

Abstract
N-Nitrosamines are a class of compounds notorious both for the potent carcinogenicity of many of its members and for their widespread occurrence throughout the human environment, from air and water to our diets and drugs. Considerable effort has been dedicated to understanding N-nitrosamines as contaminants, and methods for their prevention, remediation, and detection are ongoing challenges. Understanding the chemistry of N-nitrosamines will be key to addressing these challenges. To facilitate such understanding, we focus in this Perspective on the structure, reactivity, and synthetic applications of N-nitrosamines with an emphasis on alkyl N-nitrosamines. The role of N-nitrosamines as water contaminants and the methods for their detection are also discussed.
Graphical Abstract

N-Nitrosamines (or simply “nitrosamines”) are a class of compounds sharing the general structure 1 (Chart 1), where the amine moiety may be derived from any organic secondary amine. Many nitrosamines, particularly those with structure 2 (e.g., N-nitrosodimethylamine [NDMA], Chart 1), are known to be carcinogenic to animals and are reasonably anticipated to be human carcinogens.1–3 Scheme 1 shows the mechanism of carcinogenicity for 2. Nitrosamine 2 first undergoes enzymatic α-hydroxylation with cytochrome P450 and subsequently forms the dealkylated primary nitrosamine. The unstable primary nitrosamine further decomposes to diazonium 3, a DNA alkylating agent.2,4,5 The resulting DNA damage can lead to cancer.
Chart 1.

Some N-Nitrosamines
Scheme 1.

Transformation of Nitrosamine 2 into Diazonium 3 via Enzymatic α-Hydroxylation
In recent years, nitrosamines have drawn increased attention after several popular medications were discovered to contain unacceptable levels of nitrosamines, resulting in recalls6 and new regulatory guidance.7 However, concern over nitrosamine exposure is much older than these latest recalls. Although now used only as a research chemical, NDMA was previously used for a number of industrial applications.3,8 NDMA initially only drew concern for its high toxicity,9 but in 1956 it was reported to be carcinogenic to rats10 and scrutiny soon turned toward other nitrosamines. Now, we know that many nitrosamines are carcinogenic1–3 and they can be found throughout the human environment, from air, water, and soil to foods, drinks, and drugs.2,7,8 Those new to the problem of nitrosamines will find a number of quite recent reviews on nitrosamines as contaminants (particularly NDMA in water),11–15 but scarce discussion of nitrosamines as organic chemicals. However, an understanding of the chemistry of nitrosamines will be essential to addressing many of the challenges of nitrosamine contamination, especially with regard to detection and remediation. Herein, we address nitrosamines from an organic chemistry perspective with an emphasis on alkyl nitrosamines (i.e., 2) since those are generally more likely to be carcinogenic.1,2 We discuss first the structure and reactivity of nitrosamines, and then briefly cover their role as water contaminants and the methods for their detection.
THE STRUCTURE OF NITROSAMINES
Although nitrosamines are typically depicted as structure 1a, their actual structures and chemical reactivity are heavily influenced by the zwitterionic resonance structure 1b (Figure 1). Rotation about the N–N bond is hindered as a result of its significant double bond character, with a barrier of ~23 kcal/mol for NDMA and other acyclic dialkylnitrosamines.16–18 This is slightly higher than the barrier to rotation about the analogous C–N bond of N,N-dimethylformamide (DMF) in water (22 kcal/mol).19 This hindered rotation and the planarity of the CαCαNNO moiety20,21 result in magnetic nonequivalence of the N-substituents in symmetric nitrosamines (e.g., NDMA).16,22 For the simplest dialkylnitrosamine, NDMA, the N–N and N–O bond lengths are 1.344 and 1.235 Å in the gas phase, respectively.20 In single crystals of NDMA at low temperature, intermolecular interactions stabilize increased polar character (e.g., 1b), resulting in a shortened N–N bond (1.320 Å) and an elongated N–O bond (1.260 Å).21
Figure 1.

Two major resonance contributors of nitrosamines.
REACTIONS OF NITROSAMINES IN THE GROUND STATE
Although N-nitrosamines are not commonly employed in organic synthesis, their reactivity has been studied both for synthetic applications and for understanding the mechanisms of their carcinogenicity. Herein, we describe some reactions and synthetic applications of nitrosamines. While we particularly focus on chemistry relevant to alkyl nitrosamines, much of it is also applicable to other nitrosamine derivatives.
Protonation and Protolytic Denitrosation.
Consistent with the resonance contributors in Figure 1, nitrosamines are most basic at oxygen.23,24 For NDMA and other dialkylnitrosamines, the pKa is less than 1 for the O-protonated species (4, Chart 2).25 Hydroxydiazenium salt 4 is the only protonated form seen in NMR spectra of nitrosamines in acid,22,24 and it is predicted to be more stable by 16 kcal/mol (for R1 = R2 = Me) than the amino-N-protonated isomer (5, Chart 2).26 The N-protonated species 5 has not been directly observed, although it may occur in kinetically relevant amounts at pH 1–2.27 5 is sometimes evoked to explain reactions of nitrosamines in acid.5 One such reaction is the denitrosation of nitrosamines in acidic conditions, yielding the corresponding secondary amines.22,28,29 A common mechanism for this decomposition is shown in Scheme 2, wherein 5 is formed and subsequently denitrosated by attack of a nucleophile.5 Protolytic denitrosation can be accelerated by the addition of nucleophiles such as bromide, thiocyanate, and thiourea to the reaction.29,30
Chart 2.
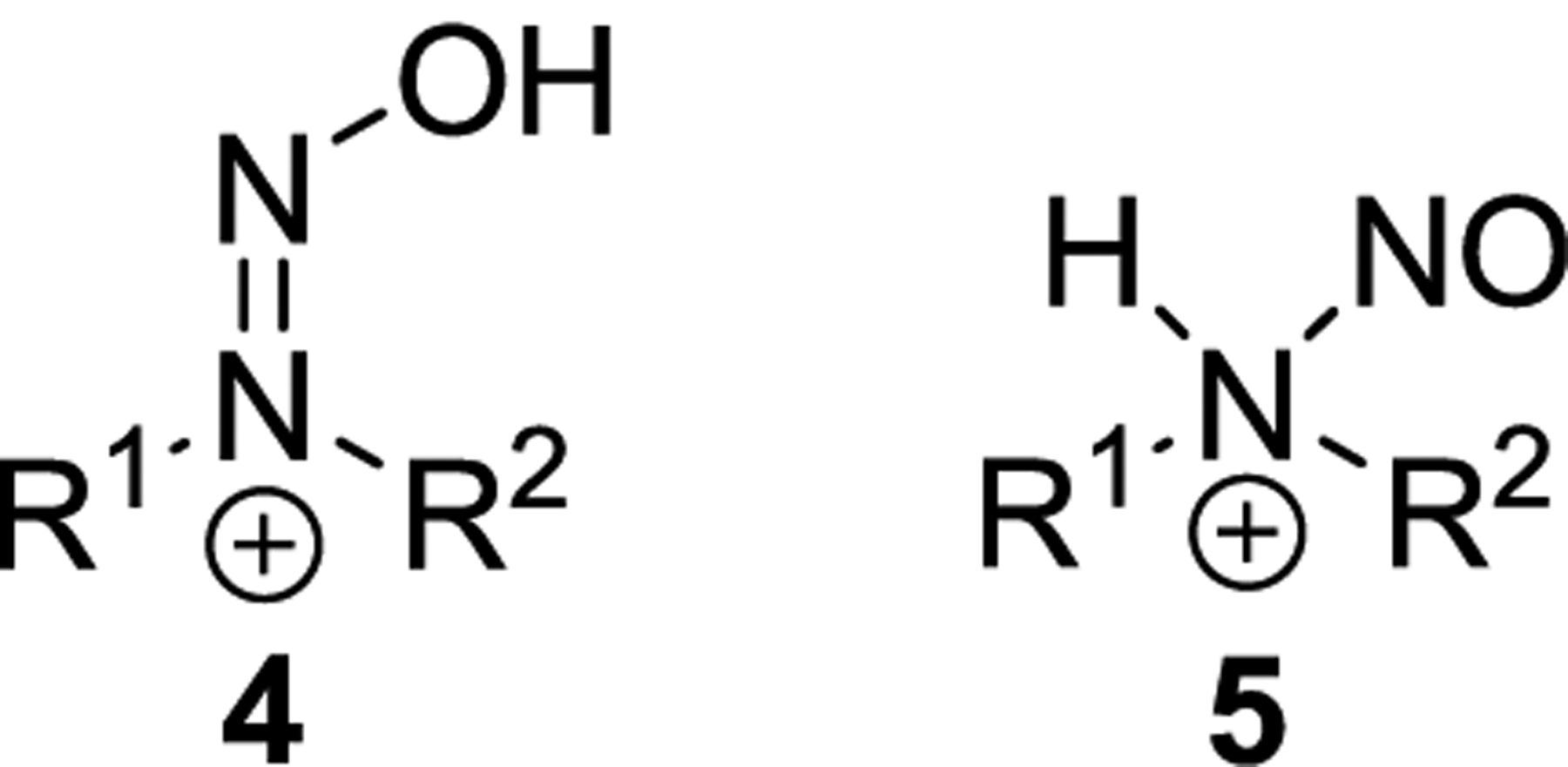
O-Protonated (4) and N-Protonated (5) Isomers of the Nitrosamine Conjugate Acid
Scheme 2.

Proposed Mechanism for Nitrosamine Denitrosation in Acid via the N-Protonated Conjugate Acid (5)
Reactions with Electrophiles: Synthesis and Reactivity of Alkoxydiazenium Salts.
Nitrosamines can react through their oxygen with various electrophiles to form O-substituted hydroxydiazenium salts (i.e., O-substituted derivatives of 4). The electrophile is most commonly an alkylating agent, forming alkoxydiazenium 6,31–34 but O-trimethylsilyl (7) and O-triflyl (8) derivatives of NDMA have also been prepared (Chart 3).35 Trialkyloxonium salts,31–33 dimethyl sulfate,31,36 alkyl fluorosulfonates,34 and alkyl halides with silver perchlorate32,35 have all been used to O-alkylate nitrosamines. These cations 6–8 are themselves electrophilic and generally the parent nitrosamine is recovered after nucleophilic attack (e.g., O-dealkylation of 6, Scheme 3),33–35 although in a few cases N-dealkylation34,35 or attack at the O-attached nitrogen occur instead.36–38 O-Triflyl derivative 8, formed from NDMA and triflic anhydride, undergoes N-dealkylation to methylate toluene and give a mixture of xylenes.35 Curiously, 3,4-dichlorothiophenol with either the O-methylated salt of N-nitrosodiethylamine (6a) or the O-ethylated salt of N-nitrosomethylethylamine (6b) in organic solvent resulted in the same product distribution, indicating both O- and N-dealkylation had occurred (Scheme 4), whereas aniline and 3,4-dichloroaniline only gave O-dealkylation products, and thiophenol and phenol showed no reaction at all.34 The mechanism is unclear, particularly for the formation of nitrosamine products after N-dealkylation.
Chart 3.

O-Alkyl (6), O-Trimethylsilyl (7), and O-Triflyl (8) Hydroxydiazenium Derivatives
Scheme 3.

O-Dealkylation of Alkoxydiazenium 6
Scheme 4.

Reaction of Alkoxydiazenium Salts 6a,b with 3,4-Dichlorothiophenol
For alkoxydiazeniums of form 9 (a subset of 6), some reactions are proposed to begin with deprotonation of the α-position, giving zwitterion 10 (Scheme 5), which behaves as structures 10b or 10c in subsequent reactions.33 Those reactions are reviewed in detail elsewhere,33 but as that review is in German, we will discuss the chemistry here briefly. Reaction of 9 with carboxylates passes through cyclic species 11 and produces either carbonylazo compound 12 (R1 = aryl, tert-butyl) or acylhydrazone 13 (R1 = Me, Et, i-Pr, Bn, etc.) (Scheme 5).39,40 Fused 1,2,4-triazolium cations (e.g., 14) are formed from the cyclization of 10 (R3 = H) and nitrogen heterocycles, while cyclization with Schiff bases produces 15 (Scheme 6).41,42 Treatment of 9 (R3 = H) with sodium hydroxide yields trans-α-hydroxydialkyldiazene 16.43,44 In that transformation, the migration of −CH(OH)R2 likely occurs through decomposition and intermolecular recombination of intermediate species (Scheme 6).44
Scheme 5.

Deprotonation of Alkoxydiazenium 9 and Subsequent Reaction with a Carboxylate
Scheme 6.

Formation of 1,2,4-Triazolium or trans-α-Hydroxydialkyldiazene Products from Alkoxydiazenium 9
α-Lithiated Nitrosamines.
The α-protons of nitrosamines are more acidic than those of the corresponding secondary amines,45 allowing them to be easily lithiated at the α-position with lithium diisopropylamide (LDA).46 These α-lithiated nitrosamines 17 can be utilized in reactions with various electrophiles as synthetic equivalents of secondary amine α-carbanions (Figure 2).46,47 Seebach and co-workers have reported a one-pot procedure for the α-substitution of secondary amines via formation of the nitrosamines. In it, the secondary amine is nitrosated with ethyl nitrite, lithiated with LDA, reacted with the desired electrophile, and then denitrosated by either Raney nickel-catalyzed hydrogenolysis alone48,49 or reduction with lithium aluminum hydride (LAH) followed by hydrogenolysis with Raney nickel.50 Other procedures recover the substituted amine via denitrosation in acid.51–53 After the first addition of electrophile, the lithiation-substitution sequence can be repeated without an intervening workup.52,54
Figure 2.

