

Abstract
Background:
Helicobacter pylori infection leads to regulatory T-cell (Treg) induction in infected mice, which contributes to H. pylori immune escape. However, the mechanisms responsible for H. pylori induction of Treg and immune tolerance remain unclear. We hypothesized DC-produced TGF-β may be responsible for Treg induction and immune tolerance.
Materials and Methods:
To test this hypothesis, we generated TGF-βΔDC mice (CD11c+ DC-specific TGF-β deletion) and assessed the impact of DC-specific TGF-β deletion on DC function during Helicobacter infection in vitro and in vivo. To examine the T-cell independent DC function, we crossed TGF-βΔDC mice onto Rag1KO background to generate TGF-βΔDCxRag1KO mice.
Results:
When stimulated with H. pylori, TGF-βΔDC BMDC/splenocyte cocultures showed increased levels of proinflammatory cytokines and decreased levels of anti-inflammatory cytokines compared to control, indicating a proinflammatory DC phenotype. Following 6 months of H. felis infection, TGF-βΔDC mice developed more severe gastritis and a trend towards more metaplasia compared to TGF-βfl/fl with increased levels of inflammatory Th1 cytokine mRNA and lower gastric H. felis colonization compared to infected TGF-βfl/fl mice. In a T-cell deficient background using TGF-βΔDCxRag1KO mice, H. felis colonization was significantly lower when DC-derived TGF-β was absent, revealing a direct, innate function of DC in controlling H. felis infection independent of Treg induction.
Conclusions:
Our findings indicate that DC-derived TGF-β mediates Helicobacter-induced Treg response and attenuates the inflammatory Th1 response. We also demonstrated a previously unrecognized innate role of DC controlling Helicobacter colonization via a Treg independent mechanism. DC TGF-β signaling may represent an important target in the management of H. pylori.
Introduction
Helicobacter pylori is the most common bacterial infection in humans worldwide and is present in more than half the world’s population. Infection is more common in developing countries, affecting up to 80% of individuals, and is thought to be related to poor hygienic conditions1,2. Interestingly, the prevalence of H. pylori infection is inversely correlated with atopic dermatitis3, asthma4–6, IBD7,8, and rheumatoid arthritis9, which is hypothesized to be related to the hygiene hypothesis or immunomodulatory effects of the bacterium itself10–12.
H. pylori is a gram-negative bacterium capable of colonizing the stomach and leading to chronic infection, contributing to the development of peptic ulcer disease, atrophic gastritis, MALT lymphoma, and gastric adenocarcinoma, which is the third leading cause of cancer mortality worldwide13. Though infected individuals generate a robust immune response, failure to eradicate the organism is common14.
Several mechanisms behind this immune evasion and subsequent persistent infection have been proposed. These include antigenic variation, modulation of adhesion to gastric epithelial cells, evasion of pattern recognition, direct inhibition of T cell proliferation via vacA, and induction of a Treg response that counters T cell immunity15,16. The evidence supporting Treg expansion is particularly robust; patients with H. pylori infection have demonstrated elevated levels of CD4+CD25+ Tregs in the gastric and duodenal mucosa compared to non-infected patients17, and there is a correlation between Foxp3+ Tregs and degree of H. pylori colonization18. Additionally, depletion of CD25+Foxp3+ Tregs in H. pylori-infected mice leads to increased gastric inflammation and reduced bacterial colonization19. Local gastric mucosal infection with H. pylori in mice has also been associated with the appearance of peripherally induced Tregs in the lung20. We previously showed that H. pylori alters the DC-polarized Th17/Treg balance toward a Treg-biased response, which suppresses the effective induction of H. pylori-specific Th17 immunity21. Treg depletion in a genetic model has resulted in significant inflammatory immune response and spontaneous H. pylori clearance22. However, the specific mechanisms behind the induction of Treg differentiation in H. pylori infection are not well understood.
Emerging evidence demonstrates that dendritic cells (DCs) are involved in the response to H. pylori infection23. We have shown that DCs are recruited to the gastric mucosa after H. pylori infection21,24. In another study, DC-depleted neonatally infected mice showed a significant reduction in H. pylori CFUs compared to TGF-βfl/fl infected mice25. DC-depleted mice infected with H. pylori also display more severe gastritis and generate stronger Th1 and Th17 responses26.
DCs are a rich source of TGF-β, which modulates T cell regulation and differentiation27. TGF-β is an important immunomodulator for T cell regulation and differentiation, inducing Treg as well as Th17 differentiation28,29. H. pylori specific immune tolerance requires TGF-β signaling, and mice with a dominant-negative form of the TGF-β receptor II have demonstrated impaired Treg induction and immune tolerance22. Hence, we hypothesized that DC-derived TGF-β mediates Treg induction, which conveys host immune tolerance in response to H. pylori infection.
To test this hypothesis, we generated DC-specific TGF-β knockout C57BL6 mice (TGF-βΔDC) to demonstrate that this group of DC TGF-β deficient mice exhibit more severe mucosal inflammation and have a lower degree of bacterial colonization. In vitro studies using BMDCs from these TGF-βΔDC mice showed increased levels of pro-inflammatory cytokines following stimulation with H. pylori compared to control. To evaluate whether TGF-β can induce immune tolerance independent of Treg response, we crossed TGF-βΔDC mice onto Rag1 KO background and generated TGF-βΔDCxRag1KO double KO mice. Our studies indicate that DC-derived TGF-β plays an important role in the induction of Treg and attenuation of inflammatory Th1 response following Helicobacter infection. Also, TGF-β may modulate immune tolerance independent of Treg, suggesting an innate component to TGF-β signaling.
Methods
Mice
Mice (B6.C-Tg(itgax-cre)1–1Reiz/J, TGF-βtm2.1Doe/J, and Rag1KO) were purchased from Jackson Laboratory for breeding. We used the Cre/lox system to generate DC-specific TGF-β1 knockout C57BL6 mice (TGF-βΔDC) by crossing cCD11c-cre mice with TGF-β1 flox-ex6 mice (Jackson Lab) and generated TGF-βΔDC-Rag1KO mice by crossing TGF-βΔDC with Rag1KO (Jackson Lab) mice. TGF-β1 flox-ex6 mice served as the TGF-βfl/fl control. All animals were housed in the animal maintenance facility at the University of Michigan Health System. This research was undertaken with the approval of the Committee on Use and Care of Animals at the University of Michigan. Mouse genotypes were confirmed by quantitative PCR using mouse tails.
Media and cytokines
For all cell cultures, a complete medium consisted of RPMI-1640 (Sigma, Milwaukee, WI) with 10% heat-inactivated fetal calf serum (ISC Biosciences, Kaysville, UT), 2 mM added Glutamine (4 mM total), and 100 U/mL Penicillin-Streptomycin. The following recombinant cytokines (R&D Systems, Minneapolis, MN) were diluted in complete medium: mGM-CSF (10 ng/mL) and IL-4 (10 ng/mL) for BMDC.
