

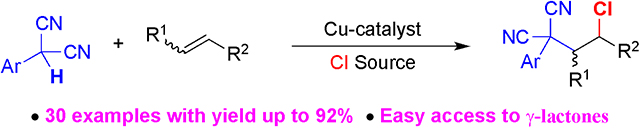
Abstract
A copper-catalyzed carbochlorination of alkenes with aryl malononitriles and chloride is disclosed. This net oxidative transformation proceeds with activated and unactivated alkenes with moderate to excellent yields. Mechanism experiments suggest addition of the malononitrile radical to form a secondary carbon radical which is intercepted by a chloride source. The resultant products can be transformed into biologically important γ-lactones in one further step.
Graphical Abstract
Owing to the high abundance of alkenes, their functionalization to efficiently generate complex molecular scaffolds is a very attractive transformation. In this regard, transition metal catalyzed functionalization of alkenes has been extensively studied in recent years.1 Among different approaches, the addition of radicals to alkenes represents a versatile entry to many adducts with the most work focused on carbon-carbon bond formation. Similarly, carbohalogenation reactions of alkenes have also been investigated.2 These reactions generate valuable organic synthons.3 Despite the advancements in this field, the majority of transformations are restricted to intramolecular alkene functionalization/cyclization.
Single-electron oxidation of a-carbonyls to carbon-centered radicals using transition metals is a well-established strategy.4 Using alkenes as a radical acceptor, vicinally functionalized motifs can be accessed in the presence of a suitable radical trap.5 Early work by the Jahn group showed that electron-rich lithium malonates undergo an intramolecular oxidative carbo-chlorination with tethered alkenes (Scheme 1a).6 They also reported an efficient synthesis of terpenoid analogues through the radical cyclization of ester enolates.7 Similarly, the Burton group demonstrated the oxidative cyclization of 4-pentenylmalonates to form lactones in presence of manganese acetate.8 Recently, the Lin group reported an electrochemical manganese catalyzed chloro-alkylation of alkenes (Scheme 1b).9 However, reactions with aryl malononitriles were unsuccessful or resulted in addition of two equivalents of the malononitrile to the alkene in this electrochemical system. Despite the breadth of these studies, only limited work has been done with malononitriles and the carbon center radicals generated by oxidation of a-aryl malononitriles have not been investigated in alkene functionalization.
Scheme 1.
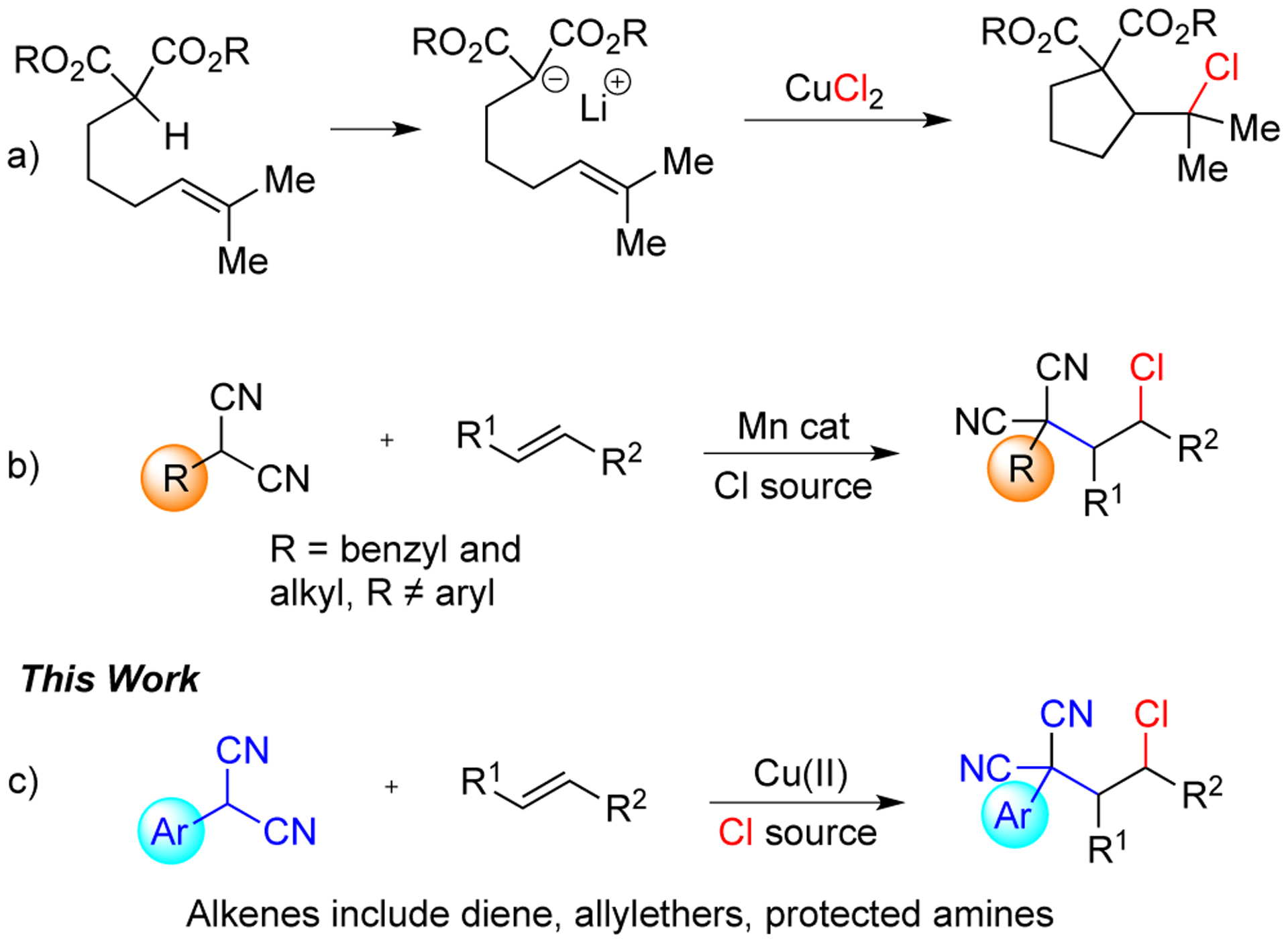
Known Alkene Carbohalogenation Reactions vs This Work
Nitriles are common functional groups occurring in natural products and bioactive molecules.10 In addition, they can be transformed to other important functional groups like aldehydes, esters, amides etc.11 Therefore, reactions that incorporate these groups are highly desirable. Previously our group discovered the oxidative coupling of toluene and alkylnitriles with aryl malononitriles, oxindoles, and related substrates.12 These reactions are believed to proceed by the formation of dimer intermediate that is in equilibrium with the reactive species, the captodative radical monomer, in solution at higher temperatures.13 Inspired by the importance of the nitrile functional group, we proposed to utilize the captodative radical generated from the aryl malononitriles in alkene functionalization. In this work we described a copper-catalyzed alkene carbochlorination reaction. Mechanistic studies were also performed and the synthetic utility was highlighted by post-modification of the product.
