

Abstract
Several native and engineered heat‐stable DNA polymerases from a variety of sources are used as powerful tools in different molecular techniques, including polymerase chain reaction, medical diagnostics, DNA sequencing, biological diversity assessments, and in vitro mutagenesis. The DNA polymerase from the extreme thermophile, Thermus scotoductus strain K1, (TsK1) was expressed in Escherichia coli, purified, and characterized. This enzyme belongs to a distinct phylogenetic clade, different from the commonly used DNA polymerase I enzymes, including those from Thermus aquaticus and Thermus thermophilus. The enzyme demonstrated an optimal temperature and pH value of 72–74°C and 9.0, respectively, and could efficiently amplify 2.5 kb DNA products. TsK1 DNA polymerase did not require additional K+ ions but it did need Mg2+ at 3–5 mM for optimal activity. It was stable for at least 1 h at 80°C, and its half‐life at 88 and 95°C was 30 and 15 min, respectively. Analysis of the mutation frequency in the amplified products demonstrated that the base insertion fidelity for this enzyme was significantly better than that of Taq DNA polymerase. These results suggest that TsK1 DNA polymerase could be useful in various molecular applications, including high‐temperature DNA polymerization.
Keywords: DNA polymerase, fidelity, heterologous expression, PCR, thermostability, Thermus
The DNA polymerase from the extreme thermophile Thermus scotoductus strain K1 (TsK1) was expressed in Escherichia coli, purified, and characterized. The enzyme demonstrated an optimal temperature and pH value of 72–74°C and 9.0, respectively, and could efficiently amplify 2.5 kb DNA products. TsK1 DNA polymerase did not require additional K+ ions but it did need Mg2+ at 3–5 mM for optimal activity. The base insertion fidelity for this enzyme was significantly better than that of Taq DNA polymerase.

1. INTRODUCTION
DNA polymerase is an omnipresent enzyme that synthesizes complementary DNA strands from an existing template in living cells. DNA polymerases are widely used for a variety of molecular techniques that rely on DNA manipulation, including polymerase chain reaction (PCR), molecular cloning, sequencing, DNA labeling, and mutagenesis among others (Gibbs et al., 2009; Killelea et al., 2009; Coulther et al., 2019). Several thermostable DNA polymerases have been isolated and studied in prokaryotes (Cho et al., 2012; Kim et al., 2007; Lee et al., 2009; Terpe, 2013; Zhang et al., 2015). These enzymes are grouped into eight families: A, B, C, D, X, Y, RT, and AEP based on their amino acid sequences (Killelea et al., 2009; Coulther et al., 2019). Most thermostable DNA polymerases primarily used in PCR procedures belong to the A‐ and B‐family polymerases from thermophilic bacteria and archaea, respectively. Each thermostable DNA polymerase has its own set of unique characteristics, including thermostability, extension rate, fidelity, processivity, specificity, resistance to contaminants and inhibitors, modified nucleotide selection, ability to bypass damage, nuclease activity, and strand displacement activity. The distinctive properties of each DNA polymerase can be used to create unique reagents (Aschenbrenner & Marx, 2017; Coulther et al., 2019; Reha‐Krantz et al., 2014). This means that the search for novel DNA polymerases has been a major focus for the last couple of decades. A‐type polymerases from the genus Thermus are the most frequently used in molecular biology and include the commonly used Taq DNA polymerase from T. aquaticus. Several polymerases with similarities to Taq have been mined from other Thermus species, including Tfi from T. filiformis, Tfl from T. flavus, Tbr from T. brockianus, Tca from T. caldophilus, and Tth from T. thermophilus. Slight amino acid sequence differences between polymerase enzymes can result in dramatic changes to their biochemical characteristics, suggesting that it is possible to mine for novel polymerases with improved functionality (Gibbs et al., 2009). Other A‐type polymerases have been isolated from Thermotoga spp., including Tma polymerase from T. maritima and Tne from T. neapolitana (Spibida et al., 2017). Despite the number of available enzymes, the growing applications of molecular biology mean that there is still a demand for novel enzymes, and this is where the field of applied science might facilitate continued improvements.
Here, the expression, purification, and characterization of a recombinant DNA polymerase I from Thermus scotoductus strain K1 (TsK1 DNA polymerase) originating from a geothermal spring in Karvachar, Nagorno‐Karabakh (Saghatelyan et al., 2015) is described.
