

Abstract
Background
Fibrosis is the final common pathway to intestinal failure in Crohn’s disease, but no medical therapies exist to treat intestinal fibrosis. Activated myofibroblasts are key effector cells of fibrosis in multiple organ systems, including the intestine. AXL is a receptor tyrosine kinase that has been implicated in fibrogenic pathways involving myofibroblast activation. We aimed to investigate the AXL pathway as a potential target for the treatment of intestinal fibrosis.
Methods
To establish proof of concept, we first analyzed AXL gene expression in 2 in vivo models of intestinal fibrosis and 3 in vitro models of intestinal fibrosis. We then tested whether pharmacological inhibition of AXL signaling could reduce fibrogenesis in 3 in vitro models of intestinal fibrosis. In vitro testing included 2 distinct cell culture models of intestinal fibrosis (matrix stiffness and TGF-β1 treatment) and a human intestinal organoid model using TGF-β1 cytokine stimulation.
Results
Our findings suggest that the AXL pathway is induced in models of intestinal fibrosis. We demonstrate that inhibition of AXL signaling with the small molecule inhibitor BGB324 abrogates both matrix-stiffness and transforming growth factor beta (TGF-β1)–induced fibrogenesis in human colonic myofibroblasts. AXL inhibition with BGB324 sensitizes myofibroblasts to apoptosis. Finally, AXL inhibition with BGB324 blocks TGF-β1-induced fibrogenic gene and protein expression in human intestinal organoids.
Conclusions
The AXL pathway is active in multiple models of intestinal fibrosis. In vitro experiments suggest that inhibiting AXL signaling could represent a novel approach to antifibrotic therapy for intestinal fibrosis such as in Crohn’s disease.
Keywords: inflammatory bowel disease, Crohn’s disease, fibrosis, myofibroblast, AXL
INTRODUCTION
Crohn’s disease (CD) affects over 775,000 Americans, and its incidence is rising worldwide.1, 2 Intestinal fibrosis is the final step to intestinal failure in patients with CD. Fibrotic disease is characterized by excessive accumulation of extracellular matrix (ECM) components in the tissue, causing severe dysfunction of the involved organ. In the intestine, fibrosis leads to strictures, followed by obstruction. The resulting increased luminal pressure, combined with inflammation and weakening of the intestinal wall, leads to penetrating complications such as fistulas and abscesses.
Intestinal obstruction and penetrating complications lead to surgery in more than 64% of all patients with CD,3 incurring total annual medical costs of greater than $3.6 billion per year in the United States.4 Many medications effectively treat inflammation in CD, but no medical therapies exist to treat intestinal fibrosis.5 The treatment of intestinal fibrosis is currently limited to surgical intervention or endoscopic balloon dilation.6 Understanding the pathogenesis of intestinal fibrosis and the development of antifibrotic therapies represent an area of great need and opportunity.
Activated myofibroblasts are key effector cells of fibrosis in CD and play a critical role in wound healing via expression of contractile proteins and production of ECM glycoproteins.7 Myofibroblast activation is mediated by several mechanisms including profibrogenic cytokines and mechanical stress.8, 9 TGF-β1 is a key profibrotic cytokine in the pathology of CD in humans and in animal models of intestinal fibrosis.10, 11 In isolated intestinal myofibroblasts, profibrotic signaling occurs through the canonical TGF-β/SMAD-mediated pathway.7, 12 TGF-β1 is expressed in many tissues and by many cell types, with complex regulation and different actions depending on the tissue type.8, 13 Macrophages are an important source of TGF-β1, and the direct interaction of macrophages with myofibroblasts has been shown to activate myofibroblasts in a profibrotic manner.14 Elegant in vitro studies have demonstrated that the ECM itself maintains a store of latent TGF-β1 that is released by the contraction of activated myofibroblasts and could play an important role in the rapid response to wound healing.9 Further, colonic myofibroblasts grown on stiff substrate are activated into a fibrogenic phenotype.15 The combination of TGF-β1 cytokine stimulation and subsequent ECM stiffness in the intestinal wall creates a self-propagating cycle of fibroblast activation,16 continued deposition of ECM, and myofibroblast contraction.
Intestinal fibrosis in CD is believed to result from chronic inflammation; however, the underlying mechanisms are yet to be fully elucidated.11 Contributions from humoral factors, cellular interactions, environmental factors, and the properties of the ECM have all been implicated in the pathogenesis of intestinal fibrosis.11 In CD strictures, tissue fibrosis is linked to alterations in TGF-β1 signaling and pathological activation of intestinal myofibroblasts.17 Despite the role of inflammation in initiating intestinal fibrosis, mouse models have shown that only very early amelioration of inflammation can prevent the development of intestinal fibrosis, and fibrosis can progress in the absence of inflammation.16
Though the role of TGF-β1 in fibrosis is well known, there is growing evidence for the involvement of AXL signaling in myofibroblast activation.18 AXL is a member of the TAM (TYRO 3, AXL, and MER) family of receptor tyrosine kinases, and AXL inhibitors are currently in clinical trials as an adjunct to chemotherapy for the treatment of a number of solid and hematologic cancers.19 The TAM family has been implicated in inflammatory bowel disease.20 AXL signaling is involved in epithelial-mesenchymal transition (EMT)21 and mediates numerous cellular responses including adhesion,22 migration and invasion,23 mechanosensing and contraction,24 proliferation and survival,25 and cytoskeletal remodeling.23 AXL signaling has been implicated in fibrosis of the liver18 and kidney.26 Recent studies suggest that AXL could be both an upstream inducer and a downstream effector of the EMT phenotype.27 The role of AXL downstream of TGF-β1 has been shown in both hepatocellular carcinoma28 and breast carcinoma.29 Expression of AXL in response to TGF-β1 is observed in noncancerous conditions of the pancreas and skin.30
AXL has been implicated in cytokine-mediated fibrogenesis and may also play a role in autopropagation of fibrogenic pathways through mechanotransduction and regulation of cell adhesion.20, 31 AXL has been shown to recruit FAK and Src to affect adhesion in schwannoma.32 AXL is involved in protection of endothelial cells from apoptosis in response to laminar shear stress in blood vessel walls and is present in molecular complexes that function in mechanotransduction.31 As further evidence for a role of AXL in mechanotransduction, AXL is a molecular checkpoint involved in myofibroblast focal adhesion maturation and sensing substrate stiffness via the traction force response to substrate rigidity.22 Other groups have shown that inhibiting AXL reverses EMT21 and that targeting AXL reduces liver fibrosis by hepatic stellate cell inactivation.18 These data suggest that targeting AXL could be an effective strategy for developing antifibrotic drugs for multiple other organ systems, including the intestine.
The small molecule BGB324, (formerly R428), is a highly selective and potent inhibitor of AXL kinase protein. This molecule, (BGB324), is a potent inhibitor of AXL, with an EC50/IC50 equal to 14nmol/L. It is also highly specific, with one article demonstrating a 10- to 100-fold higher selectivity vs most kinases out of 133 tested, including a 50-fold and 100-fold higher selectivity for AXL over fellow members Mer and Tyro3, respectively.33
We aimed to better understand if AXL is involved in the mediation of intestinal fibrosis and if inhibition of AXL is a potential therapeutic target for intestinal fibrosis. We evaluated whether AXL is upregulated in models of intestinal fibrosis. We then investigated the effect of AXL inhibition with the small molecule inhibitor BGB324 in in vitro models of intestinal fibrosis.
MATERIALS AND METHODS
Cell Culture
Human colonic fibroblast CCD-18Co cells (CRL-1459) were obtained from ATCC (Bethesda, MD). Cells were cultured in α-MEM (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS) and subcultured weekly. Cells between passage 3 and 10 were used in experiments.
Rodent Models of Intestinal Fibrosis
AXL gene expression analysis was performed on colonic tissue from 2 in vivo rodent models of intestinal fibrosis, the first using the rat 2, 4, 6 trinitrobenzenesulfonic (TNBS) enema model, and the second using mice infected with Salmonella typhimurium as previously described by our group.16, 34–37 The rat TNBS model consists of repeated weekly TNBS enemas with recovery and is a thoroughly characterized and often utilized model of intestinal fibrosis.34, 37, 38 The mouse S. typhimurium model utilizes treatment with streptomycin followed by a short infection with S. typhimurium to induce intestinal fibrosis, followed by levofloxacin elimination of the S. typhimurium; it is also well characterized and used previously by our group.16, 35, 36
TGF-β1 Fibrogenesis Model
Low-passage (passages 2–7) colonic myofibroblasts (CCD-18co cells) were seeded at 5 × 104 cells/mL on multiwell tissue culture plates (12-well, 1 mL/well for gene expression; 6-well, 3 mL/well for protein expression). Cells were serum-starved overnight before stimulation with 0.05 ng/mL of TGF-β1 (R&D Systems, Minneapolis, MN), cotreated +/- AXL inhibitor BGB324 (R428, Selleck Chemicals, Houston, TX), and diluted from a 10-mM stock solution prepared in dimethyl sulfoxide (DMSO). The maximum concentration of DMSO in any in vitro experiment was ≤0.1%. Serum starvation was performed to reduce the effect of serum on markers of fibroblast differentiation when treated with TGF-β1.39 Cells were harvested for gene expression analysis at 24 hours and for protein expression analysis at 48 hours. In the multiday TGF-β1 treatment experiments, media was changed daily to replenish the active TGF-β1 and the AXL inhibitor.