Lithionitrosamines 17 can react with various electrophiles. Denitrosation reveals the α-substituted secondary amine.
A variety of electrophiles can be added to lithionitrosamines (17). Reactions with alkyl halides, aldehydes, ketones, and a variety of carboxylic acid derivatives work well.47,51,54,55 The product of 17 and α,β-unsaturated ketones depends on the structure of the ketone. The 1,2-adduct is produced with cyclohexenone, while reaction with chalcone yields a mixture of the 1,2- and 1,4-adducts.46 Heteroatom-substituted products can be obtained from reaction of 17 with disulfides, diselenides, trimethylsilyl chloride, and trimethyltin chloride (Chart 4).56 Reaction of 17 (R3 = H) with aryl nitriles gives cyclization to form 1,2,3-triazoles 18 (Scheme 7),57 wherein the highest yields are obtained using a twofold excess of 17.58 Tetrahydro-1,2,3,4-tetrazine oxides 19 are produced by the dimerization of lithiated dialkylnitrosamines (Scheme 8),46,59,60 and a radical mechanism has been proposed.60 Oxides 19 can be reduced to the corresponding tetrahydrotetrazines with trimethyl phosphite.46,59,60
Chart 4.

Heteroatom-Substituted Nitrosamines
Scheme 7.

Cyclization of Aryl Nitriles and 17 to Give 1,2,3-Triazoles 18
Scheme 8.

Dimerization of Dialkyl 17
Reactions with Organolithium and Grignard Reagents.
N-Nitrosamines are weakly electrophilic at the nitroso-nitrogen and are susceptible to nucleophilic attack at that site by organolithium and Grignard reagents (R3M, M = MgX or Li).61–64 The initial product is an unstable oxyhydrazine (20), which undergoes elimination to form either hydrazone 21 (R3 = −CH2R only)61,62,64 or azomethine imine 22 (Scheme 9).63,64 Elimination from 20a (M = MgX) can occur before quenching with protic solvent.64 Because 22 is susceptible to further nucleophilic attack, α-substituted hydrazine 23 is often isolated after workup when Grignard reagents are used (Scheme 9).64 When the Grignard reagent used is phenylmagnesium bromide, N-phenyl hydrazine 24 is produced by a formal reduction of 20a (Scheme 9).61,62,64 In reactions with phenyllithium or tert-butyllithium, elimination from 20b (M = Li) only occurs after quenching with protic solvent.63 When 20b is quenched with water, the only product observed is the head-to-tail dimer of 22 (25, Scheme 9). When ethanol is used, the solvent adduct 26 (Scheme 9) is briefly observable by NMR, but it is in equilibrium with 22 and ultimately decomposes to 25.63 Similar to other azomethine imines,65 22 can undergo 1,3-dipolar cycloadditions with dipolarophiles.63
Scheme 9.

Nucleophilic Attack at the Nitroso-N by Organometallic Reagents and Subsequent Reactions
Reduction to 1,1-Disubstituted Hydrazines.
NDMA was once used as a starting material in the industrial manufacture of 1,1-dimethylhydrazine, a rocket fuel.66 The reduction of nitrosamines (1) to 1,1-disubstituted hydrazines (27, Figure 3) has been known since the late 19th century67 and can be effected with a wide variety of reaction conditions.68 Reduction of nitrosamines with zinc dust in acetic acid is perhaps the oldest method,67 and dialkyl 27 can be isolated as the hydrochloride salt in fair yields by this method.69 Catalytic hydrogenation was of interest for industrial purposes, and Pd/C, Pt/C, and Rh/C all yield the corresponding hydrazines, although the yield of 27 is strongly dependent on reaction conditions and the identity of 1.70,71 The yield of 27 by catalytic hydrogenation is improved by the inclusion of a salt (e.g., NH4OAc, CaCl2) in the reaction mixture.71 Hydride reduction using LAH generally provides fair to good yields of 1,1-dialkyl or 1,1-alkylaryl 27,50,72–74 but diarylnitrosamines are easily over-reduced to give the parent amine and ammonia73 unless the reaction conditions are carefully controlled.75 Reduction of 1 can be effected with sodium metal in either absolute ethanol or ethanol in liquid ammonia, but the yield is lower than with LAH for many substrates.72 TiCl4 with magnesium powder in dichloromethane/diethyl ether solution forms a reducing Ti(II) reagent that reduces a variety of nitrosamines to hydrazines in excellent yields (>90% typically), though the reagent must be made fresh for each reaction.76 Electrolytic reduction of dialkylnitrosamines proceeds smoothly in acidic ethanol, but much lower yields were obtained with nitrosamines bearing aryl substituents.77 In all of these conditions, denitrosation to the parent amine is also observed.
Figure 3.

Reduction of nitrosamines (1) to hydrazines (27).
Other Reactions of Nitrosamines.
Nitrosamines can be oxidized to their corresponding nitramines. The nitramine (28) can be obtained in good yield after treatment of 1 with either nitric acid78 or peroxytrifluoroacetic acid,79 although by different mechanisms. Peroxytrifluoroacetic acid oxidizes the existing nitroso moiety (Scheme 10a), while nitric acid effects the exchange of the nitroso and nitro groups (Scheme 10b).80
Scheme 10.

Oxidation of 15N-Labeled Nitrosamine 1 to (a) Labeled Nitramine 28 with Peroxytrifluoroacetic Acid and (b) Unlabeled 28 with Nitric Acid
We have focused this section on the chemistry of alkyl nitrosamines. The reactions of N-nitrosamines bearing other substituents (e.g., N-nitrosoenamines) or having additional reactive moieties are outside the scope of this review, but such reactions have been reviewed previously.81 Nitrosamine-metal complexes have also been reviewed elsewhere,82 and Chart 5 shows possible binding modes in such complexes. Through the formation of cyclic ortho-metalated complexes (Chart 5, right), the N-nitroso moiety can act as an easily cleavable directing group in metal-catalyzed C–H activation of N-nitrosoaniline derivatives.83–89 For completeness, we mention here the Fischer-Hepp rearrangement of aryl nitrosamines because it is relevant to N-aryl-N-alkyl nitrosamines. In acidic conditions, aryl nitrosamines (e.g., N-nitrosoaniline 29) undergo rearrangement to the corresponding p-nitroso products (e.g., 30, Figure 4).90 The exact mechanism of the Fischer-Hepp rearrangement is unknown, and both inter- and intramolecular mechanisms have been proposed.5 Because the rearrangement is performed in acidic conditions, denitrosation is increasingly competitive with increasing acidity.91 The rearrangement occurs with a variety of aromatic substituents.5
Chart 5.

Metal-Binding Modes between Nitrosamines and Metals (M)
Figure 4.

Fischer-Hepp rearrangement.
PHOTOCHEMISTRY
Photolysis.
Since the 1930s, it has been known that UV irradiation of NDMA and other N-nitrosamines induces fragmentation of their N–N bond.92 Excitation of NDMA to a singlet excited state by either its π → π* (S0 → S2) or n → π* (S0 → S1) transition leads to photolytic cleavage, although it can be reformed by recombination of the produced nitric oxide (NO) and dimethylamino radical 31 (Scheme 11).93,94 For NDMA in water, the π → π* and n → π* transitions appear in the absorption spectrum as bands at λmax = 228 nm (ε = 7378 M−1cm−1) and 332 nm (ε = 109 M−1cm−1), respectively, and other dialkylnitrosamines have similar characteristics.95,96 Photolysis of gaseous NDMA displays a fragmentation quantum yield of 1,93 whereas the quantum yields for nitrosamines in solution are much lower (Φ ≤ 0.3), presumably as a result of solvent cages that promote the back reaction shown in Scheme 11. The reported solution-phase quantum yields vary and can be affected by solution pH, the presence of oxygen, and the initial nitrosamine concentration.96–100 The variations arise, at least in part, from the effect of photolysis conditions on the multiple mechanistic pathways involved in nitrosamine photolysis, which as a result of their complex environmental dependence are not fully understood. We describe below the range of mechanisms and the products of NDMA, which is the most studied N-nitrosamine and serves as a general guide for the expected reactivity of other dialkyl analogues.
Scheme 11.

Photolysis of NDMA by N–N Bond Homolysis and the Thermal Recombination Back Reaction
The proposed pathways of nitrosamine photolysis in solution are depicted for NDMA in Figure 5, and for simplicity some secondary reactions have been excluded. In contrast to the gas phase wherein photolytic breakdown proceeds by a mechanism analogous to path A (homolysis) in Figure 5,93,94 in the solution phase NDMA* can also be protonated (33*) and decomposed by nucleophilic nitrite attack (path B)98,101 or water (hydrolysis, path C).26,102 Alternatively, 33* can rearrange (path D) to the unstable N-protonated isomer (34), which then homolytically fragments.26,102,103 Fragmentation of 34 is very different from what is observed in path A, wherein NO and 31 can recombine to regenerate NDMA.23,99 Specifically, this homolysis is effectively irreversible because the recombination of NO and aminium radical 35 would produce high-energy species 34, which is energetically unfavorable.26,103 Thus, the decomposition of 33* (via paths B–D) is not expected to lead to significant reformation of NDMA.103
Figure 5.

Proposed mechanisms of N-nitrosamine photolysis in solution using NDMA as a representative nitrosamine.
Paths A–D are consistent with experimental observations of the effect of pH on photolysis. Although the rate of nitrosamine photolysis increases with decreasing pH in aqueous solutions in air,95,104,105 the quantum yield of NDMA decomposition by photolysis is constant (Φ ≈ 0.3) over an extended range of acidity, pH 2–8.100,101 However, the decomposition quantum yields drop precipitously to below 0.1 between pH 8–9 and continue to slowly decrease with increasing alkalinity above pH 9.101 This pH dependence suggests that the pKa of 33* is 8–9, above which point decomposition from NDMA* dominates.101 Additionally, the quantum yields obtained above pH 9 in water are similar to those obtained in neutral aprotic solvent for other dialkylnitrosamines, which also decompose from the neutral excited state (e.g., NDMA*).99,106 A dependence on the initial nitrosamine concentration has also been observed in aprotic solvent. This feature is expected for decomposition by path A: at higher initial concentrations, 31 has an increased probability of hydrogen atom abstraction from unreacted NDMA rather than recombination with NO.99 In contrast to aqueous solutions, the photolysis quantum yields in aprotic solutions are significantly improved by the addition of acid,23,106,107 presumably because paths B–D become accessible.
Path E in Figure 5 illustrates how NDMA* and 33* can decompose through single-electron transfer to O2 with subsequent fragmentation of the oxidized species.101 This mechanism was proposed as part of an explanation to account for differences in quantum yield between solutions photolyzed under O2 and those under N2. In accord with this branch point in the mechanistic possibilities, a distinct decrease in quantum yield is observed with increasing pH under N2 atmosphere.101 The decreased decomposition quantum yield when photolysis is run under inert atmosphere has been noted earlier for solutions in acetonitrile but was attributed to O2 reacting with radicals in path A to hinder reformation of the nitrosamine under air and not investigated further.99 The difference between air and inert atmosphere vanishes when only the S0 → S1 (~332 nm, n → π*) transition is excited;96,101 our analysis is that this is likely the result of electron transfer and/or other reactions stemming from a longer lived T1 state that can result from intersystem crossing from the S2 state. Superoxide radical (O2•−)101 and a species with hydroxyl radical-like reactivity (suggested to be peroxynitrite, ONOO−)105 have been detected in the photolysis of aqueous NDMA. These results are consistent with the occurrence of path E, however additional investigation is still needed to confirm the existence of this pathway and identify the secondary decomposition reaction products. There are also other oxygen reaction pathways in addition to path E. In particular, irradiated nitrosamines have been found to undergo oxygen-atom exchange with dissolved O2 and to also produce the corresponding nitramines. These transformations plausibly occur via the formation and collapse of a nitrosamine peroxide intermediate.108,109
Although the initial products of the photolysis pathways in Figure 5 differ, a number of the secondary reactions result in similar final products. In aqueous solutions of NDMA, the major products are methylamine (MA), dimethylamine (DMA), nitrite, nitrate, formaldehyde, and formic acid.95,98,101 MA and formaldehyde are produced by the hydrolysis of imine 32 (32+), and the latter is oxidized to formic acid. In nonaqueous solutions without acid, the imine is observed instead of its hydrolysis products.23,110 In acidic solutions with relatively high concentrations of nitrosamine (≥~50 mM), the corresponding amidoxime (36, Scheme 12) is also obtained.23,110,111 Although there is no single agreed-upon mechanism for 36’s formation,101,111 there is evidence that this amidoxime is produced by an intermolecular reaction rather than an intramolecular rearrangement.110 The amidoxime product is not typically observed in more modern product studies of nitrosamine photolysis, which are often conducted in dilute aqueous solutions (≤1 mM nitrosamine).95,96,98 However, an amidoxime has been proposed as an intermediate in the formation of N-methylformamide during NDMA photolysis at low concentrations in water.101
Scheme 12.