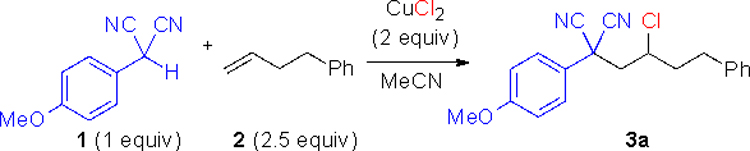
We began our investigation by combining 2 equiv copper chloride (CuCl2), 4-phenyl-1-butene, and 2-p-methoxyphenyl (PMP) malononitrile in acetonitrile. After heating at 100 °C for 24 h, the alkene functionalized product 3a was isolated in 86% yield (Table 1, entry 1). Shorter reaction times resulted in incomplete consumption of the malononitrile. Lesser amounts of CuCl2 decreased the yield (entries 2 and 3). Lowering the reaction temperature to 60 °C was ineffective giving low yield (entry 4). Other metal salts like FeCl3 and CuCl proved ineffective (entry 5). Acetonitrile was found to be the best solvent relative to others (entries 6 and 7). The alkene was used in excess to obtain better reaction rates, and excess alkene could be recovered. Water-free conditions were superior as undried solvents led to lower yields.
Table 1:
Reaction Development
 | ||||
|---|---|---|---|---|
| entry | change to conditions | T (°C) | t (h) | yield 3a (%)b |
| 1 | None | 100 | 24 | 88 (86) |
| 2 | CuCl2 (1.0 equiv) | 100 | 24 | 42 |
| 3 | CuCl2 (1.5 equiv) | 100 | 24 | 63 |
| 4 | None | 60 | 24 | 20 |
| 5 | FeCl3 or CuCl instead of CuCl2 | 100 | 24 | 0 |
| 6 | DMSO instead of MeCN | 100 | 24 | 40 |
| 7 | DMF instead of MeCN | 100 | 24 | 44 |
Reaction conditions: 1 (0.10 mmol), 2 (0.25 mmol,), CuCl2 (0.2 mmol), MeCN (1 mL), 100 °C, 24 h.
Yields as judged by 1HNMR spectrocscopy with CH2Br2 as internal standard. Yield in parentheses is the isolated yield.
With the optimized conditions, the scope of this reaction was examined (Scheme 2). Initially, the same aryl malononitrile (PMP malononitrile) was surveyed and was found to provide moderate to excellent yields with different unactivated alkenes (3a-3s). Notably, straight chain aliphatic alkenes proceed in good yield (3l- 3m). The regioselectivity was confirmed from an X-ray crystal structure of 3d which showed addition of the carbon and chlorine to the terminal and internal positions of the alkene, respectively. Unlike, the previously reported electrochemical alkene carbochlorination,9 this transformation worked well with a diene selectively functionalizing only one double bond (3n). The reaction was able to tolerate functional groups including ester (3o) and N-protected amines (3p). In addition, internal alkenes also provided moderate yields of products and good diastereoselectivity (3q-3s). The X-ray crystal structure of 3s indicates the formation of the trans-isomer as the major diastereomer. Other electron-donating and electron-withdrawing groups on the aryl of the malononitrile gave rise to excellent yields (3t-3u).
Scheme 2. Substrate Scope of Alkene Carbochlorinationa.