2. MATERIALS AND METHODS
2.1. Source of enzyme
The enzyme was from T. scotoductus strain K1, which was isolated from a geothermal spring located in Karvachar, Nagorno‐Karabakh. The draft genome sequence of T. scotoductus K1 was deposited in the DBJ/EMBL/GenBank database under the RefSeq assembly accession no. GCF_001294665.1 (Saghatelyan et al., 2015). A phylogenetic tree depicting the evolutionary distance between TsK1 DNA polymerase (Accession no. MW080815) and other Thermus spp. polymerases was constructed based on the JTT matrix model using the maximum likelihood method (Jones et al., 1992) in MEGA X software (Kumar et al., 2018). The polI codons were optimized (GenScript) to maximize expression in E. coli while maintaining the original amino acid sequence. The codon‐optimized gene was then synthesized by GenScript (https://www.genscript.com/).
2.2. Cloning
A pUC57‐Mini plasmid harboring the codon‐optimized polI sequence, 2512 bp, was used to facilitate the downstream cloning experiments completed using the FX (fragment exchange) system (Geertsma & Dutzler, 2011). The expression vector p7xC3H (6999 bp) (Addgene, LGC Standards), which contained a T7 promoter, a C‐terminal 3C protease cleavage site, and a C‐terminal 10× His tag, was used as the expression vector. Expression constructs were identified and maintained using kanamycin resistance conferred by the vector.
2.3. Protein expression and purification
1 L of 2×YT (yeast extract and tryptone) broth supplemented with 50 μg/ml kanamycin was used to culture E. coli BL21 (DE3) harboring the C‐10 recombinant expression plasmid at 37°C until an OD600 of 0.4 was obtained. Expression was induced by adding isopropyl β‐D‐1‐thiogalactopyranoside to 0.4 mM and further cultivation at 20 °C for another 16 h (at 200 rpm). Cell harvesting was performed by centrifugation followed by resuspension in buffer R (20 mM NaCl, 50 mM Tris‐HCl pH 7.5, 8% glycerol). The cells were then disrupted by lysozyme and freezing‐thawing, which was followed by sonication. The lysate was then heated at 70 °C for 20 min to precipitate most of the heat‐labile host proteins, and cell wall and insoluble debris were removed by further centrifugation at 7482 g, 4°C for 30 min. The supernatant was filtered using Whatman filters and applied to a HisTalon gravity column (Clontech Laboratories, Inc.), pre‐equilibrated with the same buffer. The column was further washed with buffer R containing 10 mM imidazole. Elution was then performed using buffer R containing 50 mM imidazole. Purification efficiency was evaluated by sodium dodecyl sulfate‐polyacrylamide gel electrophoresis (SDS‐PAGE) (Lee et al., 2009), and the fractions containing the highest concentrations of the target protein were merged. Gel‐filtration on a PD‐10 column (GE Healthcare) was used to remove the excess of imidazole. The purified fractions were then dialyzed against storage buffer (20 mM Tris‐HCl, 40 mM KCl, 0.1 mM DTT, 0.1 mM EDTA, 0.5% Tween‐20, 0.5% Nonidet P40, and 50% glycerol) (Gibbs et al., 2009) and stored at −20°C for further investigation. The final protein concentrations were measured using a Qubit Assay kit (Life Technologies).
2.4. Activity assay
Polymerase activity was evaluated using a method described previously (Choi et al., 2006) with minor modifications. Briefly, the standard reaction mixture (50 μL) contained 20 mM Tris‐HCl (pH 7.5); 40 mM KCl; 2 mM MgCl2; 100 mM dCTP, dATP, and dGTP each; 10 mM dTTP; 0.5 μCi [3H] thymidine 5’‐triphosphate (2.59–3.33 TBq/mmol; PerkinElmer); 1.25 μg activated calf thymus DNA; and 0.5 μL TsK1 DNA polymerase solution. The mixture was incubated at 70°C for 10 min, and the reaction was terminated by placing the mixture on ice and adding 0.5 M EDTA. Subsequently, an aliquot was applied onto a DE81 filter‐paper disc (23 mm; Whatman), which was then dried on a heat block, washed with 0.5 M sodium phosphate (pH 7.0) buffer for 10 min and in 70% (v/v) ethanol for 5 min, and dried. The radioactivity incorporated in the dried filter‐paper disc was then measured using a Tri‐Carb 2900 TR Liquid Scintillation Analyser (PerkinElmer).
To determine the optimal pH, three buffer systems with different buffering capacities (MOPS‐NaOH [pH 6.0–8.0], Tris‐HCl [pH 8.0–9.5], and glycine‐NaOH [pH 9.0–10.0] at 25°C) were tested using this assay system.
To determine the optimal temperature, the reaction mix containing the optimal buffer was incubated at various temperatures (45–80°C) for 10 min and then subjected to the above‐described activity assay.
The influence of K+ ions on polymerase activity was determined by adding various concentrations (from 0 to 200 mM) of KCl to the basic reaction mixture and the dependence of the polymerase on divalent cations (Mg2+, Mn2+) was determined using various concentrations (from 0 to 20 and from 0 to 10 mM, respectively) of MgCl2 and MnCl2.
