

Abstract

A ball-milling enabled tertiary amine catalyzed aza-Morita–Baylis–Hillman reaction is reported. The reaction process does not require solvent, has significantly shorter reaction times than previous methods and is reported on a range of imines and acrylate Michael acceptors across than 26 examples. A 12-fold scaled-up example is also reported as well as experimental comparisons to solution-based experiments and neat-stirred reactions.
Keywords: Mechanochemistry, Organocatalysis, Metal-free, Solvent-free, Reaction methodology
Short abstract
A ball-milling enabled tertiary amine catalyzed aza-Morita−Baylis−Hillman reaction is reported.
Introduction
The Morita–Baylis–Hillman (MBH) reaction is a powerful transformation, enabling access to highly functionalized allylic alcohols starting from aldehydes and low-molecular-weight Michael acceptors, e.g., α,β-unsaturated ketones. It was first described by Morita in 1968,1 followed by Baylis and Hillman in 1972.2−5 The main difference between the reports was that Morita utilized a tertiary phosphine (PCy3 or PBu3) as an organocatalyst, whereas Baylis and Hillman utilized a tertiary amine (diazabicyclo[2.2.2]octane (DABCO)) as an organocatalyst (Scheme 1A,B). This is an attractive process, as it is a highly atom economical C–C bond forming reaction and has therefore undergone significant development since these initial reports. Such developments include the incorporation of enantioselectivity,6−9 and an aza variant by substituting the aldehyde/ketone for an imine (Scheme 1B).10 This aza-MBH reaction has been the focus of more recent work, due to the reaction products being desirable β-amino acid derivatives.11 Developments in the aza-MBH reaction have included both imines derived from aldehydes (aldimines)12−14 and ketones (ketimines)15−17 being utilized, and many enantioselective examples, also.18−20 The MBH reaction is not without its limitations; however, with long reaction times (days), use of toxic solvents, and elevated temperatures being common; particularly for more challenging substrates.21 Hence, there is a clear opportunity to improve the overall efficiency of an otherwise attractive C–C bond forming process.
Scheme 1. Transitioning from Solution-Based to Mechanochemical MBH and Aza-MBH Processes: Opportunities for Organocatalysts under Milling Conditions, Tertiary Amine Catalysis—Morita–Baylis–Hillman Reaction (and Aza Variant), Mechanochemical MBH Reaction (Mack), and Mechanochemical Aza-MBH Reaction.

One potential way to improve the chemical processes is to explore the use of alternative reactor technologies. We hypothesized that the technique of ball-milling and mechanochemistry may offer complementary advantages to organocatalysis in general and note that there are very few reports of tertiary amine catalysis by ball-milling (Scheme 1A). Mechanochemistry uses mechanical energy to initiate reactivity within chemical bonds22 and can take place within the solid state, with no or very limited solvent present. Hence, mechanochemical processes operate at very high concentrations of reagents.
The combination of these factors can lead to significantly reduced reaction times, lowering of reaction temperature, and even altered selectivity compared to traditional solution-based chemistry.23−31 In the field of organocatalysis by ball-milling, Bolm and co-workers have shown that reactivity induced by chiral secondary amine organocatalysis operates well under mechanochemical conditions.32 More recently, our group demonstrated that N-heterocyclic carbenes (NHCs) could be catalytically active under milling conditions and facilitate the Stetter and Benzoin reactions mechanochemically.33 Tertiary amine organocatalysis, another key organocatalytic mode, has not been well explored under mechanochemical conditions. The most notable report comes from Mack and co-workers, where they have demonstrated a significantly reduced reaction time of the MBH reaction under solvent-free, ball-milling conditions.34 They were able to achieve full conversion of starting materials in as little as 30 min, which is a huge improvement over solution-based examples, where reaction times up to 2 days are commonplace (Scheme 1C). However, when more challenging substrates were investigated, the yield suffered, and the reaction time was greatly increased. Given that the field of ball-milling has advanced in understanding since the pioneering report from Mack (in 2007), we were intrigued if new understanding would permit a more broadly applicable approach and sought to develop an aza-MBH reaction system under mechanochemical conditions.
Herein, we report the application of ball-milling mechanochemistry to the aza-MBH reaction of imines and Michael acceptors (Scheme 1D).