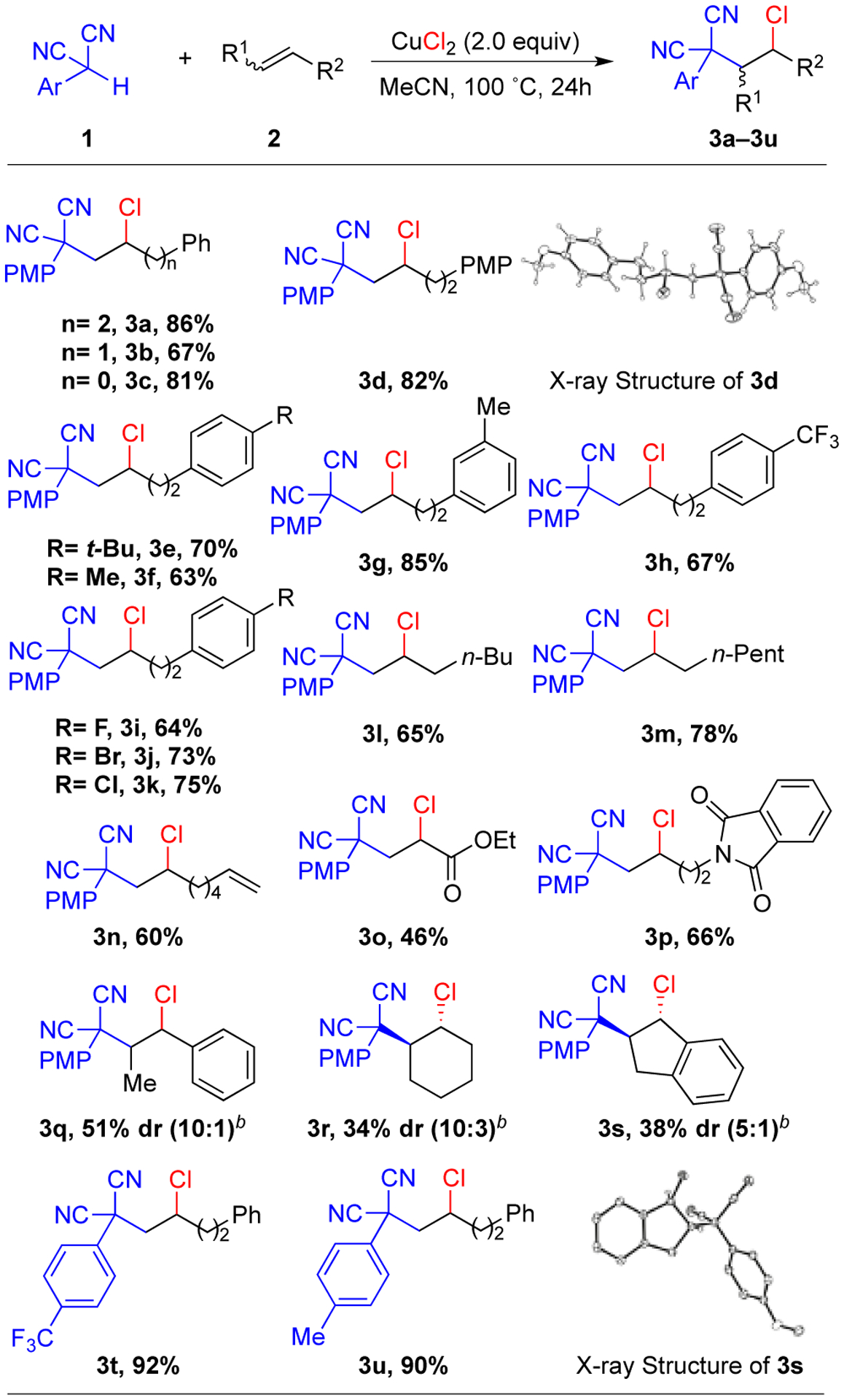
aReaction conditions: 1 (0.2 mmol), 2 (0.5 mmol, 2.5 equiv), CuCl2 (0.4 mmol, 2.0 equiv), MeCN (1 mL), 100 °C, 24 h. b80 °C.
Further studies were undertaken to determine if the reaction was feasible with a catalytic amount of copper. Upon screening oxidants and chloride sources, it was discovered that K2S2O8 and KCl were the best oxidant and chloride source respectively affording 68% isolated yield of the carbochlorinated product in presence of 15 mol % Cu(OTf)2 and 20 mol % 4,4’-di-tert-butyl-2,2’-bipyridine (dtbbpy) as ligand (See Supporting Information for details). In the absence of copper, no product was observed indicating that the K2S2O8 oxidant alone is not sufficient for the reaction to occur. Acetonitrile remained the best solvent.
Like the stoichiometric version, this catalytic process was effective with diverse alkenes and aryl malononitriles (Scheme 3). Again excess alkene was utilized to obtain sufficient rates; upon complete consumption of the limiting malononitrile, the balance of the alkene was recovered as both the unreacted form and the dichlorinated adduct (see below). The reaction can employ allyl phenyl ether (4b-4c). Again, only one double bond of a diene substrate was functionalized (4e), with the remainder of the diene being recovered or undergoing dichlorination of just one double bond. Notably, the reaction conditions are compatible with thio groups (4e); thioethers are often not well-tolerated in oxidative or copper-catalyzed processes. Electron-withdrawing and electron-donating groups on the malononitriles had no adverse effect(4a-4i). A trisubstituted alkene, 1-methylcyclohexene resulted mainly in the malononitrile dimer with trace product. The reaction of vinyl pyridine resulted in polymerization and <10% carbonchlorination. With 1-phenyl-1,3-butadiene, a mixture of products was observed that could not be separated by column chromatography. With aryl cyanoacetates instead of an aryl malononitrile, the reaction with styrene shows moderate yield; however, the product could not be purified. As aryl malononitrile can be transformed to the cyanoacetates, this avenue was not investigated further. A larger scale reaction to form 3a proceeded in lower yield (1 mmol, 46%) presumably due to the heterogeneity of the reaction mixture (KCl, K2S2O8 not fully soluble).
Scheme 3. Substrate Scope under Catalytic Conditionsa.