Formation of an Amidoxime (36) during Nitrosamine Photolysis
In aqueous solutions, the solution pH and initial concentration of NDMA significantly affect the distribution of photolysis products. In neutral and acidic conditions, DMA is the favored product when the initial concentration of NDMA is greater than 10−4 M,95 but MA is increasingly favored at lower, more environmentally relevant concentrations (≤10−5 M).96,98 DMA and nitrite formation are both maximized at approximately pH 4, and the product distribution can be diverted toward DMA at other pHs by the addition of nitrite.98,101 This is consistent with the occurrence of path B in Figure 5, which both consumes and produces nitrite (through the decomposition of N2O3) and is favored by higher concentrations of nitrite in solution (a consequence of higher initial NDMA concentration). Higher initial NDMA concentrations also facilitate DMA production by hydrogen abstraction from unreacted NDMA (or another species) by 31 and 35. Hydrogen abstraction from NDMA also leads to MA formation by hydrolysis of 32 (32+). In alkaline aqueous solutions, MA is the major amine product.98,101 Under nitrogen atmosphere, formation of N-methylformamide has been observed in solutions at pH > 5, although as mentioned this product is presumed to be generated from decomposition of N-methylformamidoxime (36 where R = CH3, R′ = H).101
Photolysis of nitrosamines in acidic aqueous conditions produces more nitrite than nitrate and in alkaline conditions the ratio is reversed.101,105 Photolysis of acidic solutions initially produces both nitrite and nitrate, however the nitrate production quickly tapers off while nitrite formation continues.95,98,105 In alkaline solutions, both are produced throughout the reaction progression, but nitrate is formed at a faster rate.105 The relative amounts of nitrate and nitrite produced by photolysis of neutral solutions are less clear, and studies have reported that nitrite may be significantly favored,95 slightly favored,98 or slightly disfavored101 relative to nitrate. Stefan and Bolton found that nitrate appeared to be primarily produced as a result of oxidation of nitrite at pH 7.95 In contrast, a study by Lee and co-workers reports their simultaneous formation.98 These differences may be a consequence of experimental conditions, which used different initial nitrosamine concentrations, solution buffer systems, and light sources for photolysis. The formation mechanisms of these ions in nitrosamine photolysis have been given much less attention than the organic products. A complete exploration of the relevant solution-phase NOX chemistry is beyond the scope of this review, but Figure 5 includes several key reactions that lead to their production.101,105
The kinetics of nitrosamine photolysis are concentration-dependent. Nitrosamine photolysis in water follows first-order kinetics for low initial concentrations (<0.1 mM).95,96,100,104,112 Stefan and Bolton found that at higher initial NDMA concentrations (0.1 and 1 mM), zero-order kinetics were initially observed, followed by first-order kinetics after a significant amount of NDMA had already degraded.95 Zero-order kinetics have also been observed for the photolysis of N-nitrosopiperidine (NPIP) in acidic aqueous methanol (1:1) at high initial concentrations.111 It is unclear in the literature why zero-order kinetics are observed at these high concentrations, but it may be a consequence of poor transmission of UV light through concentrated solutions of nitrosamines. Typical studies focus on environmentally relevant concentrations in the first-order kinetics regime. For aqueous nitrosamine samples with low initial concentrations, higher rate constants are observed for more dilute solutions,95,104 but this trend is reversed in acetonitrile.99 As mentioned above, nitrosamine photolysis is faster in more acidic solutions than in neutral or basic solutions,95,104,105 and is also accelerated in O2-saturated solutions relative to N2-saturated solutions when the π → π* transition is excited.101 The structure of the nitrosamine is also important. Dialkylnitrosamines have been found to decompose much more rapidly than diarylnitrosamines in aprotic solvents, which is likely because the latter are not susceptible to hydrogen abstraction from the α position.108
Lastly, it should be noted that although Figure 5 depicts NDMA as the species which is excited, early literature proposed that the photolabile species is in fact a hydrogen-bonded nitrosamine/acid complex (37, Figure 6) and not the nitrosamine itself.23,107 This was consistent with the then-accepted (and now thoroughly disproven) photostability of the nitrosamines in neutral solvents23,107 and the fact that nitrosamines are too weakly basic (conjugate acid pKa < 1)25,113 for the O-protonated nitrosamine conjugate acid (4, e.g., ground-state 33) to play a significant role under most conditions. Complex 37 had also been described previously to explain some observed effects of solution acidity on the UV–vis absorption spectra of nitrosamines.25,114 In this view, when complex 37 is excited it behaves analogously to 33* and produces the same products as depicted in Figure 5.26,102,115 Formation of 37 is observed in cyclohexane solutions of dialkylnitrosamines with trichloroacetic acid at millimolar concentrations (~10 mM) by UV–vis spectroscopy.114 Thus, it is plausible that such complexes are involved in nitrosamine photolysis in aprotic solvents in the presence of acid. However, the role of the evoked nitrosamine/acid complex 37 is unclear in neutral or alkaline aqueous solutions, wherein the nitrosamine is primarily engaged in weaker, but omnipresent, hydrogen bonding with water.25 Dilute acids are also expected to be largely dissociated under these conditions and the hydronium ion does not appear to be a strong enough proton donor to produce a detectable spectroscopic signature for 37. Specifically, the UV–vis absorption spectra of aqueous solutions of NDMA pH 2–8 do not contain features attributed to the strongly hydrogen-bonded complex 37.25,97,101 Thus, the formation of strongly H-bonded ground-state complex 37 must not be required for photolysis because quantum yields are consistent across pH 2–8101 and hydrolysis (path C) occurs outside of acidic solutions.102 Nevertheless, perhaps because many of the early solution-phase photolysis studies were done in weakly acidic nonaqueous conditions,23,26,107,115 the nitrosamine/acid complex 37 is often evoked in modern literature descriptions of nitrosamine photolysis in aqueous solutions.
Figure 6.

Strongly hydrogen-bonded NDMA/acid (HA) complex 37.
Synthetic Applications of Nitrosamine Photochemistry.
Although fewer investigations have focused on neutral conditions, the photochemical reactions of N-nitrosamines in acidic conditions were extensively explored by Chow and co-workers in the late 1960s through the 1980s.26,103,115–127 Many of these reactions have been reviewed previously,26,118 but are covered briefly here for completeness. Chow and co-workers found that the reactions of nitrosamines under photolytic conditions with various substrates are dominated by the reactivity of the produced aminium radical, and consequently require acidic conditions.26 Upon photolysis of a nitrosamine under inert gas, the aminium radical and nitric oxide can add across unsaturated C–C bonds in a stepwise fashion to yield products bearing a tertiary amine and an adjacent C-nitroso moiety (Figure 7). These interesting products often undergo additional transformations, such as tautomerization to the corresponding oxime (Figure 7, middle and bottom).26,118,127 When the photolysis is alternatively performed under air, the products contain a nitrate ester rather than a C-nitroso group (Figure 7, top).26,120 An exception to these acidic reactions is the photoinduced nitrosation of polycyclic phenols (e.g., 1-naphthol 38) by NDMA in neutral solutions under inert gas.116 The nitrosated products undergo rearrangement and are isolated as the quinone monooximes (39, Figure 8). The authors proposed that acid was not required because the excited polycyclic phenol (e.g., 38) undergoes a proton transfer reaction with NDMA, initiating fragmentation of NDMA to the reactive fragments.
Figure 7.

Examples of nitrosamine photoaddition across unsaturated C–C bonds.
Figure 8.

Acid-free photoinduced nitrosation of 38 by NDMA. The initial nitrosated product rearranges to 39.
Additional reactivity may be accessed in neutral solutions through the nitramine products of nitrosamine photolysis. Analogous to nitrosamines, nitramines can photolyze by homolytic scission of the N–N bond to produce an amino radical (e.g., 31) and a NOx species (NO2 instead of NO in Scheme 11).128 Photolysis in various solvents of N-nitroso-N-methylaniline (40) or the p-nitro derivative in air produced ring-nitrated products 42a,b alongside the corresponding nitramine (41, Figure 9). The nitrated species were formed from photolysis of the initial nitramine product.129 Photolysis of nitramine explosives like RDX can also effect nitration of electron-rich aromatic molecules (e.g., 43a, Figure 10a)130 and olefins131 in solution. Under UV irradiation, RDX can also formally abstract hydride from 10-methyl-9,10-dihydroacridine (43b) to yield the corresponding N-methylacridinium (44, Figure 10b).132 Although these reactions have not been explicitly explored for dialkylnitrosamines, related transformations may be possible.
Figure 9.

Irradiated N-nitrosoaniline 40 produces N-nitroaniline 41, which gives ring-nitrated anilines 42a,b with continued irradiation.
Figure 10.

Photoinduced (a) nitration of 43a and (b) hydride abstraction from 43b with nitramine explosive RDX.
N-NITROSAMINES AS WATER CONTAMINANTS
N-Nitrosamines can be found throughout the environment, but it is their presence in water that has drawn the most concern. While nitrosamines may accumulate in indoor air, particularly at tanneries or manufacturing sites in the rubber and pesticide industries,8 they are short-lived in outdoor air as a result of facile degradation by sunlight photolysis and other atmospheric reactions.133,134 In water, however, NDMA and other nitrosamines are more persistent.8 Although they may naturally decompose through photolysis, this is obviously limited to sunlight-exposed surface waters and can be hindered by light-screening by other dissolved matter.96 As indicated in our above discussion of nitrosamine chemistry, they are unlikely to degrade by other reactions in the mild aqueous conditions expected in most natural or municipal waters. This stability is made all the more troubling by their formation as disinfection byproducts (DBPs) during common water treatment processes.135 Consequently, and unsurprisingly, there is a vast and growing body of literature on the formation, remediation, and prevention of nitrosamines (particularly NDMA) in water, and a number of reviews have been published on this subject.11–14 Here, we present a brief overview of nitrosamines as water contaminants, and direct readers to the cited reviews for detailed coverage of the topic.
Formation.
Although nitrosamine production is not typically a desired outcome, there are numerous conditions which can lead to the unintentional formation of nitrosamines.136 Thus, nitrosamines can be introduced into the environment through waste streams from industrial sites where they were inadvertently formed or they can form from precursors in the environment through various biological or chemical processes.8 Much of the research on the occurrence of nitrosamines in water has focused on their formation during water disinfection. Of the disinfection methods, chloramination has received the most attention in large part due to the ubiquity of the requisite organic amine precursors in dissolved organic matter.13,14 Although nitrosamine formation during chloramination has long been established to occur, the mechanism is not entirely understood and there is still some debate about whether monochloramine (NH2Cl)137–139 or dichloramine (NHCl2)135,140,141 is the species most responsible for nitrosamine formation. The importance of the latter is supported by evidence that minimizing dichloramine during chloramination reduces nitrosamine formation.142,143 Figure 11 outlines the proposed formation pathways with each chloramine from a secondary amine as a model precursor. In short, the amine precursor attacks the electrophilic chloramine to produce an unsymmetrically substituted hydrazine derivative (45 and 45-Cl), which is subsequently oxidized to a nitrosamine.135 A wide variety of amine precursors have been shown to form nitrosamines under chloramination conditions, including both secondary and tertiary amines.13,14,135 In particular, dimethyl tertiary amines bearing a −CH2–aryl moiety (e.g., benzyl, furfuryl) (46, Chart 6) have been identified as having especially high NDMA formation potential.141,144 Notably, this group contains the pharmaceutical ranitidine (Chart 6),138,145,146 which was pulled from shelves in late 2019 after multiple lots were found to contain NDMA.6 Quaternary ammonium compounds can also contribute to nitrosamine formation, including components of consumer products147 and quaternary ammonium polymers used as coagulants in wastewater treatment (e.g., polyDADMAC, Figure 12).148,149 The quaternary ammonium cations can degrade to secondary and tertiary amines during chloramination, which can then go on to form nitrosamines.135,147 Although they produce nitrosamines in substantially lower yields than secondary and tertiary amines,147 quaternary ammonium compounds are potentially significant precursors because of their ubiquity in commercial products.
Figure 11.

Nitrosamine formation during chloramination with either (a) monochloramine or (b) dichloramine as the reactive chloramine.
Chart 6.