Matrix Stiffness Fibrogenesis Model
Polyacrylamide gels recapitulating the stiffness of normal bowel were made as previously described.15 Low-passage CCD-18co cells were seeded at 1 × 105 cells/mL in 10% FBS-containing growth media on multiwell plates containing collagen-coated acrylamide gels corresponding to soft (4.3 kPa, 0.02% bisacrylamide) matrices, pathologically stiff (28 kPa, 0.16% bisacrylamide) matrices, or on the collagen-coated plastic well bottom (which is ~106 kPa and of a magnitude stiffer than human intestine).40 Cells were allowed to attach to the matrix for 30 minutes, and then the gels were transferred to new wells. Media was aspirated off the plastic wells and replaced with low-serum (0.5% FBS) α-MEM in the presence or absence of 2-µM BGB324 (R428, Selleck Chemicals, Houston, TX). Cells were harvested for gene expression analysis at 24 hours and for protein expression analysis at 48 hours. Treatment media were not changed during the 48-hour treatment to prevent loss of floating cells. Because the 2-µM BGB324 resulted in significant (≥20%) cell detachment at 48 hours, which was expected due to its pro-apoptotic activity, the supernatant containing floating cells was collected, and cells pooled with the lysate of the adhered cells. Supernatants from all samples were collected.
Fas Ligand–mediated Apoptosis Assay
Low-passage CCD-18co cells were seeded at 1 × 105 cells/mL in 10% FBS-containing growth media on multiwell plates containing collagen-coated acrylamide gels corresponding to soft (4.3 kPa, 0.02% bisacrylamide) or pathologically stiff (28 kPa, 0.16% bisacrylamide) matrices or on collagen-coated plastic wells. Cells were allowed to attach to the matrix for 30 minutes, and then the gels were transferred to new wells. Media was aspirated off the plastic wells and replaced with low-serum (0.5% FBS) α-MEM for overnight starvation. The following day, apoptosis was induced by treatment with 100 ng/mL Fas activating antibody (clone CH11, Millipore Sigma, St. Louis, MO) in the presence or absence of 2-µM BGB324, in low-serum (0.5% FBS) α-MEM. At 5 hours, total cell protein was isolated for cleaved PARP quantitation (described in the western blot section). Because the Fas ligand (FasL) and BGB324 resulted in significant cell detachment, floating cells were collected and pooled with the adhered cells before lysis. Supernatants from all samples were collected.
Human Intestinal Organoid Culture and TGF-β1-induced Fibrogenesis Model
All work was approved by the University of Michigan Human Pluripotent Stem Cell Research Oversight Committee (HPSCRO). Human embryonic stem cells (H9, Wicell Research Institute, Madison, WI) were differentiated into human intestinal organoids (HIOs) as described previously.41, 42 Organoids with high mesenchymal cell composition were chosen, as opposed to more cyst-like organoids with high epithelial cell content. The HIOs were embedded in growth factor–reduced Matrigel (BD Biosciences, San Jose, CA, catalog #356231), overlaid with intestinal growth media as previously described,41 and later transferred to advanced DMEM/F12 media (Invitrogen, Carlsbad, CA) with 10% fetal bovine serum added. One day before initiation of TGF-β1 treatment, HIOs were serum-starved for 24 hours in serum-free advanced DMEM/F12. Serum starvation was performed to reduce the effect of serum on markers of fibroblast differentiation when treated with TGF-β1.39 The HIOs were treated with 2 ng/mL of TGF-β1 in the presence or absence of inhibitors for 4 days, with media changed daily to replenish active TGF-β1. Spironolactone (Sigma, S3378) at 250 µM, the inhibitor used as proof-of-concept in our initial study of the HIO model for testing antifibrotic drugs,43 was used as the antifibrotic control given that it has previously been shown to have potent antifibrotic effects.37, 43 Tofacitinib is a JAK inhibitor currently approved for the treatment of ulcerative colitis.44 Our group has tested tofacitinib (catalog #14703, Cell Signaling, Danvers, MA) in 3 in vitro models of intestinal fibrosis. Tofacitinib failed to significantly reduce markers of fibrogenesis in pure fibrogenic (noninflammatory) models of intestinal fibrogenesis in vitro and therefore was used as the negative control at our previously used concentration of 10 µM. Given the increased density and multicellular structure of HIOs and the Matrigel embedding, in addition to prior work from our group that empirically determined using a 4-fold higher concentration of TGF-β,43 we anticipated requiring a corresponding increase in BGB324 concentration. Initially, both 5-µM and 10-µM BGB324 concentrations were tested, with the most robust response seen with the 10-µM BGB324 (4-fold higher than in the myofibroblast models).
Human Sample Analysis
The analysis of AXL gene expression in human CD samples was performed on archived cDNA from formalin-fixed paraffin-embedded (FFPE) clinical resection tissue. Tissue was obtained from subjects undergoing resection of Crohn’s disease small bowel intestinal stricture as described previously.15 Use of these samples was approved by the University of Michigan Institutional Review Board. Samples from CD stricture (n = 8) were compared with CD surgical margin (n = 6).
Gene Expression Analysis
RNA was extracted from cells and HIOs using the RNeasy kit (Qiagen, Valencia, CA). RNAs were treated with RNase-free DNase before cDNA synthesis using the First Strand Synthesis kit (Invitrogen, Carlsbad, CA) according to manufacturer’s protocol and as previously described.15, 43 The analysis of gene expression was determined by quantitative real-time polymerase chain reaction (QPCR) of our selected genes. Analysis for collagen-1 A1 (COLA1), myosin light chain kinase (MYLK), fibronectin 1 (FN1) genes, and GAPDH was performed with the TaqMan gene expression assays (ABI, Foster City, CA). Taqman primer and probe sequences are proprietary; assay IDs for each gene evaluated are listed later on. Quantitative real-time polymerase chain reaction was performed using an Applied Biosystems Step One Plus real-time PCR system. α-Smooth muscle actin (αSMA; ACTA2) gene expression was determined with the SYBR Green assay using the following primers: ACTA2-F 5’-AATGCAGAAGGAGATCACGC-3’, ACTA2- R 5’-TCCTGTTTGCTGATCCACATC-3’, as previously described.45 For gene expression analysis of human CD samples and rodent fibrotic vs normal samples, qPCR was performed on archived cDNAs.15 An AXL-gene specific TaqMan primer was used to evaluate AXL expression. A GAPDH-specific TaqMan primer was used to repeat GAPDH gene expression normalization. The assay IDs for AXL and GAPDH are listed later on. Cycling conditions were 95°C for 10 minutes, followed by 40 cycles of 95°C for 15 seconds, and 62°C for 60 seconds. Delta-delta-Ct (ΔΔCt) values were calculated by normalizing to GAPDH expression.
QPCR Primers
This list comprises all QPCR primers used:
COL1A1: Thermo Fisher Scientific; UniGene ID: Hs.172928; Assay ID/DNA Sequence: Hs00164004_m1
MYLK: Thermo Fisher Scientific; UniGene ID: Hs.477375; Assay ID/DNA Sequence: Hs00364926_m1
FN1: Thermo Fisher Scientific; UniGene ID: Hs.203717; Assay ID/DNA Sequence: Hs01549976_m1
AXL: Thermo Fisher Scientific; UniGene ID: Hs.590970; Assay ID/DNA Sequence: Hs01064444_m1
GAPDH: Thermo Fisher Scientific; UniGene ID: NM_002046.3; Assay ID/DNA Sequence: 4352934E
Western Blot Analysis
Analysis was performed on AXL, αSMA, GAPDH, and cleaved PARP protein levels. Total protein was isolated, then separated by SDS-PAGE, and probed as previously described.15, 43, 46 To include detached cells, supernatant and washes in PBS containing protease inhibitors (Roche, Sigma) were collected. These were centrifuged at 500 x g, and the cell pellet was pooled in lysis buffer with the lysate from the adhered cells. For analysis of collagen 1 protein level, total protein was isolated as previously described.15, 35, 43 However, loading samples were prepared and boiled in nonreducing loading buffer. For transfer the gel was cut along a 60 kDa size marker. The upper half (to be probed for collagen 1) was transferred overnight at 12 V, 4°C, in transfer buffer containing 0.1% SDS. The bottom half (to be probed for GAPDH) was transferred separately at 75 minutes, 100 volts, 4°C in a standard transfer buffer (25 mM Tris-HCl, 0.2 M glycine, 20% methanol).47
Antibodies
This list comprises all antibodies used:
AXL antibody: Cell Signaling # 8661 at 1:1000 in 5% BSA/TBST
αSMA antibody: Sigma #A528 at 1:300 in 5% milk/TBST
Cleaved PARP antibody: Cell Signaling # 9541, 1:1000 in 5% milk/TBST
GAPDH antibody: Fisher Scientific, AM4300 (Clone 6c5), 1:10,000 in 5% milk/TBST
Collagen I antibody: Rockland, #600-401-103, at 1:1000 in 5% milk/TBST
Statistical Analysis
Statistical differences were determined by a 2-sided, unpaired Student t test or Mann-Whitney U test, where appropriate. Results with a P value <0.05 were considered statistically significant. Random effects meta-analysis was employed for combining distinct biological replicates where indicated in the text. Based on recent NIH emphasis on rigor of validation of experiments with immortal and unstable cell lines, we are treating each in vitro experiment as 1 independent biological replicate with random effects. Meta-analyses and graphing were performed with Rstudio (Rstudio, Inc., Boston MA; R version 3.6.1, R Foundation for Statistical Computing, Vienna, Austria.) Meta-analyses are presented of standardized mean differences (SMD) with a 95% confidence interval (CI) based on a random effects model described by Mantel and Haenszel.48 Standardized mean difference magnitude was considered small where SMD = 0.2, medium where SMD = 0.5, large where SMD = 0.8, and very large where SMD = 1.3.49
RESULTS
AXL gene and protein expression is upregulated in multiple in vitro and in vivo models of intestinal fibrosis.