Results and Discussion
Our investigations commenced with a set of model substrates, tosyl protected imine (1) and ethyl acrylate (2) with DABCO as the tertiary amine catalyst. Simply milling these reagents together in a 14 mL stainless steel jar, along with a 3 g stainless steel ball and 6 mass equiv of sand (with respect to the combined mass of starting materials) as a grinding auxiliary, for 99 min at 30 Hz afforded the aza-MBH product in 23% NMR yield and 21% isolated yield (Table 1, entry 7). Reactions were immediately quenched with acid and worked up so as to kill the catalyst activity and minimize any further reaction conversion during workup and analysis.
Table 1. Optimization of Model Aza-MBH Reaction in the Ball-Mill.

Yield determined by 1H NMR using mesitylene as internal standard; numbers in parentheses represent isolated yields.
Pleased by this initial result, we sought to fully optimize this reaction and immediately found that using 0.25 μL/mg of acetonitrile as a liquid assisted grinding (LAG) agent improved the reaction outcome to 52% NMR yield (Table 1, entry 1).35−43 Thus, we continued with the optimization and found that sodium chloride was the superior grinding auxiliary and could be reduced to 3 mass equiv (Table 1, entries 5 and 6). Next, we turned our attention to a small catalyst screen and found that both quinuclidine (78%) and 3-hydroxyquinuclidine (3-HQD, 81%, Table 1, entry 11) were more effective at catalyzing the reaction than DABCO; however, 3-HQD is around 10 times less expensive than quinuclidine; hence, this catalyst was carried forward. Notably, comparing this reaction to the same reaction system but without grinding agent afforded 57% of the desired product (Table S1, entry 12).
Finally, we conducted a LAG agent screen (Table 1, entries 12–19) across liquids with a variety of dielectric constants to elucidate whether using acetonitrile was optimal and discovered that toluene was the best liquid additive, giving the product in 85% isolated yield (Table 1, entry 14, see the Supporting Information for further optimization details). With these optimal conditions in hand, we moved on to explore the scope and limitations of this mechanochemical aza-MBH reaction.
Initial investigations into the scope of the reaction revealed that the 99 min reaction time would not be sufficiently long for all of the substrates; thus, this was extended to 3 h when needed. Pleasingly, the reaction proceeded effectively with a variety of halogen-substituted, aromatic aldimines: 4-fluoro (4; 68%), 4-chloro (7; 61%), 4-bromo (10; 51%), and 4-iodo (11; 38%) were tolerated, as well as 3-fluoro (5; 78%), chloro (8; 70%), 2-fluoro (6; 49%), and 2-chloro (9; 57%) substitution (Scheme 2A). Mildly electron donating 4-phenyl (12; 68) and 4-methyl (13; 29%) groups, along with a variety of electron withdrawing groups, including 4-nitro (16; 46%) and 4-trifluoromethyl (18; 57%), were also competent substrates. Sterically encumbered 1-naphthyl (21; 25%) also participated in the reaction, albeit at reduced yield. Heteroaromatic 2-furanyl (23; 60%) and 2-thiophenyl (24; 35%) substitution were also well tolerated. Interestingly, a cinnamyl substrate gave the 1,2-addition product (25) in moderate yield, and no competing 1,4-addition was observed. Finally, a methanesulfonamide protected imine furnished the desired product 26 in good yield. A small selection of Michael acceptors also successfully reacted under these conditions, including acrylonitrile (27) and methyl vinyl ketone (28). Interestingly, reaction with the weak Michael acceptor, acrylamide, resulted in direct addition, yielding the aminal product (29, Scheme 3B) in low yield. Incompatible substrates included strongly electron donating 4-dimethylamino (30) and 2-hydroxyl (31) substituted imines, along with sterically encumbered mesityl substrate (32) and basic nitrogen-containing pyrrole (33) (Scheme 2B). Other unsuitable substrates included alkyl aldimine (34), and acetophenone (35) and benzophenone (36) based ketimines, thus demonstrating the reduced reactivity of ketimines versus aldimines. Finally, β-substituted Michael acceptors 37 and 38, which are notoriously difficult substrates in the MBH reaction,44 were also unreactive under the milling conditions explored here. With regards to control of enantioselectivity of the reaction, we have found that use of enantioenriched 3-hydroxyquinuclidine (as its R enantiomer) delivers 2% ee in this model process (Scheme 2C). Use of β-isocupreidine, a structural relative of quinine, featuring a quinuclidine core, as catalyst, delivered an encouraging 64% ee but with 20% yield of product. However, the related and opposite handedness of this catalyst, α-isocupreine, afforded 20% yield and −26% ee (i.e., 26% ee in favor of the opposite enantiomer).