Dimethyl Tertiary Amine Bearing a −CH2–aryl Group (46) and Ranitidine, Which Contains the 46 Motif
Figure 12.
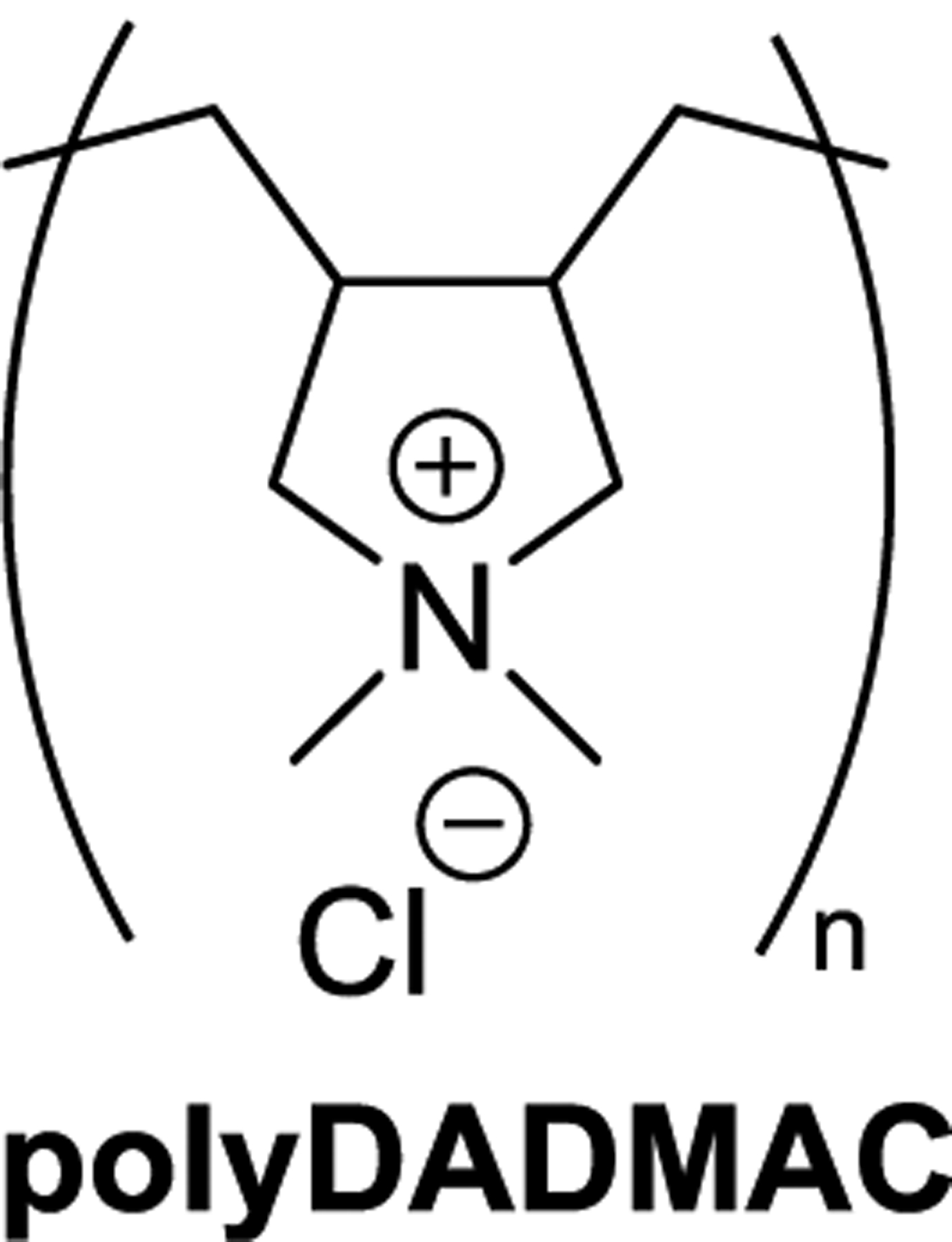
PolyDADMAC, a coagulant polymer.
Chlorination of nitrite-containing water can lead to the formation of nitrosating species and consequently result in the formation of nitrosamines during disinfection (Scheme 13).150 Both secondary150 and tertiary151,152 amines may serve as precursors. This pathway is relatively unimportant in drinking water, which typically has low nitrite concentrations, but may be significant in other water matrices with elevated amounts of nitrite and amine precursors.135,153 Outside of chlorination, it has also been demonstrated that the nitrosation of aliphatic secondary amines in water can be catalyzed by micelles formed from cationic surfactants.154 Cationic surfactants are common in a variety of consumer products and thus may often be present in municipal waste streams.
Scheme 13.

Formation of a Nitrosamine during Chlorination of Nitrite-Containing Water
Disinfection of water via ozonation has also been tied to NDMA formation, but the yields of NDMA from most precursors are very low unless ammonia and bromide levels are also elevated.14,155 During ozonation, ammonia can form hydroxylamine (NH2OH) and, in the presence of bromide, brominated nitrogenous species like bromamines (e.g., NH2Br). Analogously to the pathways depicted in Figure 11, these products can react with amines to form hydrazine derivatives which are oxidized to nitrosamines by ozone.155 A narrow subset of precursors including dimethylamine-containing hydrazines156 and, by a slightly different mechanism, sulfamides157 are converted to nitrosamines in high yields during ozonation, but these precursors are not commonly present in significant concentrations.13,14 Thus, in many water matrices, nitrosamine formation during ozonation is not expected to be significant, but in some waters (e.g., municipal wastewater) it can be quite high.158 Similarly, while treatment with chlorine dioxide produces very little NDMA (<0.1% yield) from most precursors,14 it produces considerably more from the plant regulator daminozide (~5%),159 so nitrosamine formation by this pathway may primarily be a concern for waters impacted by agricultural runoff.
Remediation and Mitigation.
Techniques for the remediation and mitigation of nitrosamines in water fall generally into one of three broad categories: destruction of nitrosamines, physical removal of nitrosamines (i.e., filtration), or prevention of nitrosamine formation. Of the first type, destruction of nitrosamines, irradiation with UV light was one of the earliest methods and it remains one of the most established.11,14,95,96,100,102,160 Direct photolysis can be effective,95,100 but the high UV fluence required for nitrosamine treatment can make this method costly.13 Additionally, direct UV photolysis does little to destroy nitrosamine precursors, including the secondary amine produced by photolysis, and so subsequent reformation of nitrosamines remains a plausible issue in UV-treated waters.13 This issue can be somewhat mitigated by the inclusion of an oxidant like ozone during UV treatment (UV/O3), which has been found to reduce the amount of secondary amine produced by photodegradation.104 Some recent work suggests that including peroxodisulfate (S2O82−) during UV treatment (UV/S2O82−) may improve the efficiency of NDMA photodegradation,160 though more work is needed to evaluate the product distribution and performance with other nitrosamines.
Other means of nitrosamine destruction exist, but none are as well-established as UV treatment. Treatment of contaminated waters with ozone/hydrogen peroxide (O3/H2O2) is more effective at degrading nitrosamines than conventional ozonation, but this technique may have limited practical application as a result of the risk of carcinogenic bromate formation under these conditions.161 Electrochemical oxidation162 and reduction163 of aqueous NDMA have been reported, and several metals have been demonstrated to catalyze the reduction of nitrosamines in water.164–166 Various methods utilizing bioremediation (e.g., bacterial degradation) of nitrosamines in water have shown promise.14 One such method, propane biosparging, has been demonstrated in the field for the in situ treatment of an NDMA-contaminated aquifer.167 Propane-oxidizing bacteria are capable of degrading NDMA, so introducing additional propane via biosparging feeds these bacteria and promotes biodegradation of NDMA.167,168
The second strategy, physical removal of nitrosamines, is often challenging. Removal of nitrosamines from water through adsorption to materials such as activated carbon is typically ineffective due to the low hydrophobicity of many nitrosamines,11 although removal does improve with increasing hydrophobicity of individual nitrosamines.169 Sand and soil filtration are also ineffective.11 Studies on the effectiveness of reverse osmosis (RO) for nitrosamine removal have found that membrane rejection of NDMA is often low and also highly variable with operating conditions, such as temperature.170,171 Rejection is better for larger nitrosamines, though there is much less information about their removal by RO because most reports focus on NDMA.170 In addition to size affecting removal by RO, nitrosamines’ ability to hydrogen bond with the membrane materials appears to facilitate their diffusion through the membrane.172 When tested with two different polyamide membranes, NDMA and N-nitrosomethylethylamine (NMEA) were rejected at a lower rate than both larger nitrosamines and also similarly sized non-nitrosamine structural analogs with reduced hydrogen bonding ability (e.g., their formamide analogs).172 The rejection of nitrosamines by polyamide RO membranes can be greatly improved through methods such as heat-treating the membranes173 or functionalizing the surface with graphene oxide,174 but both of these modifications reduce water permeability which may limit their practical application. Physical removal techniques can be paired with destructive methods. In one example, powder-activated carbon was used to adsorb NDMA to a reactive membrane, where it was then electrochemically reduced.163
Given the difficulties and costs of remediating nitrosamine-contaminated water, the adage that an ounce of prevention is worth a pound of cure rings strikingly true here. Prevention of nitrosamine formation is arguably the most important aspect of long-term control of nitrosamine contamination in many situations. Considerable research has been dedicated to methods for removing precursors or for transforming them into species with reduced nitrosamine formation potential, and many of these strategies are analogous to those mentioned above for nitrosamines.13,14 Although not particularly effective for nitrosamine removal, activated carbon can remove nitrosamine precursors and reduce the nitrosamine formation potential during chloramination.175,176 Preoxidation to destroy potential precursors can significantly decrease nitrosamine production during chloramination.177 However, a preoxidation step can itself increase the formation of other DBPs and so these trade-offs must be considered when choosing an oxidant.178 Modification of water treatment conditions can also reduce nitrosamine formation. For example, purification of the coagulant polymer polyDADMAC (Figure 12) to remove lower molecular weight fractions has been shown to reduce NDMA formation during disinfection without harming polymer performance in water treatment.179
The most effective long-term strategies for controlling nitrosamines will combine methods of eliminating existing nitrosamines with tactics to prevent their formation in the first place. There is unlikely to be a single “best” solution to the problem of environmental nitrosamines because there is no single cause for their presence. Mitigation strategies that effectively control disinfection-related contamination might do very little to reduce nitrosamines originating from other sources, and more work is needed to understand what those sources may be. Additionally, research on aqueous nitrosamines has often focused on NDMA exclusively, and this is a significant problem for the field. It has long been known that numerous nitrosamines are carcinogenic to mammals1,3,180 and there is evidence that NDMA may only make up a small fraction of the total nitrosamine content in drinking water,181 and yet there is relatively little work focusing on those other nitrosamines. These other nitrosamines may respond very differently from NDMA to remediation strategies, have different origins, and/or have different effects on human health, and future work must fill this gap in our understanding. Essential to this work will be reliable, sensitive methods for detecting nitrosamines, which are discussed in the next section.
DETECTION AND SENSING OF N-NITROSAMINES
Our ability to understand how N-nitrosamines form and spread in our environment—not only in air and water, but also in our diets and drugs—is inherently limited by our methods for detecting these contaminants. In recent decades, particular attention has been paid to quantitative detection of nitrosamines in water, motivated in part by their occurrence as byproducts of manufacturing and disinfection processes. Although there are no federal regulations on nitrosamines in water in the United States, the US Environmental Protection Agency (EPA) has set a screening level of 0.11 ng/L (0.11 ppt, 1.5 pM) for NDMA in residential tap water based on a 10−6 cancer risk.182 Similar screening levels were also set for other small dialkylnitrosamines. Several states have set their own drinking water guidelines for NDMA.66 In some states, such as Massachusetts (10 ng/L guideline), the guidelines are as much based on how much NDMA can be practically detected as they are on the cancer risk.183,184 Thus, analytical methods for aqueous nitrosamines must be highly sensitive for practical use.
At the simplest level, many of the common methods for adequately sensitive (ng/L) detection of aqueous nitrosamines are based on a similar concept: Extract the nitrosamines from water, greatly concentrate the sample in organic solvent, chromatographically separate the components, and then detect the components, often with mass spectrometry.12,15,185 Naturally, there are numerous variations within this framework, and these analytical methods for detection of aqueous nitrosamines have been reviewed elsewhere.12,185 Additionally, N-nitrosamine detection methods were recently the subject of a review by Parr and Joseph.15 Here, we will focus on those methods which leverage the chemistry of nitrosamines in their detection scheme to reduce sample preparation and instrumentation requirements.
For comparison purposes, we briefly describe here EPA Method 521, the standard EPA method for sensitive detection of volatile nitrosamines in drinking water.186 In this method, analytes are extracted from 0.5 L of water via solid phase extraction (SPE) using a cartridge of coconut charcoal. The sample is then eluted/extracted with DCM and concentrated to less than 1 mL. After the addition of an internal standard, the volume is adjusted to 1.0 mL with DCM. This concentrated sample is then analyzed by gas chromatography-tandem mass spectrometry (GC–MS/MS). Method 521 can detect NDMA and six other volatile nitrosamines at low ng/L concentrations in drinking water, with limits of detection (LODs) ranging from 0.26 to 0.66 ng/L (Table 1). Notably, the LODs for NDMA (0.28 ng/L) and N-nitrosodiethylamine (NDEA, 0.26 ng/L)186 are higher than their respective EPA screening levels for residential tap water (0.11 ng/L and 0.17 ng/L).182 However, it may be possible to obtain lower LODs with this method than were reported in the original 2004 technical document with newer, more sensitive instrumentation.
Table 1.
| N-nitrosamine | Method 521 LOD (ng/L) | screening level (ng/L) |
|---|---|---|
| N-nitrosodimethylamine (NDMA) | 0.28 | 0.11 |
| N-nitrosodiethylamine (NDEA) | 0.26 | 0.17 |
| N-nitrosomethylethylamine (NMEA) | 0.28 | 0.71 |
| N-nitrosodi-n-propylamine (NDPA) | 0.32 | 11 |
| N-nitrosodi-n-butylamine (NDBA) | 0.36 | 2.7 |
| N-nitrosopyrrolidine (NPYR) | 0.35 | 37 |
| N-nitrosopiperidine (NPIP) | 0.66 | 8.2 |
GC may also be coupled with other detection methods, such as nitrogen-phosphorus detection and chemiluminescent nitric oxide detection, for determination of nitrosamine content.12,187 Although EPA Method 521 and other GC-based methods can measure volatile nitrosamines with high sensitivity, they often struggle with nonvolatile or thermally unstable nitrosamines, such as N-nitrosodiphenylamine. Various liquid chromatography-mass spectrometry (LC–MS) methods, particularly LC–MS/MS, have been developed which are compatible with a wider range of nitrosamines.15,185 However, as exemplified by EPA Method 521, the high sensitivity of GC–MS and LC–MS methods alike come at the cost of time-consuming sample preparation and expensive instrumentation.
The latter issue, instrumentation cost, is reduced in methods which couple high-performance liquid chromatography (HPLC) with detection by UV–vis absorption or emission, rather than mass spectrometry. In one such detection scheme, pre-column acidic denitrosation of NDMA and subsequent reaction with dansyl chloride gave the fluorescent dansyl amine (Scheme 14), which was then separated by HPLC and detected by fluorimetry.188 Although choosing to perform the denitrosation and derivatization pre-column clearly risks interference by ambient dimethylamine, the authors reported that dimethylamine was only ineffectively adsorbed during SPE of aqueous samples and thus was present in insignificant amounts during the later reaction with dansyl chloride. Rather than rely on the extraction step to remove likely interferents, HPLC-absorption/emission methods more often utilize nitrosamine reactivity after chromatographic separation. The reduction of tris(2,2′-bipyridyl)ruthenium(III) (Ru(bpy)33+) by aliphatic amines generates chemiluminescence,189 and this reaction was utilized in conjunction with post-column photolytic denitrosation to detect aliphatic nitrosamines.190 The nitrite generated by nitrosamine photolysis has also been utilized in their detection: post-column UV irradiation and subsequent addition of Griess reagent (Figure 13a) allows for colorimetric measurement of nitrosamines.191,192 This concept has been applied to nitrosamines in beer, gastric juices,191 and water.192 Positive responses to the online Griess test (Figure 13b) are obtained from both nitrosamines and nitramines, but these can be distinguished from each other by comparison of observed retention times to those of analytical standards.192
Scheme 14.