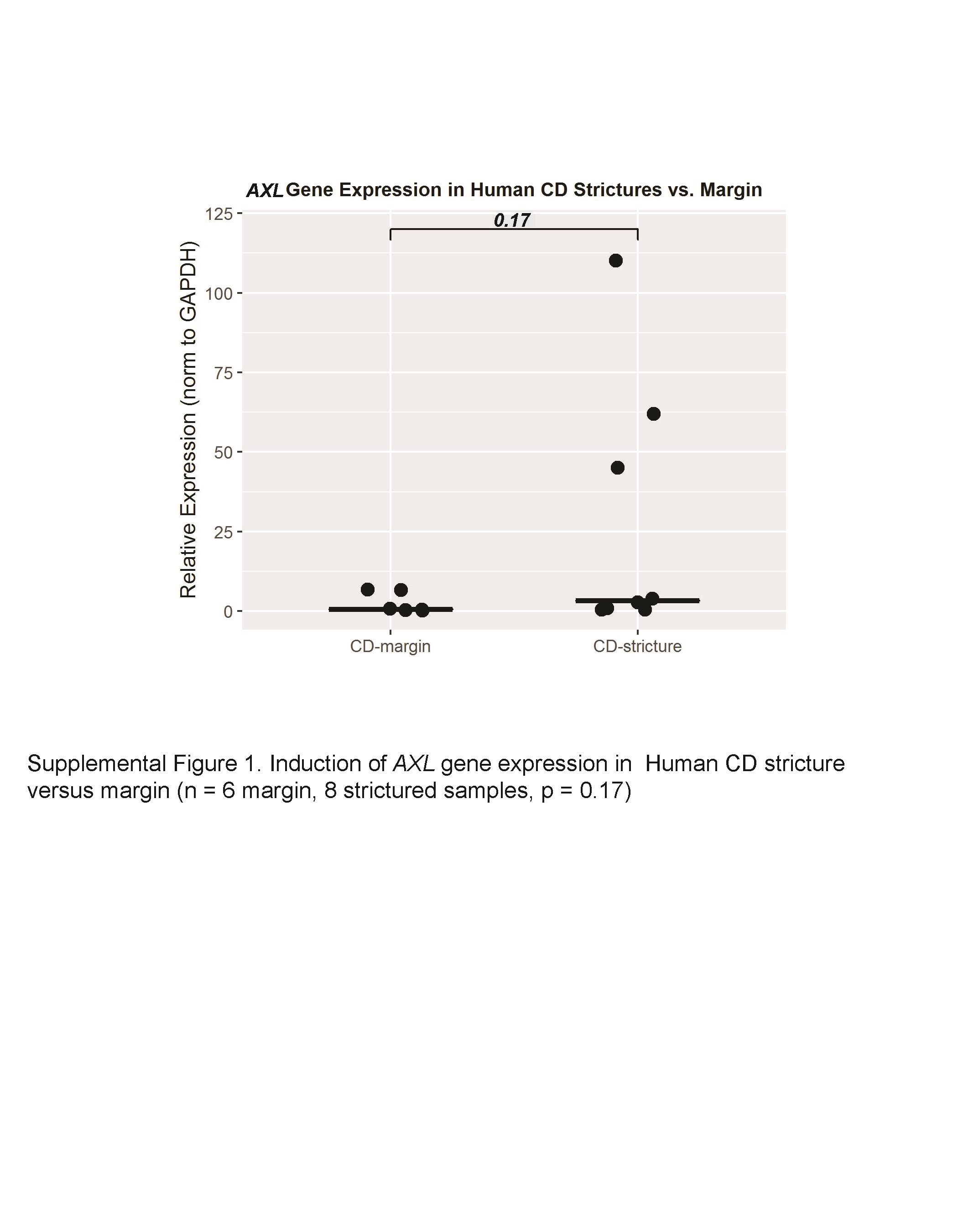
To investigate the possibility of AXL involvement in intestinal fibrosis, we first evaluated AXL gene expression in multiple models of intestinal fibrosis. Using a matrix stiffness fibrogenesis model, we found that CCD-18Co cells express a very low level of AXL gene when plated on soft (4.3 kPa) collagen-coated bisacrylamide gels, corresponding to the stiffness of physiologically normal intestine. Plating these cells on pathologically stiff (28 kPa, 0.16% bisacrylamide) matrices resulted in a 1.4-fold induction of AXL gene expression (P = 0.03; Fig. 1A). Using a TGF-β1 fibrogenesis model in CCD-18Co myofibroblasts, we found that AXL gene expression is induced 1.7-fold by TGF-β1 (P = 0.02; Fig. 1B). AXL gene expression was 2.0-fold higher in the fibrostenotic condition in both the mouse S. typhimurium and rat TNBS models (P = 0.09 and 0.001 respectively; Fig. 1C and D). Lastly, using the TGF-β1 human intestinal organoid model, we found that AXL expression increases 1.8-fold in response to TGF-β1 (P = 0.08; Fig. 1E). Preliminary experiments on a limited set of archived human tissue from CD resections were also performed. Using a Mann-Whitney U test, these data did not achieve statistical significance (z score = 1.4, P = 0.17). However, there was a bimodal distribution of AXL gene expression. In some CD strictures, AXL gene expression was unchanged compared with margin; however in others, AXL expression was markedly induced (45 to >100 fold; Supplemental Fig. 1). This may reflect the fibrotic heterogeneity of CD strictures.50 Though the AXL gene expression data is not uniformly statistically significant, there is a consistent numerical increase in AXL gene expression across multiple intestinal fibrosis models.
FIGURE 1.

Induction of AXL gene expression in multiple models of intestinal fibrosis. AXL gene expression is induced in (A) CCD-18Co stiffness model utilizing soft or pathologically stiff collagen coated acrylamide gels (n = 11 per group, P = 0.031); (B) CCD-18Co TGF-β1 model (n = 18 per group, P = 0.022); (C) mouse S. typhimurium model (n = 5 per group, P = 0.087); (D) rat TNBS model (n = 12 per group, P = 0.0015); (E) human intestinal organoids untreated vs TGF-β1 treated (n = 14 untreated, 13 treated, P = 0.077); (F) summary table of gene expression.
AXL protein expression was induced by fibrotic stimuli in both the CCD-18Co TGF-β1 and matrix stiffness models, as demonstrated by Western blot (Fig. 2A and C). AXL protein expression was induced in the matrix stiffness model of intestinal fibrosis; however, this did not reach statistical significance (n = 3). AXL protein expression was numerically increased in the TGF-β1 model of intestinal fibrosis and nearly reached significance (Fig. 2B; P = 0.059; n = 3).
FIGURE 2.

BGB324 abrogates CCD18Co myofibroblast fibrogenic protein expression. A, Western blot of fibrogenic proteins in the CCD-18Co TGF-β1 model. CCD-18Co cells were treated with TGF-β1 +/- BGB324 for 48 hours. Total cell protein was extracted for Western blot analysis. Treatment with BGB324 results in the formation of an AXL cleavage product 55 kDa in size. Treatment with 0.5, 1, and 2-μM BGB324 and TGF-β1 resulted in a dose-dependent decrease in both AXL and αSMA protein expression. A longer exposure (data not shown) revealed that 2-μM BGB324 did not completely abrogate AXL and αSMA protein expression. B, Quantification of protein expression in the CCD-18Co TGF-β1 model using ImageJ. C, Western blot of fibrogenic proteins in the matrix stiffness model. CCD18Co cells were cultured for 48 hours on multiwell plates containing soft matrices or on the collagen-coated plastic well bottom. This was done in the presence or absence of BGB324 in low-serum media. Total cell protein was extracted for Western blot analysis at 48 hours. As the matrix stiffness model requires collagen coating of gels and plastic, this precludes quantitation of collagen protein in this model. Treatment with BGB324 again results in the formation of an AXL cleavage product of 55 kDa.