Various dilutions of the enzyme solution were used in each reaction under optimal pH and temperature conditions to determine the specific activity of TsK1 DNA polymerase. One unit of TsK1 DNA polymerase was defined as the amount of enzyme needed to convert 10 pmol of [3H] TTP into an acid‐insoluble product at 72°C in 10 min.
To investigate the thermostability properties of this enzyme, we subjected the purified enzyme, without any additional stabilizers, to 75, 80, 88, and 95°C for up to 1 h. Aliquots of the enzyme were removed at 5, 15, 30, 45, and 60 min and quenched on ice. The residual activity of these samples was then determined in the optimal reaction mix as described above.
All measurements were carried out in triplicate.
2.5. Fidelity assay
The fidelity assays were performed using the blue‐white screening method described previously (Lee et al., 2009) with some modifications. Briefly, primers pUC19_F (5′‐gcatgaAAGCTTGCATGCCTGCAGGTCGAC‐3′) and pUC19_R (5′‐gcatgaCATATGCGGTGTGAAATACCGCAC‐3′), which incorporate a HindIII and NdeI site, respectively (underlined), were used to amplify a 265 bp fragment of the lacZα gene using pUC19 as a template. The primers were designed manually using the Primer3 online tool (https://www.ncbi.nlm.nih.gov/tools/primer‐blast/), and PCR was performed using TsK1 DNA polymerase, OneTaq (NEB), Fusion (Thermo Fisher), and Taq (Sigma) DNA polymerases in their optimal buffers and assayed under their optimal reaction conditions. The PCR products were then digested with HindIII and NdeI, purified, and ligated to the 2421 bp HindIII/NdeI fragment from pUC19. Each ligation mixture was then used to transform chemically competent E. coli TOP10 cells. Transformed cells were plated on LB agar plates supplemented with 100 μg/ml carbenicillin, 40 μg/ml X‐gal, and 0.3 mM IPTG. Pale blue and white colonies were formed by cells containing mutant plasmids, while blue colonies were formed by cells containing wild‐type plasmids. The percentage of white and pale blue colonies was then calculated.
2.6. PCR assays
PCRs using TsK1 DNA polymerase were then performed using optimized buffer (10 mM Tris‐HCl (pH 9.0), 50 mM KCl, 0.1% Triton X) under the optimal cycling conditions (extension at 68–72°C for 1 min/kb, annealing for 20–30 s, and denaturation at 94°C for 20–30 s). Various commercially available and manually designed (using Primer3 online tool (https://www.ncbi.nlm.nih.gov/tools/primer‐blast/)) primer sets were applied using both genomic and plasmid DNA as a template (Table 1) to create amplicons of different sizes.
Table 1.
Primers and templates used for testing the amplification ability of TsK1
| Primer | Sequence, 5′−3′ | Template | Amplicon size | Reference |
|---|---|---|---|---|
| K517r a | ATTACCGCGGCTGCTGG | Bacterial genomic DNA | 500 bp | Muyzer et al., 1993 |
| A8‐28F a | AGAGTTTGATCCTGGCTCAG | Bacterial genomic DNA | 500 bp | Edwards et al., 1989 |
| 27_F a | GAGTTTGATCCTGGCTCAG | Bacterial genomic DNA | 1.5 kb | Woese et al., 1983 |
| R13 a | AGAAAGGAGGTGATCCAGCC | Bacterial genomic DNA | 1.5 kb | Dorsch & Stackebrandt, 1992 |
| T7 a | TAATACGACTCACTATAGGG | Plasmid p7xC3H | 1929 bp | https://www.addgene.org/mol‐bio‐reference/sequencing‐primers/ |
| T7_term a | GCTAGTTATTGCTCAGCGG | Plasmid p7xC3H | 1929 bp | https://www.addgene.org/mol‐bio‐reference/sequencing‐primers/ |
| Puc19_f b | GCATGAAAGCTTGCATGCCTGCAGGTCGAC | Plasmid pUC19 | 265 bp | This work |
| Puc19_r b | GCATGACATATGCGGTGTGAAATACCGCAC | Plasmid pUC19 | 265 bp | This work |
| Tpol1_F b | ATATCATATGCTGCCGCTGTTTGAGCCGAAGG | pUC57‐Mini plasmid harboring polI gene | 2.5 kb | This work |
| Tpol1_R b | TATACTCGAGTGCCGCCTTCGCGCTCAGCCAG | pUC57‐Mini plasmid harboring polI gene | 2.5 kb | This work |
Commercially available primers,
Manually designed primers
3. RESULTS
3.1. Polymerase gene
The original TsK1 DNA polymerase gene encompassed 2496 bp and encoded an 830 amino acid protein with an estimated molecular mass of 93,613 Da. The alignment of several related amino acid sequences from various Thermus species, available in the NCBI database was used to perform phylogenetic analysis. The phylogenetic tree, rooted using Meiothermus granaticus (Figure 1), showed that TsK1 DNA polymerase was very closely related to the polymerases from T. scotoductus SA‐01 and T. antranikianii and distinct from the DNA polymerase I enzymes from T. aquaticus and T. thermophilus. Moreover, it shared 85.75% sequence similarity with native Taq polymerase.
