

Abstract
The comet assay is a versatile method for measuring DNA strand breaks in individual cells. It can also be applied to cells isolated from treated animals. In this review, we highlight advantages and limitations of this in vivo comet assay in a regulatory context. Modified versions of the standard protocol detect oxidized DNA bases and may be used to reveal sites of DNA base loss, DNA interstrand crosslinks, and the extent of DNA damage induced indirectly by reactive oxygen species elicited by chemical-induced oxidative stress. The assay is, however, at best semi-quantitative, and we discuss possible approaches to improving DNA damage quantitation and highlight the necessity of optimizing protocol standardization to enhance the comparability of results between laboratories. As a genotoxicity test in vivo, the in vivo comet assay has the advantage over the better established micronucleus erythrocyte test that it can be applied to any organ, including those that are specific targets of chemical carcinogens or those that are the first sites of contact of ingested or inhaled mutagens. We illustrate this by examples of its use in risk assessment for the food contaminants ochratoxin and furan. We suggest that improved quantitation is required to reveal the full potential of the comet assay and enhance its role in the battery of in vivo approaches to characterize the mechanisms of toxicity and carcinogenicity of chemicals and to aid the determination of safe human exposure limits.
Keywords: comet assay, DNA damage, risk assessment
Introduction
The comet assay is a method for measuring DNA strand breaks in individual cells. It is based upon the principle that fragmented DNA migrates more rapidly than intact DNA under electrophoresis through an agarose matrix. The detailed description of the technique is outside the scope of this review and is extensively illustrated in other publications [1, 2]. Briefly, single-cell suspensions in agarose are layered on microscopic slides, lysed with detergent and high molarity NaCl to disrupt membranes and remove histones, and then electrophoresed. In the presence of strand breaks, DNA migrates toward the anode forming an image resembling the tail of a comet when stained with a fluorescent dye and viewed under fluorescence microscopy (Fig. 1A). Its versatility and sensitivity have led to its application to assess DNA damage induced by chemical or physical agents in cells from numerous different organisms under a wide variety of experimental conditions [3, 4]. The comet assay is used in human monitoring studies as a biomarker of exposure to agents causing damage to DNA [5, 6] and in ecotoxicological studies in a variety of sentinel organisms [7–9]. Its application in hazard characterization under controlled laboratory conditions contributes to the understanding of the mode of action of chemicals and informs risk assessment [10–12].
Figure 1.
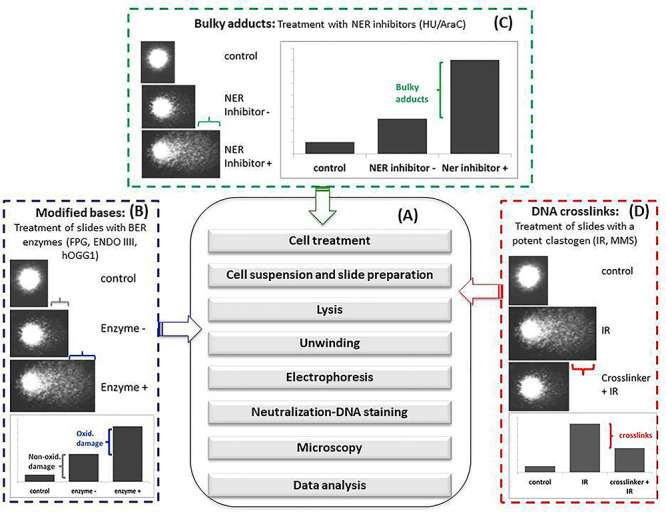
Alkaline comet assay and its variants. (A) Main steps of the standard protocol; (B) protocol modifications introduced to detect modified bases (generally oxidized bases). These modifications consist in treating slides after lysis (see arrow) with BER enzymes that produce DNA breaks at the site of modified base. The slides then undergo the other steps of the standard protocol. The increase of %TI of BER enzyme treated slides over the non-treated slides gives the amount of modified bases; (C) protocol modification introduced to detect bulky adducts. This modification consists in treating cells (in colture) (see arrow) with inhibitors of the NER enzymatic pathway in order to accumulate incomplete DNA repair sites detected as strand breaks by the comet assay. The slides then undergo the other steps of the standard protocol. The increase of %TI of NER inhibitor treated cells over the untreated cells gives the amount of bulky adducts; (D) protocol modification introduced to detect DNA crosslinks. This modification consists in increasing DNA migration by treating slides (see arrow) with a potent clastogen (generally ionizing radiations) or by potentiating electrophoresis conditions. The slides then undergo the other steps of the standard protocol. The decrease of %TI indicates the presence of DNA crosslinks.
DNA Lesions Detected by the Comet Assay
The test can be conducted under neutral or alkaline electrophoresis conditions each one enhancing the detection of different types of DNA lesions. Initially, the prevalent opinion was that under neutral conditions mostly double-strand breaks (DSBs) were revealed, whereas under alkaline conditions, a broader spectrum of lesions was detected including, in addition to DSBs, single-strand breaks (SSBs) and apurinic/apyrimidinic sites (AP sites). However, more recently, a better understanding of DNA migration processes and comet formation suggests that the spectrum of lesions detected under the different electrophoresis conditions largely overlaps [13]. DNA breaks or AP sites are continuously produced under physiological conditions by processes such as hydrolysis or damage by •OH and other free radicals. Exposure to physical and chemical agents produces numerous types of DNA lesion including damaged bases, AP sites, inter- and intrastrand crosslinks and direct strand breaks with a variety of termini [14]. In its current form the comet assay employs several modifications each designed to reveal a particular type of DNA lesion thereby extending its range of utility (Fig. 1B–D). In particular, the use of lesion-specific DNA glycosylases/endonucleases, which remove altered bases and introduce DNA AP sites or strand breaks, is widespread (Fig. 1B). The most widely used enzymes include the Escherichia coli formamidopyrimidine DNA glycosylase (Fpg) and endonuclease III (Endo III) and the mammalian counterpart of Fpg, 8-oxoguanine DNA glycosylase (OGG1) [15]. Fpg excises oxidized purines from DNA. Its substrates include the ring-opened purines 2,6-diamino-4-hydroxy-5-formamidopyrimidine and 4-6-diamino-5 formamidopyrimidine as well as 8-oxo-7,8-dihydroguanine (8-oxoGua). Endo III acts similarly to remove oxidized pyrimidines, including DNA thymine glycol and uracil glycol. The specificity of these enzymes for a particular base alteration is not absolute. Thus, Fpg and EndoIII also recognize some types of alkylation damage [16, 17], These Fpg-mediated SSBs might derive from AP sites following spontaneous hydrolysis or enzymatic excision of N7-methylguanine (N7-meGua) and N3-methyladenine (N3-meAde). In addition, alkali treatment of N7-meGua can lead to the formation of ring-opened derivatives (FaPy-Gua) that are also recognized and incised by Fpg. In contrast, the eukaryotic OGG1 is a more specific enzyme that is unable to recognize methylated bases [18]. As a consequence, no significant increase in SSBs was observed following treatment with the alkylating agent methylmethanesulphonate or ethylnitrosourea when assessed by the hOGG1 modified comet assay, whereas an increase was observed with the Fpg modified comet assay [17, 19].
In a similar approach, the uracil DNA glycosylase has been used to detect uracil (U) misincorporated in DNA [20] and the 3-meAde DNA glycosylase and AlkA to increase the sensitivity and selectivity of the comet assays against alkylated bases [21].
Many environmental carcinogens react with DNA bases to produce bulky adducts. These important DNA lesions block replication and transcription and contribute to cell-cycle arrest and cell death; they also induce mutations possibly leading to carcinogenesis [22–24]. Most of these lesions are repaired by nucleotide excision repair (NER), a repair pathway in which the incision and excision steps of lesion removal are highly coordinated to minimize the generation of persistent DNA strand break intermediates [25]. This repair strategy compromises the ability of the comet assay to detect bulky DNA adducts. To counteract this limitation, inhibitors of NER are included in the assay to permit the accumulation of incomplete DNA repair events that are detected as SSBs. DNA synthesis inhibitors aphidicolin, hydroxyurea (HU) and 1-β-D-arabinofuranosyl cytosine (AraC) have all been successfully applied to allow the detection of DNA strand breaks formed during NER of bulky DNA adducts induced by UVC-radiation and the carcinogens benzo[a]pyrene and aflatoxin B1 [26, 27] (Fig. 1C). Among nine known in vivo genotoxic agents, only one generated SSBs measurable by the standard comet assay in the absence of HU/AraC, whereas seven were positive in the presence of the inhibitors [27]. In addition, the DNA T4 endonuclease V, which repairs UV-damaged DNA, has also been used to detect cyclobutane pyrimidime dimers in the comet assay [28].