BGB324 Abrogates Fibrogenic Protein Induction in 2 in Vitro Models of Intestinal Fibrosis
Previous work in our lab has shown that protein levels of the extracellular matrix component collagen 1 and the cytoskeletal protein and marker of myofibroblast activation, αSMA, are induced in vitro in models of fibrogenesis using CCD-18Co human intestinal myofibroblasts.15, 45 In this study, treatment of CCD-18Co myofibroblasts with 0.05 ng/mL of TGF-β1 for 48 hours induced the expression of αSMA protein 5.4-fold (P = 0.00019), whereas cotreatment with increasing levels of BGB324 results in a dose-dependent abrogation of this induction (P = 0.012, 0.0015, 0.00012 vs TGF-β1 treated; Fig. 2A and B). A trend toward significance was seen with regard to induction of collagen 1 protein (P = 0.17) and subsequent abrogation by 2-μM BGB324 (P = 0.12; Fig. 2A and B). The dose-dependent decrease in αSMA and collagen expression corresponded with similar dose-dependent abrogation of AXL protein expression, with a statistically significant reduction of AXL protein expression at the 2-μM BGB324 dose (P = 0.024 vs TGF-β1 treated; Fig. 2A and B). In addition, a cleavage product around 55 kDa was observed (Fig. 2A and C), suggesting a possible mechanism of action of BGB324 by affecting AXL protein levels by inducing proteolysis. The size of this cleavage product corresponds with the cleavage product generated by the activity of α-secretase, as previously reported.51, 52
In the matrix stiffness model, culture of CCD-18Co cells for 48 hours on collagen-coated plastic (orders of magnitude stiffer than 4.3 kPa bisacrylamide gels, which correspond to the stiffness of normal human intestine) results in the induction of αSMA and AXL protein expression (Fig. 2C). Treatment with 2-µM BGB324 abrogates this induction (Fig. 2C). As this model requires collagen coating of the gels and the tissue culture substrate, we cannot accurately quantitate the collagen 1 protein in this matrix stiffness model. Similar to the TGF-β1model, treatment with BGB324 abrogates expression of the full-length AXL protein with a concurrent increase in cleaved AXL product.
The small molecule AXL inhibitor BGB324 abrogates fibrogenic gene induction in the CCD-18Co myofibroblast matrix stiffness and TGF-β1 models of intestinal fibrosis.
Having found a consistent increase in AXL gene expression across multiple models of disease, we proceeded to test whether an AXL inhibitor could abrogate fibrogenic gene expression under conditions of pathological substrate stiffness or fibrogenic cytokine (TGF-β1) treatment. We have previously used these in vitro models to demonstrate the efficacy of an inhibitor of the Rho/MRTF/SRF pathway.45, 46
Plating CCD-18Co myofibroblasts on pathologically stiff substrates results in the induction of mechanosensitive fibrogenic genes.45 Due to the high model variability observed in a series of 5 separate experiments with 3 wells (technical replicates) per treatment group (Supplemental Fig. 2), a random-effects meta-analysis of gene expression was performed. Comparing the effect of 2-µM BGB324 treatment of cells plated on plastic to untreated cells plated on plastic (matrix-stimulated), we observed very large standard mean difference effect sizes for 4 fibrogenic genes (COL1A1, MYLK, FN1, and ACTA2; Fig. 3). The largest effects of BGB324 were observed for COL1A1 (SMD = –5.6, P = 0.006) and MYLK (SMD = –7.4, P < 0.000001). Smaller yet very large effect sizes were observed for FN1 (SMD = –3.3, P < 0.000001) and ACTA2 (SMD = –3.7, P = 0.08).
FIGURE 3.

Inhibition of fibrogenic gene expression by the AXL inhibitor BGB324 in the matrix stiffness model. CCD-18Co cells were cultured for 48 hours on multiwell plates containing collagen-coated acrylamide gels corresponding to soft (4.3kPa, 0.02% bisacrylamide) matrices or on the collagen-coated plastic well bottom in the presence or absence of 2-μM BGB324. Cells were harvested for gene expression analysis at 48 hours. Total cell mRNA was extracted, and gene expression of COL1A1, FN1, MYLK, and αSMA (ACTA2) were measured using QPCR analysis. Gene expression was normalized to GAPDH expression. Data are presented as random effects meta-analysis of gene expression over 5 independent experiments comparing plastic-plated vs plastic plus 2-μM BGB324. Each experiment consisted of 3 wells (technical replicates) per treatment per group.
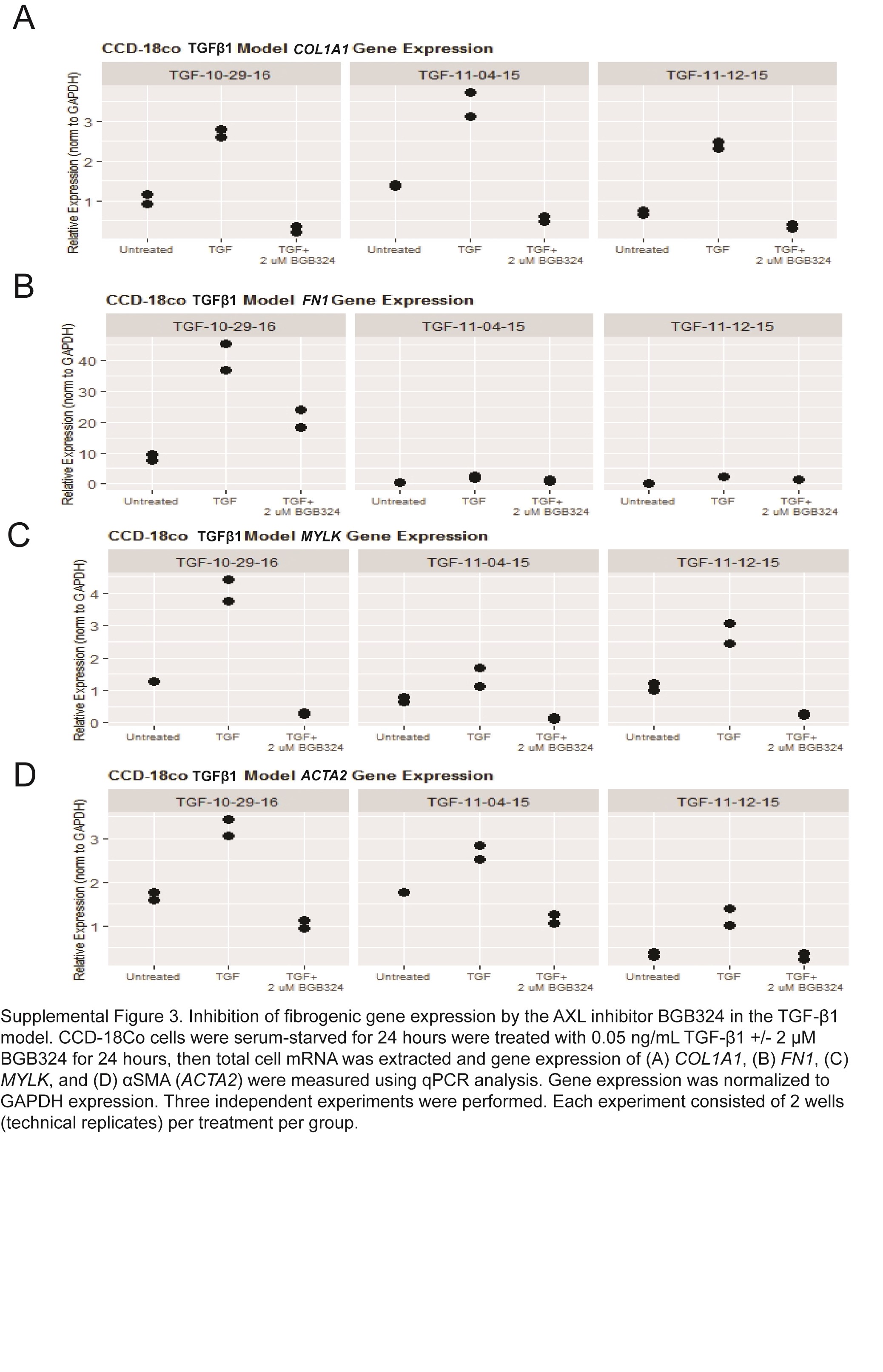
In the TGF-β1 model, treatment of CCD-18Co with TGF-β1 also results in upregulation of fibrogenic genes.45 We performed a random-effects meta-analysis of gene expression over 3 separate experiments consisting of 2 wells (technical replicates) per treatment group. Similar to the stiffness model, we observed very large SMD effect sizes for the 4 fibrogenic genes (COL1A1, MYLK, FN1, and ACTA2) in the 2-µM BGB324-treated groups vs the TGF-β1-treated samples (Fig. 4, Supplemental Fig. 3). The largest effect size was observed for COL1A1 (SMD = –8.3, P = 0.001). Very large effect sizes were observed for MYLK (SMD = –3.9, P = 0.0004), FN1 (SMD = –2.3, P = 0.013), and ACTA2 (SMD = –3.7, P = 0.0003). The relatively smaller effect size of MYLK in the TGF-β1 model compared with the stiffness model may reflect the role of MYLK in actin stress fiber contraction in the ECM.