FIGURE 1.

Phylogenetic tree showing the evolutionary relationship between TsK1 DNA polymerase and other Thermus DNA polymerases based on maximum likelihood analysis. The tree with the highest likelihood (7699.30) is shown, and the bootstrap value (1000 replicates) for each clade is shown next to each branch. The final dataset contained 824 positions. All positions containing gaps or missing data were eliminated (complete deletion option). Bar indicates 0.05 substitutions per amino acid position. Evolutionary analyses were conducted using MEGA X
3.2. Expression and purification of TsK1 DNA polymerase
The overexpression of the desired protein in E. coli was achieved successfully. SDS‐PAGE analysis indicated that the recombinant protein was approximately 94 kDa (Figure 2, lanes 1 and 2), which agrees with the molecular mass calculated based on amino acid sequence, supporting our conclusion that the correct full‐length protein was expressed in this system. In the sonicated extracts, the presence of a significant amount of 94 kDa protein (Figure 2, lane 3) suggests that the expressed protein is soluble. Reduced bands and background following heat treatment suggests successful precipitation of most host proteins (Figure 2, lane 4), and the high degree of homogeneity following affinity chromatography (Figure 2, lane 5) confirmed the presence of a highly purified protein.
FIGURE 2.

Sodium dodecyl sulfate‐polyacrylamide gel electrophoresis of TsK1 DNA polymerase purification: (1) noninduced culture, (2) induced culture, (3) sonicated extract from host cells, (4) supernatant after heat treatment, 5) purified protein, M1—Full Range Rainbow molecular‐mass marker (Amersham)
3.3. Optimal conditions for TsK1 DNA polymerase activity
We evaluated TsK1 DNA polymerase activity between 45 and 80°C and determined that the optimal temperature range for this enzyme was 68–75°C, and the highest activity was observed at 72°C (Figure 3a). An evaluation of the pH requirements for this enzyme revealed that it was most active between pH 8.5 and 9.2 in Tris‐HCl buffer, demonstrating the highest activity at pH 9.0 (Figure 3b). To study the effects of divalent cations on TsK1 DNA polymerase activity, we evaluated various concentrations of MgCl2 and MnCl2 and confirmed that the enzyme was highly dependent on Mg2+ ions. We identified that the Mg2+ ion concentration of 3–5 mM was optimal for the activity of this enzyme, whereas a concentration of >8 mM led to decreased activity. The presence of Mn2+ ions had almost no influence on polymerase activity (Figure 3d).
FIGURE 3.

Properties of TsK1 DNA polymerase. (a) Effect of temperature on TsK1 DNA polymerase activity; (b) effect of pH on TsK1 DNA polymerase activity in MOPS‐NaOH (■), Tris‐HCl (▲), and glycine‐NaOH (●) buffers; (c) effect of KCl concentration on TsK1 DNA polymerase activity; and (d) effect of the divalent cations Mg2+ (■) and Mn2+ (●) on TsK1 DNA polymerase activity. Each point represents the average of 3 measured values, and error bars represent the standard deviation between these 3 values
The influence of KCl on TsK1 DNA polymerase activity is shown in Figure 2c and demonstrates that this enzyme was active at up to 70 mM KCl and that higher concentrations of potassium reduced the enzyme activity of TsK1 DNA polymerase. The specific activity of pure TsK1 DNA polymerase was calculated to be 27 units/µg. We went on to examine the thermostability of TsK1 DNA polymerase and determined that the enzyme was stable at temperatures of 75 and 80°C and was relatively stable at 88°C. It lost half of its activity after 15 min of incubation at 95°C (Figure 4).
FIGURE 4.