DNA interstrand crosslinks (ICLs) are among the most cytotoxic DNA lesions. They block both DNA replication and transcription and inhibit recombination by preventing the separation of DNA strands. Several known or suspected carcinogens induce ICLs. ICLs repair is complex and entails different repair pathways, such as NER, structure-specific endonucleases, translesion DNA polymerases (TLS) and homologous recombination (HR). NER plays an important role in ICLs removal in quiescent cells, whereas the ICLs repair in S phase requires a more complex orchestration of multiple factors, including structure-specific endonucleases, TLS and HR [29].
The presence of ICLs retards DNA migration in the comet assay. Their presence can be inferred from the decreased migration of DNA from cells treated with a suspected ICL-inducing agent followed by a known SSBs inducing agent (generally ionizing radiation) immediately prior to electrophoresis. Alternatively, ICLs can be revealed by a reduction of the background level of DNA migration under extreme electrophoretic conditions (Fig. 1D). This approach was used to quantify ICLs induced by the photoactivated 4-hydroxymethyl-4,5,8-trimethylpsoralen (HMT) in vitro [30]. Under electrophoresis conditions that maximized DNA migration, a dose-dependent decrease of DNA migration was observed shortly after HMT treatment. However, at later times, the incisions formed during ICLs repair caused the formation of DNA strand breaks, which antagonize the migration-inhibiting effect of ICLs in the comet assay. This causes a progressive increase of DNA migration in HMT-treated samples that became equal to the untreated ones, and, at even longer times, results in a further increase of DNA migration over the untreated control. These results indicate that antagonistic phenomena acting on DNA migration should be considered when assessing DNA crosslinking by the comet assay. On one hand, DNA ICLs reduce DNA migration; on the other hand, incisions occurring during ICL repair increase DNA migration. Therefore, the extent of migration depends on the kinetics of ICL production and recognition, DNA incision and processing by repair proteins and sampling time becomes a critical variable in the experimental design. These steps in turn depend on the characteristics of the chemical and the cell physiology. These factors may underlie the relative low sensitivity of the assay in revealing DNA crosslinks and for disparate results obtained in studies assessing the genotoxicity of chemicals suspected of crosslinking. This may be particularly true in in vivo studies when the timing between exposure and cell analysis cannot be completely controlled. Indeed, the detection of crosslinking agents is not included among the purposes of the ‘In Vivo Mammalian Alkaline Comet Assay’ (OECD TG guideline 489) [31] for which ‘further work would be needed to adequately characterize the necessary protocol modifications’.
Endogenous vs Exogenous Sources of DNA Damage
It has been estimated that human genome sustains ~40,000–70,000 lesions per day [32–36]. The vast majority of these are SSBs arising from oxidation (by metabolic by-products) or base loss via glycosyl bond hydrolysis (Fig. 2). SSBs may be occasionally converted into the more dangerous DSBs (25 events per cell per day). Spontaneous DNA base loss is largely due to spontaneous depurination events, with a minor contribution from depyrimidination (10,000 and 500 lesions per cell per day, respectively). Spontaneous deamination of cytosine to U occurs around 200 times per cell per day. If uncorrected, the resulting U:G mismatches will give rise to G > A transition mutations.
Figure 2.
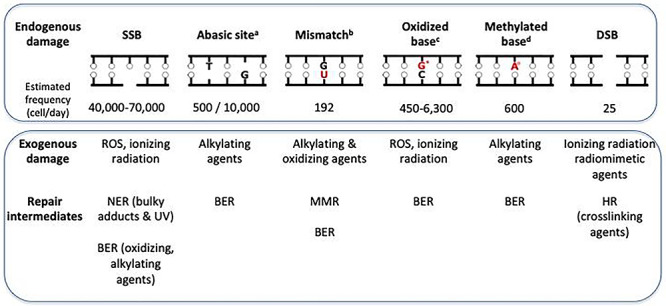
Endogenous and exogenous DNA lesions and their repair. (a) The estimated frequencies refer to apyrimidinic and apurinic sites, respectively; (b) the estimated frequency refers only to mismatches derived from deamination of cytosine to uracil (mismatches due to DNA replication errors by mammalian DNA polymerases are excluded); (c) the estimated frequencies refer only to 8-oxoGua (G*); (d) the estimated frequency refers only to N3-meAde (A°). DNA adduct frequencies are taken from references [32–36].
Oxidation is an important source of endogenous genomic damage. Reactive oxygen species (ROS) are generated during normal aerobic cellular metabolism. Depending on the analytic method used, the rate of production of DNA 8-oxoGua, the major mutagenic oxidized base, can vary from 450 to 6300 lesions per cell per day. 8-OxoGua pairs preferentially with adenine rather than cytosine and GC > TA transversions are considered to be the signature mutation of oxidative stress.
S-adenosylmethionine (SAM), a methyl group donor and a cofactor in several transmethylation reactions is also an endogenous genotoxic agent. SAM non-enzymatically methylates the ring nitrogen of DNA purines to generate around 600 N3-meAde and 4000 N7-meGua lesions per day in the human genome.
The effects of endogenous DNA damage are mitigated by DNA repair. The main repair pathways are NER, base excision repair (BER), HR, non-homologous end joining and mismatch repair (MMR) [33–37]. Most of them also protect the genome against DNA damage induced by noxious foreign chemicals (some examples are shown in Fig. 2). Exposure to some exogenous chemicals can also enhance ‘spontaneous’ DNA decay. One example of this enhancement is the accelerated depurination that accompanies modifications of DNA bases induced by alkylating agents.
One of the properties of the comet assay is the capacity of this test to detect DNA lesions derived either from direct damage to DNA or identify DNA intermediates occurring during the repair process of an initial damage. Thus, we can consider an untreated control in the comet assay as the reflection of the steady-state level of endogenous DNA lesions, including the intermediates of repair processes. In contrast, exposure to a genotoxic agent introduces a burst of additional DNA lesions that may overwhelm the DNA repair machinery possibly leading to irreversible gene or chromosome mutations. Thus, measuring an increase of DNA migration in treated samples vs. matched controls provides a marker of the agent genotoxic mode of action useful for the classification and regulation of chemical exposures.
Efforts to Improve the Comparability and Interpretation of Comet Assay Results
Since its first appearance >30 years ago [38], the comet assay has been widely used. Its low cost and versatility have led to its adoption as an assay of choice worldwide. The sheer number of laboratories using the comet assay has meant that numerous small variations to the protocol have occurred. This multitude of assay protocols presents a problem of comparability of results among laboratories. In addition, problems of intra-laboratory variability among experiments are frequent. A significant effort has been expended to identify crucial parameters that affect the performance of the comet assay and to clarify how different factors, from sample preparation to cell scoring and analysis, can influence the results [2, 13, 39–41]. Some progress has been made, and the variation among laboratories and experiments has been reduced. Despite this advance, there are currently no consensus guidelines for the application of the comet assay in in vitro or in biomonitoring studies.
The problem of intra- and inter-laboratory variability limits the direct comparison of DNA damage levels among different studies or multicentre trials. It is particularly challenging in human biomonitoring studies. To address these issues, the European Standards Committee on Oxidative DNA Damage and the European Comet Assay Validation Group (ECVAG) launched international trials focused especially on the measurement of oxidatively damaged DNA in human cells. These led to some improvements both in the standard and in the Fpg modified comet assay [42–45].