Acidic Denitrosation of NDMA and Subsequent Derivatization with Dansyl Chloride
Figure 13.
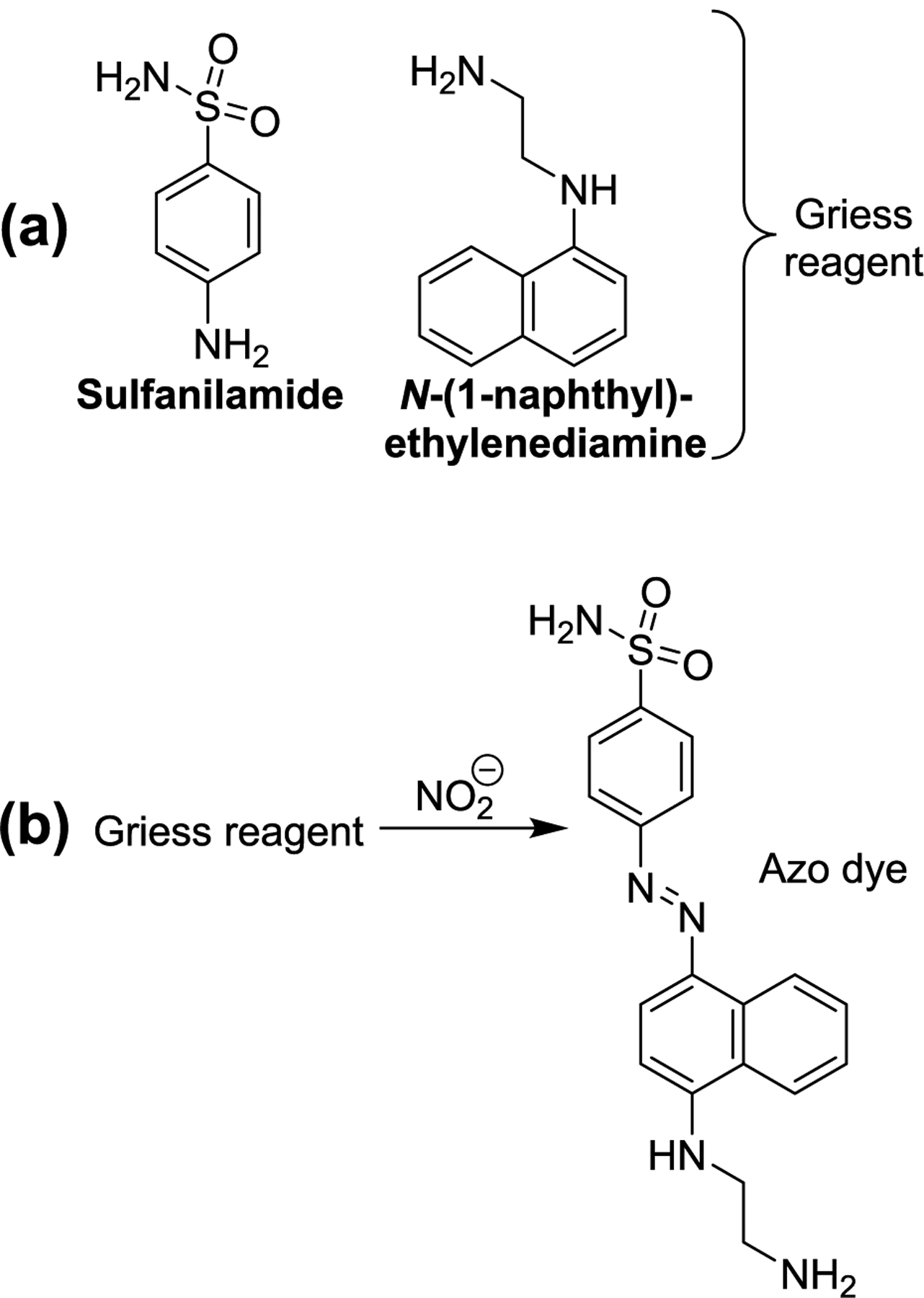
(a) Typical components of the Griess reagent and (b) their reaction with nitrite to produce a colorful azo dye. This is the basis of the Griess test for nitrite.
Although the above-described HPLC methods can detect low ng/L concentrations of nitrosamine, they all require significant preconcentration by SPE to achieve that sensitivity.188,190–192 Even if the entire detection scheme from extraction to detection is fully automated, as in the case of the Ru(bpy)33+ chemiluminescence method,190 SPE is nevertheless time-consuming and consequently the analysis of a single sample often takes at least 1 h, and sometimes much longer.192 Furthermore, activated carbon-based materials commonly used for SPE have been shown to catalyze the formation of nitrosamines from secondary amines under aerobic conditions, leading to potential errors in analysis.193–195 Without preconcentration, none of the methods mentioned thus far can achieve ng/L detection limits, which makes the HPLC-photochemical reaction-chemiluminescence (HPLC–PR–CL) method196 first reported by Kodamatani et al. in 2009 notable in the realm of HPLC-based methods of nitrosamine detection. In this method, a small volume (200 μL) of aqueous sample is directly injected for HPLC separation without preconcentration, followed by post-column photolysis to produce peroxynitrite (ONOO−). Luminol is then added and reacts with the peroxynitrite, resulting in chemiluminescence that is measured by a standard chemiluminescence detector. The complete analysis takes minutes, rather than hours, and low-ng/L detection limits are achieved for NDMA and several other nitrosamines. Interference from hypochlorite, commonly found in reclaimed wastewater, could be eliminated by brief pretreatment of the sample with ascorbic acid, and interference from residuals in ultrafiltration-treated wastewater could be controlled by reducing the injection volume to 20 μL while still achieving a method detection limit of 2 ng/L for NDMA.197 No interference is observed in the direct injection of reverse osmosis permeate, which has allowed this method to be used for online near real-time monitoring of NDMA in wastewater treated by reverse osmosis at pilot scale.198 The method has been validated for the detection of four nitrosamines in several recycled water matrices199 and performs comparably to SPE-GC–MS/MS methods.197 However, although the sensitivity is impressive when detecting NDMA, NMEA, N-nitrosomorpholine (NMOR), and N-nitrosopyrrolidine (NPYR), the method’s performance detecting NDEA and NPIP is disappointing (LODs > 15 ng/L).199
Although chromatography-based methods for nitrosamine detection are well established, the required instrumentation inherently restricts their use to specialized settings (e.g., laboratories). This limits the widespread accessibility of these methods, and hinders work requiring large numbers of water sources to be tested. Chromatography and mass spectrometry can be miniaturized, however portable devices are still highly expensive and complex to operate, and so a practical integrated device for in-field detection of these carcinogens is not possible by these current methods. The desire for a simple, rapid nitrosamine test is not new. Particularly in the 1960s and 1970s, a number of colorimetric and fluorimetric indicators were reported for the detection of nitrosamines.200 Similar to the more modern methods described above, these methods typically cleaved the nitrosamine N–N bond and then added an indicator which reacted with either an NOx fragment (e.g., detection of nitrite by Griess reagent)201,202 or the secondary amine (e.g., derivatization with dansyl chloride) to give colored or fluorescent product.203 However, these methods often had high LODs and poor selectivity, even when used as thin-layer chromatography stains rather than as stand-alone indicators.15,200 Other methods first reduced the nitrosamine to the corresponding hydrazine, which was then condensed with an aromatic aldehyde to give a colored or fluorescent hydrazone product.204–206 Those methods are also incompatible with a portable in-field detection scheme, as they require harsh reagents like LAH to reduce the nitrosamine.204
Several more modern chromatography- and mass spectrometry-free detection methods also utilize cleavage of the N–N bond. In particular, there are several methods for quantifying total N-nitrosamine (TONO) content in water that subject samples to denitrosative conditions and then detect the released nitric oxide (NO) with a commercial NO detector.207,208 This concept has also been used to measure TONO content in food, personal care products, and human bodily fluids.209 Mitch and colleagues utilize acidic triiodide to cleave the N–N bond,181,207 but these denitrosation conditions have been shown to produce the nitrosating agent nitrosyl iodide (NOI).210 NOI may simply decompose to NO in solution, but conceivably it could also nitrosate species (e.g., amines) in the sample, reducing the observed NO yield. Furthermore, successive injections of nitrosamine samples into the acidic triiodide solution resulted in a gradual reduction of the measured NO, suggesting a degradation of the reagent solution.208 Better signal reproducibility is obtained when, instead of chemical denitrosation, UV light is used to cleave the N–N bond.208 Additionally, photolysis can be performed with only a small UV light source, and thus is more compatible than chemical denitrosation for use in a portable nitrosamine detector. However, both TONO methods share several drawbacks.207,208 First, both chemical and photolytic denitrosation methods show differences in conversion efficiency among different nitrosamines, so the analysis accuracy may vary with nitrosamine composition. Second, S-nitrosothiols and nitrite also produce NO under either of these denitrosative conditions, and so pretreatment steps are required to eliminate these species prior to denitrosation. Third, without preconcentration, the detection limits of these methods are impractically high (~0.1 μM, or 7.4 μg/L as NDMA). The detection limit can be lowered with extraction and preconcentration, but extraction efficiencies can vary widely across nitrosamines of different polarities using any single extraction method.207 Not only may this skew results by total nitrosamine composition, but also preconcentration is time-consuming and ill-suited for use in in-field analysis.
To this point, we have almost exclusively discussed methods of nitrosamine detection that do not measure the nitrosamines directly, but rather detect one of the species produced by breaking the nitrosamine N–N bond. However, the products most often used to detect nitrosamines (e.g., nitrite, NO, and secondary amines) may be present independently of nitrosamines in complex samples such as environmental waters or wastewater. Products like nitrite and nitric oxide may also be formed from species other than nitrosamines under the implemented denitrosation conditions.5 These interferents can be removed through sample preparation and/or chromatographic separation, but this increases the total cost and time required to perform the analysis. It thus would be advantageous to detect nitrosamines more directly. However, there are exceedingly few nonchromatographic methods of this kind for nitrosamines in any matrix, let alone for nitrosamines in water.
For detection of aqueous NDMA, Cetoó et al. reported an impedimetric sensor consisting of molecularly imprinted polymer (MIP) particles made of cross-linked poly(methyl methacrylate) trapped in a polypyrrole matrix on a glassy carbon electrode.211 The MIP particles were formed using NDMA as the template, so when exposed to aqueous solutions only NDMA is easily trapped near the electrode surface. The sensor showed significantly smaller responses to other structurally related molecules (e.g., DMF) and the presence of these related molecules in solution did not significantly affect the sensor’s response to NDMA. Because no sample preparation is required, the entire analysis can be completed within 20 min, but a 30 min regeneration period to remove NDMA is required before the sensor can be reused. Although the speed and portability of this highly selective sensor are attractive, the LOD is only 0.85 μg/L (850 ppt) and so a preconcentration step would be needed to make this sensor practical for most water sources. A somewhat lower LOD was reported by Lin et al. for NDMA (10 nM, ~ 740 ppt) and NDEA (10 nM, ~ 1 ppb) in water through the use of surface-enhanced Raman scattering (SERS).212 The SERS substrate consisted of a zwitterionic copolymer, poly(glycidyl methacrylate-r-sulfobetaine methacrylate) (PGMA-r-PSBMA), grafted onto hexagonal gold nanorods (Au NRs). When water evaporates from the sample on the substrate, the NDMA and NDEA are held near the Au NRs by association with PGMA-r-PSBMA, allowing the detection of these small molecules with SERS. Importantly, the report only demonstrates that it is possible to detect NDMA and NDEA with Raman spectroscopy, not that this is a quantitative detection method. However, a variety of hazardous chemicals can be detected with SERS-based methods, and on-site detection is possible with portable Raman spectrometers.213,214 It is conceivable that further work could yield a sensitive and portable SERS-based nitrosamine sensor.
Although not proposed for water analysis, a conceptually interesting fluorescence-based sensor in aqueous solution was reported by Anzenbacher et al. which could recognize several nitrosamines.215 Their sensor utilized two fluorescent receptors: cucurbit[6]uril (CB[6]) derivative 47 and cucurbituril-like acyclic molecule 48 (Figure 14a). When bound to a metal ion, their fluorescence is partially quenched, and displacement of the metal by another guest either recovers or further quenches the fluorescence, depending on the identity of the new guest. The differing sizes and flexibilities of 47 and 48 affect their respective affinities for guests, and their correspondingly different changes in emission intensity can be used together to identify guests. Using Eu3+ as the metal ion, linear discriminant analysis was applied to the response of the two-probe assay and used to sort guests into one of three categories (biological amines, nitrosamines, and tobacco alkaloids). Most notably, the assay could differentiate between tobacco-specific nitrosamines N-nitrosonornicotine (NNN) and nicotine-derived nitrosamine ketone (NNK), and the structurally related tobacco alkaloids nicotine and cotinine (Figure 14b). This may be due to differences in protonation: the assay is performed at pH 3, which would protonate the alkaloids but not NNN and NNK, resulting in very different interactions with the probes. Although NDMA and NPIP were included in the two-probe qualitative assay, they were not included in the authors’ efforts to use their sensor for quantitative measurements. Of the nitrosamines, only NNN and NNK were included for quantitative studies. This is likely because these studies were done only with probe 48, which did not respond strongly to the smaller nitrosamines. The authors report LODs of 50 and 270 ppb for NNN and NNK, respectively. Although more rigorous selectivity studies are needed to evaluate the sensor, these detection limits are suitable for use with tobacco products.216 Although it is unlikely this sensor will ever be practical for water testing, the concept itself is promising if receptors are developed which respond strongly to small dialkylnitrosamines like NDMA, preferably at circumneutral pH.
Figure 14.