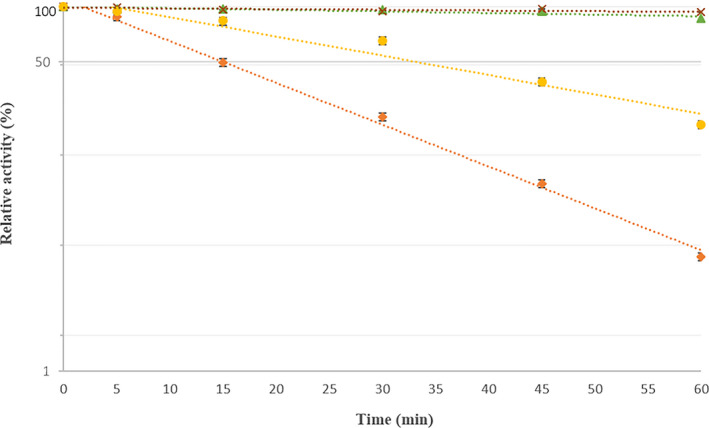
TsK1 DNA polymerase thermostability. ×Represents 75°C, ▲ represents 80°C, ● represents 88°C, ♦ represents 95°C. Data are represented on a logarithmic scale. Each point represents the average of 3 measured values, and error bars represent the standard deviation between these 3 values
3.4. TsK1 DNA polymerase fidelity
More than 33,000 colonies were counted from the TsK1 ligation mix, and approximately 5000 colonies were counted for each of the other enzymes. The proportion of mutant (white) colonies were calculated to be 1.26%, 2.40%, 2.39%, and 0.7792% for TsK1, Taq, OneTaq, and Fusion polymerase, respectively. Various mutant colonies were picked from each experiment and tested using the same primer pair used in the initial amplification. Besides colonies harboring amplicons with the correct size (265 bp), several tested colonies containing ‘double inserts’ were detected as well.
3.5. PCR by TsK1 DNA polymerase
The electrophoretogram of the PCR products amplified by using the TsK1 DNA polymerase is shown in Figure 5. Lanes 2 (500 bp) and 3 (1.5 kb) represent amplicons from bacterial genomic DNA, and lanes 1 (265 bp), 4 (1920 bp), and 5 (2.5 kb) represent products amplified from plasmid templates. The high intensities of the bands suggest that TsK1 DNA polymerase can amplify products of up to 2.5 kb with high efficiency.
FIGURE 5.

Polymerase chain reaction products amplified using TsK1 DNA polymerase. Lane 1, 265 bp; lane 2, 500 bp; lane 3, 1500 bp; lane 4, 1920 bp; lane 5, 2.5 kb; M, GeneRuler 1 kb Plus DNA ladder (New England BioLabs)
4. DISCUSSION
The expression, purification, and characterization of a DNA polymerase I from T. scotoductus K1 are described in the current study. The amino acid sequence comparison showed that this enzyme shares a high degree of similarity to DNA polymerase from T. scotoductus SA‐01. This finding was expected, as according to earlier evaluations of the 16S rRNA gene sequences from these bacteria, they showed >99% similarity (Saghatelyan et al., 2015). TsK1 DNA polymerase is relatively different from T. aquaticus DNA polymerase, which might explain the differences in the behavior of this enzyme as discussed below.
The FX cloning strategy used to clone TsK1 DNA polymerase was based on the use of type IIS restriction enzymes, which digest DNA at an exact distance from their asymmetric recognition sites. The resulting overhang is defined only based on its distance from the recognition site and not by its sequence. Following digestion, the recognition sites are physically separated from the cleavage site, minimizing cloning‐related artifacts in the open reading frame (Geertsma & Dutzler, 2011). The optimization of coding triplets facilitated the proper expression of this thermophilic enzyme in a mesophilic host, thus yielding a significant amount of soluble enzyme. Although heat treatment and further centrifugation eliminated most host proteins, some undesired proteins were still noted in the supernatant (Figure 2, lane 4). The HisTalon gravity column is a cobalt‐charged tetradentate chelator, specific for his‐tagged proteins, and has rigorous requirements for the 3D positioning of histidine residues (only contiguous histidine residues or specially arranged, surrounding histidine residues can bind the cobalt in the reactive core) compared with most commonly used nickel‐based resins. This allows more specific binding of the recombinant His‐tagged TsK1 DNA polymerase, resulting in an almost pure protein (Figure 2, lane 5).
Following optimization of buffering conditions, TsK1 DNA polymerase demonstrated the highest activity at pH 9.0 in Tris‐HCl buffer, which is different from that of Taq that prefers a less alkaline pH (Chien et al., 1976; Lawyer et al., 1993). Polymerase I from T. caldophilus GK24 requires an optimum pH of 8.7 (Park et al., 1993).
TsK1 DNA polymerase activity was rather independent of KCl at lower concentrations, although high concentrations of this salt did inhibit its enzymatic activity. Reportedly, KCl at concentrations above 100 mM inhibits Taq polymerase as well (Chien et al., 1976). Although TsK1 DNA polymerase does not appear to be dependent on KCl when using calf thymus DNA as a template, the influence of KCl may differ with different enzymes and different templates (Lawyer et al., 1993
).