To reduce the inter-laboratory variability in assay data, it has been proposed that comet assay DNA damage parameters (e.g. % DNA in tail) be transformed to the number of lesions per basepair (bp) based on calibration curves generated from cells exposed to ionizing radiation. The relationship between ionizing radiation dose and DNA SSBs was established by alkaline sucrose gradient sedimentation studies. About 0.3 strand breaks/109 dalton DNA are introduced per Gy of radiation. This approximates to 1000 breaks per diploid mammalian cell/Gy [46]. Based on this dose equivalence, cell-type-specific calibration curves were generated by different laboratories and used to calculate the number of lesions/106 bp DNA corresponding to a particular percentage of DNA in the tail. This conversion significantly reduced inter-laboratory variation in an ECVAG trial conducted among 10 laboratories, indicating that it could facilitate the comparisons between data from different research groups [45].
In a recent paper, Moller and collaborators [47] searched PubMed database for human biomonitoring studies that had used comet assays to measure oxidatively damaged DNA in nucleated blood cells. The study applied the ECVAG calibration curve [48] in which 1% DNA in tail corresponds to 0.0273 lesions/106 bp to data from the published assays. Results showed a large variation in the reported level of Fpg-sensitive sites in human white blood cells (ranges: 0.05–1.31 lesions/106 bp), whereas less variation was found for hOGG1 (0.04–0.18 lesions/106 bp). However, these data suggest that still problems exist regarding the quantitative comparison of results from different publications.
One approach to counteracting the variability among measurements is the inclusion of reference standards. These may be slides prepared from batches of cells untreated or treated with a DNA-damaging agent undergoing the same comet procedures as the analyzed samples, or even of cells, morphologically recognizable, placed on the same slide. They can be used to monitor the performance of the assay or even to normalize sample data to reference values [49, 50].
In 2007, Ueno and coworkers [51] proposed also for in vivo studies with comet assay the application of radiation-based calibration curves to transform comet parameters into number of breaks. Linear dose–effect relationships for % tail DNA in nine different organs (liver, kidney, lung, spleen, colon, urinary bladder, thymus, brain and bone marrow) of mice irradiated with 3, 6, 12 or 24 Gy of X-rays were reported. Of note, organs were collected only 3 minutes after a high dose-rate irradiation to avoid as much as possible the bias introduced by fast repair mechanisms occurring during and immediately after irradiation. The slopes of the dose–response curves varied among the organs tested: e.g., 1Gy X-rays increased the % tail DNA above the respective background level by 1.8% in the liver and 0.6% in bone marrow. This finding suggested differences in the radiosensitivity of nuclear DNA and DNA repair capacity among organs and pointed to the need of obtaining organ-specific calibration curves for comet assay in vivo.
Although scientifically sound, these calibration approaches have not been extensively applied probably because their most rigorous applications would require for each laboratory producing its own cell-type-specific curves, and not all laboratories have the necessary expertise or instrumentation.
A confounding factor for the interpretation of comet assay results might be the induction of aspecific cell toxicity irrespectively of DNA damage. In fact, cell toxicity may result in the appearance of highly damaged comets (the so-called hedgehogs) that could lead to false-positive results. For this reason, it is strongly advised to record this category of comets separately from the other ones and not include them in the calculation of the assay parameters. However, because their mechanism of formation is unclear, they generally are not used as marker of toxicity [31]. To assist the investigators in the recognition and classification of this type of cells, an atlas of comet assay images has been published by the Japanese Environmental Mutagen Society [52].
The Standardization of in vivo Comet Assay and Its Application Within Regulatory Test Batteries
Although initially developed as an in vitro test, the versatility of the comet assay makes it extremely attractive for in vivo studies. In particular, a genotoxicity assay that could be employed to assess DNA damage in tissues of toxicological relevance or in site-of-contact organs was needed for studies of human risk assessment.
Between 2006 and 2013, the Japanese Center for the Validation of Alternative Methods led a project aimed at the validation of the liver and stomach comet assays [53], which, eventually, culminated in the release in late 2014 of the OECD TG 489 ‘In Vivo Mammalian Alkaline Comet Assay’ [31]. The project methodology and results were extensively reported in ref. [53]. The assays were conducted on 40 coded chemicals with known genotoxic and carcinogenic activity. Necrosis and apoptosis were evaluated in parallel in the target tissues by histopathological analysis as markers of acute cytotoxicity. The comet assay proved to be reasonably sensitive with 6 of 19 genotoxic carcinogens yielding negative results. Some of the apparent inconsistencies could be rationalized when the modes of action for genotoxicity and/or carcinogenicity were taken into consideration. For example, considering that the genotoxicity of busulfan is predominantly via DNA crosslinking formation, it is not surprising that this chemical was not detected by the standard comet assay that has a limited sensitivity for this type of DNA lesions. A good specificity of the comet assay was shown by the finding that only 2 of 21 chemicals that were either genotoxic non-carcinogens or non-genotoxic carcinogens yielded positive results.
Even before its formal validation [31], the test had been introduced by several national and international regulatory agencies as an integral part of the genotoxicity testing strategies, which generally consists of multi-tier approaches comprising in vitro and in vivo tests.
Since 2011, the ‘Guidance on Genotoxicity Testing and Data Interpretation for Pharmaceuticals Intended for Human Use’ included the comet assay in liver as an additional in vivo assay, further to micronucleus (MN) assay in rodent hematopoietic cells, for substances that induce gene mutations in vitro. This guideline to the selection of genotoxicity tests for new pharmaceuticals has since been adopted worldwide [54].
In Europe, the ‘European Community Regulation on chemicals and their safe use’ introduced in 2007, requires companies manufacturing or importing chemical substances into the European Union in quantities ≥1 ton/year to register these substances with the European Chemicals Agency (ECHA) [55]. ECHA requirements for registration depend on the tonnage and the potential mutagenicity of a substance is among the information required for its registration. The strategies to provide this information are based on the use of different pieces of information, including non-testing data (Structure activity relationship, quantitative structure activity relationship and read-across approaches) and results from in vitro and in vivo testing. Information from a bacterial gene mutation test is required for all the tonnage groups. A negative result with this test is sufficient for the registration of substances in the low-level tonnage group. For compounds in higher tonnage groups, a second in vitro MN or chromosome aberration test is needed. In case of positive results, an in vivo follow-up is required. Depending on the type of damage elicited in vitro (gene mutation or chromosomal damage) Transgenic Rodent (TGR) Somatic and Germ Cell Gene Mutation Assays [56], comet assay [31], MN test [57] or chromosome aberration test [58] would be considered as appropriate follow-up tests.
Genotoxicity testing strategies applicable to food and animal feed safety assessment have been defined by the European Food Safety Authority (EFSA) [59, 60]. A step-wise approach is recommended for the generation and evaluation of data on genotoxic potential. This begins with a battery of in vitro tests, comprising a bacterial reverse mutation assay and an in vitro MN assay. In case of positive in vitro results, review of the available relevant literature data on the test substance and, where necessary, appropriate in vivo studies are recommended. Suitable in vivo tests are considered the mammalian erythrocyte MN test, the TGR assay and the comet assay. A single positive test is sufficient to classify the compound as an in vivo genotoxin. Both a negative systemic genotoxicity test (i.e. MN test) and a negative test on site-of-contact or metabolically competent organ (i.e. TGR or comet assays in gastrointestinal (GI) tract and liver) are deemed necessary to classify a compound as non-genotoxic in vivo.
The in vivo genotoxicity tests can detect different kind of damage and each has its own advantages and limitations. The MN test detects clastogenic and aneuploidogenic events in bone marrow erythrocyte precursors. Its main limitation is that the compound or its metabolite(s) have to reach the bone marrow in a sufficient concentration to induce DNA damage. On the other hand, both TGR and comet assays can be performed on different organs, including the target organs for toxicity and carcinogenicity. TGR assay mainly detects gene mutations, whereas the comet assay detects DNA lesions, including those produced during DNA repair. Although both MN and TGR assays reveal irreversibly fixed DNA alterations, the comet assay reveals transient lesions potentially leading to irreversible damage. In a comparative review of group 1 carcinogens selected from the International Agency for Research on Cancer, a 90% sensitivity of comet assay (any organ) was reported, higher than the 70% sensitivity achieved by MN test [61]. A working group on in vivo genotoxicity testing strategies convened on the occasion of the seventh International Workshop on Genotoxicity Testing [62] compared the ability of comet and TGR assays (any organ) to detect chemical carcinogens. The study revealed that more carcinogens tested positive in the comet assay than in the TGR assay. The same study noted that some non-carcinogens also tested positive in the comet assay. This lower specificity reflected indirect mechanisms of DNA breakage driven by toxicity, oxidative stress or extreme pharmacology. Eventually, the working group concluded that the comet assay in the liver or GI tract was as suitable as the TGR assay as a follow-up approach after evidence of in vitro genotoxicity.