Scheme 2. Scope of the Ball-Milling Enabled Aza-MBH Reaction: Substrate Scope, Unsuccessful Substrates under Current Conditions, and Exploring Enantioselective Control.
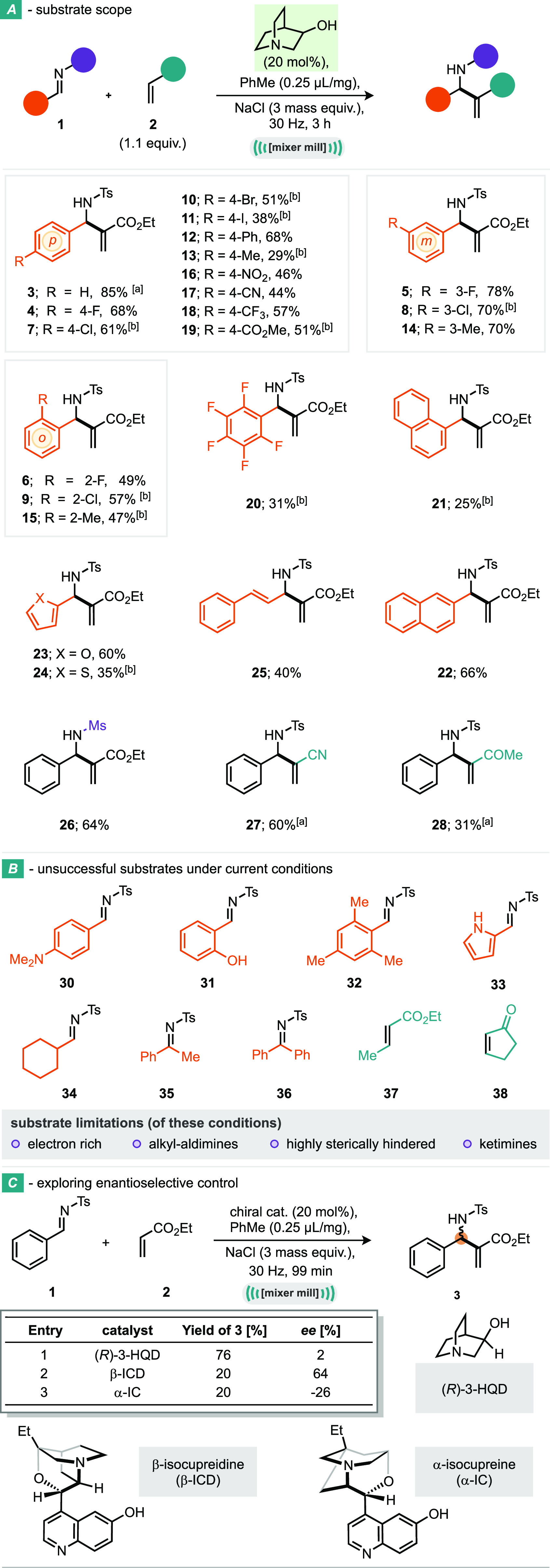
99 min reaction.
40 mol % catalyst. Reaction conditions: imine (0.25 mmol), Michael acceptor (0.28 mmol), 3-quinuclidinol catalyst (20 mol %), toluene (25 μL), NaCl (300 mg), 14 mL jar, 1 × 3 g stainless steel ball, 3 h, under air in mixer mill. Isolated yields reported.
Scheme 3. Application to Aldehydes and Scaleup Example: Application to Aldehyde, Acrylamide as Michael Acceptor—Direct Addition to Imine, Increased Scale, and Comparison of Reaction Techniques.