(a) Receptors in Anzenbacher and co-workers’ two-probe fluorimetric assay, CB[6] derivative 47 and cucurbituril-like molecule 48. (b) Tobacco-specific nitrosamines and tobacco alkaloids that could be differentiated by the assay.
In the final sensor we will discuss, the ability of N-nitrosamines to bind to metal centers and form stable complexes82 is leveraged for detection of dialkylnitrosamines in air.217 This single-walled carbon nanotube (SWCNT)-based chemiresistive sensor, reported by Swager and co-workers, utilizes a cobalt(III) tetraphenylporphyrin (49, Figure 15a) as a selector for nitrosamines. The SWCNTs were covalently functionalized with 4-pyridyl groups, which were used to anchor the metalloporphyrin (Figure 15b). Nitrosamines in the air can coordinate to the Co(III) center through their oxygen (e.g., Chart 5, left side) and this interaction produces an increase in resistance. The sensor was found to be highly selective for the tested dialkylnitrosamines (NDMA, NDEA, and NDBA) over common volatile organic compounds, which produced significantly smaller responses. Critical for real-world use, the sensor was not significantly affected by humidity. The potential utility of this sensor for distributed air monitoring was demonstrated by integrating the sensor device into a commercial sensing node, which enabled online detection of NDMA at ppb levels. The LOD was 1 ppb for all three nitrosamines. Although nitrosamines are not expected to significantly accumulate in outdoor air, 1 ppb is well below levels of nitrosamines that have been observed indoors in industrial settings.218
Figure 15.

(a) Cobalt(III) tetraphenylporphyrin (49) used by Swager and co-workers as a selector for nitrosamines. (b) 49 anchored to the SWCNT through a 4-pyridyl group covalently attached to the nanotube’s surface.
While selective, sensitive nitrosamine detection has been achievable in laboratory settings for many years through techniques such as GC–MS/MS, these methods are labor- and time-intensive and require expensive instrumentation. To facilitate on-site/in-field testing, more work is needed to leverage the chemistry of nitrosamines into robust, selective, and sensitive sensors, particularly for aqueous nitrosamines. Furthermore, sensors are needed which are inexpensive to produce and easy to use so that they may be used by nonscientists in communities affected by nitrosamine pollution (i.e., citizen science).
FINAL REMARKS AND OUTSTANDING CHALLENGES
Here, we have provided a primer on the chemistry of nitrosamines, their role as water pollutants, and the methods for their detection. Although work has often focused on NDMA in recent years, it must be emphasized that nitrosamines are a diverse group of chemicals unified by a markedly simple structure, the N–N=O group. That diversity complicates both removal and detection efforts because individual nitrosamines can vary widely in terms of traits like size, shape, and hydrophilicity. However, their shared N-nitroso moiety is a challenging functional handle for practical applications because of its relative stability under mild conditions. That core N–N=O structure can be formed from a wide range of precursors under numerous different conditions, making the prevention of nitrosamines a multi-faceted problem with no single solution. At the root of these different sides to the nitrosamine problem—prevention, removal, detection—is the chemistry of nitrosamines, and so an understanding of nitrosamine chemistry is essential to developing effective mitigation processes for environmental and public health. In particular, leveraging the chemical behavior of these carcinogens will enable the development of better sensors and better extraction materials.
ACKNOWLEDGMENTS
This work was supported by the National Institute of Environmental Health Sciences Superfund Basic Research Program, National Institutes of Health, P42 ES027707. J.C.B. was supported by the National Science Foundation Graduate Research Fellowship under Grant No. 1122374. We thank the staff at the MIT Libraries for their invaluable help in obtaining remote access to references during the COVID-19 pandemic.
Biographies
Biographies

Jessica C. Beard received her B.A. in chemistry from Northwestern University in 2017. Later that year, she joined the Swager group at the Massachusetts Institute of Technology, where she is currently a chemistry Ph.D. candidate. Her work focuses on the development of optically responsive indicators for the detection of aqueous contaminants.