Generation of bone marrow-derived DCs
BMDCs from TGF-βfl/fl or TGF-βΔDC mice were derived using mouse GM-CSF (10 ng/mL) and IL-4 (10 ng/mL) as previously described19 except BMDCs were cultured with serum free RPMI1640 to exclude exogenous serum TGF-β. and cultured with RPMI1640 containing 10% fetal bovine serum (FBS) BMDCs were harvested and enriched (106 cells/mL) by gradient centrifugation using OptiPrep density solution (Sigma, St. Louis, MO) according to the manufacturer’s instructions on day 6. For H. pylori-stimulated BMDC experiments, 1 × 106 cells/mL of BMDCs were plated in a 12 well plate, treated with 107CFU/mL H. pylori (DC to H. pylori ratio of 1 to 10), 107CFU/mL Escherichia coli (E. coli) (DC to E. coli ratio of 1 to 10), PBS, or E. coli lipopolysaccharide (LPS). After overnight (18h) culture, the supernatant was harvested and TGF- β was measured using ELISA.
Helicobacter culture and infection
H. pylori SS1 was cultured on Campylobacter-selective agar (BD Diagnostics, Bedford, MA, USA) for 3 days in a humidified microaerophilic chamber at 37°C (BBL Gas System, with CampyPak Plus packs, BD Biosciences San Jose, CA) as previously described21.
H. felis was cultured in sterile-filtered Brucella broth (BD, Franklin Lakes, NJ) with 10% FBS (Atlanta Biologicals, Lawrenceville, GA) using the GasPak™ EZ Campy Container System (BD) at 37°C at an agitation rate of 150 rpm for 3–5 days. The cultures were spun down at 2700 rpm at room temperature, and the pellets resuspended in Brucella broth plus 10% FBS (Thermo Fisher Scientific, Houston, TX). Bacteria were counted using a hemocytometer by diluting the cells 1∶100 in 9∶1 HBSS/Formalin solution. TGF-βΔDC, TGF-βfl/fl, TGF-βΔDCxRag1KO, and control TGF-βΔDC mice were gavaged 3 times over 5 days with 108 CFU H. felis in 100 μL of Brucella broth.
Animal studies
After 6 months infection with H. felis, the mice were euthanized. The stomach was removed and analyzed. In addition, splenocytes from TGF-βfl/fl or TGF-βΔDC mice were cocultured for 18h with BMDCs from uninfected control mice and 107 CFU/mL H. felis. The splenocyte-to-BMDC ratio was 10 to 1. After 72 h, the supernatant was collected and IL-12p70, IFN-γ, and TNF-α levels were measured by ELISA (eBioscience/BD Biosciences, San Diego, CA/San Jose, CA). Splenocytes were collected and the percentages of CD4+FoxP3+ T cells (Treg) were measured by fluorescence-activated cell sorting (FACS).
Histological scoring
The stomachs of mice were removed and two adjacent full-thickness longitudinal strips were removed from the lesser and greater curvatures of the stomach and fixed in formalin for histologic analysis. The specimens were scored according to previously published protocol30. Briefly, 200x microscopic fields were scored individually for the presence or absence of each of the following 4 histological criteria: 1) polymorphonuclear leukocytes neutrophilic (PMN) infiltration, 2) mononuclear infiltration, 3) follicles, and 4) epithelial metaplasia. The gastritis score is defined as the the sum of the percentage of 200x microscopic fields with PMN, mononuclear infiltration, and follicles. The percentage of 200x microscopic fields with epithelial metaplasia was also measured.
Extraction of RNA, reverse transcription, and quantitative real-time polymerase chain reaction (RT-PCR)
Total RNA from stomach samples was prepared using the RNeasy Mini Kit (QIAGEN, Hilden, Germany). Samples were reverse-transcribed using iScript™ cDNA Synthesis Kit (BIO-RAD, Hercules, California). Expression of H. felis, TGF-β, TNF-α, IFN-γ, IL-12, IL-6, IL-1β, IL-10, and HPRT RNA was measured using iQ™SYBR Green Supermix Kit obtained from BIO-RAD. Primers are shown in Table 1. Finally, quantitation of relative differences in expression was calculated using the comparative 2−ΔΔCT method31.
Table 1.
Primers and annealing temperatures used for the amplification of each gene.
| Gene | Primer(5’-3’) | Annealing temperature |
|---|---|---|
| HPRT | F:5’-AGGACCTCTCGAAGTGTTGGATAC-3’ R:5’-AACTTGCGCTCATCTTAGGCTTTG-3’ |
65 |
| IL-6 | F:5’-GAGGATACCACTCCCAACAGACC-3’ R:5’-AAGTGCATCATCGTTGTTCATACA-3’ |
65 |
| IL-10 | F:5’-AGTGGAGCAGGTGAAGAGTG-3’ R:5’-TTCGGAGAGAGGTACAAACG-3’ |
58 |
| IFN-γ | F:5’TCAAGTGGCATAGATGTGGAAGAA--3’ R:5’-TGGCTCTGCAGGATTTTCATG-3’ |
65 |
| FoxP3 | F:5’-TCTCCAGGTTGCTCAAAGTC-3’ R:5’-GCAGAAGTTGCTGCTTTAGG-3’ |
58 |
| TNF-α | F:5’-CATCTTCTCAAAATTCGAGTGACAA-3’ R:5’-TGGGAGTAGACAAGGTACAACCC-3’ |
65 |
| TGF-β | F: 5’-GCTACCATGCCAACTTCTGT-3’ R: 5’-CGTAGTAGACGATGGGCAGT-3’ |
58 |
Statistical analysis
The results were evaluated using unpaired Student’s t-tests (Mean±SEM). Statistics were performed in the GraphPad Prism program suite (GraphPad Software, Inc., La Jolla, CA). Significant values were indicated as follows: *P < 0.05, **P < 0.01, and ***P < 0.001.
RESULTS
TGF-βΔDC DCs produce diminished TGF-β and exhibit an inflammatory phenotype
We previously showed that BMDCs produced TGF-β at homeostasis as well as when exposed to H. pylori 21, suggesting DC production of TGF-β may contribute to immune tolerance in H. pylori infection. To test this hypothesis, we generated a DC-specific TGF-β knockout murine model (Figure 1A). We verified DC-specific TGF-β depletion by comparing BMDC TGF-β production in TGF-βfl/fl vs TGF-βΔDC mice (Figure 1B).
Figure 1.