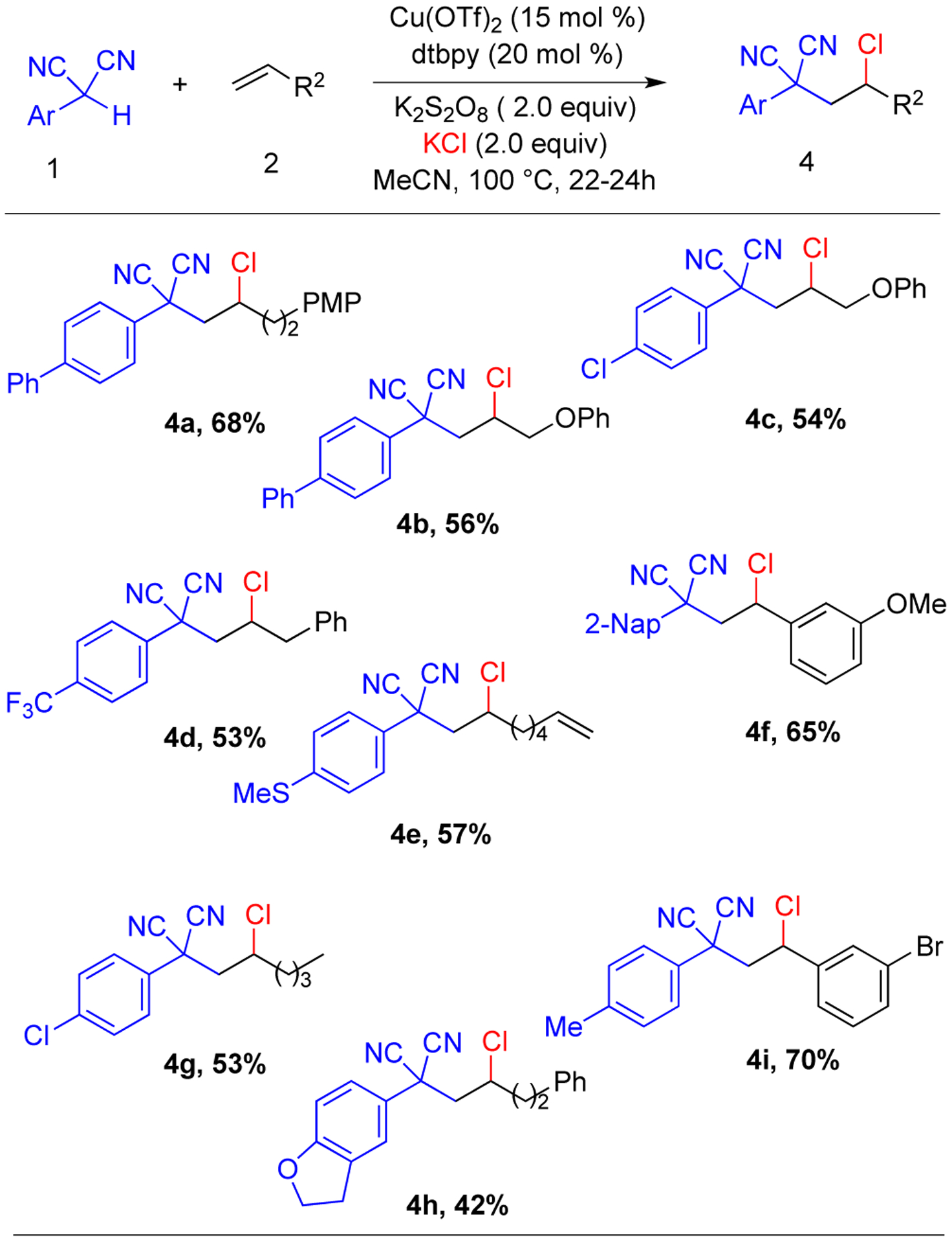
aReaction conditions: 1 (0.2 mmol), 2 (0.5 mmol, 2.5 equiv), Cu(OTf)2 (0.03 mmol), dtbbpy (0.04 mmol), K2S2O8 (0.4 mmol), KCl (0.4 mmol), MeCN (1 mL), 100 °C, 20–24 h.
To determine the possible reaction pathway, the aryl malononitriles dimers were investigated as intermediates since aryl malononitriles are known to readily dimerize under oxidative conditions.12 In addition, dimers formed in the absence of alkene and were observed in reactions with alkene when they were halted prior to completion. With dimer 5 in place of the aryl malononitrile under the standard conditions, 36% yield of product 3a was obtained indicating that dimer could be an intermediate in this reaction (Scheme 4a). A control reaction without the aryl malononitrile formed (3,4-dichlorobutyl)benzene 6 in 34% yield (Scheme 4b). To determine if this dihalide is an intermediate,14 compound 6 was subjected to the standard reaction conditions in place of the alkene. With or without copper salts, no product formed indicating that an SN2 displacement of the terminal chloride by the malononitrile anion is unlikely (Scheme 4c).