Timothy M. Swager is a Professor of Chemistry at the Massachusetts Institute of Technology. He obtained his B.S. from Montana State University in 1983 and Ph.D. from Caltech in 1988, studying under Robert H. Grubbs. His research program is focused materials chemistry with emphasis on systems that advance analytical science.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Lijinsky W Structure-Activity Relations in Carcinogenesis by N-Nitroso Compounds. Cancer Metastasis Rev. 1987, 6, 301–356. [DOI] [PubMed] [Google Scholar]
- (2).Tricker AR; Preussmann R Carcinogenic N-Nitrosamines in the Diet: Occurrence, Formation, Mechanisms and Carcinogenic Potential. Mutat. Res., Genet. Toxicol 1991, 259, 277–289. [DOI] [PubMed] [Google Scholar]
- (3).National Toxicology Program. N-Nitrosamines Report on Carcinogens, 14th ed.; Research Triangle Park, NC, 2016. [Google Scholar]
- (4).Hecht SS Tobacco Carcinogens, Their Biomarkers and Tobacco-Induced Cancer. Nat. Rev. Cancer 2003, 3, 733–744. [DOI] [PubMed] [Google Scholar]
- (5).Williams DLH Nitrosation Reactions and the Chemistry of Nitric Oxide, 1st ed.; Elsevier BV: Amsterdam, The Netherlands, 2004. [Google Scholar]
- (6).Krietsch Boerner L The Lurking Contaminant. C&EN 2020, 98, 27–31. [Google Scholar]
- (7).U.S. Food and Drug Administration. Control of Nitrosamine Impurities in Human Drugs Guidance for Industry; USFDA, 2020. [Google Scholar]
- (8).Agency for Toxic Substances and Disease Registry (ATSDR). Toxicological Profile for N-Nitrosodimethylamine; ATSDR: Atlanta, GA, 1989. [PubMed] [Google Scholar]
- (9).Barnes JM; Magee PN Some Toxic Properties of Dimethylnitrosamine. Occup. Environ. Med 1954, 11, 167–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Magee PN; Barnes JM The Production of Malignant Primary Hepatic Tumours in the Rat by Feeding Dimethylnitrosamine. Br. J. Cancer 1956, 10, 114–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Mitch WA; Sharp JO; Trussell RR; Valentine RL; Alvarez-Cohen L; Sedlak DL N-Nitrosodimethylamine (NDMA) as a Drinking Water Contaminant: A Review. Environ. Eng. Sci 2003, 20, 389–404. [Google Scholar]
- (12).Nawrocki J; Andrzejewski P Nitrosamines and Water. J. Hazard. Mater 2011, 189, 1–18. [DOI] [PubMed] [Google Scholar]
- (13).Krasner SW; Mitch WA; McCurry DL; Hanigan D; Westerhoff P Formation, Precursors, Control, and Occurrence of Nitrosamines in Drinking Water: A Review. Water Res. 2013, 47, 4433–4450. [DOI] [PubMed] [Google Scholar]
- (14).Sgroi M; Vagliasindi FGA; Snyder SA; Roccaro P N-Nitrosodimethylamine (NDMA) and Its Precursors in Water and Wastewater: A Review on Formation and Removal. Chemosphere 2018, 191, 685–703. [DOI] [PubMed] [Google Scholar]
- (15).Parr MK; Joseph JF NDMA Impurity in Valsartan and Other Pharmaceutical Products: Analytical Methods for the Determination of N-Nitrosamines. J. Pharm. Biomed. Anal 2019, 164, 536–549. [DOI] [PubMed] [Google Scholar]
- (16).Looney CE; Phillips WD; Reilly EL Nuclear Magnetic Resonance and Infrared Study of Hindered Rotation in Nitrosamines. J. Am. Chem. Soc 1957, 79, 6136–6142. [Google Scholar]
- (17).Harris RK; Spragg RA Vapour-Phase Nuclear Magnetic Resonance Studies of Hindered Internal Rotation: Dimethylnitrosamine. Chem. Commun 1967, No. 7, 362–363. [Google Scholar]
- (18).Lunazzi L; Cerioni G; Ingold KU Conformational Studies by Dynamic NMR. V. The Stereodynamics of Hindered Aliphatic Hydrazones. J. Am. Chem. Soc 1976, 98, 7484–7488. [Google Scholar]
- (19).Wiberg KB; Rablen PR; Rush DJ; Keith TA Amides. 3. Experimental and Theoretical Studies of the Effect of the Medium on the Rotational Barriers for N,N-Dimethylformamide and N,N-Dimethylacetamide. J. Am. Chem. Soc 1995, 117, 4261–4270. [Google Scholar]
- (20).Rademacher P; Stølevik R; Lüttke W Structure of Nitroso- and Nitro-dimethylamine. Angew. Chem., Int. Ed. Engl 1968, 7, 806. [Google Scholar]
- (21).Krebs B; Mandt J Kristallstruktur Des N-Nitrosodimethylamins. Chem. Ber 1975, 108, 1130–1137. [Google Scholar]
- (22).Olah GA; Donovan DJ; Keefer LK Carcinogen Chemistry. I. Reactions of Protonated Dialkylnitrosamines Leading to Alkylating and Aminoalkylating Agents of Potential Metabolic Significance. J. Natl. Cancer Inst 1975, 54, 465–472. [PubMed] [Google Scholar]
- (23).Burgess EM; Lavanish JM Photochemical Decomposition of N-Nitrosamines. Tetrahedron Lett. 1964, 5, 1221–1226. [Google Scholar]
- (24).Kuhn SJ; McIntyre JS Reactions of Amides and Related Compounds: III. N.M.R. Investigation of the Protonation of N,N-Dimethylnitrosoamine, N,N-Diethylnitrosoamine, and N-Nitrosopiperidine. Can. J. Chem 1966, 44, 105–109. [Google Scholar]
- (25).Layne WS; Jaffe HH; Zimmer H Basicity of N-Nitrosamines. II. Aqueous Sulfuric Acid Solutions. J. Am. Chem. Soc 1963, 85, 1816–1820. [Google Scholar]
- (26).Chow YL Nitrosamine Photochemistry: Reactions of Aminium Radicals. Acc. Chem. Res 1973, 6, 354–360. [Google Scholar]
- (27).Keefer LK; Hrabie JA; Hilton BD; Wilbur D Nitrogen Protonation of N-Nitrosodimethylamine. J. Am. Chem. Soc 1988, 110, 7459–7462. [Google Scholar]
- (28).Zahradník R Über N-Nitrosoderivate Sekundärer Amine I. Kinetik Und Mechanismus Der Zersetzung in Stark Saurem Medium. Collect. Czech. Chem. Commun 1958, 23, 1529–1539. [Google Scholar]
- (29).Biggs ID; Williams DLH Kinetics and Mechanism of the Fischer-Hepp Rearrangement and Denitrosation. Part V. The Mechanism of Denitrosation. J. Chem. Soc., Perkin Trans 2 1975, No. 2, 107–111. [Google Scholar]
- (30).Hallett G; Williams DLH The Reactivity of Thiourea, Alkylthioureas, Cysteine, Glutathione, S-Methylcysteine, and Methionine towards N-Methyl-N-Nitrosoaniline in Acid Solution. J. Chem. Soc., Perkin Trans 2 1980, No. 4, 624–627. [Google Scholar]
- (31).Schmidpeter A Reaktionen von Nitrosaminen Mit Elektrophilen, I. Die Alkylierung von Nitrosaminen. Tetrahedron Lett. 1963, 4, 1421–1424. [Google Scholar]
- (32).Hünig S; Büttner G; Cramer J; Geldern L; Hansen H; Lücke E Alkoxy-diazenium-Salze, I. Synthese Und Allgemeine Eigenschaften. Chem. Ber 1969, 102, 2093–2108. [Google Scholar]
- (33).Hünig S Aus Der Chemie Reaktiver Stickstoffhaltiger Zwischenstoffe. Helv. Chim. Acta 1971, 54, 1721–1747. [Google Scholar]
- (34).Michejda CJ; Koepke SR O-Alkylation of N-Nitrosamines. Transalkylation of N-Nitrosamines and Formation of Electrophilic Intermediates. IARC Sci. Publ 1982, 41, 451–457. [PubMed] [Google Scholar]
- (35).Ohannesian L; Keefer LK Displacement of O- Versus N-Substituents from Nitrosamine-Derived Diazenium Ions by Three Divergent Mechanisms. Tetrahedron Lett. 1988, 29, 2903–2906. [Google Scholar]
- (36).Hafner K; Wagner K Cyclopentadienone-Hydrazone. Angew. Chem., Int. Ed. Engl 1963, 2, 740. [Google Scholar]
- (37).Shustov GV; Tavakalyan NB; Shustova LL; Chervin II; Kostyanovskii RG Geminal Systems. 13. Synthesis and Aminoaminating Ability of N,N-Di-Tert-Alkyl-N′-Alkoxydiazenium Salts. Bull. Acad. Sci. USSR, Div. Chem. Sci 1980, 29, 765–770. [Google Scholar]
- (38).Stöldt E; Kreher R N-Alkoxydiazenium-Salze Mit Substituierten α-Stellungen. Chem. Ber 1983, 116, 819–822. [Google Scholar]
- (39).Eicher T; Hünig S; Hansen H The Synthesis of Carbonylazo Compounds by the Reaction of Alkoxydiazenium Salts with Carboxylates. Angew. Chem., Int. Ed. Engl 1967, 6, 699. [Google Scholar]
- (40).Eicher T; Hünig S; Hansen H Alkoxy-Diazenium-Salze, III. Acyldiazene Durch Reaktion Mit Carboxylaten. Chem. Ber 1969, 102, 2889–2899. [Google Scholar]
- (41).Eicher T; Hünig S; Nikolaus P Synthesis of s-Triazolium Cations by Reaction of Alkoxydiazenium Salts with Nitrogen Heterocycles. Angew. Chem., Int. Ed. Engl 1967, 6, 699–700. [Google Scholar]
- (42).Eicher T; Hünig S; Hansen H; Nikolaus P Alkoxy-Diazenium-Salze, IV. Reaktion Mit N-Heterocyclen Und Schiffschen Basen Zu s-Triazolium-Salzen. Chem. Ber 1969, 102, 3159–3175. [Google Scholar]
- (43).Büttner G; Hünig S Alkoxy-Diazenium-Salze, VI. Reaktion Mit Hydroxyl-Ionen Zu trans-α-Hydroxy-Dialkyl-Diazenen. Chem. Ber 1971, 104, 1088–1103. [Google Scholar]
- (44).Büttner G; Hünig S Alkoxy-Diazenium-Salze, VII. Aufklärung Der Bildung von trans-α-Hydroxy-Dialkyldiazenen Und Deren Unabhangige Synthese. Chem. Ber 1971, 104, 1104–1117. [Google Scholar]
- (45).Keefer LK; Fodor CH Facile Hydrogen Isotope Exchange as Evidence for an α-Nitrosamino Carbanion. J. Am. Chem. Soc 1970, 92, 5747–5748. [Google Scholar]
- (46).Seebach D; Enders D Umpolung of Amine Reactivity. Nucleophilic α-(Secondary Amino)-Alkylation via Metalated Nitrosamines. Angew. Chem., Int. Ed. Engl 1975, 14, 15–32. [Google Scholar]
- (47).Seebach D; Enders D Lithiierte Methylnitrosamine Nucleophile α-sek.-Aminomethylierung. Chem. Ber 1975, 108, 1293–1320. [Google Scholar]
- (48).Enders D; Pieter R; Renger B; Seebach D Nucleophilic α-sec-Aminoalkylation: 2-(Diphenylhydroxymethyl)pyrrolidine. Org. Synth 1978, 58, 113. [Google Scholar]
- (49).Enders D; Hassel T; Pieter R; Renger B; Seebach D Reductive Denitrosation of Nitrosamines to Secondary Amines with Hydrogen/Raney Nickel. Synthesis 1976, No. 8, 548–550. [Google Scholar]
- (50).Seebach D; Wykypiel W Safe One-Pot Carbon-Carbon Bond Formation with Lithiated Nitrosamines Including Denitrosation by Sequential Reduction with Lithium Aluminium Hydride and Raney-Nickel. Synthesis 1979, No. 6, 423–424. [Google Scholar]
- (51).Seebach D; Enders D Metalation at Primary, Secondary, and Tertiary Carbon Atoms in Open-Chain and Cyclic Nitrosamines. Angew. Chem., Int. Ed. Engl 1972, 11, 1101–1102. [Google Scholar]
- (52).Barton DHR; Bracho RD; Gunatilaka AAL; Widdowson DA Phenol Oxidation and Biosynthesis. Part XXV. New Syntheses of Bis-(2-Arylethyl)Amines of Biosynthetic Importance. J. Chem. Soc., Perkin Trans 1 1975, No. 6, 579. [Google Scholar]
- (53).Fraser RR; Passannanti S Synthesis of 2,5-Dialkylpyrrolidines via Metallated Nitrosamines, A Constituent of Fire Ant Venom. Synthesis 1976, No. 8, 540–541. [Google Scholar]
- (54).Seebach D; Enders D; Renger B Lithiierung Und Elektrophile Substitution an α-Methylengruppen von Nitrosaminen Umpolung Der Reaktivität Sekundärer Amine. Chem. Ber 1977, 110, 1852–1865. [Google Scholar]
- (55).Seebach D; Enders D C-C-Bond Formation in the α-Position to Nitrogen in Secondary Amines. Lithiodimethylnitrosamine. Angew. Chem., Int. Ed. Engl 1972, 11, 301–302. [Google Scholar]
- (56).Seebach D; Enders D Synthesis of α-Heterosubstituted Nitrosamines. Novel Test Substances for Cancer and Mutagenesis Research? J. Med. Chem 1974, 17, 1225–1227. [DOI] [PubMed] [Google Scholar]
- (57).Seebach D; Enders D Reaction of Metalated Nitrosamines with Nitriles. A New Method for Preparation of υ-Triazoles. Angew. Chem., Int. Ed. Engl 1972, 11, 1102–1103. [Google Scholar]
- (58).Seebach D; Enders D; Dach R; Pieter R Reaktion Metallierter Nitrosamine Mit Nitrilen Eine Neue Methode Zur Darstellung von υ-Triazolen. Chem. Ber 1977, 110, 1879–1886. [Google Scholar]
- (59).Seebach D; Enders D; Renger B; Brügel W Isolation and Identification of v-Tetrazine Derivatives on Decomposition of Nitrosamine Anions. Angew. Chem., Int. Ed. Engl 1973, 12, 495. [Google Scholar]
- (60).Seebach D; Dach R; Enders D; Renger B; Jansen M; Brachtel G 1,4,5,6-Tetrahydro-v-Tetrazin-Derivate. Helv. Chim. Acta 1978, 61, 1622–1647. [Google Scholar]
- (61).Wieland H; Fressel H Versuche Zur Darstellung von Derivaten Des Oxy-Hydrazins. Ber. Dtsch. Chem. Ges 1911, 44, 898–904. [Google Scholar]
- (62).Michejda CJ; Schluenz RW Reactions of N-Nitrosamines with Grignard and Lithium Reagents. J. Org. Chem 1973, 38, 2412–2415. [Google Scholar]
- (63).Farina PR; Tieckelmann H Some Reactions of Organolithium Compounds with Nitrosamines. J. Org. Chem 1973, 38, 4259–4263. [Google Scholar]
- (64).Farina PR; Tieckelmann H Reactions of Grignard Reagents with Nitrosamines. J. Org. Chem 1975, 40, 1070–1074. [Google Scholar]
- (65).Nájera C; Sansano JM; Yus M 1,3-Dipolar Cycloadditions of Azomethine Imines. Org. Biomol. Chem 2015, 13, 8596–8636. [DOI] [PubMed] [Google Scholar]
- (66).U.S. Environmental Protection Agency. Technical Fact Sheet - N-Nitroso-Dimethylamine (NDMA); United States Environmental Protection Agency, 2017. [Google Scholar]
- (67).Fischer E Ueber Die Hydrazinverbindungen Der Fettreihe. Ber. Dtsch. Chem. Ges 1875, 8, 1587–1590. [Google Scholar]