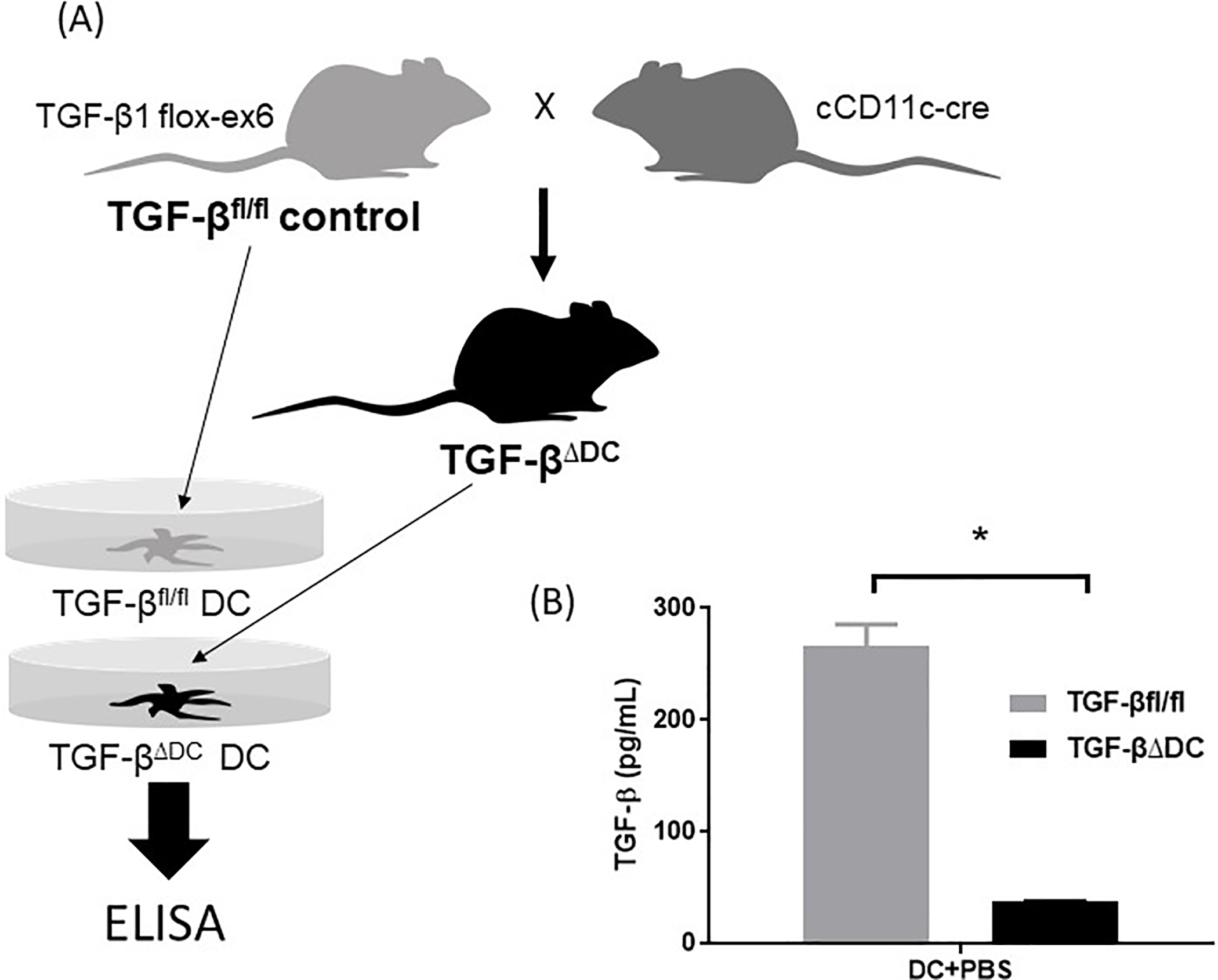
Generation of TGF-βΔDC mice, which are TGF-β-deficient. (A) TGF-βΔDC mice were generated by crossing cD11c-cre mice with TGF-β1 flox-ex6 mice, which were used as the TGF-βfl/fl control. (B) BMDCs derived from wt (TGF-βfl/fl) vs TGF-βΔDC mice were cultured for 18h and supernatant TGF-β was quantified using enzyme-linked immune absorbent assay (ELISA), confirming deficient TGF-β production in the TGF-βΔDC DCs. DC = dendritic cells, PBS = phosphate-buffered saline. Results are shown as mean ± S.E.M. *P < 0.05.
When stimulated with PBS, H. pylori, E. coli, or LPS in vitro, TGF-βΔDC BMDC and splenocyte coculture supernatant contained markedly lower levels of TGF-β than control TGF-βfl/fl BMDC coculture supernatant (Figure 2A). Proinflammatory cytokine levels were significantly higher in the TGF-βΔDC group when stimulated with H. pylori, E. coli, and LPS (Figure 2B). Anti-inflammatory IL-10 levels were decreased in the TGF-βΔDC group compared to control when stimulated with H. pylori, E. coli, and LPS (Figure 2C). Overall, this decrease in anti-inflammatory cytokine levels and an increase in proinflammatory cytokine levels indicates a proinflammatory DC phenotype.
Figure 2.
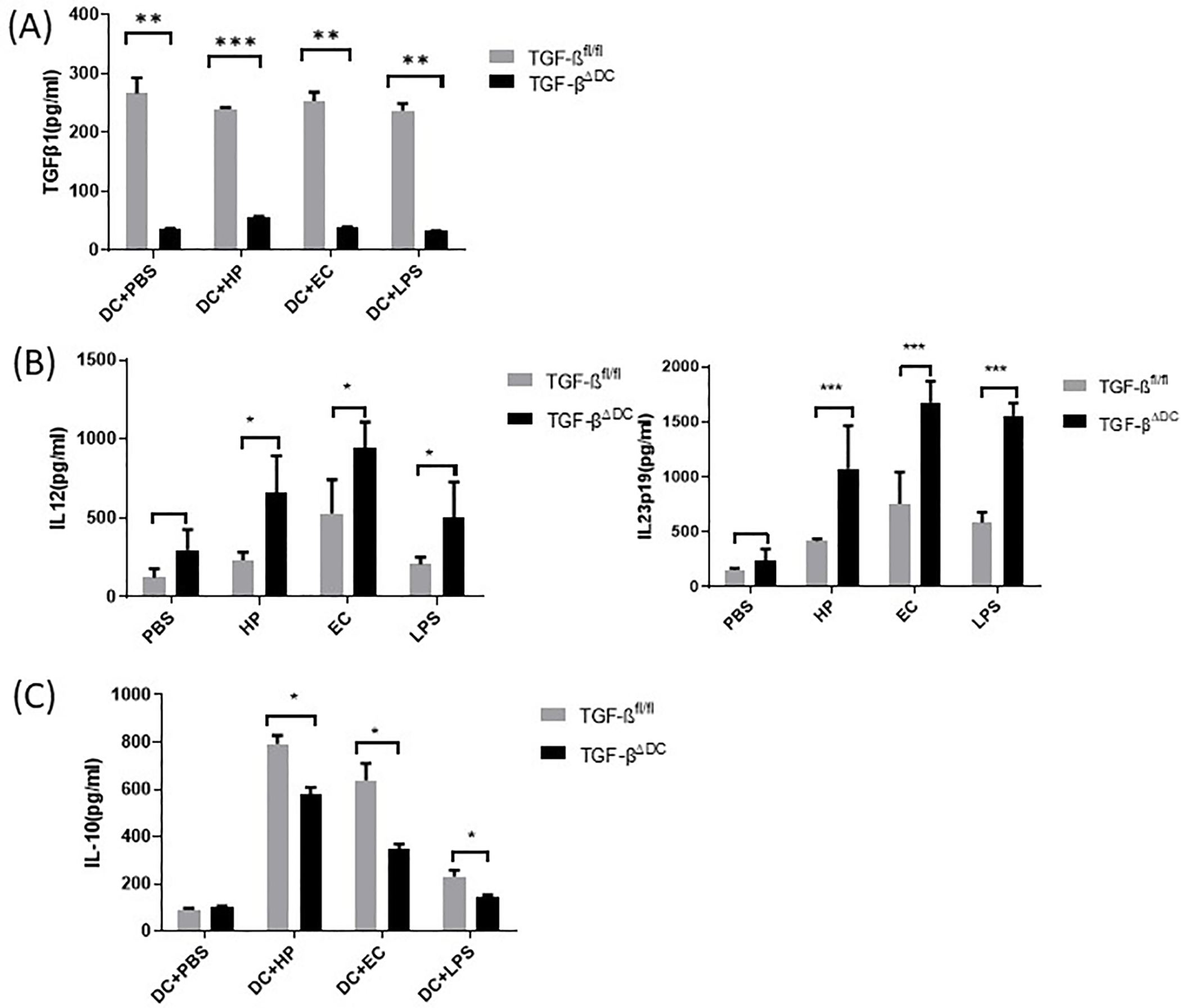
TGF-βΔDC bone marrow-derived DCs produce diminished TGF-β and exhibit an inflammatory phenotype. BMDCs derived from control (TGF-βfl/fl) vs TGF-βΔDC mice were cultured for 18h with H. pylori (107 CFU H. pylori to DC ratio 1:10), E. coli (107 CFU, E. coli to DC ratio 1:10), PBS, or LPS. Supernatant levels of (A) TGF-β were measured. When these BMDCs were cocultured with splenocytes, (B) IL-23p19 and IL-12 as well as (C) IL-10 were measured via ELISA. Data are representative of the results of three independent experiments. DC = dendritic cells, PBS = phosphate-buffered saline, EC = E. coli, HP = H. pylori. Results are shown as mean ± S.E.M. *P < 0.05, ** P<0.01, ***P<0.001.