FIGURE 4.

Inhibition of fibrogenic gene expression by the AXL inhibitor BGB324 in the TGF-β1 model. CCD-18Co cells were serum-starved for 24 hours and were treated with 0.05 ng/mL TGF-β1 +/- 2-μM BGB324 for 24 hours; then the total cell mRNA was extracted, and gene expression of COL1A1, FN1, MYLK, and αSMA (ACTA2) were measured using QPCR analysis. Data are presented as a random effects meta-analysis of gene expression over 3 independent experiments comparing TGF-β1-treated vs TGF-β1 plus 2-μM BGB324. Each experiment consisted of 2 wells (technical replicates) per treatment per group.
BGB324 Sensitizes CCD-18CO Human Intestinal Myofibroblasts to FasL-mediated Apoptosis
Using the myofibroblast matrix stiffness model, we show that inhibition of AXL signaling with BGB324 sensitizes CCD-18Co human intestinal myofibroblasts to FasL-mediated apoptosis (Fig. 5). Increased levels of cleaved poly (ADP-ribose) polymerase (PARP) protein correspond to apoptosis induction. Intestinal myofibroblasts show low-level PARP cleavage in response to Fas ligand alone. Treatment of CCD-18Co with 2-µM BGB324 sensitizes the myofibroblasts to apoptosis when treated with FasL, resulting in a dramatic and statistically significant induction of cleaved PARP protein (Fig. 5A). Additional matrix stiffness experiments (n = 3) confirmed BGB324-mediated cleaved-PARP induction (Fig. 5B, P = 0.0012 to 0.014).
FIGURE 5.

BGB324 sensitizes CCD18Co cells to apoptosis. CCD18Co cells underwent induction of apoptosis with Fas ligand in the presence or absence of 2-μM BGB324. Total cell protein was extracted for Western blot analysis at 5 hours. Plating on stiff matrix (28kPa) or plastic conferred resistance to apoptosis, and this resistance was abrogated with BGB324 treatment. A, Representative gel, total n = 3. B, Quantification of protein expression using ImageJ.
BGB324 Abrogates Fibrogenic Gene and Protein Induction in TGF-Β1-Treated Human Intestinal Organoids (HIO)
We have previously demonstrated that pluripotent stem cell–derived HIOs recapitulate the 3-dimensional, multicell-type structure of the human intestine because they possess both epithelium and a diverse mesenchyme, including myofibroblasts.41 Further, we have previously demonstrated that TGF-β1 treatment induces a robust fibrotic response in HIOs.43 Three independent experiments were performed on separate days using new HIOs derived from the same cell lines for each independent experiment. Each independent experiment consisted of 3 wells (technical replicates) per treatment group of a 24-well plate. Each well contained multiple (3–4) Matrigel droplets that contained 5 to 10 HIOs per drop.
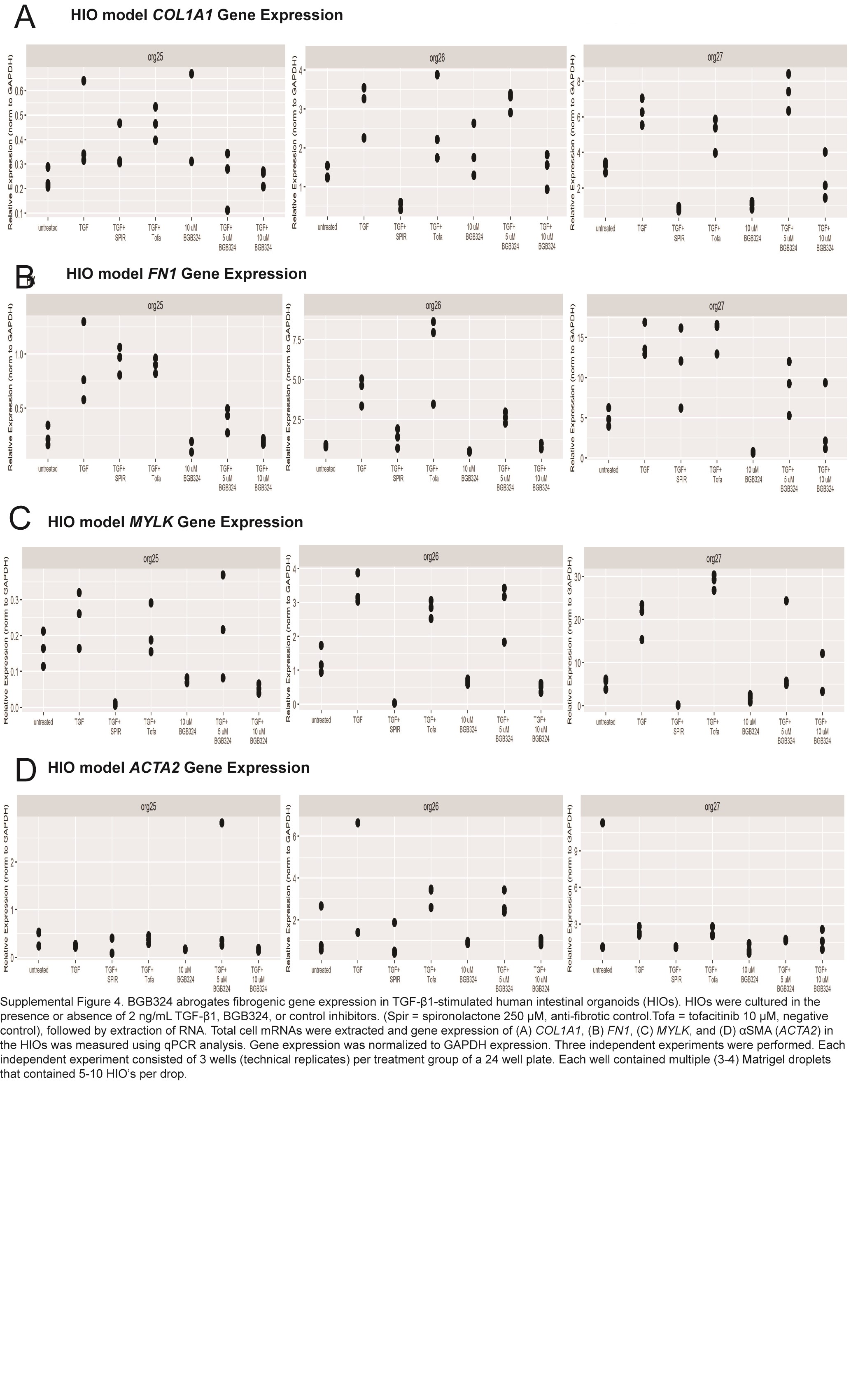
In each independent experiment, treatment with TGF-β1 induced expression of fibrogenic genes (COL1A1, MYLK, FN1, and ACTA2) compared with untreated HIOs. Cotreatment with tofacitinib (negative control) and TGF-β1 did not abrogate fibrogenic gene expression. In contrast, cotreatment with spironolactone (antifibrotic control) and TGF-β1 abrogated expression of COL1A1 5.5-fold (P = 0.015) and MYLK >100-fold (P = 0.038). Spironolactone reduced ACTA2 expression but did not reach significance (P = 0.11). Expression of FN1 was unaffected by spironolactone (P = 0.50; Supplemental Fig. 4).
To elucidate the effects of BGB324 while controlling for differences in the population of diverse cell types within the HIOs, we performed a random effects meta-analysis of the effect of BGB324 cotreatment with TGF-β1 compared with TGF-β1 treatment alone. Treatment with 10-µM BGB324 demonstrated a significant reduction in expression of 4 fibrogenic genes (COL1A1, MYLK, FN1, and ACTA2) vs the TGF-β1- treated samples (Fig. 6A). Large effect sizes were observed for COL1A1 (–1.9, P = 0.001), MYLK (–3.1, P = 0.0001), FN1 (–2.6, P = 0.0001), and ACTA2 (–1.1, P = 0.026). The magnitude of the effect sizes in the HIOs (which contain multiple cell types) was lower than in both CCD-18co myofibroblast models, suggesting a true biological effect, as myofibroblast cells are a minority of cells within HIOs.43
FIGURE 6.