Table 1 reports some examples of the performance of in vivo standard alkaline comet assays in identifying non-genotoxic and genotoxic chemicals characterized by different mechanisms of action.
Table 1.
Results of standard alkaline comet assays following in vivo exposure to chemicals in relation to their mechanisms of genotoxicitya
| Chemical (CAS N°) | IARCb | Main mechanism of action | In vivo standard comet assay | References |
|---|---|---|---|---|
| 1,2-Dimethylhydrazine dihydrochloride (306-37-6) | 2A | DNA alkylation | Positive | [53] |
| Methyl methanesulfonate (66-27-3) | 2A | DNA alkylation | Positive | [53] |
| 2-Acetylaminofluorene (53-96-3) | Not classified | Bulky adducts | Negative | [53] |
| Benzo[a]pyrene (50-32-8) | 1 | Bulky adducts | Positive | [61] |
| Acetaldehyde (75-07-0) | 2B | DNA and DNA-protein crosslinks | Positive | [61] |
| Hexamethylphosphoramide (680-31-9) | 2B | DNA-protein crosslinks | Negative | [62] |
| Busulfan (55-98-1) | 1 | DNA crosslinks | Negative | [53] |
| Cisplatin (15663-27-1) | 2A | DNA crosslinks | Positive | [53] |
| 1,2-Dibromoethane (106-93-4)) | Not classified | DNA crosslinks | Positive | [53] |
| Etoposide (33419-42-0) | 1 | Topoisomerase inhibitor | Positive | [61] |
| Hydroquinone (123-31-9) | 3 | Mitotic spindle interaction | Negative | [53] |
| 5-Fluorouracil (51-21-8) | 3 | DNA synthesis inhibitor | Negative | [53] |
| Cadmium chloride (10108-64-2) | Not classified | Oxidative stress | Positive | [53] |
| Di(2-ethylhexyl) phthalate (117-81-7) | 2B | Endocrine disruptor | Negative | [53] |
| Chloroform (67-66-3) | 2B | Liver toxicity | Positive | [53] |
| Ethanol (64-17-5) | 1 | Non-genotoxic | Negative | [53, 61] |
| Trichloroethylene (79-01-6) | 1 | Non-genotoxic | Negative | [61, 62] |
aThe positive/negative classification according to the quoted reference(s) does not take into account the organ(s) tested and does not exclude that different results may be obtained in other published experiments.
bInternational agency for research on cancer classification.
Another genotoxicity assay that detects the initial events of the DNA damage response is based on the phosphorylation of histone H2AX (γ-H2AX). This is assessed by the formation of microscopically visible foci analyzed by flow cytometry or by western blotting. The assay was originally proposed in the field of radiation biology as a sensitive assay for detecting DSBs [63]. More recently, it has been validated with some promising results against the in vitro comet assay for the detection of chemical mutagens [64, 65]. More limited data have been collected so far in vivo. Thus, additional validation studies are recommended in view of the potential application of the assay in multiple organs and to verify the possible superior performance for the detection of some classes of compounds such as the crosslinkers [64].
Based on their complementarity, a combination of multiple tests is suggested to provide an added value [61]. Also, the combination of in vivo tests assessing different endpoints in different tissues in the same animal is encouraged by regulatory authorities in light of 3R principle. The International workshops on genotoxicity testing working group [62] concluded that a combination of the MN test in bone marrow and the liver comet assay was adequate to detect in vivo mutagens or genotoxic carcinogens. For orally administered compounds, a comet assay in a single GI site was recommended.
Quantitative Approaches for the Assessment of a Genotoxic Risk
In the last 10 years, improvements in the determination and interpretation of dose–effect relationships [66] have led to a more quantitative approach to the characterization of in vivo genotoxicity. At present, regulatory authorities determine health-based guidance values (HBGV) and margin of exposure (MoE) largely on experimental cancer and toxicity data. More recently, however, gene and chromosome mutations have been advocated as a bona fide, quantifiable toxicological endpoint which can inform risk assessment [67].
DNA damage, as measured by the comet assay, could be regarded as a too transient and heterogeneous endpoint to lend itself to such analyses, but it has not totally escaped attention. A quantitative analysis of toxicological data is generally based on mathematical modeling of the dose–response relationships that allow establishing the so-called ‘point of departure’ (POD), which is a point on the dose–response curve roughly corresponding to an estimated low effect level or no effect level. The best method to establish PODs is still a matter of debate. A recent paper that analyzed data sets obtained by the comet, the MN, the TGR and the Pig-a gene mutation assays came to the conclusion that PODs must be assay specific and based on historical control data [68]. Interestingly, in the case of a model monofunctional alkylating compound (temozolomide), a close concordance of PODs across all these assays was shown [69], suggesting that, for this chemical and most likely other monofunctional alkylating agents, the comet assay was as suitable to determine POD values as were the assays for irreversible gene or chromosome mutations. In 2018, the UK Committee on Mutagenicity regretted that an evaluation of the use of comet assay data in quantitative analyses had not yet been undertaken [70]. Surely, for DNA tail intensity to be included among the mutagenicity endpoints deserving a more quantitative analysis, further progress needs to be made toward test standardization and data interpretation.
Case Studies: Interpretation of Results
Currently, EFSA guidelines recommend the use of an MoE approach for substances that are both genotoxic and carcinogenic [71]. The MoE provides a comparison between the observed experimental data and the environmental level of interest. The aim is to help decide on acceptable or tolerable level of exposure taking into account the risk management options available. An MoE of 10,000 or higher is considered to be of low concern from a public health point of view.
However, the Scientific Committee Opinion on genotoxicity testing strategies [59, 60] describes some circumstances under which genotoxicity might occur only at doses resulting in saturation of detoxification pathways. Examples include mutagens that might act through a threshold mechanism. Thus, the mutagenic potential of oxidants depends on their capability to overcome the physiological cellular defense against ROS. Other examples are substances that interact with molecular targets other than DNA (e.g. alterations of DNA polymerases, topoisomerases, DNA repair and spindle proteins). In such cases, establishing an HBGV (Tolerable Daily Intake, Tolerable Weekly Intake, etc.) might be possible.
Here, we review two examples of how the comet assay provides an important tool in evaluating the genotoxicity associated with exposures to food contaminants [ochratoxin A (OTA) and furan] [72, 73]. In particular, we show how comet assays can help to clarify the mechanisms underlying the genotoxic properties of chemical contaminants, e.g. whether these are the consequence of a direct, DNA-reactive mode of action (formation of chemical-specific covalent adducts) or are due to an indirect production of DNA damage (induction of oxidative stress, damage to DNA repair proteins, etc.). We highlight the difficulties associated with the interpretation of the results of the comet assays and stress the importance of considering all the available data, including mechanistic studies, for a proper risk assessment of food contaminants.
Ochratoxin A
OTA is a mycotoxin produced by Aspergillus and Penicillium fungi and is found as a contaminant in various foods. It causes kidney tumors in rodents. However, the mechanisms of genotoxicity are unclear and both direct (DNA reactive) and indirect genotoxic and non-genotoxic modes of action have been proposed.
In vitro exposure of mammalian cells to OTA induced both gene mutations [74] and chromosomal damage [75]. Several observations suggested that at least some of these in vitro genotoxic effects are secondary to oxidative stress induced by OTA. These included high levels of ROS and of 8-oxoGua in DNA [76, 77] as well as comet assays showing an increase in SSBs following incubation with the Fpg/EndoII enzymes [78].