TGF-βΔDC mice infected with H. felis develop more severe gastritis compared to infected TGF-βfl/fl control mice
Next, we infected the TGF-βΔDC mice and TGF-βfl/fl mice with H. felis (108 CFU/mL H. felis via gavage 3 times over 5 days). H. felis was used as it produces more severe gastritis in mice and achieves higher levels of colonization compared to H. pylori32–34. Our data show that after 6 months of H. felis infection, TGF-βΔDC mice developed more severe gastritis compared to control TGF-βfl/fl mice, as evidenced by increased neutrophils, gland distortion, and metaplasia. Gastric TGF-β mRNA expression was confirmed to be significantly decreased in the TGF-βΔDC mice compared to wildtype (Figure 3A). Representative micrographs of gastric histology are shown in Figure 3B. The gastritis score for the TGF-βΔDC group was 2.7 fold higher than in the control group (p<0.01) (Figure 3C), showing that in the absence of DC-TGF-β, infected mice developed more severe gastritis compared to control. Additionally, there was a trend towards increased metaplasia in the TGF-βΔDC mice although values that did not reach statistical significance (p=0.11) (Figure 3D). These findings indicate DC-derived TGF-β plays a role in modulating gastric inflammation and likely subsequent metaplasia in Helicobacter infection.
Figure 3.
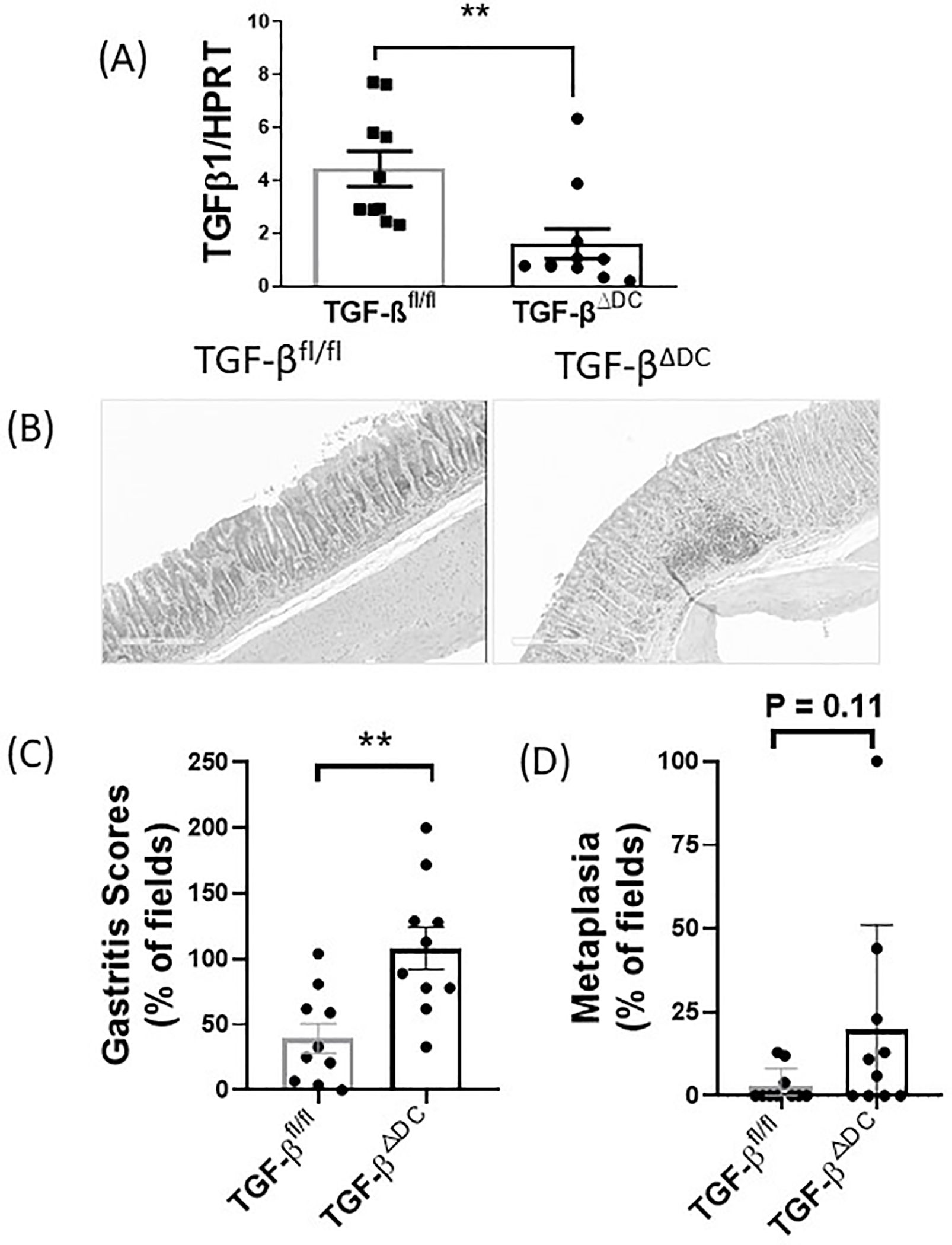
TGF-βΔDC mice infected with H. felis show reduced gastric TGF-β expression and develop more severe gastritis compared to infected control (TGF-βfl/fl) mice. TGF-βΔDC or TGF-βfl/fl C57BL/6 mice (n = 10 per group) were orally challenged with H. felis (108 CFU/mL) 3 times over 5 days starting on day 0. Stomach samples were analyzed after sacrifice at 6 months. Gastritis scores were determined in a blinded fashion. (A) TGF-β expression as measured via quantitative PCR. (B) Micrographs of gastric histology. (C) Gastritis score and (D) metaplasia in stomach samples from wt (TGF-βfl/fl) vs TGF-βΔDC mice infected with H. felis. Results are shown as mean ± S.E.M. **P < 0.01.