The application of optimal reaction conditions to an aldehyde substrate, similar to Mack’s system, was also investigated. By reacting 4-nitrobenzaldehyde (39) with ethyl acrylate (2), under our standard conditions, the desired product (40) was obtained in 57% isolated yield (Scheme 3A), which is a notable decrease when compared to the conditions developed by Mack for this aldehyde with methyl methacrylate (reported as >98%) and highlights the need for two different reactions systems for different substrates. This reaction can be scaled up from our standard 0.25 mmol reaction to a 3 mmol reaction (12-fold). This was simply achieved by increasing the quantity of all reagents, the LAG, and the grinding auxiliary but by also increasing the jar size from 14 to 25 mL and the mass of the ball from 3 to 12 g (Scheme 3C). In this manner, 0.86 g of product was delivered (3) in an 80% isolated yield.
A comparison study of reactors has been made for this reaction (Scheme 3D). Running the parent reaction system in solvent (1 mL of toluene) without any grinding auxiliary, it can be seen that the reaction is relatively slow, as expected based on all of the previous solution-based literature. However, what is perhaps somewhat surprising is how fast this reaction is under “neat-stirring” conditions (orange line). Three different permutations were explored for this reaction, (a) with both added toluene and NaCl, (b) with only added NaCl, and (c) with neither added toluene or NaCl. Permutation c was the most efficient and is the data shown in the graph. When the neat-stirred data are compared to the ball-milled reactor data, it can be seen that for the first hour the data track closely with each other and then diverge. This divergency is likely attributable to both temperature effects and mixing effects. Over the course of the reaction, the milled reactor will increase in temperature until it reaches close to a steady-state temperature; typically, this is in the region 35–50 °C, dependent on the reaction and filling degrees. This is in stark contrast to the neat-stirred reaction which was run at room temperature (i.e., ∼20 °C). With regards to mixing, the fragmentation of reactant particles that are coated in product is key for driving reactions forward. Such fragmentation permits unreacted starting materials to meet throughout the course of the reaction and are necessary when materials are not fully dissolved and/or solvated. The smashing and shearing forces applied by a ball-mill are clearly significantly larger than those from a stirrer bar and thus could contribute to the superior reaction rates shown here. We have also calculated some green metrics of this reaction: for both the solution and milled versions, including E factor, mass intensity, reaction mass efficiency, and molar efficiency. With regards to E factor, the milled process produces approximately 3 times less waste per mass of product than the solution reactions (4.5 and 14.5, respectively), and the molar efficiency is twice as good in the case of the milled reaction (4% milled and 2% solution). Mass intensity and reaction mass efficiency also point towards similar outcomes. Notably, all of our calculations include NaCl, which is environmentally benign but necessary for an effective milled reaction process. Omission of NaCl from green metric calculations leads to even more favorable outcomes for the presently developed process.
Conclusions
In conclusion, the organocatalytic aza-MBH reaction has been explored by ball-milling, with good substrate scope. This reaction process does not require the use of solvent and is complete within 3 h. Preliminary experiments into control of enantioselectivity were promising with β-isocupreidine delivering 64% ee. The reaction was also successfully scaled by 12-fold. Finally, comparisons of this reaction process to both the solvent-based and stirring without solvent systems highlight the increased efficiency of the ball-milled reaction and point towards superior mixing compared to the neat stirring approach.
Acknowledgments
We gratefully acknowledge the School of Chemistry, Cardiff University, for generous support and the EPSRC-funded Bath/Bristol/Cardiff Catalysis Centre for Doctoral Training (M.T.J.W. EP/L016443/1).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acssuschemeng.0c07320.
Details on reaction optimization, experimental procedures, spectral data of all compounds, HPLC traces, and green metric calculations (PDF)
The authors declare no competing financial interest.
Notes
Information about the data that underpins the results presented in this article, including how to access them, can be found in the Cardiff University data catalogue under: http://doi.org/10.17035/d.2020.0120616585.