In rodents, OTA induces a narrow spectrum of chromosomal damage concentrated in the portion of the kidney that is also target for OTA carcinogenesis (the kidney outer medulla). DNA damage includes chromosome hypercondensation, abnormally separated chromatids, multipolar mitotic spindles, endoreduplications, polyploidy and aneuploidy [79]. OTA is also a weak inducer of gene mutations in rats and mice [80–83]. The mutations are detected in rats after a short exposure to a carcinogenic OTA dose and are specifically restricted to the cancer target site. The mutational spectrum identified large deletions as the main mutagenic events, with no apparent increase in the GC > TA transversions typically associated with oxidative DNA damage. In addition, no changes in the levels of DNA 8-oxoGua were observed in the outer medulla [80–83]. Thus, OTA-induced mutations in rat and mouse kidneys are clearly not a simple consequence of oxidative DNA damage. The molecular mechanisms underlying in vivo OTA genotoxicity remain unclear since the formation of OTA induced DNA adducts is controversial [84, 85]; their chemical nature remains undefined and the reported levels are extremely low (20-70 × 10−9 nucleotides) [85].
The results obtained by in vivo comet assays indicate that OTA exposure increases SSBs levels in the kidney [79, 86] but also in non-target tissues such as liver and blood [79]. In some studies OTA-induced SSBs were increased by DNA digestion with Fpg/EndoIII [86].
In summary, the detection of mutations in the target tissue pointed to a genotoxic mechanism of OTA-induced carcinogenesis, and, despite some evidence of oxidative stress, the mutational spectrum suggested that DNA oxidation was not involved. The comet assay provided supportive evidence in vitro and in vivo regarding the induction of both oxidation- and non-oxidation- mediated DNA damage. Unfortunately, the analysis of DNA adducts yielded inconclusive results hampering the definitive classification of OTA as a genotoxic carcinogen. This example shows how complex it can be in some cases to resolve the complexity of a chemical mode of action even when a full set of data with reliable and sensitive methods is available.
Furan
Furan is a volatile compound formed in food during thermal processing. It induces cholangiofibrosis in rats and hepatocellular adenomas/carcinomas in mice [73]. Furan is metabolized by cytochrome P450 2E1 (CYP2E1) to the reactive metabolite cis-but-2-ene-1,4-dialdehyde (BDA). In vitro studies indicate that BDA-induced ethano DNA adducts are unstable and can be transformed into substituted etheno-acetaldehyde adducts. These secondary lesions retain an aldehyde group and therefore have the potential to form ICLs [87]. The in vitro genotoxic properties of BDA are clearly manifested by its ability to induce gene mutations both in bacteria [88, 89] and in mammalian cells [90].
In vitro BDA was reported to be positive in the alkaline elution assay with Chinese hamster ovary cells for both DNA strand breaks and crosslinks induction at millimolar concentrations [91], whereas a comet assay on BDA-treated L5178Y tk+/− cells showed a dose-dependent increase only in SSBs, mostly evident at toxic doses [90].
The greatest degree of uncertainty concerns the carcinogenic mode of action of furan. Is furan directly or indirectly genotoxic? What is the contribution of oxidative DNA damage to its tumorigenicity? A very low level of furan-induced DNA adducts was observed in vivo, and these were not identical to BDA-induced DNA adducts [92]. It is also unclear whether furan induces gene mutations in vivo [93, 94]. Nevertheless, there is convincing evidence that chronic exposure to furan induces micronuclei, chromosome aberrations and DSBs in proliferating, but not in quiescent, splenocytes from mice and rats. DNA lesions responsible for these effects remain undefined, and it is unclear whether the observed chromosomal instability is due to direct damage to DNA (formation of ICLs or DNA adducts) or is the consequence of secondary events associated with oxidative stress-induced DNA damage.
A similarly complex scenario can also be derived from the results of the in vivo comet assays which reveal that furan-induced DNA damage is dependent on treatment protocol and dose. Thus, no induction of SSBs and/or ICLs was observed in the liver of mice chronically exposed per os to furan (28-days, 2-15 mg/kg bw), whereas both types of DNA lesion were increased after a single acute oral dose (250 mg/kg bw) [95, 96]. These data suggest that high levels of DNA lesions must be introduced into DNA to be revealed by the ‘modified’ comet assay, most likely because of the limited sensitivity of this assay in identifying DNA ICLs.
The kinetics of comet assays on the liver of mice chronically exposed to furan revealed that SSBs appeared rapidly after exposure and disappeared within a few hours post-treatment [97]. This observation might explain some of the negative results observed in comet assays in which liver cells were analyzed at late times post-treatment [98]. In addition, they also suggest that repair of oxidized DNA bases might underlie this set of rapidly formed and resealed SSBs. Indeed, the increased number of SSBs following digestion with Fpg and EndoIII of liver DNA from furan treated animals confirms the presence of oxidatively damaged DNA. Finally, increased levels of DNA 8-oxoGua (and ROS) have been observed in liver and blood of furan-treated rats and mice [99–100].
In the absence of a clear identification and detection of DNA adducts, comet assay results were useful to evidence, at least at high dose level, the formation of ICLs, a lesion with the highest potential of causing chromosomal aberrations. At the same time also oxidative DNA damage was demonstrated. It was out of comet capability the assessment of the relative weight of each mechanism in the formation of chromosomal aberrations. Moreover, in vivo comet assays proved to be useful for searching DNA damage in tumor target and non-target organs, demonstrating specific induction of DNA lesions in the liver, the target organ for furan-induced carcinogenesis.
Concluding Remarks
Comet assay is a versatile genotoxicity test that measures DNA breaks or AP sites induced directly by physical and chemical agents or produced during repair processes of primary lesions. The assay has several assets but also some limitations as summarized in Table 2. One of its great assets is the applicability to any type of cells, cycling and not cycling, which translates into the possibility of using it to test the effects in a variety of tumor targets and site-of-contact organs of experimental rodents. Indeed, comet assay data may contribute to characterize the mode of action of suspect chemical carcinogens, showing the possible induction of DNA damage at tumor target sites. Furthermore, one comet assay variant has been developed to detect an increase of oxidized DNA bases. This assay may contribute to assess the relative weights of DNA damage induced directly by covalent binding of chemicals to DNA vs DNA damage induced indirectly by an increase of ROS elicited by chemical-induced oxidative stress.
Table 2.
Pros and cons of in vivo comet assay within a regulatory context
| Pros | Cons |
|---|---|
| Applicable to virtually all animal models | Measurement of transient DNA lesion instead of irreversible gene and chromosomal mutations |
| No specific rodent strain requirement | Relative increase of readouts vs. controls instead of absolute numbers of mutations |
| Applicable to multiple organs | Limited standardization of assay procedures |
| Possibility to be integrated with other complementary short term genotoxicity assays in the same animals | Sensitive to indirect genotoxicity mechanisms linked to toxicity and cellular stress |
| Low cost | Insensitive to some genotoxicity modes of action (aneugenicity, DNA crosslinking) |
Despite these capacities, comet assay still suffers of important limitations that limit its usefulness. The assay conditions have not yet been stringently established and this has made sometimes difficult to compare results obtained by different laboratories. This might be an issue especially for a test that measures not an irreversible genotoxic effect, like micronuclei or gene mutations, but a transient, dynamic, still repairable DNA damage. Improved test standardization would also enhance the applicability of the assay in the field of human biomonitoring, helping to dissect the main variables influencing the results (e.g. sex, age, life style and genetics of DNA repair) and to better exploit studies of human exposed cohorts for risk assessment.
In addition, comet assay data do not return a number of genetic alterations, but a variation of parameters with respect to untreated controls. This is why proposals have been made to calibrate these parameters vs. number of DNA breaks, using ionizing radiation dose–effect relationships, but this promising approach needs to be further developed. When these issues are improved, comet assay will display its full potential within a complementary battery of in vivo tests, from organ-specific DNA adduct measurement to erythrocyte MN frequency, to characterize the mechanisms of toxicity and carcinogenicity of chemicals, which are increasingly considered to set human exposure limits.
Acknowledgements
The authors thank Peter Karran for manuscript revision. MB is currently a member of the CONTAM panel of EFSA. E.C. is currently a member of the Working Group on flavorings of EFSA.