TGF-βΔDC mice infected with H. felis display elevated Th1 cytokine production and decreased H. felis-specific Treg response and gastric colonization.
We next examined the in vivo TGF-βΔDC mouse cytokine response to H. felis infection vs control TGF-βfl/fl mouse cytokine response. Stomach samples taken after euthanasia at 6 months showed higher levels of IFN-γ, TNF-α, and IL-12 compared to the levels in the control TGF-βfl/fl group (Figure 4A), indicating a stronger Th1 response. In the TGF-βΔDC mice, levels of IFN-γ, TNF-α, and IL-12 were significantly higher than levels observed in the control group (p<0.05) (Figure 4A). Moreover, the spleens of H. felis-infected TGF-βΔDC mice showed a decreased H. felis-specific Treg response compared to control TGF-βfl/fl spleens (Figure 4B, p<0.05). Also, we determined that increased gastritis severity and inflammatory cytokine production were associated with decreased H. felis colonization. We quantified gastric H. felis mRNA using RT-PCR on the stomach samples from infected mice to measure colonization. Infected TGF-βΔDC mice had a lower degree of gastric H. felis mRNA compared to infected TGF-βfl/fl mice (Figure 4C) indicating lower colonization in the knockout mice. These data support the hypothesis that TGF-β plays an important role in immune tolerance leading to persistent Helicobacter infection.
Figure 4.
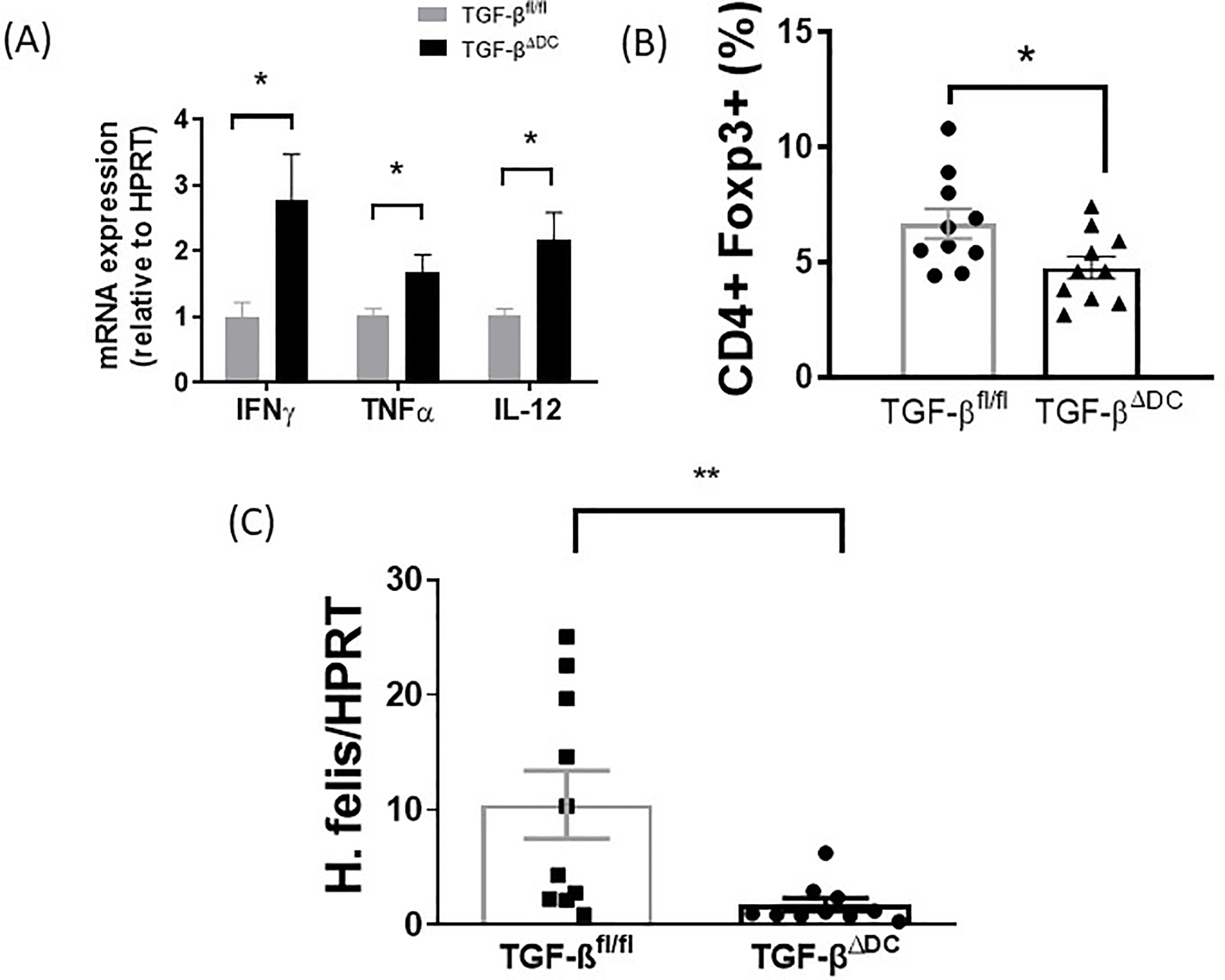
H. felis-infected TGF-βΔDC mice show increased Th1 responses, decreased Treg responses, and increased H. felis colonization compared to control. After chronic H. felis infection of 6-month duration, stomachs from TGF-βfl/fl and TGF-βΔDC mice were removed. Splenocytes from these mice were cocultured with BMDCs from uninfected control mice and 107 CFU/mL H. felis. After 18h, expression of (A) IFNγ, TNFα, and IL-12 mRNA was measured via qPCR. (B) Splenocytes were stimulated with H. felis lysate and H. felis-specific CD4+FoxP3+ T cells via flow cytometry, and (C) H. felis mRNA via quantitative PCR (n=10 mice per group). Results are shown as mean ± S.E.M.* P<0.05, ***P<0.0001.
TGF-βΔDCxRag1KO mice (DC-TGF-β deficient and T cell-deficient) display lower degree of gastric H. felis colonization compared to Rag1 KO mice
To investigate whether TGF-β acts solely via modulation of the adaptive immune response to induce immune tolerance to H. felis, generated TGF-βΔDCxRag1KO mice by crossing T and B cell-deficient Rag1KO mice with TGF-βΔDC mice (Figure 5A). TGF-βfl/fl mice served as the control. We compared H. felis colonization after 6 months in these two groups. As T cells are required for gastritis, neither group of mice displayed evidence of significant histological gastritis (data not shown and previously reported35) and inflammatory cytokines mRNA (IFN-γ, IL-6, IL-1β, IL-10, and TNF-α) measured were not significantly different between the two groups (Figure 5B). However, independent of gastric inflammation, the infected DKO (TGF-βΔDCxRag1KO) mice had lower levels of H. felis mRNA in the gastric tissue compared to infected Rag1KO mice (p<0.05, Figure 5C). This suggests that while Treg response contributes to Helicobacter immune tolerance, DC-derived TGF-β has an additional innate role independent of Treg expansion or modification of T cell response which supports Helicobacter colonization.