Scheme 4.
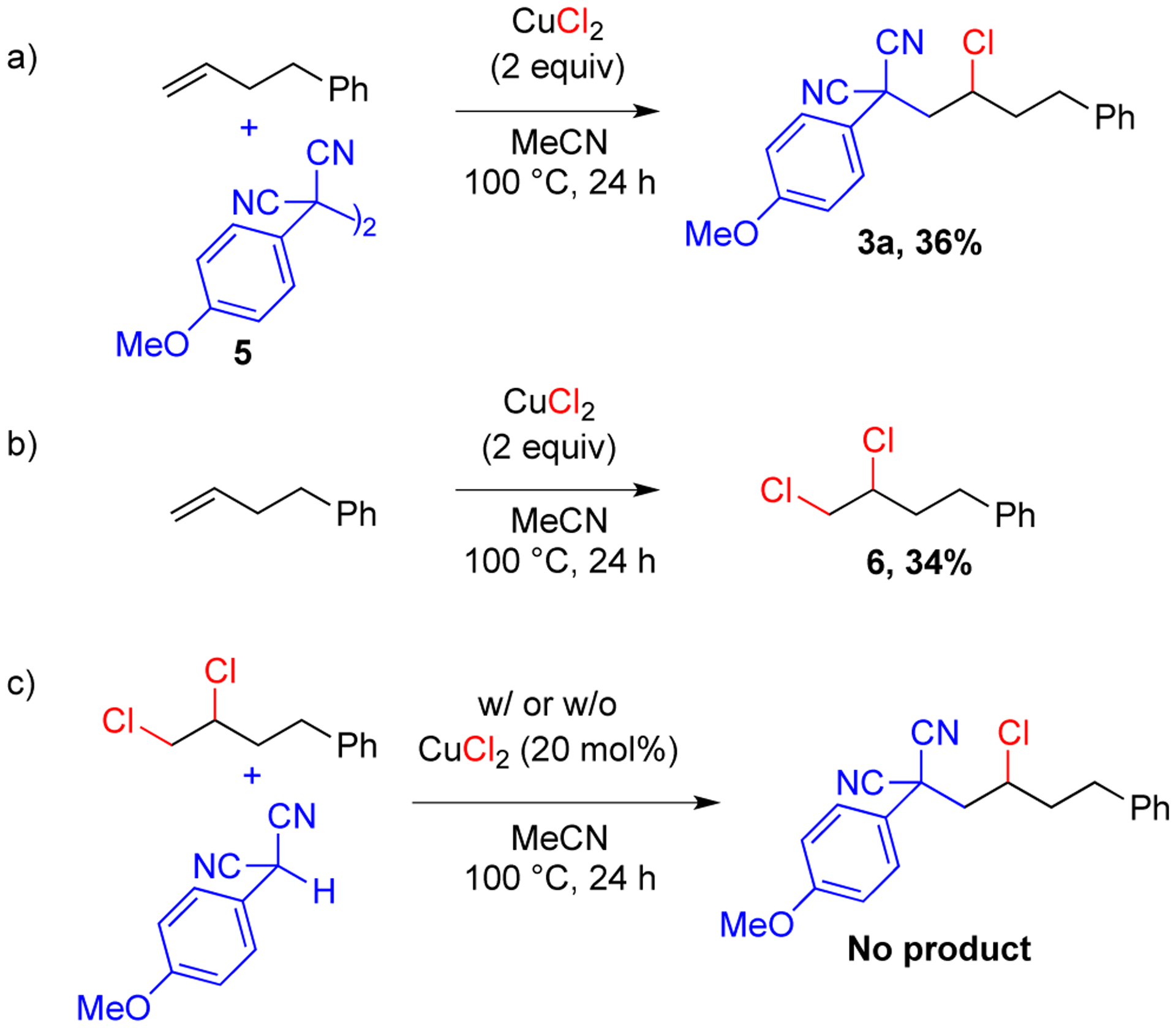
Control Experiments
Both the aryl malononitrile radical (see above) and the aryl malononitrile cation can form under oxidizing conditions. To determine which is involved, the addition to radical probe, 2-(vinylcyclopropyl)benzene, was undertaken (Scheme 5). Under catalytic condition, 24% yield of the cyclopropyl ring opening product 7 was observed (Scheme 5a). Stoichiometric copper conditions gave rise to diene 9 (Scheme 5b). The diene could be formed after the cyclopropane ring opening followed by dehydrochlorination. In support of this hypothesis, diene 9 was observed upon subjecting 7 to condition B. The direct carbochlorination product 8 was not observed under either set of conditions. These results are consistent with addition of aryl malononitrile radical to the alkene forming a secondary alkyl radical that can be intercepted by the copper catalyst.
Scheme 5. Radical Probe Experiments.
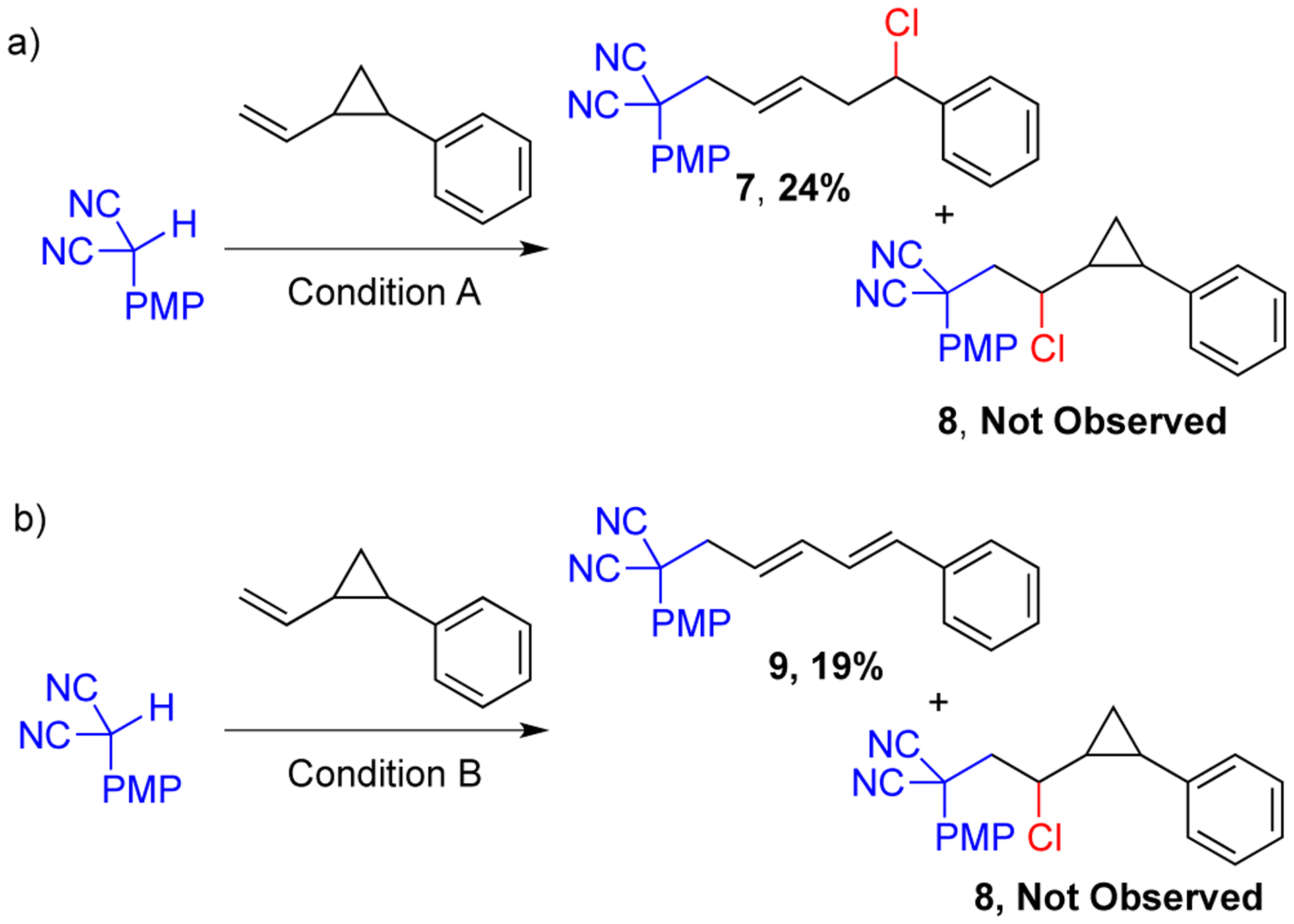
Condition A = Cu(OTf)2 (15 mol %), dtbbpy (20 mol %), K2S2O8 (2 equiv) KCl (2 equiv), MeCN, 100 °C, 22 h. Condition B = CuCl2 (2 equiv), MeCN, 100 °C, 24 h.