Supplementary Material
References
- Morita K.; Suzuki Z.; Hirose H. A Tertiary Phosphine-catalyzed Reaction of Acrylic Compounds with Aldehydes. Bull. Chem. Soc. Jpn. 1968, 41, 2815–2815. 10.1246/bcsj.41.2815. [DOI] [Google Scholar]
- Baylis A. B.; Hillman M. E. D.. German Patent 2155113, 1972; Chem. Abstr. 1972, 77, 34174q. [Google Scholar]
- Basavaiah D.; Rao A.; Satyanarayana T. Recent advances in the Baylis-Hillman reaction and applications. Chem. Rev. 2003, 103, 811–891. 10.1021/cr010043d. [DOI] [PubMed] [Google Scholar]
- Declerck V.; Martinez J.; Lamaty F. Aza-Baylis-Hillman reaction. Chem. Rev. 2009, 109, 1–48. 10.1021/cr068057c. [DOI] [PubMed] [Google Scholar]
- Ma G.; Jiang J.; Shi M.; Wei Y. Recent extensions of the Morita-Baylis-Hillman reaction. Chem. Commun. 2009, 5496–5514. 10.1039/b909405a. [DOI] [PubMed] [Google Scholar]
- Iwabuchi Y.; Nakatani M.; Yokoyama N.; Hatakeyama S. Chiral amine-catalyzed asymmetric Baylis-Hillman reaction: A reliable route to highly enantiomerically enriched (α-methylene-β-hydroxy)esters. J. Am. Chem. Soc. 1999, 121, 10219–10220. 10.1021/ja992655+. [DOI] [Google Scholar]
- Wei Y.; Shi M. Recent advances in organocatalytic asymmetric morita-baylis-hillman/aza-morita-baylis-hillman reactions. Chem. Rev. 2013, 113, 6659–6690. 10.1021/cr300192h. [DOI] [PubMed] [Google Scholar]
- Hu F.; Shi M. The highly enantioselective catalytic aza-Morita–Baylis–Hillman reaction. Org. Chem. Front. 2014, 1, 587–595. 10.1039/C4QO00026A. [DOI] [Google Scholar]
- Pellissier H. Recent developments in the asymmetric organocatalytic Morita–Baylis–Hillman reaction. Tetrahedron 2017, 73, 2831–2861. 10.1016/j.tet.2017.04.008. [DOI] [Google Scholar]
- Campi E. M.; Holmes A.; Perlmutter P.; Teo C. A Simple, Mild Synthesis of 3-Aryl-2-methylidene-3-(p-tolylsulfonylamino)propanoates. Aust. J. Chem. 1995, 48, 1535–1540. 10.1071/CH9951535. [DOI] [Google Scholar]
- Fülöp F. The chemistry of 2-aminocyclopentanecarboxylic acid. Stud. Nat. Prod. Chem. 2000, 22, 273–306. 10.1016/S1572-5995(00)80028-9. [DOI] [Google Scholar]
- Shi M.; Zhao G. Baylis-Hillman reactions of N-arylidenediphenylphosphinamides with methyl vinyl ketone, methyl acrylate, and acrylonitrile. Tetrahedron Lett. 2002, 43, 4499–4502. 10.1016/S0040-4039(02)00813-4. [DOI] [Google Scholar]
- Shi M.; Xu Y. Lewis base effects in the Baylis-Hillman reaction of imines with methyl vinyl ketone. Eur. J. Org. Chem. 2002, 2002, 696–701. . [DOI] [Google Scholar]
- Xu Y.; Shi M. Highly Efficient aza-Baylis-Hillman Reaction of N-Tosylated Imines with MVK, Acrolein, and Phenyl Acrylate or α-Naphthyl Acrylate: Lewis Base Effects and A Convenient Method to Synthesize α,β-Unsaturated β-Amino Carbonyl Compounds. J. Org. Chem. 2004, 69, 417–425. 10.1021/jo035103p. [DOI] [PubMed] [Google Scholar]
- Hu F.; Wei Y.; Shi M.; Pindi S.; Li G. Asymmetric catalytic aza-Morita–Baylis–Hillman reaction for the synthesis of 3-substituted-3-aminooxindoles with chiral quaternary carbon centers. Org. Biomol. Chem. 2013, 11, 1921–1924. 10.1039/c3ob27495k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y.; Li J.-L.; Zhou Q.; Dong L.; Chen Y. Enantioselective Aza-Morita–Baylis–Hillman Reaction with Ketimines and Acrolein Catalyzed by Organic Assemblies. Chem. - Eur. J. 2013, 19, 9447–9451. 10.1002/chem.201301558. [DOI] [PubMed] [Google Scholar]