BGB324 abrogates fibrogenic gene and protein expression in TGF-β1-stimulated human intestinal organoids. A, BGB324 abrogates fibrogenic gene expression in TGF-β1-stimulated HIOs. Human intestinal organoids were cultured in the presence or absence of TGF-β1 and BGB324 followed by extraction of RNA. Total cell mRNAs were extracted and gene expression of COL1A1, FN1, MYLK, and αSMA (ACTA2) in the HIOs was measured using QPCR analysis. Gene expression was normalized to GAPDH expression. Data are presented as random effects meta-analyses of gene expression over 3 independent experiments (ORG-25, 26, 27). Each independent experiment consisted of 3 wells (technical replicates) per treatment group of a 24-well plate. Each well contained multiple (3–4) Matrigel droplets that contained 5 to 10 HIOs per drop. B, HIOs cultured in the presence or absence of TGF-β1, BGB324, or control inhibitors. Total cell protein was isolated for Western blot analysis. An αSMA Western blot which is representative of 4 separate experiments is shown. Cotreatment of HIOs with 10 μM BGB324 reduces the αSMA protein expression induced by TGF-β1. Abbreviations: Spir, spironolactone, antifibrotic control; Tofa, tofacitinib, negative control.
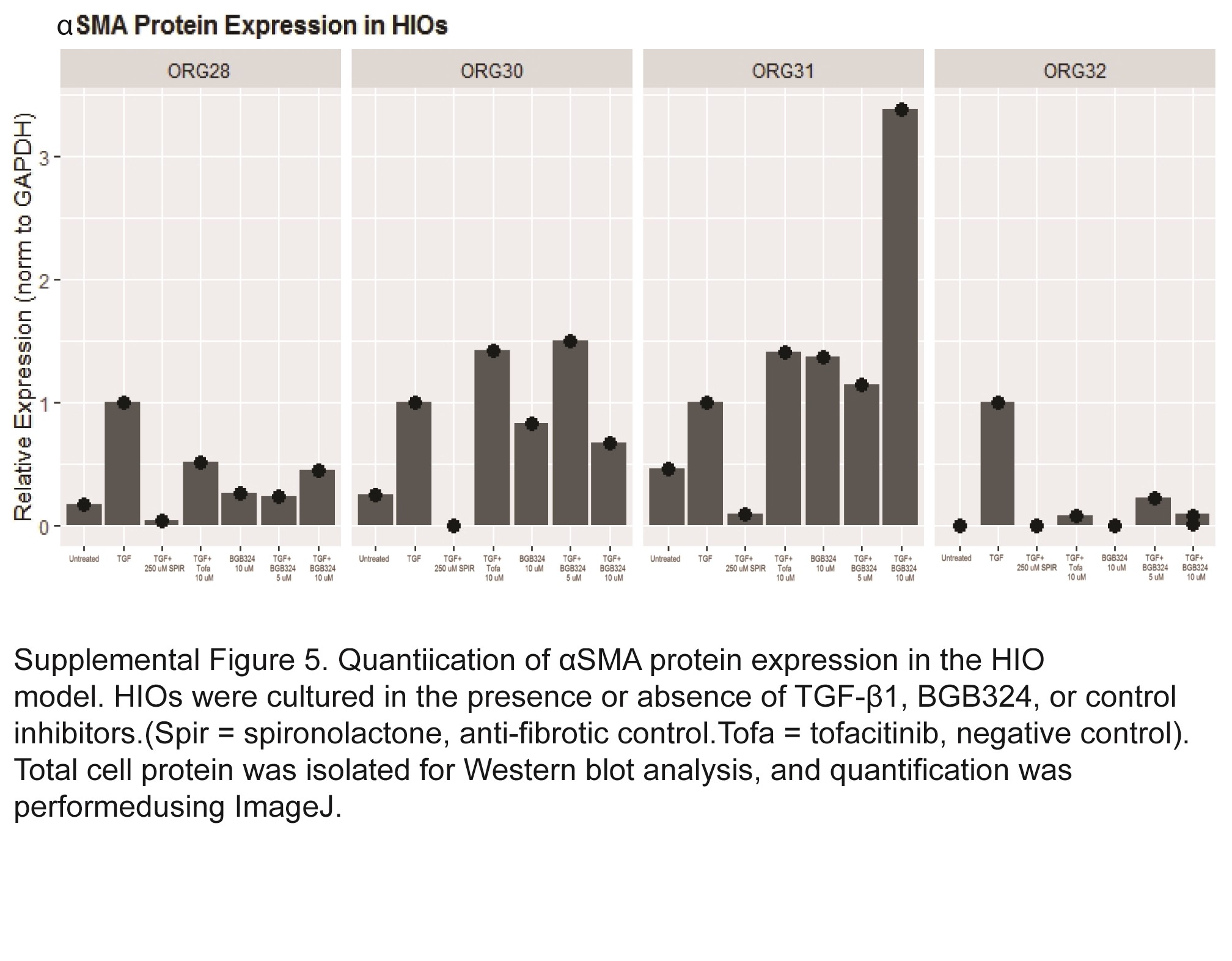
Protein expression of αSMA was induced in HIOs by treatment with TGF-β1 at 2 ng/mL for 4 days. Effects of cotreatment with 10-µM BGB324 was compared with cotreatment of TGF-β1 with negative (tofacitinib) and positive (spironolactone) controls (Fig. 6B). Spironolactone profoundly repressed TGF-β1 induction of αSMA, whereas tofacitinib had no effect. Cotreatment with 5- or 10-µM BGB324 attenuated αSMA expression (Fig. 6B, quantification using ImageJ was also performed and is presented in Supplemental Fig. 5).
DISCUSSION
Our findings suggest that AXL kinase is potentially a druggable target for antifibrotic intervention in intestinal fibrosis. We demonstrate that the AXL gene expression seems to increase in in vitro cytokine and matrix-based myofibroblast models, a multicellular model (HIOs), and 2 rodent models of intestinal fibrosis. Moderate induction of AXL protein is also observed in all 3 in vitro models. We demonstrate that blocking AXL signaling with the small molecule inhibitor BGB324 abrogates fibrogenic gene and protein expression in response to both cytokine and mechanical stimuli. This antifibrotic efficacy of BGB324 is also demonstrated in a 3-dimensional, multicell-type HIO model of intestinal fibrosis.
Inhibition of AXL signaling also resulted in fibroblast sensitization to FasL-induced apoptosis, as evidenced by induction of cleaved PARP expression with FasL and BGB324 cotreatment. Fas-mediated apoptosis results in cleavage of proteins such as PARP.53 One of the hallmarks of fibrosis is excessive and uncontrolled wound healing due to the pathological persistence of myofibroblasts that express αSMA and produce collagen.7, 54 Activated myofibroblasts are resistant to apoptosis, which is thought to be a key cause for their persistence in the stiff microenvironment.55 The significant PARP induction we saw in response to FasL with BGB324 supports a possible a role for AXL in intestinal fibrosis that involves apoptosis resistance in addition to profibrotic gene and protein expression.
The cell culture models in which the antifibrotic efficacy of BGB324 was tested consist of myofibroblasts, the primary effector cell of intestinal fibrosis. These models are reductionist, and their advantage is the ability to focus specifically on the effect of fibrogenic stimuli and putative antifibrotic drugs on these cells in isolation. These 2-dimensional cultures are limited because they are missing the complexity of the human gut. Recapitulating the antifibrotic effect of BGB324 in the human intestinal organoid model, with its 3-dimensional structure and multicell type composition, provides further evidence that the efficacy of this drug could translate to humans.
This work and work done by others show that inhibiting AXL is potentially antifibrotic.18, 21 Therapies targeting the AXL pathway have already shown promise for human use in cancer therapy and are undergoing clinical trials as enhancers of chemotherapy.56 Although 2 drugs (nintedanib and pirfenidone) have been approved as antifibrotics in pulmonary fibrosis,57 no antifibrotic drugs exist for use in intestine or any organ other than the lung. Previous studies in our lab showed promising in vitro antifibrotic results with spironolactone, but treatment with spironolactone in rodent models resulted in increased intestinal inflammation and mortality.37 These results demonstrate AXL inhibition as one of a select few mechanisms that has shown antifibrotic efficacy in human intestinal organoids.43
In an effort to investigate AXL as a target in human disease, we performed a pilot analysis of AXL gene expression in a limited sample of CD strictures and the adjacent margins. AXL gene expression was low in all margin samples. However, AXL gene expression was strongly upregulated in a subset of the CD strictures. We theorize that our observed findings could be the result of both our limited sample size and the inherent variability in stricture pathophysiology, as Crohn’s strictures can have both an inflammatory and fibrotic component.50, 58 More investigation into the contribution of the AXL pathway in human intestinal strictures is needed, such as testing of AXL expression in a larger number of human samples and immunohistochemistry with fluorescence in situ hybridization on formalin-fixed paraffin-embedded samples.
Modest increases in AXL expression in our models and inconclusive preliminary human data represent a limitation of our study. Though the data from our models are indeed modest, the uniform increase in AXL expression across all models makes this preliminary evidence more compelling. Each model has a very distinct underlying mechanism to recapitulate fibrosis, and increases of variable strength and significance were seen across all models we tested.
Significant limitations of all 3 of the in vitro model systems tested include the lack of a functioning immune system and a sterile microenvironment devoid of microbiota. These factors are believed to play a role in the pathogenesis of Crohn’s disease, and these effects are not represented in our reductionist models. An additional significant limitation is that although the AXL pathway is a mediator of fibrosis in other organ systems,59 it has yet to be validated as mediator of intestinal fibrosis in humans in vivo. This will be an important step in the development of any therapy targeting AXL.