Figure 5.

TGF-βΔDCxRag1KO double knockout mice (TGF-β deficient and T cell-deficient) show a lower degree of gastric H. felis colonization independent of inflammatory cytokine levels. After chronic 6-month H. felis infection, the stomachs from Rag1KO and Rag1KO/TGF-βΔDC mice were removed (n=10 mice per group). (A) Schematic representation TGF-βΔDCxRag1KO generation. (B) Gastric cytokine levels including IFN-γ, IL-6, IL-1β, IL-10, and TNF-ɑ were measured via quantitative PCR and were not significantly different between the Rag1KO and TGF-βΔDCxRag1KO mice. (C) H. felis mRNA was measured via quantitative PCR to assess colonization (n=10 mice per group). Results are shown as mean ± S.E.M. * P<0.05.
DISCUSSION
H. pylori colonizes half of the world’s population and most of those infected are asymptomatic. However, H. pylori infection can cause decades-long gastritis. This long term infection and chronic inflammation result in the development of peptic ulcer disease, gastric adenocarcinoma, and MALT lymphoma36–38. Despite persistent gastric inflammation with vigorous humoral and cellular immune responses, humans frequently fail to clear the bacterium and colonization persists for life unless treated. This failure to eradicate H. pylori has been attributed to ineffective host immune response and the induction of immune tolerance.
DCs are recruited to the gastric epithelium during H. pylori infection21,24,39. These antigen-presenting cells can migrate from the peripheral tissue to the draining lymph node or spleen with the captured antigen, where they present the antigen to naïve T-cells and initiate host immunity40. As such, they function as a link between the innate and adaptive immune responses. Depending on the local environment and costimulatory signals, DCs may activate cytotoxic/helper T cells and B cells41. They also help maintain immunologic tolerance to self and commensal bacteria by presenting these antigens in the absence of inflammatory cytokines42. We have previously shown that dendritic cells are recruited to the gastric mucosa following H. pylori infection and that H. pylori can induce tolerogenic programming of DCs to inhibit the host immune response21,24. Using a mouse model of H. pylori infection, we showed that H. pylori DNA downregulates DC production of pro-inflammatory cytokines IL-12 and type 1 interferon43. This may be mediated by increased frequency of an immunoregulatory sequence, TTTAGGG, which likely activates the DNA-sensing TLR-9 signaling pathway44. In addition to its DNA, H. pylori cell wall LPS activates DC TLR-2 to inhibit Th1 immunity and induce immune tolerance45. However, the mediators behind this immunoregulatory function have not been fully elucidated.
Since TGF-β induces naïve T cell differentiation into Foxp3+ regulatory T cells, we hypothesized that TGF-β produced by BMDCs is the key mediator in Treg activation and inhibition of the immune response, leading to the immune tolerance commonly observed in H. pylori infection. To test this hypothesis, we generated DC-specific TGF-β knockout mice and verified the successful knockdown of TGF-β from BMDCs in vitro. When infected with H. felis, these mice developed more severe gastritis accompanied by enhanced Th1 response with marked elevation in IFN-γ and TNF-α production. They also displayed 77% lower colonization compared to wild type mice. The spleen from these TGF-βΔDC mice had a 29% decrease in FoxP3+Tregs compared to wildtype. Taken together, these in vivo and in vitro studies showed that BMDC-derived TGF-β plays an important role in H. pylori infection by modulating gastric inflammation and inducing Treg differentiation, leading to immune tolerance and Helicobacter persistence. This observation is consistent with the known immunomodulatory roles of TGF-β in suppressing effector T cell proliferation and inducing Treg differentiation46,47.
Following H. pylori infection, TGF-β production is upregulated in many cells, including gastric fibroblasts, FoxP3+Tregs, macrophages, and DCs20,48,49. In this study, we demonstrated a clear role for DC-derived TGF-β in Treg expansion. Based on our observations and other findings reported in the literature50–52, we propose that following H. pylori infection, DCs migrate to peripheral lymphoid tissue, release TGF-β, stimulate Treg induction, and thus influence systemic immunity, which may lead to a reduction of inflammatory Th1 cytokines and enhanced colonization.
To examine whether DC-derived TGF-β acts solely by affecting T cell differentiation to induce immune tolerance following H. pylori infection, we generated double knock out mice by crossing TGF-βΔDC with Rag1KO and infected these DKO mice with H. felis. We reasoned that if DC-derived TGF-β acts to induce immune tolerance via Treg induction, the degree of H. felis colonization would be similar between the TGF-βΔDCxRag1KO and Rag1KO mice. As expected, we did not detect gastritis in either group of mice because T cells are required for the development of mucosal inflammation. IFN-γ, IL-6, IL-1β, IL-10, and TNF-ɑ mRNA levels were not significantly different between the Rag1KO and TGF-βΔDCxRag1KO mice, supporting the absence of gastritis. However, we observed a lower H. felis colonization in the TGF-βΔDCxRag1KO mice compared to the Rag1KO mice. This suggests that in addition to acting on adaptive immunity, DC-derived TGF-β may also exert its effects via a T cell-independent pathway. This reveals a direct innate immune function of DCs in the response to Helicobacter infection. It is conceivable that DC-derived TGF-β may act via autocrine signaling pathways that further upregulate DC TGF-β expression, and may have wide-ranging effects on the innate immune populations similar to the effect of Tregs in suppressing innate lymphoid cells53,54. Additionally, TGF-β has been shown to suppress TLR signaling and inhibit myeloid cell activation55,56. These possibilities would be worthwhile targets for investigation, though are beyond the scope of our current study.
In conclusion, our findings demonstrate that DC-derived TGF-β mediates Treg response in H. pylori infection, resulting in an attenuated Th1 inflammatory response. Using a double knockout mouse model, we also demonstrated a previously unrecognized innate role of DCs orchestrating response to Helicobacter colonization via a Treg-independent mechanism.
ACKNOWLEDGEMENTS
This study was funded in full by National Institutes of Health, grant number R01 DK087708–01 (JYK), P30-DK034933–35 (University of Michigan Center for GI Research), and funding from the Department of Internal Medicine, Michigan Medicine, University of Michigan.
Footnotes
CONFLICT OF INTEREST STATEMENT
The authors have no relevant competing interests.