- Takizawa S.; Rémond E.; Arteaga F. A.; Yoshida Y.; Sridharan V.; Bayardon J.; Jugé S.; Sasai H. P-chirogenic organocatalysts: application to the aza-Morita–Baylis–Hillman (aza-MBH) reaction of ketimines. Chem. Commun. 2013, 49, 8392–8394. 10.1039/c3cc44549f. [DOI] [PubMed] [Google Scholar]
- Shi M.; Xu Y. Catalytic, asymmetric Baylis-Hillman reaction of imines with methyl vinyl ketone and methyl acrylate. Angew. Chem., Int. Ed. 2002, 41, 4507–4510. . [DOI] [PubMed] [Google Scholar]
- Shi M.; Chen L.; Li C. Chiral phosphine Lewis bases catalyzed asymmetric aza-Baylis-Hillman reaction of N-sulfonated imines with activated olefins. J. Am. Chem. Soc. 2005, 127, 3790–3800. 10.1021/ja0447255. [DOI] [PubMed] [Google Scholar]
- Abermil N.; Masson G.; Zhu J. Highly enantioselective aza Morita-Baylis-Hillman reaction catalyzed by bifunctional β-isocupreidine derivatives. J. Am. Chem. Soc. 2008, 130, 12596–12597. 10.1021/ja805122j. [DOI] [PubMed] [Google Scholar]
- Fort Y.; Berthe M. C.; Caubere P. The ‘Baylis-Hillman Reaction’ mechanism and applications revisited. Tetrahedron 1992, 48, 6371–6384. 10.1016/S0040-4020(01)88227-2. [DOI] [Google Scholar]
- Horie K.; Barón M.; Fox R. B.; He J.; Hess M.; Kahovec J.; Kitayama T.; Kubisa P.; Maréchal E.; Mormann W.; Stepto R. F. T.; Tabak D.; Vohlídal J.; Wilks E. S.; Work W. J. Definitions of terms relating to reactions of polymers and to functional polymeric materials. Pure Appl. Chem. 2004, 76, 889–906. 10.1351/pac200476040889. [DOI] [Google Scholar]
- Howard J. L.; Cao Q.; Browne D. L. Mechanochemistry as an emerging tool for molecular synthesis: what can it offer?. Chem. Sci. 2018, 9, 3080–3094. 10.1039/C7SC05371A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen J.; Mack J. Mechanochemistry and organic synthesis: from mystical to practical. Green Chem. 2018, 20, 1435–1443. 10.1039/C7GC03797J. [DOI] [Google Scholar]
- Do J.-L.; Friščić T. Mechanochemistry: A Force of Synthesis. ACS Cent. Sci. 2017, 3, 13–19. 10.1021/acscentsci.6b00277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez J. G.; Bolm C. Altering Product Selectivity by Mechanochemistry. J. Org. Chem. 2017, 82, 4007–4019. 10.1021/acs.joc.6b02887. [DOI] [PubMed] [Google Scholar]
- Eguaogie O.; Vyle J. S.; Conlon P. F.; Gilea M. A.; Liang Y. Mechanochemistry of nucleosides, nucleotides and related materials. Beilstein J. Org. Chem. 2018, 14, 955–970. 10.3762/bjoc.14.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metro T.-X.; Martinez J.; Lamaty F. 1,1′-Carbonyldiimidazole and Mechanochemistry: A Shining Green Combination. ACS Sustainable Chem. Eng. 2017, 5, 9599–9602. 10.1021/acssuschemeng.7b03260. [DOI] [Google Scholar]
- Tan D.; Garcia F. Main group mechanochemistry: from curiosity to established protocols. Chem. Soc. Rev. 2019, 48, 2274–2292. 10.1039/C7CS00813A. [DOI] [PubMed] [Google Scholar]
- Friščić T.; Motillo C.; Titi H. M. Mechanochemistry for Synthesis. Angew. Chem., Int. Ed. 2020, 59, 1018–1029. 10.1002/anie.201906755. [DOI] [PubMed] [Google Scholar]