Contributor Information
Eugenia Cordelli, Territorial and Production Systems Sustainability Department, Health Protection Technology Division, ENEA, CR Casaccia, Via Anguillarese 301, Rome 00123, Italy.
Margherita Bignami, Department of Environment and Health, Istituto Superiore di Sanità, Viale Regina Elena 299, Rome 00161, Italy.
Francesca Pacchierotti, Territorial and Production Systems Sustainability Department, Health Protection Technology Division, ENEA, CR Casaccia, Via Anguillarese 301, Rome 00123, Italy.
Conflict of Interest Statement
None declared.
References
- 1. Azqueta A, Collins AR. The essential comet assay: a comprehensive guide to measuring DNA damage and repair. Arch Toxicol 2013;87:949–68. [DOI] [PubMed] [Google Scholar]
- 2. Azqueta A, Muruzabal D, Boutet-Robinet E et al. Technical recommendations to perform the alkaline standard and enzyme-modified comet assay in human biomonitoring studies. Mutat Res 2019;843:24–32. [DOI] [PubMed] [Google Scholar]
- 3. Møller P Genotoxicity of environmental agents assessed by the alkaline comet assay. Basic Clin Pharmacol Toxicol 2005;96:1–42. [PubMed] [Google Scholar]
- 4. Speit G, Vasquez M, Hartmann A. The comet assay as an indicator test for germ cell genotoxicity. Mutat Res 2009;681:3–12. [DOI] [PubMed] [Google Scholar]
- 5. Costa-Amaral IC, Carvalho LVB, Santos MVC et al. Environmental assessment and evaluation of oxidative stress and genotoxicity biomarkers related to chronic occupational exposure to benzene. Int J Environ Res Public Health 2019;16:2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ladeira C, Smajdova L. The use of genotoxicity biomarkers in molecular epidemiology: applications in environmental, occupational and dietary studies. AIMS Genet 2017;4:166–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. de Lapuente J, Lourenço J, Mendo SA et al. The comet assay and its applications in the field of ecotoxicology: a mature tool that continues to expand its perspectives. Front Genet 2015;6:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Santos CL, Pourrut B, Ferreira de Oliveira JM. The use of comet assay in plant toxicology: recent advances. Front Genet 2015;6:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gajski G, Žegura B, Ladeira C et al. The comet assay in animal models: from bugs to whales - (part 1 invertebrates). Mutat Res 2019;779:82–113. [DOI] [PubMed] [Google Scholar]
- 10. Crebelli R, Caiola S, Conti L et al. Can sustained exposure to PFAS trigger a genotoxic response? A comprehensive genotoxicity assessment in mice after subacute oral administration of PFOA and PFBA. Regul Toxicol Pharmacol 2019;106:169–77. [DOI] [PubMed] [Google Scholar]
- 11. Cordelli E, Keller J, Eleuteri P et al. No genotoxicity in rat blood cells upon 3- or 6-month inhalation exposure to CeO2 or BaSO4 nanomaterials. Mutagenesis 2017;32:13–22. [DOI] [PubMed] [Google Scholar]
- 12. Booth ED, Rawlinson PJ, Maria Fagundes P et al. Regulatory requirements for genotoxicity assessment of plant protection product active ingredients, impurities, and metabolites. Environ Mol Mutagen 2017;58:325–44. [DOI] [PubMed] [Google Scholar]
- 13. Collins AR, Oscoz AA, Brunborg G et al. The comet assay: topical issues. Mutagenesis 2008;23:143–51. [DOI] [PubMed] [Google Scholar]
- 14. Abbotts R, Wilson DM 3rd. Coordination of DNA single strand break repair. Free Radic Biol Med 2017;107:228–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Collins AR Measuring oxidative damage to DNA and its repair with the comet assay. Biochim Biophys Acta 1840;2014:794–800. [DOI] [PubMed] [Google Scholar]
- 16. David SS, Williams SD. Chemistry of glycosylases and endonucleases involved in base-excision repair. Chem Rev 1998;98:1221–61. [DOI] [PubMed] [Google Scholar]
- 17. Speit G, Schütz P, Bonzheim I et al. Sensitivity of the FPG protein towards alkylation damage in the comet assay. Toxicol Lett 2004;146:151–8. [DOI] [PubMed] [Google Scholar]
- 18. Rosenquist TA, Zharkov DO, Grollman AP. Cloning and characterization of mammalian 8-oxoguanine DNA glycosylase. Proc Natl Acad Sci U S A 1997;94:7429–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Smith CC, O’Donovan MR, Martin EA. hOGG1 recognizes oxidative damage using the comet assay with greater specificity than FPG or ENDOIII. Mutagenesis 2006;21:185–90. [DOI] [PubMed] [Google Scholar]
- 20. Duthie SJ, McMillan P. Uracil misincorporation in human DNA detected using single cell gel electrophoresis. Carcinogenesis 1997;18:1709–14. [DOI] [PubMed] [Google Scholar]
- 21. Collins A, Dusinska M, Horska A. Detection of alkylation damage in human lymphocyte DNA with the comet assay. Acta Biochim Pol 2001;48:611–4. [PubMed] [Google Scholar]
- 22. Henkler F, Stolpmann K, Luch A. Exposure to polycyclic aromatic hydrocarbons: bulky DNA adducts and cellular responses. Experientia Suppl 2012;101:107–31. [DOI] [PubMed] [Google Scholar]
- 23. Baird WM, Hooven LA, Mahadevan B. Carcinogenic polycyclic aromatic hydrocarbon-DNA adducts and mechanism of action. Environ Mol Mutagen 2005;45:106–14. [DOI] [PubMed] [Google Scholar]
- 24. Veglia F, Matullo G, Vineis P. Bulky DNA adducts and risk of cancer: a meta-analysis. Cancer Epidemiol Biomarkers Prev 2003;12:157–60. [PubMed] [Google Scholar]
- 25. Staresincic L, Fagbemi AF, Enzlin JH et al. Coordination of dual incision and repair synthesis in human nucleotide excision repair. EMBO J 2009;28:1111–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gedik CM, Ewen SW, Collins AR. Single-cell gel electrophoresis applied to the analysis of UV-C damage and its repair in human cells. Int J Radiat Biol 1992;62:313–20. [DOI] [PubMed] [Google Scholar]
- 27. Ngo LP, Owiti NA, Swartz C et al. Sensitive CometChip assay for screening potentially carcinogenic DNA adducts by trapping DNA repair intermediates. Nucleic Acids Res 2020;48:e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Collins AR, Mitchell DL, Zunino A et al. UV-sensitive rodent mutant cell lines of complementation groups 6 and 8 differ phenotypically from their human counterparts. Environ Mol Mutagen 1997;29:152–60. [PubMed] [Google Scholar]
- 29. Hashimoto S, Anai H, Hanada K. Mechanisms of interstrand DNA crosslink repair and human disorders. Genes and Environment 2016;38:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rothfuss A, Grompe M. Repair kinetics of genomic interstrand DNA cross-links: evidence for DNA double-strand break-dependent activation of the Fanconi anemia/BRCA pathway. Mol Cell Biol 2004;24:123–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. OECD (2016), Test No. 489: In Vivo Mammalian Alkaline Comet Assay, OECDGuidelines for the Testing of Chemicals, Section 4, OECD Publishing, Paris. doi: 10.1787/9789264264885-en. [DOI] [Google Scholar]
- 32. Lindahl T Instability and decay of the primary structure of DNA. Nature 1993;362:709–16. [DOI] [PubMed] [Google Scholar]
- 33. Lindahl T, Barnes D. Repair of endogenous DNA damage. Cold Spring Harb Symp Quant Biol 2000;65:127–33. [DOI] [PubMed] [Google Scholar]
- 34. Nakamura J, Mutiu E, Sharma V et al. The endogenous exposome. DNA Repair 2014;19:3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tubbs A, Nussenzweig A. Endogenous DNA damage as a source of genomic instability in cancer. Cell 2017;168:644–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Collins AR, Cadet J, Möller L et al. Are we sure we know how to measure 8-oxo-7,8-dihydroguanine in DNA from human cells? Arch Biochem Biophys 2004;423:57–65. [DOI] [PubMed] [Google Scholar]