Divalent cations are necessary for polymerization. Here, we demonstrated that the TsK1 DNA polymerase activity was dependent on Mg2+ to some extent. This is in agreement with the literature, which suggests that Mg2+ is a critical component for polymerases (Choi et al., 2006). For TsK1 DNA polymerase, the optimal concentration of Mg2+ is 3–5 mM, in contrast to that for Taq (10 mM), recombinant Taq (2–4 mM), and Tca (12 mM) (Chien et al., 1976; Lawyer et al., 1993; Park et al., 1993).
The optimal temperature for TsK1 DNA polymerase was shown to be 72–74°C, which is comparable with other Taq‐like polymerases (Lawyer et al., 1993; Park et al., 1993) and the lower polymerization activity described at 80°C could also be explained by the template (activated calf thymus DNA) denaturation at higher temperatures, as the TsK1 DNA polymerase was stable at 80°C.
Although the half‐life of TsK1 DNA polymerase (without the use of additional stabilizers) at 95°C (15 min) is lower than that of Taq (Lawyer et al., 1993), it is close to another commercially available rTaq (20 min) polymerase (Gibbs et al., 2009) suggesting that it could still be used in PCR based applications. Furthermore, this characteristic could be enhanced through the application of directed mutagenesis techniques. Some polymerases from Thermus strains show lower thermostability than TsK1 DNA polymerase; these include Wai28A.1, NMX2A.1, Fiji3A.1, and OHA.2 with a half‐life of 4, 3.5, 6, and 2.5 min, respectively, at 95°C. However, these enzymes also show reduced PCR amplification efficiency compared with Taq (Gibbs et al., 2009). In contrast, TsK1 DNA polymerase demonstrated high amplification efficiency using various templates (Figure 5). Of the other Thermus polymerases, only Tca polymerase has been shown to exhibit high thermostability (70 min at 95°C in the presence of gelatin) (Park et al., 1993).
Numerous dilations to the basic PCR approach have been reported and respective enzymes with various degrees of similarity to Taq polymerase have been mined from other Thermus spp. (Gibbs et al., 2009). Gibbs and colleagues compared six recombinant polymerases originating from different Thermus species, which were selected based on their degree of divergence, and demonstrated that all of these enzymes retained similar biochemical characteristics. Moreover, some studies suggest that the other polymerases originating from Thermus spp. can be more efficient in certain cases of PCR in contrast to Taq. For instance, it has been reported, that the reverse transcriptase activity of a recombinant DNA polymerase from T. thermophilus was 100‐fold higher than the RT activity of T. aquaticus DNA pol, although these two strains are closely related (Gibbs et al., 2009). These and other results indicate that few or even one residue variations of polymerases may have a dramatic influence on their biochemical specifications.
PCR itself is a complicated reaction and depends on several factors including polymerase activity, template type, and the concentration of other components. KCl facilitates the elongation of the primer‐DNA complex, and the activity of thermostable DNA polymerases is promoted by K+ ions which guard the negatively charged DNA backbone. This binding decreases the electrostatic repulsion between the two DNA strands and facilitates preferential elongation. During PCR, K+ ions have an optimum promoting effect on DNA polymerases at a concentration of about 50 mM (van Pelt‐Verkuil et al., 2008). Thus, a TsK1 DNA polymerase buffer containing 50 mM KCl and 3 mM MgCl2 was shown to be optimal for the amplification of 2.5 kb products using various templates (Figure 5).
Our results suggest that TsK1 DNA polymerase is twice as accurate as Taq but approximately 1.6 times less accurate than Fusion polymerase. However, Fusion polymerase is approximately 52 times more accurate than Taq (http://tools.thermofisher.com/content/sfs/brochures/phusion‐dna‐polymerase‐brochure.pdf). These results can be explained by the limitations of the method used. First, the fragment size amplified by these enzymes was relatively small (265 bp), thus reducing the chance of an incorrect inclusion. Next, the examination of a few mutant colonies indicated the presence of so‐called double inserts, which resulted in defective lacZ, but these inserts may not be mutated. Also, several ‘silent’ mutations may occur and produce active lacZ but still harbor a mutation. However, some studies using similar methods for fidelity testing indicated mutation frequencies of 2.3, 1.3, 0.85, 3, and 2.3% for rTaq, OHA.2, Wai28A.1, NMX2A.1, and Fiji3A.1 polymerases, respectively (Gibbs et al., 2009). Even with these limitations, we can still confidently assert that TsK1 DNA polymerase is at least 2 times more accurate than Taq but further investigations are needed to confirm this finding.
5. CONCLUSION
Although Taq is the most commonly used polymerase enzyme, there are several other enzymes from other strains of Thermus that exhibit properties similar to Taq polymerase. Here, we describe a novel DNA polymerase from the newly isolated extreme thermophilic T. scotoductus K1. TsK1 DNA polymerase was cloned and expressed using an E. coli host, then purified and characterized. Optimization of the expression and purification conditions yielded a high concentration of recombinant TsK1 DNA polymerase, and this enzyme was shown to exhibit high PCR efficiency. This suggests that TsK1 DNA polymerase may be a good candidate for the development of novel molecular biology methodologies.