The potential for DMSO vehicle contributing to our findings is a limitation of this study. The maximum amount of DMSO exposure (ie, the HIO experiments) was 0.1%. Earlier work in our lab demonstrated that DMSO did not have a significant effect on this set of gene or protein expression. We did not, however, use DMSO in our controls for these data in an effort to reduce complexity of dose-response experiments. The effect of tofacitinib as a negative control, which was constituted in the same DMSO vehicle, would support a negligible effect of DMSO in our HIO model. Additionally, the stability of GAPDH in our QPCR data for these experiments supports that the levels of DMSO used are nontoxic. Though the likelihood of DMSO significantly contributing to our findings is low, lack of internal controls is a limitation of our study.
The potential for off-target effects of BGB324 to influence our findings is another limitation of our study. In cell-based assays, BGB324 demonstrated high selectivity for AXL, with the only additional receptor affected within a 35-fold IC50 being vascular endothelial growth factor receptor 2 (VEGFR2).33 This is expressed almost exclusively in endothelial cells.60 There are no endothelial cells in our cell culture assays, and our HIOs are similarly devoid of endothelial cells.61 Although the specificity of this inhibitor does support AXL inhibition as the most likely mechanism for our findings, we did not perform experiments to evaluate for possible off-target effects of BGB324, particularly the potential for effects on off-target tyrosine kinases. To confirm our findings, further investigation into this pathway is needed utilizing alternative AXL modulation techniques such as genetic modification or antibody-based methods.
An important theoretical limitation to the use of an antifibrotic that inhibits AXL in the context of an inflammatory disease is the risk of potentiating inflammation. AXL and the closely related Mer tyrosine kinase (MERTK) are expressed on macrophages and dendritic cells, where they play an anti-inflammatory role.62 The anti-inflammatory role of AXL is seen in macrophages and dendritic cells of the intestinal lamina propria. Work by Bosurgi et al in AXL and Mer knockout mice using a dextran sulfate sodium induced colitis model suggests that AXL could have a role in preventing colonic inflammation and inflammation-associated cancer.63 Potential future use of an AXL inhibitor as an antifibrotic in Crohn’s disease would likely require maintenance cotherapy with a potent anti-inflammatory therapy.
An additional consideration in the development of any antifibrotic drug is the delicate relationship between fibrosis and healing. The balance of profibrotic signals and antifibrotic signals is critical for reconstitution of damaged tissue, with scarring or failure of wound healing resulting from signals that cause dysregulation in favor of one direction or the other. In that regard, pathways that are thought to be more downstream and related to smaller adjustments in the process make attractive targets that are less likely to have overpowering effects on healing. Because of this, inhibition of a TAM (Tyro3, AXL, and Mer) kinase such as AXL could be ideal. The TAM receptors evolved relatively late and are thought to play “fine-tuning” but nonessential roles in the immunity of complex organisms.64 As a secondary concern for clinical wound healing, it is possible that a potent antifibrotic therapy would interfere with the healing of surgical wounds and bowel anastomoses, which might require discontinuation for a period before any surgical intervention.
Testing in animal models is needed not only to further evaluate the antifibrotic potential of targeting this pathway but also assess potential for pro-inflammatory effects. Animal models will also inform whether dosing levels or timing strategy can be manipulated to overcome potential detrimental pro-inflammatory effects of AXL blockade. Coculture methods could be effective in evaluating for pro-inflammatory potential of AXL inhibitors in immune cells such as macrophages.
CONCLUSION
The lack of antifibrotic therapeutics is a critical unsolved problem in CD. Our results suggest that the AXL pathway may be an important mediator of intestinal fibrosis. Targeting AXL in vitro with a small molecule inhibitor abrogates fibrogenesis. These results suggest that AXL inhibition represents a promising potential therapeutic target for the treatment of intestinal fibrosis in Crohn’s disease that warrants further investigation.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Author Contribution: CS, ER, LJ, JB, and PH drafted the manuscript. CS, ER, LJ, JS, and PH contributed to the conceptualization and planning of ideas and experiments. CS, ER, LJ, SH, JS, and PH contributed to the planning and performing experiments. CS, ER, LJ, JB, KC, JS, and PH contributed to the interpretation of data. CS, ER, LJ, JB, and PH contributed to the drafting of manuscript. All authors critically reviewed the article. All authors had access to the study data, and all authors reviewed and approved the final manuscript.
Supported by: University of Michigan Center for the Discovery of New Medicines, Michigan Drug Discovery. NIDDK, R01 DK 118154, recipient PDRH;
NIDDK, T32 DK 094775, recipient CAS.
Conflicts of Interest: PH has received consulting fees from AbbVie, Amgen, Genentech, JBR Pharma and Lycera. All other authors report no conflicts of interest.
REFERENCES
- 1. Shivashankar R, Tremaine WJ, Harmsen WS, et al. . Incidence and prevalence of Crohn’s disease and ulcerative colitis in Olmsted County, Minnesota from 1970 through 2010. Clin Gastroenterol Hepatol. 2017;15:857–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ng SC, Shi HY, Hamidi N, et al. . Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet. 2018;390:2769–2778. [DOI] [PubMed] [Google Scholar]
- 3. Dhillon S, Loftus EV, Tremaine WJ, et al. . The natural history of surgery for Crohn’s disease in a population-based cohort from Olmsted County, Minnesota. Am J Gastroenterol. 2005;100:S305–S305. [Google Scholar]
- 4. Kappelman MD, Rifas-Shiman SL, Porter CQ, et al. . Direct health care costs of Crohn’s disease and ulcerative colitis in US children and adults. Gastroenterology. 2008;135:1907–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fiocchi C, Lund PK. Themes in fibrosis and gastrointestinal inflammation. Am J Physiol Gastrointest Liver Physiol. 2011;300:G677–G683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lan N, Stocchi L, Delaney CP, et al. . Endoscopic stricturotomy vs ileocolonic resection in the treatment of ileocolonic anastomotic strictures in Crohn’s disease. Gastrointest Endosc. 2019;90:259–268. [DOI] [PubMed] [Google Scholar]
- 7. Powell DW, Mifflin RC, Valentich JD, et al. . Myofibroblasts. II. Intestinal subepithelial myofibroblasts. Am J Physiol. 1999;277:C183–C201. [DOI] [PubMed] [Google Scholar]
- 8. Hinz B. Tissue stiffness, latent TGF-beta1 activation, and mechanical signal transduction: implications for the pathogenesis and treatment of fibrosis. Curr Rheumatol Rep. 2009;11:120–126. [DOI] [PubMed] [Google Scholar]
- 9. Wipff PJ, Rifkin DB, Meister JJ, et al. . Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J Cell Biol. 2007;179:1311–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ma Y, Guan Q, Bai A, et al. . Targeting TGF-beta1 by employing a vaccine ameliorates fibrosis in a mouse model of chronic colitis. Inflamm Bowel Dis. 2010;16:1040–1050. [DOI] [PubMed] [Google Scholar]
- 11. Bettenworth D, Rieder F. Pathogenesis of intestinal fibrosis in inflammatory bowel disease and perspectives for therapeutic implication. Dig Dis. 2017;35:25–31. [DOI] [PubMed] [Google Scholar]
- 12. Shi Y, Massagué J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. [DOI] [PubMed] [Google Scholar]
- 13. Robertson IB, Rifkin DB. Regulation of the bioavailability of TGF-β and TGF-β-related proteins. Cold Spring Harb Perspect Biol. 2016;8:a021907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lodyga M, Cambridge E, Karvonen HM, et al. . Cadherin-11-mediated adhesion of macrophages to myofibroblasts establishes a profibrotic niche of active TGF-β. Sci Signal. 2019;12. [DOI] [PubMed] [Google Scholar]
- 15. Johnson LA, Rodansky ES, Sauder KL, et al. . Matrix stiffness corresponding to strictured bowel induces a fibrogenic response in human colonic fibroblasts. Inflamm Bowel Dis. 2013;19:891–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Johnson LA, Luke A, Sauder K, et al. . Intestinal fibrosis is reduced by early elimination of inflammation in a mouse model of IBD: impact of a “Top-Down” approach to intestinal fibrosis in mice. Inflamm Bowel Dis. 2012;18:460–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Di Sabatino A, Jackson CL, Pickard KM, et al. . Transforming growth factor beta signalling and matrix metalloproteinases in the mucosa overlying Crohn’s disease strictures. Gut. 2009;58:777–789. [DOI] [PubMed] [Google Scholar]