- 37. Dogliotti E, Bignami M. DNA repair and genetic instability. Endogenous and exogenous sources of damage and hereditary syndromes In: Wild CP, Weiderpass E, Stewart BW (eds). World Cancer Report 2020. International Agency for Research on Cancer, 170–80. [Google Scholar]
- 38. Singh NP, McCoy MT, Tice RR et al. A simple technique for quantitation of low levels of DNA damage in individual cells. Exp Cell Res 1988;175:184–91. [DOI] [PubMed] [Google Scholar]
- 39. Azqueta A, Gutzkow KB, Brunborg G et al. Towards a more reliable comet assay: optimising agarose concentration, unwinding time and electrophoresis conditions. Mutat Res 2011;724:41–5. [DOI] [PubMed] [Google Scholar]
- 40. Enciso JM, Gutzkow KB, Brunborg G. et al. Standardisation of the in vitro comet assay: influence of lysis time and lysis solution composition on the detection of DNA damage induced by X-rays. Mutagenesis 2018; 33: 25-30. [DOI] [PubMed] [Google Scholar]
- 41. Glei M, Schneider T, Schlörmann W. Comet assay: an essential tool in toxicological research. Arch Toxicol 2016;90:2315–36. [DOI] [PubMed] [Google Scholar]
- 42. Ersson C, Møller P, Forchhammer L et al. An ECVAG inter-laboratory validation study of the comet assay: inter-laboratory and intra-laboratory variations of DNA strand breaks and FPG-sensitive sites in human mononuclear cells. Mutagenesis 2013;28:279–86. [DOI] [PubMed] [Google Scholar]
- 43. Forchhammer L, Johansson C, Loft S et al. Variation in the measurement of DNA damage by comet assay measured by the ECVAG inter-laboratory validation trial. Mutagenesis 2010;25:113–23. [DOI] [PubMed] [Google Scholar]
- 44. Forchhammer L, Ersson C, Loft S et al. Inter-laboratory variation in DNA damage using a standard comet assay protocol. Mutagenesis 2012;27:665–72. [DOI] [PubMed] [Google Scholar]
- 45. Johansson J, Møller P, Forchhammer L et al. An ECVAG trial on assessment of oxidative damage to DNA measured by the comet assay. Mutagenesis 2010;25:25–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ahnstrom G, Erixon K. Measurement of strand breaks by alkaline denaturation and hydroxyapatite chromatography In: Friedberg EC, Hanawalt PC (eds). DNA Repair. A Laboratory Manual of Research Procedures. New York: Marcel Dekker, 1981, 403–18. [Google Scholar]
- 47. Møller P, Jantzen K, Løhr M et al. Searching for assay controls for the Fpg- and hOGG1-modified comet assay. Mutagenesis 2018;33:9–19. [DOI] [PubMed] [Google Scholar]
- 48. Møller P, Cooke MS, Collins A et al. Harmonising measurements of 8-oxo-7,8-dihydro-2′-deoxyguanosine in cellular DNA and urine. Free Radic Res 2012;46:541–53. [DOI] [PubMed] [Google Scholar]
- 49. Collins AR, El Yamani N, Lorenzo Y et al. Controlling variation in the comet assay. Front Genet 2014;5:359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zainol M, Stout J, Bowman KJ et al. ECVAG introducing a true internal standard for the comet assay to minimize intra- and inter-experiment variability in measures of DNA damage and repair. Nucleic Acids Res 2009. doi: 10.1093/nar/gkp826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ueno S, Kashimoto T, Susa N et al. Assessment of DNA damage in multiple organs of mice after whole body X-irradiation using the comet assay. Mutat Res 2007;634:135–45. [DOI] [PubMed] [Google Scholar]
- 52.Mammalian Mutagenesis Study Group of the Environmental Mutagen Society of Japan. 2014. Atlas of Comet Assay Images. Tokyo, Japan: Scientist Press Co., Ltd. [Google Scholar]
- 53. Uno Y, Kojima H, Omori T et al. JaCVAM-organized international validation study of the in vivo rodent alkaline comet assay for detection of genotoxic carcinogens: II. Summary of definitive validation study results. Mutat Res Genet Toxicol Environ Mutagen 2015;786-788:45–76. [DOI] [PubMed] [Google Scholar]
- 54. ICH 2011. International Conference on Harmonisation of technical requirements for registration of pharmaceuticals for human use ICH harmonised tripartite guideline. Guidance on genotoxicity testing and data interpretation for pharmaceuticals intended for human use. S2(R1). Current Step 4 version dated 9 November 2011.
- 55. ECHA (European Chemical Agency) Guidance on Information Requirements and Chemical Safety Assessment, Chapter R.7a: Endpoint Specific Guidance, 2017, Version 6.0 https://echa.europa.eu/documents/10162/13632/information_requirements_r7a_en.pdf/e4a2a18f-a2bd-4a04-ac6d-0ea425b2567f (8 December 2020, date last accessed).
- 56. OECD , 2020. OECD Guideline for the Testing of Chemicals: Transgenic Rodent Somatic and Germ Cell Gene Mutation Assays (TG 488). 10.1787/9789264203907-en (8 December 2020, date last accessed). [DOI]
- 57. OECD (2016), Test No. 474: Mammalian Erythrocyte Micronucleus Test, OECD Guidelines for the Testing of Chemicals, Section 4, OECD Publishing, Paris. doi: 10.1787/9789264264762-en. [DOI] [Google Scholar]
- 58. OECD (2016), Test No. 475: Mammalian Bone Marrow Chromosomal Aberration Test, OECD Guidelines for the Testing of Chemicals, Section 4, OECD Publishing, Paris. doi: 10.1787/9789264264786-en. [DOI] [Google Scholar]
- 59. EFSA Scientific Committee, 2011 Scientific opinion on genotoxicity testing strategies applicable to food and feed safety assessment. EFSA J 2011;9:2379. [Google Scholar]
- 60. EFSA Scientific Committee Clarification of some aspects related to genotoxicity assessment. EFSA J 2017;15:5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bhagat J Combinations of genotoxic tests for the evaluation of group 1 IARC carcinogens. J Appl Toxicol 2018;38:81–99. [DOI] [PubMed] [Google Scholar]
- 62. Kirkland D, Uno Y, Luijten M et al. In vivo genotoxicity testing strategies: report from the 7th international workshop on genotoxicity testing (IWGT). Mutat Res 2019;847:403035. [DOI] [PubMed] [Google Scholar]
- 63. Golfier S, Jost G, Pietsch H et al. Dicentric chromosomes and gamma-H2AX foci formation in lymphocytes of human blood samples exposed to a CT scanner: a direct comparison of dose response relationships. Radiat Prot Dosimetry 2009;134:55–61. [DOI] [PubMed] [Google Scholar]
- 64. Nikolova T, Marini F, Kaina B. Genotoxicity testing: comparison of the γH2AX focus assay with the alkaline and neutral comet assays. Mutat Res 2017;822:10–8. [DOI] [PubMed] [Google Scholar]
- 65. Kopp B, Khoury L, Audebert M. Validation of the γH2AX biomarker for genotoxicity assessment: a review. Arch Toxicol 2019;93:2103–14. [DOI] [PubMed] [Google Scholar]
- 66. Gollapudi BB, Johnson GE, Hernandez LG et al. Quantitative approaches for assessing dose-response relationships in genetic toxicology studies. Environ Mol Mutagen 2013;54:8–18. [DOI] [PubMed] [Google Scholar]
- 67. Heflich RH, Johnson GE, Zeller A et al. Mutation as a toxicological endpoint for regulatory decision-making. Environ Mol Mutagen 2020;61:34–41. [DOI] [PubMed] [Google Scholar]
- 68. Zeller A, Duran-Pacheco G, Guérard M. An appraisal of critical effect sizes for the benchmark dose approach to assess dose-response relationships in genetic toxicology. Arch Toxicol 2017;91:3799–807. [DOI] [PubMed] [Google Scholar]
- 69. Guérard M, Johnson G, Dertinger S et al. Dose-response relationship of temozolomide, determined by the Pig-a, comet, and micronucleus assay. Arch Toxicol 2017;91:2443–53. [DOI] [PubMed] [Google Scholar]
- 70. COM (Committee on Mutagenicity of Chemicals in Food, Consumer Products and the Environment) 2018. Statement 2018/S1: Statement on the Quantitative Approaches to the Assessment of Genotoxicity Data. https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/704462/COM_statement_on_quantitative_approaches.pdf (8 December 2020, date last accessed).