CONFLICT OF INTEREST
None declared.
AUTHOR CONTRIBUTION
Ani Saghatelyan: Conceptualization (equal); Formal analysis (equal); Visualization (lead); Writing‐original draft (lead); Writing‐review & editing (equal). Hovik Panosyan: Conceptualization (equal); Formal analysis (equal); Funding acquisition (equal); Project administration (equal); Supervision (equal); Writing‐review & editing (equal). Armen Trchounian: Writing‐review & editing (supporting). Nils‐Kaare Birkeland: Conceptualization (equal); Formal analysis (equal); Funding acquisition (equal); Project administration (equal); Supervision (equal); Writing‐review & editing (lead).
ETHICS STATEMENT
None required.
ACKNOWLEDGEMENTS
We are grateful to Dr. Antonio García‐Moyano at the University of Bergen, for his valuable technical and methodological suggestions and to Dr. Birte Töpper at the University of Bergen, for her assistance in the laboratory. This work was supported by the CPEA‐LT‐2016/10095 and CPEA‐LT‐2017/10061 grants from the Eurasia program of the Norwegian Agency for International Cooperation and Quality Enhancement in Higher Education (Diku) and by the RA MES State Committee for Science, within the scope of the 15T‐1F399 and 18T‐1F261 research projects.
DATA AVAILABILITY STATEMENT
The nucleotide sequence of the synthetic codon‐optimized polI gene encoding the TsK1DNA polymerase is available in GenBank under accession number MW080815: https://www.ncbi.nlm.nih.gov/nuccore/MW080815
REFERENCES
- Aschenbrenner, J. , & Marx, A. (2017). DNA polymerases and biotechnological applications. Current Opinion in Biotechnology, 48, 187–195. 10.1016/j.copbio.2017.04.005 [DOI] [PubMed] [Google Scholar]
- Chien, A. , Edgar, D. B. , & Trela, J. M. (1976). Deoxyribonucleic acid polymerase from the extreme thermophile Thermus aquaticus . Journal of Bacteriology, 127(3), 1550–1557. 10.1128/JB.127.3.1550-1557.1976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho, S. S. , Kim, K. P. , Lee, K. K. , Youn, M. H. , & Kwon, S. T. (2012). Characterization and PCR application of a new high‐fidelity DNA polymerase from Thermococcus waiotapuensis . Enzyme and Microbial Technology, 51(6–7), 334–341. 10.1016/j.enzmictec.2012.07.017 [DOI] [PubMed] [Google Scholar]
- Choi, J. J. , Nam, K. H. , Min, B. , Kim, S. J. , Soll, D. , & Kwon, S. T. (2006). Protein trans‐splicing and characterization of a split family B‐type DNA polymerase from the hyperthermophilic archaeal parasite Nanoarchaeum equitans . Journal of Molecular Biology, 356(5), 1093–1106. 10.1016/j.jmb.2005.12.036 [DOI] [PubMed] [Google Scholar]
- Coulther, T. A. , Stern, H. R. , & Beuning, P. J. (2019). Engineering polymerases for new functions. Trends in Biotechnology, 37(10), 1091–1103. 10.1016/j.tibtech.2019.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsch, M. , & Stackebrandt, E. (1992). Some modifications in the procedure of direct sequencing of PCR amplified 16S rDNA. Journal of Microbiological Methods, 16(4), 271–279. 10.1016/0167-7012(92)90017-X [DOI] [Google Scholar]
- Edwards, U. , Rogall, T. , Blöcker, H. , Emde, M. , & Böttger, E. C. (1989). Isolation and direct complete nucleotide determination of entire genes. Characterization of a gene coding for 16S ribosomal RNA. Nucleic Acids Research, 17(19), 7843–7853. 10.1093/nar/17.19.7843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geertsma, E. R. , & Dutzler, R. (2011). A versatile and efficient high‐throughput cloning tool for structural biology. Biochemistry, 50(15), 3272–3278. 10.1021/bi200178z [DOI] [PubMed] [Google Scholar]
- Gibbs, M. D. , Reeves, R. A. , Mandelman, D. , Mi, Q. , Lee, J. , & Bergquist, P. L. (2009). Molecular diversity and catalytic activity of Thermus DNA polymerases. Extremophiles, 13(5), 817–826. 10.1007/s00792-009-0269-8 [DOI] [PubMed] [Google Scholar]
- Jones, D. T. , Taylor, W. R. , & Thornton, J. M. (1992). The rapid generation of mutation data matrices from protein sequences. Computer Applications in the Biosciences, 8(3), 275–282. 10.1093/bioinformatics/8.3.275 [DOI] [PubMed] [Google Scholar]