Based on the above studies and the literature,15 a possible reaction pathway is outlined in Scheme 6. Initially, the aryl malononitrile oxidizes to form dimer, which is known to be in equilibrium with the carbon center radical A at higher temperature.12 This radical A adds to the terminal carbon of the alkene to generate secondary radical B. This radical combines with the Cu(II) catalyst to provide Cu(III) intermediate C. Reductive elimination from C forms product and generates Cu(I), which can be reoxidized to Cu(II). However, the formation of product through a Cu(II) to Cu(I) pathway could also be possible.16 A pathway involving further oxidation of radical B to a cation is also plausible. To interrogate this possibility, an oxidation resistant nucleophile that could only trap the carbocation, not the radical B, was investigated. Thus, KCN was used in place of KCl under the conditions from Scheme 3 with para-methoxyphenylmalononitrile in combination with both styrene, which can form a stabilized radical/carbocation, and 4-phenyl-1-butene, which would not. Under these conditions, only malononitrile dimer was observed and no alkene addition adducts were seen, which points away from a carbocation mechanism.17
Scheme 6.
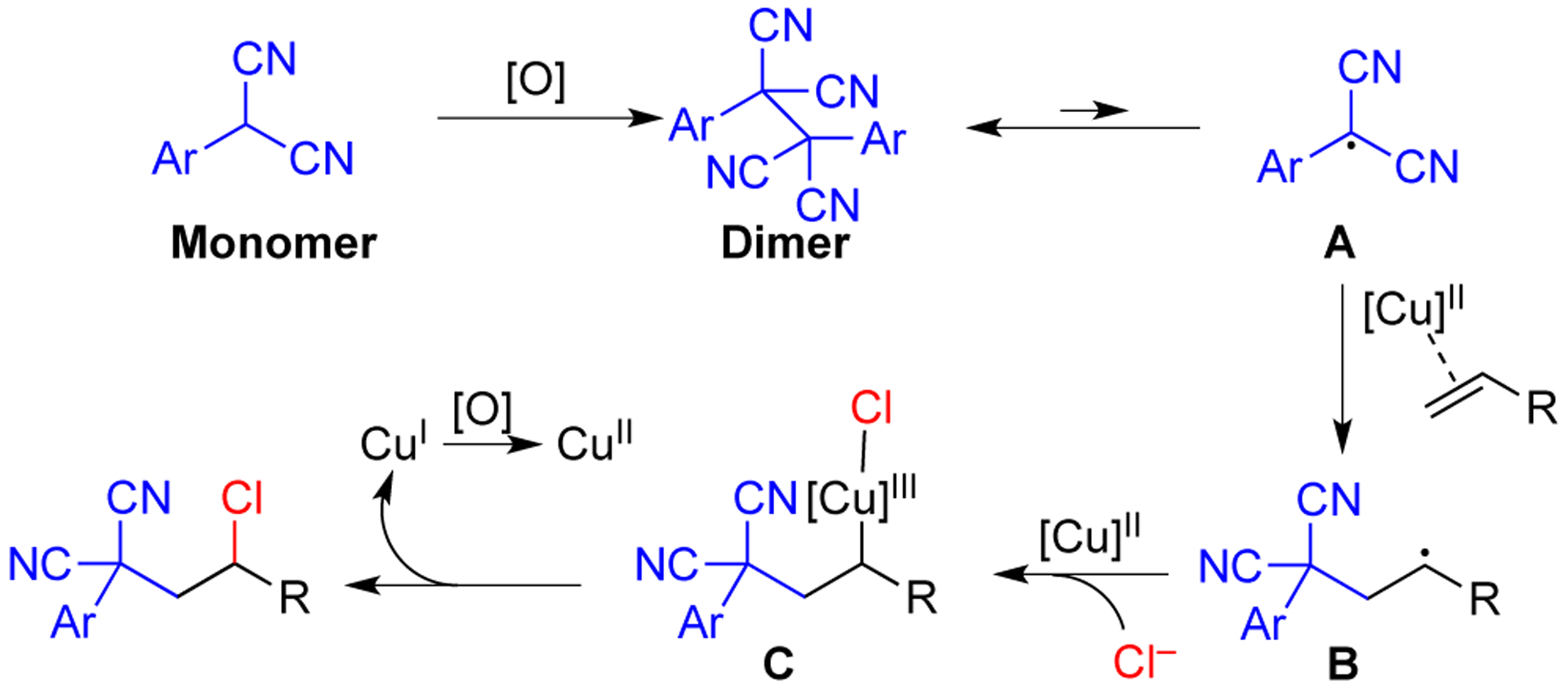
Proposed Mechanism
To highlight the synthetic utility of this reaction, further modifications of the carbochlorinated products were undertaken. Using precedented procedures,18 selective hydrolyses of one of the nitrile groups in the products 3c and 3a were performed to generate amides 10 and 12, respectively (Scheme 7). Further cyclization of the amide oxygen onto the chloride formed γ-lactones 11 and 13. Thus, the reaction products from this method allow access to the γ-lactone backbone with quaternary centers.19
Scheme 7.