- 71. EFSA Scientific committee; scientific opinion on the applicability of the margin of exposure approach for the safety assessment of impurities which are both genotoxic and carcinogenic in substances added to food/feed. EFSA J 2012;10:2578. [Google Scholar]
- 72. EFSA CONTAM Panel (EFSA Panel on Contaminants in the Food Chain), Schrenk D, Bodin L et al. Scientific opinion on the risk assessment of ochratoxin a in food. EFSA J 2020 2020;18:6113. [Google Scholar]
- 73. EFSA CONTAM Panel (EFSA Panel on Contaminants in the Food Chain), Knutsen HK, Alexander J et al. Scientific opinion on the risks for public health related to the presence of furan and methylfurans in food. EFSA J 2017 2017;15:5005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Palma N, Cinelli S, Sapora O et al. Ochratoxin A-induced mutagenesis in mammalian cells is consistent with the production of oxidative stress. Chem ResToxicol 2007;20:1031–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Mosesso P, Cinelli S, Pinero J et al. In vitro cytogenetic results supporting a DNA nonreactive mechanism for ochratoxin A, potentially relevant for its carcinogenicity. Chem Res Toxicol 2008;21:1235–43. [DOI] [PubMed] [Google Scholar]
- 76. Ali R, Mittelstaedt RA, Shaddock JG et al. Comparative analysis of micronuclei and DNA damage induced by Ochratoxin A in two mammalian cell lines. Mutat Res 2011;723:58–64. [DOI] [PubMed] [Google Scholar]
- 77. Liu J, Wang Y, Cui J et al. Ochratoxin A induces oxidative DNA damage and G1 phase arrest in human peripheral blood mononuclear cells in vitro. Toxicol Lett 2012;211:164–71. [DOI] [PubMed] [Google Scholar]
- 78. Ali R, Guo X, Lin H et al. Mutant frequency in comparison to oxidative DNA damage induced by ochratoxin A in L5178Y tk+/− (3.7.2C) mouse lymphoma cells. Drug Chem Toxicol 2014;37:227–32. [DOI] [PubMed] [Google Scholar]
- 79. Mally A, Pepe G, Ravoori S et al. Ochratoxin A causes DNA damage and cytogenetic effects but no DNA adducts in rats. Chem Res Toxicol 2005;18:1253–61. [DOI] [PubMed] [Google Scholar]
- 80. Hibi D, Suzuki Y, Ishii Y et al. Site-specific in vivo mutagenicity in the kidney of gpt delta rats given a carcinogenic dose of ochratoxin A. Toxicol Sci 2011;122:406–14. [DOI] [PubMed] [Google Scholar]
- 81. Kuroda K, Hibi D, Ishii Y et al. Ochratoxin A induces DNA double-strand breaks and large deletion mutations in the carcinogenic target site of gpt delta rats. Mutagenesis 2014;29:27–36. [DOI] [PubMed] [Google Scholar]
- 82. Kuroda K, Hibi D, Ishii Y et al. Role of p53 in the progression from ochratoxin A-induced DNA damage to gene mutations in the kidneys of mice. Toxicol Sci 2015;144:65–76. [DOI] [PubMed] [Google Scholar]
- 83. Hibi D, Kijima A, Suzuki Y et al. Effects of p53 knockout on ochratoxin A-induced genotoxicity in p53-deficient gpt delta mice. Toxicology 2013;304:92–9. [DOI] [PubMed] [Google Scholar]
- 84. Mally A, Volkel W, Amberg A et al. Functional, biochemical, and pathological effects of repeated oral administration of ochratoxin A to rats. Chem Res Toxicol 2005;18:1242–52. [DOI] [PubMed] [Google Scholar]
- 85. Mantle PG, Faucet-Marquis V, Manderville RA et al. Structures of covalent adducts between DNA and ochratoxin A: a new factor in debate about genotoxicity and human risk assessment. Chem Res Toxicol 2010;23:89–98. [DOI] [PubMed] [Google Scholar]
- 86. Domijan AM, Zeljezic D, Kopjar N et al. Standard and Fpg-modified comet assay in kidney cells of ochratoxin A- and fumonisinB (1)-treated rats. Toxicology 2006;222:53–9. [DOI] [PubMed] [Google Scholar]
- 87. Halila S, Velasco T, De Clercq P et al. Fine-tuning furan toxicity: fast and quantitative DNA interchain cross-link formation upon selective oxidation of a furan containing oligonucleotide. Chem Commun 2005;21:936–8. [DOI] [PubMed] [Google Scholar]
- 88. Peterson LA, Naruko KC, Predecki DP. A reactive metabolite of furan, cis-2-butene-1,4-dial, is mutagenic in the Ames assay. Chem Res Toxicol 2000;13:531–4. [DOI] [PubMed] [Google Scholar]
- 89. Byrns MC, Vu CC, Neidigh JW et al. Detection of DNA adducts derived from the reactive metabolite of furan, cis-2-butene-1,4-dial. Chem Res Toxicol 2006;19:414–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Kellert M, Brink A, Richter I et al. Tests for genotoxicity and mutagenicity of furan and its metabolite cis-2-butene-1,4-dial in L5178Y Tk+/− mouse lymphoma cells. Mutat Res 2008;657:127–32. [DOI] [PubMed] [Google Scholar]
- 91. Marinari UM, Ferro M, Bassi AM et al. DNA-damaging activity of biotic and 2247 xenobiotic aldehydes in chinese hamster ovary cells. Cell Biochem Funct 1984;2:243–8. [DOI] [PubMed] [Google Scholar]
- 92. Neuwirth C, Mosesso P, Pepe G et al. Furan carcinogenicity: DNA binding and genotoxicity of furan in rats in vivo. Mol Nutr Food Res 2012;56:1363–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Terrell AN, Huynh M, Grill AE et al. Mutagenicity of furan in female big blue B6C3F1 mice. Mutat Res Genet Toxicol and Environ Mutagen 2014;770:46–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Hibi D, Yokoo Y, Suzuki Y et al. Lack of genotoxic mechanisms in early-stage furan-induced hepatocellular tumorigenesis in gpt delta rats. J Appl Toxicol 2017;37:142–9. [DOI] [PubMed] [Google Scholar]
- 95. Leopardi P, Cordelli E, Villani P et al. Assessment of in vivo genotoxicity of the rodent carcinogen furan: evaluation of DNA damage and induction of micronuclei in mouse splenocytes. Mutagenesis 2010;25:57–62. [DOI] [PubMed] [Google Scholar]
- 96. Cordelli E, Leopardi P, Villani P et al. Toxic and genotoxic effects of oral administration of furan in mouse liver. Mutagenesis 2010;25:305–14. [DOI] [PubMed] [Google Scholar]
- 97. Ding W, Petibone DM, Latendresse JR et al. In vivo genotoxicity of furan in F344 rats at cancer bioassay doses. Toxicol Appl Pharmacol 2012;261:164–71. [DOI] [PubMed] [Google Scholar]
- 98. McDaniel LP, Ding W, Dobrovolsky VN et al. Genotoxicity of furan in big blue rats. Mutat Res 2012;742:72–8. [DOI] [PubMed] [Google Scholar]
- 99. Hickling KC, Hitchcock JM, Oreffo V et al. Evidence of oxidative stress and associated DNA damage, increased proliferative drive, and altered gene expression in rat liver produced by the cholangiocarcinogenic agent furan. Toxicol Pathol 2010;38:230–43. [DOI] [PubMed] [Google Scholar]
- 100. Wang E, Chen F, Hu X et al. Protective effects of apigenin against furan-induced toxicity in mice. Food Funct 2014;5:1804–12. [DOI] [PubMed] [Google Scholar]