal translocation breakpoints using long-read sequencing. MethodsX. 2019;6:2499–2503. doi: 10.1016/j.mex.2019.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jeck WR, Lee J, Robinson H, Le LP, Iafrate AJ, Nardi V. A nanopore sequencing-based assay for rapid detection of gene fusions. J Mol Diagn. 2019;21:58–69. doi: 10.1016/j.jmoldx.2018.08.003. [DOI] [PubMed] [Google Scholar]
- 9.Merker JD, Wenger AM, Sneddon T, Grove M, Zappala Z, Fresard L, Waggott D, Utiramerur S, Hou Y, Smith KS, Montgomery SB, Wheeler M, Buchan JG, Lambert CC, Eng KS, Hickey L, Korlach J, Ford J, Ashley EA. Long-read genome sequencing identifies causal structural variation in a Mendelian disease. Genet Med. 2018;20:159–163. doi: 10.1038/gim.2017.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang S, Liang F, Lei C, Wu J, Fu J, Yang Q, Luo X, Yu G, Wang D, Zhang Y, Lu D, Sun X, Liang Y, Xu C. Long-read sequencing and haplotype linkage analysis enabled preimplantation genetic testing for patients carrying pathogenic inversions. J Med Genet. 2019;56:741–749. doi: 10.1136/jmedgenet-2018-105976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miao H, Zhou J, Yang Q, Liang F, Wang D, Ma N, Gao B, du J, Lin G, Wang K, Zhang Q. Long-read sequencing identified a causal structural variant in an exome-negative case and enabled preimplantation genetic diagnosis. Hereditas. 2018;155:32. doi: 10.1186/s41065-018-0069-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chow JFC, Cheng HHY, Lau EYL, Yeung WSB, Ng EHY. Distinguishing between carrier and noncarrier embryos with the use of long-read sequencing in preimplantation genetic testing for reciprocal translocations. Genomics. 2020;112:494–500. doi: 10.1016/j.ygeno.2019.04.001. [DOI] [PubMed] [Google Scholar]
- 13.Wei S, Weiss ZR, Gaur P, Forman E, Williams Z. Rapid preimplantation genetic screening using a handheld, nanopore-based DNA sequencer. Fertil Steril. 2018;110:910–6.e2. doi: 10.1016/j.fertnstert.2018.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang W, Liu Y, Wang L, Wang H, Ma M, Xu M, Xu X, Gao ZY, Duan J, Cram DS, Yao Y. Clinical application of next-generation sequencing in preimplantation genetic diagnosis cycles for Robertsonian and reciprocal translocations. J Assist Reprod Genet. 2016;33:899–906. doi: 10.1007/s10815-016-0724-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Basha B, Mularo F, Cook JR. Design, validation, and clinical implementation of a gap-polymerase chain reaction method for α-thalassemia genotyping using capillary electrophoresis. Hemoglobin. 2017;41:124–130. doi: 10.1080/03630269.2017.1327868. [DOI] [PubMed] [Google Scholar]
- 16.Ou Z, Chen Z, Yin M, Deng Y, Liang Y, Wang W, et al. Re-analysis of whole blastocysts after trophectoderm biopsy indicated chromosome aneuploidy. Hum Genomics. 2020. 10.1186/s40246-019-0253-z. [DOI] [PMC free article] [PubMed]
- 17.Galanello R, Cao A. Gene test review. Alpha-thalassemia. Genet Med. 2011;13:83–8. [DOI] [PubMed]
- 18.Wang L, Shen J, Cram DS, Ma M, Wang H, Zhang W, et al. Preferential selection and transfer of euploid noncarrier embryos in preimplantation genetic diagnosis cycles for reciprocal translocations. Fertil Steril. 2017;108:620–7.e4. [DOI] [PubMed]
- 19.Kuliev A, Rechitsky S, Verlinsky O, Tur-Kaspa I, Kalakoutis G, Angastiniotis M, et al. Preimplantation diagnosis and HLA typing for haemoglobin disorders. Reprod BioMed Online. 2005;11:362–70. [DOI] [PubMed]
- 20.Rechitsky S, Pakhalchuk T, San Ramos G, Goodman A, Zlatopolsky Z, Kuliev A. First systematic experience of preimplantation genetic diagnosis for single-gene disorders, and/or preimplantation human leukocyte antigen typing, combined with 24-chromosome aneuploidy testing. Fertil Steril. 2015;103:503–12. [DOI] [PubMed]
- 21.Escribà MJ, Vendrell X, Peinado V. Segmental aneuploidy in human blastocysts: a qualitative and quantitative overview. Reprod Biol Endocrinol. 2019;17:76. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.