- 18. Bárcena C, Stefanovic M, Tutusaus A, et al. . Gas6/AXL pathway is activated in chronic liver disease and its targeting reduces fibrosis via hepatic stellate cell inactivation. J Hepatol. 2015;63:670–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gay CM, Balaji K, Byers LA. Giving AXL the axe: targeting AXL in human malignancy. Br J Cancer. 2017;116:415–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rothlin CV, Leighton JA, Ghosh S. Tyro3, AXL, and MERTK receptor signaling in inflammatory bowel disease and colitis-associated cancer. Inflamm Bowel Dis. 2014;20:1472–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wu F, Li J, Jang C, et al. . The role of AXL in drug resistance and epithelial-to-mesenchymal transition of non-small cell lung carcinoma. Int J Clin Exp Pathol. 2014;7:6653–6661. [PMC free article] [PubMed] [Google Scholar]
- 22. Prager-Khoutorsky M, Lichtenstein A, Krishnan R, et al. . Fibroblast polarization is a matrix-rigidity-dependent process controlled by focal adhesion mechanosensing. Nat Cell Biol. 2011;13:1457–1465. [DOI] [PubMed] [Google Scholar]
- 23. Abu-Thuraia A, Gauthier R, Chidiac R, et al. . AXL phosphorylates Elmo scaffold proteins to promote Rac activation and cell invasion. Mol Cell Biol. 2015;35:76–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yang B, Lieu ZZ, Wolfenson H, et al. . Mechanosensing controlled directly by tyrosine kinases. Nano Lett. 2016;16:5951–5961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Axelrod H, Pienta KJ. AXL as a mediator of cellular growth and survival. Oncotarget. 2014;5:8818–8852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liu F, Zhuang S. Role of receptor tyrosine kinase signaling in renal fibrosis. Int J Mol Sci. 2016;17:972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rankin EB, Giaccia AJ. The receptor tyrosine kinase AXL in cancer progression. Cancers. 2016;8:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Reichl P, Dengler M, van Zijl F, et al. . AXL activates autocrine transforming growth factor-β signaling in hepatocellular carcinoma. Hepatology. 2015;61:930–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li Y, Jia L, Liu C, et al. . AXL as a downstream effector of TGF-β1 via PI3K/Akt-PAK1 signaling pathway promotes tumor invasion and chemoresistance in breast carcinoma. Tumour Biol. 2015;36:1115–1127. [DOI] [PubMed] [Google Scholar]
- 30. Bauer T, Zagórska A, Jurkin J, et al. . Identification of AXL as a downstream effector of TGF-β1 during Langerhans cell differentiation and epidermal homeostasis. J Exp Med. 2012;209:2033–2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. D’Arcangelo D, Ambrosino V, Giannuzzo M, et al. . AXL receptor activation mediates laminar shear stress anti-apoptotic effects in human endothelial cells. Cardiovasc Res. 2006;71:754–763. [DOI] [PubMed] [Google Scholar]
- 32. Ammoun S, Provenzano L, Zhou L, et al. . AXL/Gas6/NFκB signalling in schwannoma pathological proliferation, adhesion and survival. Oncogene. 2014;33:336–346. [DOI] [PubMed] [Google Scholar]
- 33. Holland SJ, Pan A, Franci C, et al. . R428, a selective small molecule inhibitor of AXL kinase, blocks tumor spread and prolongs survival in models of metastatic breast cancer. Cancer Res. 2010;70:1544–1554. [DOI] [PubMed] [Google Scholar]
- 34. Kim K, Johnson LA, Jia C, et al. . Noninvasive ultrasound elasticity imaging (UEI) of Crohn’s disease: animal model. Ultrasound Med Biol. 2008;34:902–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Johnson LA, Rodansky ES, Moons DS, et al. . optimisation of intestinal fibrosis and survival in the mouse S. Typhimurium model for antifibrotic drug discovery and preclinical applications. J Crohns Colitis. 2017;11:724–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Higgins PD, Johnson LA, Luther J, et al. . Prior Helicobacter pylori infection ameliorates Salmonella typhimurium-induced colitis: mucosal crosstalk between stomach and distal intestine. Inflamm Bowel Dis. 2011;17:1398–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Johnson LA, Govani SM, Joyce JC, et al. . Spironolactone and colitis: increased mortality in rodents and in humans. Inflamm Bowel Dis. 2012;18: 1315–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Morris GP, Beck PL, Herridge MS, et al. . Hapten-induced model of chronic inflammation and ulceration in the rat colon. Gastroenterology. 1989;96:795–803. [PubMed] [Google Scholar]
- 39. Siani A, Khaw RR, Manley OW, et al. . Fibronectin localization and fibrillization are affected by the presence of serum in culture media. Sci Rep. 2015;5:9278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Discher DE, Smith L, Cho S, et al. . Matrix mechanosensing: from scaling concepts in ‘Omics data to mechanisms in the nucleus, regeneration, and cancer. Annu Rev Biophys. 2017;46:295–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Spence JR, Mayhew CN, Rankin SA, et al. . Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature. 2011;470:105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. McCracken KW, Howell JC, Wells JM, et al. . Generating human intestinal tissue from pluripotent stem cells in vitro. Nat Protoc. 2011;6:1920–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rodansky ES, Johnson LA, Huang S, et al. . Intestinal organoids: a model of intestinal fibrosis for evaluating anti-fibrotic drugs. Exp Mol Pathol. 2015;98: 346–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sandborn WJ, Su C, Panes J. Tofacitinib as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2017;377:496–497. [DOI] [PubMed] [Google Scholar]
- 45. Johnson LA, Rodansky ES, Haak AJ, et al. . Novel Rho/MRTF/SRF inhibitors block matrix-stiffness and TGF-β-induced fibrogenesis in human colonic myofibroblasts. Inflamm Bowel Dis. 2014;20:154–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Johnson LA, Sauder KL, Rodansky ES, et al. . CARD-024, a vitamin D analog, attenuates the pro-fibrotic response to substrate stiffness in colonic myofibroblasts. Exp Mol Pathol. 2012;93:91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mantel N, Haenszel W. Statistical aspects of the analysis of data from retrospective studies of disease. J Natl Cancer Inst. 1959;22:719–748. [PubMed] [Google Scholar]
- 49. Cohen J. Statistical Power Analysis for the Behavioral Sciences. 2nd ed.Hillsdale, N.J.: L. Erlbaum Associates; 1988. [Google Scholar]
- 50. Dillman JR, Stidham RW, Higgins PD, et al. . Ultrasound shear wave elastography helps discriminate low-grade from high-grade bowel wall fibrosis in ex vivo human intestinal specimens. J Ultrasound Med. 2014;33:2115–2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lu Y, Wan J, Yang Z, et al. . Regulated intramembrane proteolysis of the AXL receptor kinase generates an intracellular domain that localizes in the nucleus of cancer cells. FASEB J. 2017;31:1382–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. O’Bryan JP, Fridell YW, Koski R, et al. . The transforming receptor tyrosine kinase, AXL, is post-translationally regulated by proteolytic cleavage. J Biol Chem. 1995;270:551–557. [DOI] [PubMed] [Google Scholar]
- 53. Cousens LP, Goulette FA, Darnowski JW. JAK-mediated signaling inhibits Fas ligand-induced apoptosis independent of de novo protein synthesis. J Immunol. 2005;174:320–327. [DOI] [PubMed] [Google Scholar]
- 54. Gabbiani G. The myofibroblast in wound healing and fibrocontractive diseases. J Pathol. 2003;200:500–503. [DOI] [PubMed] [Google Scholar]
- 55. Huang SK, Horowitz JC. Outstaying their welcome: the persistent myofibroblast in IPF. Austin J Pulm Respir Med. 2014;1:3. [PMC free article] [PubMed] [Google Scholar]
- 56. Shen Y, Chen X, He J, et al. . AXL inhibitors as novel cancer therapeutic agents. Life Sci. 2018;198:99–111. [DOI] [PubMed] [Google Scholar]
- 57. Richeldi L, Collard HR, Jones MG. Idiopathic pulmonary fibrosis. Lancet. 2017;389:1941–1952. [DOI] [PubMed] [Google Scholar]
- 58. Chan WPW, Mourad F, Leong RW. Crohn’s disease associated strictures. J Gastroenterol Hepatol. 2018;33:998–1008. [DOI] [PubMed] [Google Scholar]
- 59. Bellan M, Cittone MG, Tonello S, et al. . Gas6/TAM System: A Key Modulator of the Interplay between Inflammation and Fibrosis. Int J Mol Sci. 2019;20:5070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Huang J, Zhu H, Wang X, et al. . The patterns and expression of KDR in normal tissues of human internal organs. J Mol Histol. 2011;42:597–603. [DOI] [PubMed] [Google Scholar]
- 61. Wells JM, Spence JR. How to make an intestine. Development. 2014;141:752–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sheridan C. First AXL inhibitor enters clinical trials. Nat Biotechnol. 2013;31: 775–776. [DOI] [PubMed] [Google Scholar]
- 63. Bosurgi L, Bernink JH, Delgado Cuevas V, et al. . Paradoxical role of the proto-oncogene AXL and Mer receptor tyrosine kinases in colon cancer. Proc Natl Acad Sci U S A. 2013;110:13091–13096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Davra V, Kimani SG, Calianese D, et al. . Ligand activation of TAM family receptors-implications for tumor biology and therapeutic response. Cancers. 2016;8:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.