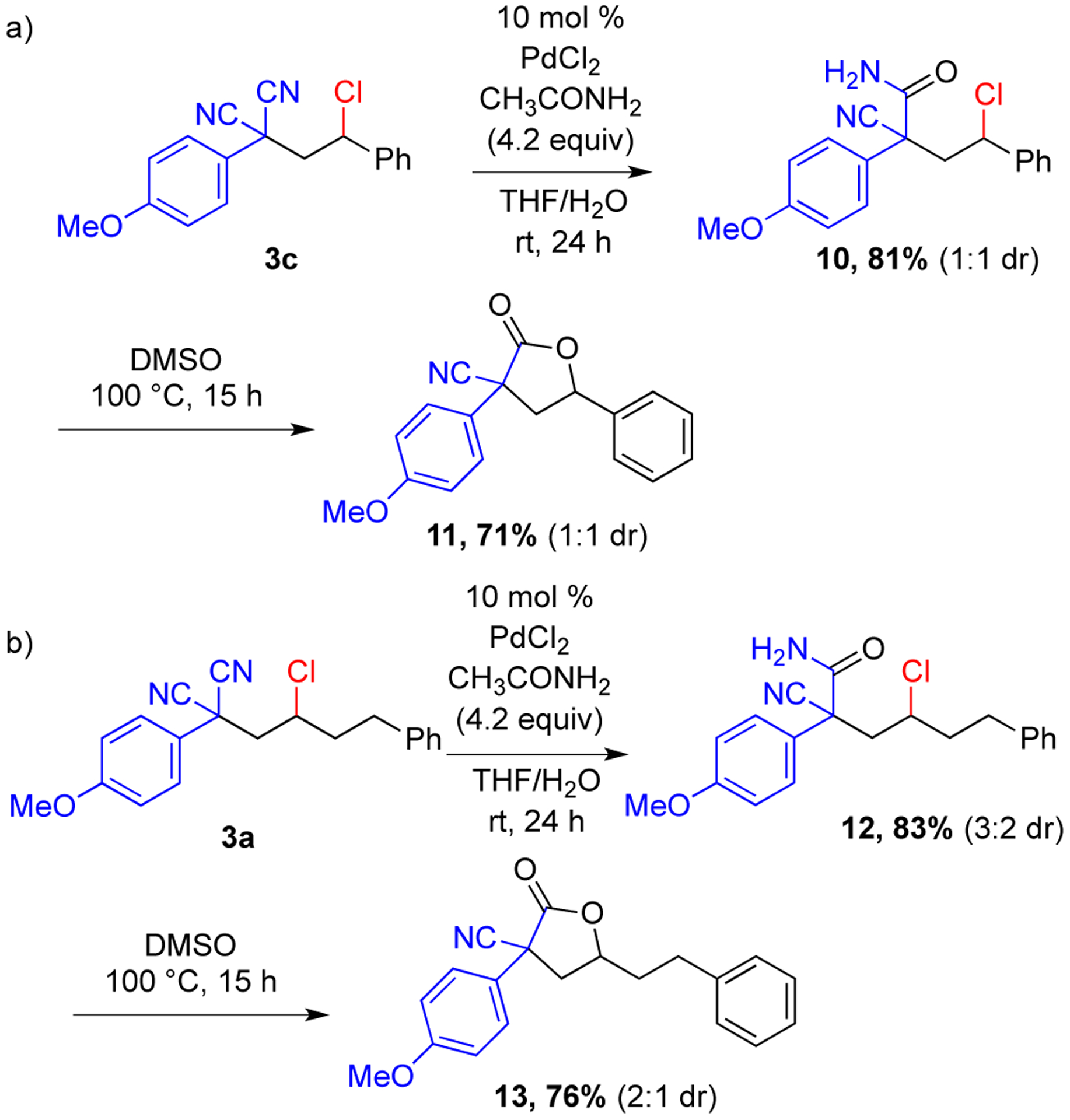
Post-Modification of Alkene Carbochlorination Products
In summary, a copper-catalyzed carbochlorination of alkenes has been developed. A carbon-centered radical generated from oxidation of aryl malononitrile is intercepted by an alkene followed by chloride allowing a three-component coupling. A simple, inexpensive chloride source KCl, is employed. Highlights of the method include the ability to monofunctionalized dienes and conditions that are orthogonal to halides and thioanisoles, the latter of which normally do not tolerate oxidative conditions. Mechanism experiments suggests the formation of radical intermediate after addition of the malonyl radical which further recombines with chloride in a copper-mediated process. The products can be readily transformed to form γ-lactones with highly substituted carbon centers.
Supplementary Material
ACKNOWLEDGMENT
We are grateful to the NSF (CHE1764298) and the NIH (R35 GM131902) for financial support of this research. Partial instrumentation support was provided by the NIH and NSF (1S10RR023444, 1S10RR022442, CHE-0840438, CHE-0848460, 1S10OD011980, CHE-1827457, 3R01GM118510-03S1, 3R01GM087605-06S1) as well as the Vagelos Institute for Energy Science and Technology. Dr. Charles W. Ross III (UPenn) is acknowledged for obtaining accurate mass data. We are grateful to Dr. Patrick Carroll (UPenn) for obtaining crystal structures.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website. FAIR Data is available as Supporting Information for Publication and includes the primary NMR FID files for compounds: [3a-u, 4a-i, 6, 7, 9–13]. See FID for Publication for additional information.
Experimental procedures and spectral data (PDF).
Accession Codes
CCDC 2046520 and 2046521 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, U.K.; fax: +44 1223 336033
The authors declare no competing financial interest.