- Pérez-Venegas M.; Juaristi E. Mechanochemical and Mechanoenzymatic Synthesis of Pharmacologically Active Compounds: A Green Perspective. ACS Sustainable Chem. Eng. 2020, 8, 8881–8893. 10.1021/acssuschemeng.0c01645. [DOI] [Google Scholar]
- Rodríguez B.; Bruckmann A.; Bolm C. A Highly Efficient Asymmetric Organocatalytic Aldol Reaction in a Ball Mill. Chem. - Eur. J. 2007, 13, 4710–4722. 10.1002/chem.200700188. [DOI] [PubMed] [Google Scholar]
- Nicholson W. I.; Seastram A. C.; Iqbal S. A.; Reed-Berendt B. G.; Morrill L. C.; Browne D. L. N-Heterocyclic Carbene Acyl Anion Organocatalysis by Ball-Milling. ChemSusChem 2020, 13, 131–135. 10.1002/cssc.201902346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack J.; Shumba M. Rate enhancement of the Morita–Baylis–Hillman reaction through mechanochemistry. Green Chem. 2007, 9, 328–330. 10.1039/B612983H. [DOI] [Google Scholar]
- Karki S.; Friščić T.; Jones W.; Motherwell W. D. S. Screening for Pharmaceutical Cocrystal Hydrates via Neat and Liquid-Assisted Grinding. Mol. Pharmaceutics 2007, 4, 347–354. 10.1021/mp0700054. [DOI] [PubMed] [Google Scholar]
- Fernandes J. A.; Sardo M.; Marfa L.; Choquesillo-Lazarte D.; Masciocchi N. X-ray and NMR Crystallography Studies of Novel Theophylline Cocrystals Prepared by Liquid Assisted Grinding. Cryst. Growth Des. 2015, 15, 3674–3683. 10.1021/acs.cgd.5b00279. [DOI] [Google Scholar]
- Jung S.; Choi I.; Kim I. W. Liquid-Assisted Grinding to Prepare a Cocrystal of Adefovir Dipivoxil Thermodynamically Less Stable than Its Neat Phase. Crystals 2015, 5, 583–591. 10.3390/cryst5040583. [DOI] [Google Scholar]
- Mukherjee A.; Rogers R. D.; Myerson A. S. Cocrystal formation by ionic liquid-assisted grinding: case study with cocrystals of caffeine. CrystEngComm 2018, 20, 3817–3821. 10.1039/C8CE00859K. [DOI] [Google Scholar]
- Chen L.; Regan M.; Mack J. The Choice Is Yours: Using Liquid-Assisted Grinding To Choose between Products in the Palladium-Catalyzed Dimerization of Terminal Alkynes. ACS Catal. 2016, 6, 868–872. 10.1021/acscatal.5b02001. [DOI] [Google Scholar]
- Howard J. L.; Sagatov Y.; Repusseau L.; Schotten C.; Browne D. L. Controlling reactivity through liquid assisted grinding: the curious case of mechanochemical fluorination. Green Chem. 2017, 19, 2798–2802. 10.1039/C6GC03139K. [DOI] [Google Scholar]
- Howard J. L.; Sagatov Y.; Browne D. L. Mechanochemical electrophilic fluorination of liquid β-ketoesters. Tetrahedron 2018, 74, 3118–3123. 10.1016/j.tet.2017.11.066. [DOI] [Google Scholar]
- Jiang Z. J.; Li Z. H.; Yu J. B.; Su W. K. Liquid-Assisted Grinding Accelerating: Suzuki–Miyaura Reaction of Aryl Chlorides under High-Speed Ball-Milling Conditions. J. Org. Chem. 2016, 81, 10049–10055. 10.1021/acs.joc.6b01938. [DOI] [PubMed] [Google Scholar]
- Howard J. L.; Brand M. C.; Browne D. L. Switching Chemoselectivity: Using Mechanochemistry to Alter Reaction Kinetics. Angew. Chem., Int. Ed. 2018, 57, 16104–16108. 10.1002/anie.201810141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y.; Shi M. Aza-Baylis–Hillman reaction of N-tosylated imines with β-substituted α,β-unsaturated esters. Tetrahedron 2006, 62, 461–475. 10.1016/j.tet.2005.09.058. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.