- Killelea T., Ralec C., Bossé A., Henneke G. (2014). PCR performance of a thermostable heterodimeric archaeal DNA polymerase. Frontiers in Microbiology, 5, 10.3389/fmicb.2014.00195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, Y. J. , Lee, H. S. , Bae, S. S. , Jeon, J. H. , Lim, J. K. , Cho, Y. , Nam, K. H. , Kang, S. G. , Kim, S. J. , Kwon, S. T. , & Lee, J. H. (2007). Cloning, purification, and characterization of a new DNA polymerase from a hyperthermophilic archaeon, Thermococcus sp. NA1. Journal of Microbiology and Biotechnology, 17(7), 1090–1097. [PubMed] [Google Scholar]
- Kumar, S. , Stecher, G. , Li, M. , Knyaz, C. , & Tamura, K. (2018). MEGA X: Molecular evolutionary genetics analysis across computing platforms. Molecular Biology and Evolution, 35(6), 1547–1549. 10.1093/molbev/msy096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawyer, F. C. , Stoffel, S. , Saiki, R. K. , Chang, S. Y. , Landre, P. A. , Abramson, R. D. , & Gelfand, D. H. (1993). High‐level expression, purification, and enzymatic characterization of full‐length Thermus aquaticus DNA polymerase and a truncated form deficient in 5’ to 3’ exonuclease activity. PCR Methods and Applications, 2(4), 275–287. 10.1101/gr.2.4.275 [DOI] [PubMed] [Google Scholar]
- Lee, J. I. , Kil, E.‐J. , Song, J.‐G. , Kim, Y. J. , Choi, J. J. , Shim, H. , & Kwon, S.‐T. (2009). Characterization and PCR optimization of the thermostable family B DNA polymerase from Thermococcus guaymasensis . Enzyme and Microbial Technology, 45(2), 103–111. 10.1016/j.enzmictec.2009.05.003 [DOI] [Google Scholar]
- Muyzer, G. , de Waal, E. C. , & Uitterlinden, A. G. (1993). Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction‐amplified genes coding for 16S rRNA. Applied and Environmental Microbiology, 59(3), 695–700. 10.1128/aem.59.3.695-700.1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, J. H. , Kim, J. S. , Kwon, S. T. , & Lee, D. S. (1993). Purification and characterization of Thermus caldophilus GK24 DNA polymerase. European Journal of Biochemistry, 214(1), 135–140. 10.1111/j.1432-1033.1993.tb17905.x [DOI] [PubMed] [Google Scholar]
- Reha‐Krantz, L. J. , Woodgate, S. , & Goodman, M. F. (2014). Engineering processive DNA polymerases with maximum benefit at minimum cost. Frontiers in Microbiology, 5, 380 10.3389/fmicb.2014.00380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saghatelyan A., Poghosyan L., Panosyan H., Birkeland N.‐K. (2015). Draft Genome Sequence of Thermus scotoductus Strain K1, Isolated from a Geothermal Spring in Karvachar, Nagorno Karabakh. Genome Announcements, 3, (6), 10.1128/genomea.01346-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spibida, M. , Krawczyk, B. , Olszewski, M. , & Kur, J. (2017). Modified DNA polymerases for PCR troubleshooting. Journal of Applied Genetics, 58(1), 133–142. 10.1007/s13353-016-0371-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terpe, K. (2013). Overview of thermostable DNA polymerases for classical PCR applications: from molecular and biochemical fundamentals to commercial systems. Applied Microbiology and Biotechnology, 97(24), 10243–10254. 10.1007/s00253-013-5290-2 [DOI] [PubMed] [Google Scholar]
- van Pelt‐Verkuil, E. , van Belkum, A. , & Hays, J. P. (2008). Principles and technical aspects of PCR amplification. Springer; 10.1007/978-1-4020-6241-4 [DOI] [Google Scholar]
- Woese, C. R. , Gutell, R. , Gupta, R. , & Noller, H. F. (1983). Detailed analysis of the higher‐order structure of 16S‐like ribosomal ribonucleic acids. Microbiological Reviews, 47(4), 621–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, L. , Kang, M. , Xu, J. , & Huang, Y. (2015). Archaeal DNA polymerases in biotechnology. Appled Microbiology and Biotechnology, 99(16), 6585–6597. 10.1007/s00253-015-6781-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The nucleotide sequence of the synthetic codon‐optimized polI gene encoding the TsK1DNA polymerase is available in GenBank under accession number MW080815: https://www.ncbi.nlm.nih.gov/nuccore/MW080815