REFERENCES
- 1.Hooi JKY, Lai WY, Ng WK, et al. Global Prevalence of Helicobacter pylori Infection: Systematic Review and Meta-Analysis. Gastroenterology. 2017;153(2):420–429. doi: 10.1053/j.gastro.2017.04.022 [DOI] [PubMed] [Google Scholar]
- 2.Salih BA. Helicobacter pylori Infection in Developing Countries: The Burden for How Long? Saudi J Gastroenterol Off J Saudi Gastroenterol Assoc. 2009;15(3):201–207. doi: 10.4103/1319-3767.54743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Herbarth O, Bauer M, Fritz GJ, et al. Helicobacter pylori colonisation and eczema. J Epidemiol Community Health. 2007;61(7):638–640. doi: 10.1136/jech.2006.046706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen Y, Blaser MJ. Helicobacter pylori Colonization Is Inversely Associated with Childhood Asthma. J Infect Dis. 2008;198(4):553–560. doi: 10.1086/590158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reibman J, Marmor M, Filner J, et al. Asthma Is Inversely Associated with Helicobacter pylori Status in an Urban Population. PLOS ONE. 2008;3(12):e4060. doi: 10.1371/journal.pone.0004060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Q, Yu C, Sun Y. The association between asthma and Helicobacter pylori: a meta-analysis. Helicobacter. 2013;18(1):41–53. doi: 10.1111/hel.12012 [DOI] [PubMed] [Google Scholar]
- 7.Castaño-Rodríguez N, Kaakoush NO, Lee WS, Mitchell HM. Dual role of Helicobacter and Campylobacter species in IBD: a systematic review and meta-analysis. Gut. 2017;66(2):235–249. doi: 10.1136/gutjnl-2015-310545 [DOI] [PubMed] [Google Scholar]
- 8.Luther J, Dave M, Higgins PDR, Kao JY. Association between Helicobacter pylori infection and inflammatory bowel disease: a meta-analysis and systematic review of the literature. Inflamm Bowel Dis. 2010;16(6):1077–1084. doi: 10.1002/ibd.21116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Radić M Role of Helicobacter pylori infection in autoimmune systemic rheumatic diseases. World J Gastroenterol. 2014;20(36):12839–12846. doi: 10.3748/wjg.v20.i36.12839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arnold IC, Dehzad N, Reuter S, et al. Helicobacter pylori infection prevents allergic asthma in mouse models through the induction of regulatory T cells. J Clin Invest. 2011;121(8):3088–3093. doi: 10.1172/JCI45041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lankarani KB, Honarvar B, Athari SS. The Mechanisms Underlying Helicobacter Pylori-Mediated Protection against Allergic Asthma. Tanaffos. 2017;16(4):251–259. [PMC free article] [PubMed] [Google Scholar]
- 12.Kyburz A, Müller A. Helicobacter pylori and Extragastric Diseases. Curr Top Microbiol Immunol. 2017;400:325–347. doi: 10.1007/978-3-319-50520-6_14 [DOI] [PubMed] [Google Scholar]
- 13.Ishaq S, Nunn L. Helicobacter pylori and gastric cancer: a state of the art review. Gastroenterol Hepatol Bed Bench. 2015;8(Suppl1):S6–S14. [PMC free article] [PubMed] [Google Scholar]
- 14.Abadi ATB. Strategies used by helicobacter pylori to establish persistent infection. World J Gastroenterol. 2017;23(16):2870–2882. doi: 10.3748/wjg.v23.i16.2870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cooke CL, Huff JL, Solnick JV. The role of genome diversity and immune evasion in persistent infection with Helicobacter pylori. FEMS Immunol Med Microbiol. 2005;45(1):11–23. doi: 10.1016/j.femsim.2005.04.002 [DOI] [PubMed] [Google Scholar]
- 16.Sansonetti PJ, Di Santo JP. Debugging how bacteria manipulate the immune response. Immunity. 2007;26(2):149–161. doi: 10.1016/j.immuni.2007.02.004 [DOI] [PubMed] [Google Scholar]
- 17.Lundgren A, Strömberg E, Sjöling A, et al. Mucosal FOXP3-expressing CD4+ CD25high regulatory T cells in Helicobacter pylori-infected patients. Infect Immun. 2005;73(1):523–531. doi: 10.1128/IAI.73.1.523-531.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kandulski A, Wex T, Kuester D, et al. Naturally occurring regulatory T cells (CD4+, CD25high, FOXP3+) in the antrum and cardia are associated with higher H. pylori colonization and increased gene expression of TGF-beta1. Helicobacter. 2008;13(4):295–303. doi: 10.1111/j.1523-5378.2008.00612.x [DOI] [PubMed] [Google Scholar]
- 19.Rad R, Brenner L, Bauer S, et al. CD25+/Foxp3+ T cells regulate gastric inflammation and Helicobacter pylori colonization in vivo. Gastroenterology. 2006;131(2):525–537. doi: 10.1053/j.gastro.2006.05.001 [DOI] [PubMed] [Google Scholar]
- 20.Altobelli A, Bauer M, Velez K, Cover TL, Müller A. Helicobacter pylori VacA Targets Myeloid Cells in the Gastric Lamina Propria To Promote Peripherally Induced Regulatory T-Cell Differentiation and Persistent Infection. mBio. 2019;10(2). doi: 10.1128/mBio.00261-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kao JY, Zhang M, Miller MJ, et al. Helicobacter pylori immune escape is mediated by dendritic cell-induced Treg skewing and Th17 suppression in mice. Gastroenterology. 2010;138(3):1046–1054. doi: 10.1053/j.gastro.2009.11.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arnold IC, Lee JY, Amieva MR, et al. Tolerance rather than immunity protects from Helicobacter pylori-induced gastric preneoplasia. Gastroenterology. 2011;140(1):199–209. doi: 10.1053/j.gastro.2010.06.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bimczok D, Clements RH, Waites KB, et al. Human primary gastric dendritic cells induce a Th1 response to H. pylori. Mucosal Immunol. 2010;3(3):260–269. doi: 10.1038/mi.2010.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kao JY, Rathinavelu S, Eaton KA, et al. Helicobacter pylori-secreted factors inhibit dendritic cell IL-12 secretion: a mechanism of ineffective host defense. Am J Physiol Gastrointest Liver Physiol. 2006;291(1):G73–81. doi: 10.1152/ajpgi.00139.2005 [DOI] [PubMed] [Google Scholar]
- 25.Otsu S, Gotoh K, Yamashiro T, et al. Transfer of antigen-pulsed dendritic cells induces specific T-Cell proliferation and a therapeutic effect against long-term Helicobacter pylori infection in mice. Infect Immun. 2006;74(2):984–993. doi: 10.1128/IAI.74.2.984-993.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oertli M, Sundquist M, Hitzler I, et al. DC-derived IL-18 drives Treg differentiation, murine Helicobacter pylori-specific immune tolerance, and asthma protection. J Clin Invest. 2012;122(3):1082–1096. doi: 10.1172/JCI61029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seeger P, Musso T, Sozzani S. The TGF-β superfamily in dendritic cell biology. Cytokine Growth Factor Rev. 2015;26(6):647–657. doi: 10.1016/j.cytogfr.2015.06.002 [DOI] [PubMed] [Google Scholar]
- 28.Yoshimura A, Muto G. TGF-β function in immune suppression. Curr Top Microbiol Immunol. 2011;350:127–147. doi: 10.1007/82_2010_87 [DOI] [PubMed] [Google Scholar]