REFERENCES
- (1).For review in alkene difunctionalization see,; Lan XW; Wang NX; Xing Y Recent Advances in Radical Difunctionalization of Simple Alkenes. European J. Org. Chem 2017, 2017, 5821–5851. [Google Scholar]
- (2).For examples of metal catalyzed carbohalogenation of alkenes see.,; (a) Newman SG; Howell JK; Nicolaus N; Lautens M Palladium-Catalyzed Carbohalogenation: Bromide to Iodide Exchange and Domino Processes. J. Am. Chem. Soc 2011, 133, 14916–14919. [DOI] [PubMed] [Google Scholar]; (b) Jia X; Petrone DA; Lautens M A Conjunctive Carboiodination: Indenes by a Double Carbopalladation- Reductive Elimination Domino Process. Angew. Chem. Int. Ed 2012, 51, 9870–9872. [DOI] [PubMed] [Google Scholar]; (c) Ouyang J; Su X; Chen Y; Yuan Y; Li Y Copper-Catalyzed Carbochlorination or Carbobromination via Radical Cyclization of Aryl Amines. Tetrahedron Lett. 2016, 57, 1438–1441. [Google Scholar]; (d) Yoon H; Marchese AD; Lautens M Carboiodination Catalyzed by Nickel. J. Am. Chem. Soc 2018, 140, 10950–10954. [DOI] [PubMed] [Google Scholar]; (e) Goto M; Maejima S; Yamaguchi E; Itoh A Regioselective Carboiodination of Styrenes: N-Iodosuccinimide Affords Complete Reaction Regioselectivity. Asian J. Org. Chem 2020, 9, 210–213. [Google Scholar]
- (3).Cristina Silva Costa D Additions to Non-Activated Alkenes: Recent Advances. Arab. J. Chem 2020, 13, 799–834. [Google Scholar]
- (4).Schmittel M; Haeuseler A One-Electron Oxidation of Metal Enolates and Metal Phenolates. J. Organomet. Chem 2002, 661, 169–179. [Google Scholar]
- (5).For seminal work see:; Kharasch MS; Skell PS; Fisher P Reactions of Atoms and Free Radicals in Solution. XII. The Addition of Bromo Esters to Olefins. J. Am. Chem. Soc 1948, 70, 1055–1059. [Google Scholar]
- (6).Jahn U; Hartmann P; Dix I; Jones PG Lithium Malo-nate Enolates as Precursors for Radical Reactions - Con-venient Induction of Radical Cyclizations with Either Radical or Cationic Termination. European J. Org. Chem 2001, 2001, 3333–3355. [Google Scholar]
- (7).Jahn U; Hartmann P; Kaasalainen E Efficient Oxidative Radical Cyclizations of Ester Enolates with Carbocation Desilylation as Termination: Synthesis of Cyclopentanoid Monoterpenes and Analogues. Org. Lett 2004, 6, 257–260 [DOI] [PubMed] [Google Scholar]
- (8).Logan AWJ; Parker JS; Hallside MS; Burton JW Manganese(III) Acetate Mediated Oxidative Radical Cyclizations. Toward Vicinal All-Carbon Quaternary Stereocenters. Org. Lett 2012, 14, 2940–2943. [DOI] [PubMed] [Google Scholar]
- (9).Fu N; Shen Y; Allen AR; Song L; Ozaki A; Lin S Mn-Catalyzed Electrochemical Chloroalkylation of Alkenes. ACS Catal. 2019, 9, 746–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).(a) Fleming FF Nitrile-Containing Natural Products. Nat. Prod. Rep, 1999,16, 597–606. [Google Scholar]; (b) Fleming FF; Yao L; Ravikumar PC; Funk L; Shook BC Nitrile-Containing Pharmaceuticals: Efficacious Roles of the Nitrile Pharmacophore. J. Med. Chem 2010, 53, 7902–7917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Fatiadi AJ Preparation and Synthetic Applications of Cyano Compounds In: Patai S and Rappaport Z, Eds., Wiley, New York: (1983). [Google Scholar]; (b) Rappoport ZR In: Chemistry of the Cyano group, John Wiley and Sons, London, 1970, pp 121–312. [Google Scholar]
- (12).(a) Curto JM; Kozlowski MC Chemoselective Activation of Sp 3 vs Sp 2 C − H Bonds with Pd(II). J. Am. Chem. Soc 2015, 137, 18–21. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hong G; Nahide PD; Neelam UK; Amadeo P; Vijeta A; Curto JM; Hendrick CE; Vangelder KF; Kozlowski MC Bonds : Oxidative Coupling To Form Quaternary Centers. ACS Catal. 2019, 9, 3716–3724. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Hong G; Nahide PD; Kozlowski MC Cyanomethylation of Substituted Fluorenes and Oxindoles with Alkyl Nitriles Org. Lett 2020, 22, 1563–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Frenette M; Aliaga C; Font-Sanchis E; Scaiano JC Bond Dissociation Energies for Radical Dimers Derived from Highly Stabilized Carbon-Centered Radicals. Org. Lett 2004, 6, 2579–2582. [DOI] [PubMed] [Google Scholar]
- (14).Maejima S; Yamaguchi E; Itoh A Trans-Diastereoselective Syntheses of γ-Lactones by Visible Light-Iodine-Mediated Carboesterification of Alkenes. ACS Omega 2019, 4, 4856–4870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).a) Yu H, Li Z, Bolm C, Org. Lett 2018, 20, 2076–2079; [DOI] [PubMed] [Google Scholar]; b) Chen C, Xu X-H, Yang B, Qing F-L, Org. Lett 2014, 16, 3372–3375. [DOI] [PubMed] [Google Scholar]
- (16).A pathway involving a Cu(I) to Cu(II) cycle is also possible:; Zhang W, Wang F, McCann SD, Wang D, Chen P, Stahl SS, Liu G. Enantioselective cyanation of benzylic C-H bonds via copper-catalyzed radical relay. Science. 2016, 353, 1014–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17). We thank a reviewer for both suggesting the carbocation possibility as well as the experiment to differentiate it.
- (18).Maffioli SI; Marzorati E; Marazzi A Mild and Reversible Dehydration of Primary Amides with PdCl2 in Aqueous Acetonitrile. Org. Lett 2005, 7, 5237–5239. [DOI] [PubMed] [Google Scholar]
- (19).(a) Hoffmann HMR; Rabe J Synthesis and Biological Activity of α-Methylene-γ-butyrolactones. Angew. Chem. Int. Ed 1985, 24, 94–110. [Google Scholar]; (b) Chadwick M; Trewin H; Gawthrop F; Wagstaff C Sesquiterpenoids Lactones: Benefits to Plants and People. Int. J. Mol. Sci 2013, 14, 12780–12805. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.