- 29.Konkel JE, Zhang D, Zanvit P, et al. Transforming Growth Factor-β Signaling in Regulatory T Cells Controls T Helper-17 Cells and Tissue-Specific Immune Responses. Immunity. 2017;46(4):660–674. doi: 10.1016/j.immuni.2017.03.015 [DOI] [PubMed] [Google Scholar]
- 30.Eaton KA, Danon SJ, Krakowka S, Weisbrode SE. A reproducible scoring system for quantification of histologic lesions of inflammatory disease in mouse gastric epithelium. Comp Med. 2007;57(1):57–65. [PubMed] [Google Scholar]
- 31.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods San Diego Calif. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- 32.Poh AR, O’Donoghue RJJ, Ernst M, Putoczki TL. Mouse models for gastric cancer: Matching models to biological questions. J Gastroenterol Hepatol. 2016;31(7):1257–1272. doi: 10.1111/jgh.13297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee A, Fox JG, Otto G, Murphy J. A small animal model of human Helicobacter pylori active chronic gastritis. Gastroenterology. 1990;99(5):1315–1323. doi: 10.1016/0016-5085(90)91156-z [DOI] [PubMed] [Google Scholar]
- 34.Cantorna MT, Balish E. Inability of human clinical strains of Helicobacter pylori to colonize the alimentary tract of germfree rodents. Can J Microbiol. 1990;36(4):237–241. doi: 10.1139/m90-041 [DOI] [PubMed] [Google Scholar]
- 35.Roth KA, Kapadia SB, Martin SM, Lorenz RG. Cellular immune responses are essential for the development of Helicobacter felis-associated gastric pathology. J Immunol Baltim Md 1950. 1999;163(3):1490–1497. [PubMed] [Google Scholar]
- 36.Wroblewski LE, Peek RM, Wilson KT. Helicobacter pylori and gastric cancer: factors that modulate disease risk. Clin Microbiol Rev. 2010;23(4):713–739. doi: 10.1128/CMR.00011-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Correa P, Piazuelo MB. Helicobacter pylori Infection and Gastric Adenocarcinoma. US Gastroenterol Hepatol Rev. 2011;7(1):59–64. [PMC free article] [PubMed] [Google Scholar]
- 38.Dunne C, Dolan B, Clyne M. Factors that mediate colonization of the human stomach by Helicobacter pylori. World J Gastroenterol. 2014;20(19):5610–5624. doi: 10.3748/wjg.v20.i19.5610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sebrell TA, Hashimi M, Sidar B, et al. A Novel Gastric Spheroid Co-culture Model Reveals Chemokine-Dependent Recruitment of Human Dendritic Cells to the Gastric Epithelium. Cell Mol Gastroenterol Hepatol. 2019;8(1):157–171.e3. doi: 10.1016/j.jcmgh.2019.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Andres S, Schmidt H-MA, Mitchell H, Rhen M, Maeurer M, Engstrand L. Helicobacter pylori defines local immune response through interaction with dendritic cells. FEMS Immunol Med Microbiol. 2011;61(2):168–178. doi: 10.1111/j.1574-695X.2010.00761.x [DOI] [PubMed] [Google Scholar]
- 41.Chang S-Y, Ko H-J, Kweon M-N. Mucosal dendritic cells shape mucosal immunity. Exp Mol Med. 2014;46(3):e84–e84. doi: 10.1038/emm.2014.16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Audiger C, Rahman MJ, Yun TJ, Tarbell KV, Lesage S. The Importance of Dendritic Cells in Maintaining Immune Tolerance. J Immunol Baltim Md 1950. 2017;198(6):2223–2231. doi: 10.4049/jimmunol.1601629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Luther J, Owyang SY, Takeuchi T, et al. Helicobacter pylori DNA decreases pro-inflammatory cytokine production by dendritic cells and attenuates dextran sodium sulphate-induced colitis. Gut. 2011;60(11):1479–1486. doi: 10.1136/gut.2010.220087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Owyang SY, Luther J, Owyang CC, Zhang M, Kao JY. Helicobacter pylori DNA’s anti-inflammatory effect on experimental colitis. Gut Microbes. 2012;3(2):168–171. doi: 10.4161/gmic.19181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sun X, Zhang M, El-Zataari M, et al. TLR2 mediates Helicobacter pylori-induced tolerogenic immune response in mice. PloS One. 2013;8(9):e74595. doi: 10.1371/journal.pone.0074595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Travis MA, Sheppard D. TGF-β activation and function in immunity. Annu Rev Immunol. 2014;32:51–82. doi: 10.1146/annurev-immunol-032713-120257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fu S, Zhang N, Yopp AC, et al. TGF-beta induces Foxp3 + T-regulatory cells from CD4 + CD25 - precursors. Am J Transplant Off J Am Soc Transplant Am Soc Transpl Surg. 2004;4(10):1614–1627. doi: 10.1111/j.1600-6143.2004.00566.x [DOI] [PubMed] [Google Scholar]
- 48.Krzysiek-Maczka G, Wrobel T, Targosz A, et al. Helicobacter pylori-activated gastric fibroblasts induce epithelial-mesenchymal transition of gastric epithelial cells in vitro in a TGF-β-dependent manner. Helicobacter. 2019;24(5):e12653. doi: 10.1111/hel.12653 [DOI] [PubMed] [Google Scholar]
- 49.Rahimian G, Sanei MH, Shirzad H, et al. Virulence factors of Helicobacter pylori vacA increase markedly gastric mucosal TGF-β1 mRNA expression in gastritis patients. Microb Pathog. 2014;67–68:1–7. doi: 10.1016/j.micpath.2013.12.006 [DOI] [PubMed] [Google Scholar]
- 50.Harris PR, Wright SW, Serrano C, et al. Helicobacter pylori gastritis in children is associated with a regulatory T-cell response. Gastroenterology. 2008;134(2):491–499. doi: 10.1053/j.gastro.2007.11.006 [DOI] [PubMed] [Google Scholar]
- 51.Alam MS, Kurtz CC, Rowlett RM, et al. CD73 is expressed by human regulatory T helper cells and suppresses proinflammatory cytokine production and Helicobacter felis-induced gastritis in mice. J Infect Dis. 2009;199(4):494–504. doi: 10.1086/596205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Raghavan S, Suri-Payer E, Holmgren J. Antigen-specific in vitro suppression of murine Helicobacter pylori-reactive immunopathological T cells by CD4CD25 regulatory T cells. Scand J Immunol. 2004;60(1–2):82–88. doi: 10.1111/j.0300-9475.2004.01447.x [DOI] [PubMed] [Google Scholar]
- 53.Kullberg MC, Jankovic D, Feng CG, et al. IL-23 plays a key role in Helicobacter hepaticus-induced T cell-dependent colitis. J Exp Med. 2006;203(11):2485–2494. doi: 10.1084/jem.20061082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Maloy KJ, Salaun L, Cahill R, Dougan G, Saunders NJ, Powrie F. CD4+CD25+ T(R) cells suppress innate immune pathology through cytokine-dependent mechanisms. J Exp Med. 2003;197(1):111–119. doi: 10.1084/jem.20021345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Letterio JJ, Roberts AB. Regulation of immune responses by TGF-beta. Annu Rev Immunol. 1998;16:137–161. doi: 10.1146/annurev.immunol.16.1.137 [DOI] [PubMed] [Google Scholar]
- 56.Sanjabi S, Oh SA, Li MO. Regulation of the Immune Response by TGF-β: From Conception to Autoimmunity and Infection. Cold Spring Harb Perspect Biol. 2017;9(6). doi: 10.1101/cshperspect.a022236 [DOI] [PMC free article] [PubMed] [Google Scholar]