- (68).Ohme R; Zubek A Synthesen Des Hydrazins Und Seiner Alkylderivate. Z. Chem 1968, 8, 41–51. [Google Scholar]
- (69).Hatt HH Unsym.-Dimethylhydrazine Hydrochloride. Org. Synth 1936, 16, 22–24. [Google Scholar]
- (70).Klager K; Wilson EM; Helmkamp GK Catalytic Production of N,N-Dimethylhydrazine. Ind. Eng. Chem 1960, 52, 119–120. [Google Scholar]
- (71).Smith GW; Thatcher DN Catalytic Hydrogenation of Nitrosamines to Unsymmetrical Hydrazines. Ind. Eng. Chem. Prod. Res. Dev 1962, 1, 117–120. [Google Scholar]
- (72).Zimmer H; Audrieth LF; Zimmer M; Rowe RA The Synthesis of Unsymmetrically Disubstituted Hydrazines. J. Am. Chem. Soc 1955, 77, 790–793. [Google Scholar]
- (73).Schueler FW; Hanna C A Synthesis of Unsymmetrical Dimethyl Hydrazine Using Lithium Aluminum Hydride. J. Am. Chem. Soc 1951, 73, 4996. [Google Scholar]
- (74).Hanna C; Schueler FW The Reaction of Disubstituted Nitrosamines with Lithium Aluminum Hydride. J. Am. Chem. Soc 1952, 74, 3693–3694. [Google Scholar]
- (75).Poirier RH; Benington F Reduction of N-Nitrosodiphenylamine to unsym-Diphenylhydrazine by Lithium Aluminum Hydride. J. Am. Chem. Soc 1952, 74, 3192. [Google Scholar]
- (76).Entwistle ID; Johnstone RAW; Wilby AH Metal-Assisted Reactions—Part 11: Rapid Reduction of N-Nitrosoamines to N,N-Disubstituted Hydrazines; the Utility of Some Low-Valent Titanium Reagents. Tetrahedron 1982, 38, 419–423. [Google Scholar]
- (77).Iversen PE Organic Electrosyntheses. III. Reduction of N-Nitrosamines. Acta Chem. Scand 1971, 25, 2337–2340. [Google Scholar]
- (78).Gafurov RG; Korepin AG; Sogomonyan EM; Salakhova AN; Eremenko LT 3-Fluoro-3,3-Dinitro-1-Aminopropane in the Mannich Reaction. Bull. Acad. Sci. USSR, Div. Chem. Sci 1972, 21, 1823–1824. [Google Scholar]
- (79).Emmons WD Peroxytrifluoroacetic Acid. I. The Oxidation of Nitrosamines to Nitramines. J. Am. Chem. Soc 1954, 76, 3468–3470. [Google Scholar]
- (80).Gafurov RG; Sogomonyan EM; Eremenko LT Conversion of Nitrosamines to Nitramines. Bull. Acad. Sci. USSR, Div. Chem. Sci 1973, 22, 2764. [Google Scholar]
- (81).Saavedra JE Recent Synthetic Applications of N-Nitrosamines and Related Compounds. Org. Prep. Proced. Int 1987, 19, 83–159. [Google Scholar]
- (82).Lee J; Chen L; West AH; Richter-Addo GB Interactions of Organic Nitroso Compounds with Metals. Chem. Rev 2002, 102, 1019–1065. [DOI] [PubMed] [Google Scholar]
- (83).Liu B; Fan Y; Gao Y; Sun C; Xu C; Zhu J Rhodium(III)-Catalyzed N-Nitroso-Directed C–H Olefination of Arenes. High-Yield, Versatile Coupling under Mild Conditions. J. Am. Chem. Soc 2013, 135, 468–473. [DOI] [PubMed] [Google Scholar]
- (84).Gao T; Sun P Palladium-Catalyzed N-Nitroso-Directed C–H Alkoxylation of Arenes and Subsequent Formation of 2-Alkoxy-N-Alkylarylamines. J. Org. Chem 2014, 79, 9888–9893. [DOI] [PubMed] [Google Scholar]
- (85).Liang Y; Jiao N Cationic Cobalt(III) Catalyzed Indole Synthesis: The Regioselective Intermolecular Cyclization of N-Nitrosoanilines and Alkynes. Angew. Chem., Int. Ed 2016, 55, 4035–4039. [DOI] [PubMed] [Google Scholar]
- (86).Hu X; Chen X; Shao Y; Xie H; Deng Y; Ke Z; Jiang H; Zeng W Co(III)-Catalyzed Coupling-Cyclization of Aryl C–H Bonds with α-Diazoketones Involving Wolff Rearrangement. ACS Catal. 2018, 8, 1308–1312. [Google Scholar]
- (87).Song X; Gao C; Li B; Zhang X; Fan X Regioselective Synthesis of 2-Alkenylindoles and 2-Alkenylindole-3-Carboxylates through the Cascade Reactions of N-Nitrosoanilines with Propargyl Alcohols. J. Org. Chem 2018, 83, 8509–8521. [DOI] [PubMed] [Google Scholar]
- (88).Wu Y; Pi C; Cui X; Wu Y Rh(III)-Catalyzed Tandem Acylmethylation/Nitroso Migration/Cyclization of N-Nitrosoanilines with Sulfoxonium Ylides in One Pot: Approach to 3-Nitrosoindoles. Org. Lett 2020, 22, 361–364. [DOI] [PubMed] [Google Scholar]
- (89).Zhao G; Zhu M; Provot O; Alami M; Messaoudi S Synthesis of 2,3-Substituted β-N-Glycosyl Indoles through C–H Activation/Annulation Process under Rh(III)-Catalysis. Org. Lett 2020, 22, 57–61. [DOI] [PubMed] [Google Scholar]
- (90).Fischer O; Hepp E Zur Kenntniss Der Nitrosamine. Ber. Dtsch. Chem. Ges 1886, 19, 2991–2995. [Google Scholar]
- (91).Biggs ID; Williams DLH Kinetics and Mechanism of the Fischer-Hepp Rearrangement and Denitrosation. Part VII. Reactions at High Acidity. J. Chem. Soc., Perkin Trans 2 1976, No. 5, 601–605. [Google Scholar]
- (92).Bamford CH A Study of the Photolysis of Organic Nitrogen Compounds. Part I. Dimethyl- and Diethyl-Nitrosoamines. J. Chem. Soc 1939, 12–17. [Google Scholar]
- (93).Geiger G; Huber JR Photolysis of Dimethylnitrosamine in the Gas Phase. Helv. Chim. Acta 1981, 64, 989–995. [Google Scholar]
- (94).Geiger G; Stafast H; Brühlmann U; Huber JR Photodissociation of Dimethylnitrosamine. Chem. Phys. Lett 1981, 79, 525–528. [Google Scholar]
- (95).Stefan MI; Bolton JR UV Direct Photolysis of N-Nitrosodimethylamine (NDMA): Kinetic and Product Study. Helv. Chim. Acta 2002, 85, 1416–1426. [Google Scholar]
- (96).Plumlee MH; Reinhard M Photochemical Attenuation of N-Nitrosodimethylamine (NDMA) and Other Nitrosamines in Surface Water. Environ. Sci. Technol 2007, 41, 6170–6176. [DOI] [PubMed] [Google Scholar]
- (97).Ho T-FL; Bolton JR; Lipczynska-Kochany E Quantum Yields for the Photodegradation of Pollutants in Dilute Aqueous Solution: Phenol, 4-Chlorophenol and N-Nitrosodimethylamine. J. Adv. Oxid. Technol 1996, 1, 170–178. [Google Scholar]
- (98).Lee C; Choi W; Kim YG; Yoon J UV Photolytic Mechanism of N-Nitrosodimethylamine in Water: Dual Pathways to Methylamine versus Dimethylamine. Environ. Sci. Technol 2005, 39, 2101–2106. [DOI] [PubMed] [Google Scholar]
- (99).Fiz N; Usero JL; Casado J Photolysis of N-Nitrosamines in Neutral Media. Int. J. Chem. Kinet 1993, 25, 341–351. [Google Scholar]
- (100).Sharpless CM; Linden KG Experimental and Model Comparisons of Low- and Medium-Pressure Hg Lamps for the Direct and H2O2 Assisted UV Photodegradation of N-Nitrosodimethylamine in Simulated Drinking Water. Environ. Sci. Technol 2003, 37, 1933–1940. [DOI] [PubMed] [Google Scholar]
- (101).Lee C; Choi W; Yoon J UV Photolytic Mechanism of N-Nitrosodimethylamine in Water: Roles of Dissolved Oxygen and Solution pH. Environ. Sci. Technol 2005, 39, 9702–9709. [DOI] [PubMed] [Google Scholar]
- (102).Polo J; Chow YL Efficient Photolytic Degradation of Nitrosamines. J. Natl. Cancer Inst 1976, 56, 997–1001. [DOI] [PubMed] [Google Scholar]
- (103).Chow YL; Wu ZZ; Lau MP; Yip RW On the Singlet and Triplet Excited States of Nitrosamines. J. Am. Chem. Soc 1985, 107, 8196–8201. [Google Scholar]
- (104).Xu B; Chen Z; Qi F; Ma J; Wu F Comparison of N-Nitrosodiethylamine Degradation in Water by UV Irradiation and UV/O3: Efficiency, Product and Mechanism. J. Hazard. Mater 2010, 179, 976–982. [DOI] [PubMed] [Google Scholar]
- (105).Kwon BG; Kim JO; Namkung KC The Formation of Reactive Species Having Hydroxyl Radical-like Reactivity from UV Photolysis of N-Nitrosodimethylamine (NDMA): Kinetics and Mechanism. Sci. Total Environ 2012, 437, 237–244. [DOI] [PubMed] [Google Scholar]
- (106).Gowenlock BG; Pfab J; Williams GC Quantum Yields for the Photolysis of Some Nitrosamines in Solution. J. Chem. Res., Synop 1978, 362–363. [Google Scholar]
- (107).Chow Y-L Photolysis of N-Nitrosoamines. Tetrahedron Lett. 1964, 5, 2333–2338. [Google Scholar]
- (108).Oliveira MS; Ghogare AA; Abramova I; Greer EM; Prado FM; Di Mascio P; Greer A Mechanism of Photochemical O-Atom Exchange in Nitrosamines with Molecular Oxygen. J. Org. Chem 2015, 80, 6119–6127. [DOI] [PubMed] [Google Scholar]
- (109).Ghogare AA; Debaz CJ; Silva Oliveira M; Abramova I; Mohapatra PP; Kwon K; Greer EM; Prado FM; Valerio HP; Di Mascio P; et al. Experimental and DFT Computational Insight into Nitrosamine Photochemistry - Oxygen Matters. J. Phys. Chem. A 2017, 121, 5954–5966. [DOI] [PubMed] [Google Scholar]
- (110).Axenrod T; Milne GWA Photochemistry of Nitrosamines: Mechanism Studies. Tetrahedron 1968, 24, 5775–5783. [Google Scholar]
- (111).Chow YL Photochemistry of Nitroso Compounds in Solution. V. Photolysis of N-Nitrosodialkylamines. Can. J. Chem 1967, 45, 53–62. [Google Scholar]
- (112).Shah AD; Dai N; Mitch WA Application of Ultraviolet, Ozone, and Advanced Oxidation Treatments to Washwaters to Destroy Nitrosamines, Nitramines, Amines, and Aldehydes Formed during Amine-Based Carbon Capture. Environ. Sci. Technol 2013, 47, 2799–2808. [DOI] [PubMed] [Google Scholar]
- (113).Lee C; Lee Y; Schmidt C; Yoon J; von Gunten U Oxidation of Suspected N-Nitrosodimethylamine (NDMA) Precursors by Ferrate (VI): Kinetics and Effect on the NDMA Formation Potential of Natural Waters. Water Res. 2008, 42, 433–441. [DOI] [PubMed] [Google Scholar]
- (114).Layne WS; Jaffe HH; Zimmer H Basicity of N-Nitrosamines. I. Non-Polar Solvents. J. Am. Chem. Soc 1963, 85, 435–438. [Google Scholar]
- (115).Chow YL; Lau MP; Perry RA; Tam JNS Photoreactions of Nitroso Compounds in Solution. XX. Photo-reduction, Photoelimination, and Photoaddition of Nitrosamines. Can. J. Chem 1972, 50, 1044–1050. [Google Scholar]
- (116).Chow YL; Wu Z-Z Light-Induced Self-Nitrosation of Polycyclic Phenols with Nitrosamine. Excited State Proton Transfer. J. Am. Chem. Soc 1987, 109, 5260–5267. [Google Scholar]
- (117).Chow YL; Richard H; Lockhart RW Addition Reactions of Aminium Radicals: Oxidative and Non-Oxidative Photoaddition of Nitrosoamines to Non-Conjugated Polyenes. J. Chem. Soc., Perkin Trans 1 1982, 1 (9), 1419–1428. [Google Scholar]
- (118).Chow YL; Colon CJ; Chang DWL; Pillay KS; Lockhart RL; Tezuka T Nitrosamine Photolysis as a Synthetic Method: The Addition of Aminium Radicals to Unsaturated Carbon-Carbon Bonds. Acta Chem. Scand 1982, 36, 623–634. [Google Scholar]
- (119).Chow YL Photo-Addition of N-Nitrosodialkylamines to Cyclohexene. Can. J. Chem 1965, 43, 2711–2716. [Google Scholar]
- (120).Chow YL; Tam JNS; Colón CJ; Pillay KS Photoreactions of Nitroso Compounds in Solution. XXIII. A Novel Oxidative Photoreaction of Nitrosamines and Nitrosamides. Can. J. Chem 1973, 51, 2469–2476. [Google Scholar]
- (121).Lau MP; Cessna AJ; Chow YL; Yip RW Flash Photolysis of N-Nitrosopiperidine. The Reactive Transient. J. Am. Chem. Soc 1971, 93, 3808–3809. [Google Scholar]
- (122).Chow YL; Colón CJ Photochemistry of Nitroso Compounds in Solution. IX. Steric Effects in the Photoaddition of N-Nitrosodialkylamines to Olefins. Magnetic Non-Equivalence of Methylene Protons Caused by a Distant Asymmetry. Can. J. Chem 1967, 45, 2559–2568. [Google Scholar]
- (123).Chow YL; Chen SC; Chang DWL Photoreactions of Nitroso Compounds in Solution. XIX Stereochemistry of Nitrosamine Photoaddition to Olefin. Can. J. Chem 1971, 49, 3069–3071. [Google Scholar]
- (124).Chow YL; Colón CJ; Quon HH; Mojelsky T Photoreactions of Nitroso Compounds in Solution. XXII. Stereo-chemical Consequences of the Photoaddition of N-Nitrosopiperidine to Conjugated Acyclic Dienes: A Nuclear Magnetic Resonance Study of Enone Oximes. Can. J. Chem 1972, 50, 1065–1077. [Google Scholar]
- (125).Chow YL; Chen SC; Pillay KS; Perry RA Photoreactions of Nitroso Compounds in Solution. XXI. Stereo-chemical Course of Photoaddition of Nitrosamines to Some Cyclic Olefins and Conformational Isomers Generated by A 1,3-Interaction. Can. J. Chem 1972, 50, 1051–1064. [Google Scholar]
- (126).Mojelsky T; Chow YL Polar Effects of the Aminium Radical Addition to Styrenes. J. Am. Chem. Soc 1974, 96, 4549–4554. [Google Scholar]
- (127).Chow YL; Chang DWL C-Nitroso-Enamines. J. Chem. Soc. D 1971, No. 1, 64. [Google Scholar]
- (128).Peyton GR; LeFaivre MH; Maloney SW CERL Technical Report for U.S. Army Corps of Engineers: Verification of RDX Photolysis Mechanism; Champaign, IL, 1999. [Google Scholar]
- (129).Gowenlock BG; Pfab J; Young VM Photolysis of Some N-Nitroso- and N-Nitro-Anilines in Solution. J. Chem. Soc., Perkin Trans 2 1997, No. 5, 915–919. [Google Scholar]
- (130).Andrew TL; Swager TM Detection of Explosives via Photolytic Cleavage of Nitroesters and Nitramines. J. Org. Chem 2011, 76, 2976–2993. [DOI] [PubMed] [Google Scholar]
- (131).Wang C; Huang H; Bunes BR; Wu N; Xu M; Yang X; Yu L; Zang L Trace Detection of RDX, HMX and PETN Explosives Using a Fluorescence Spot Sensor. Sci. Rep 2016, 6, 25015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (132).Andrew TL; Swager TM A Fluorescence Turn-on Mechanism to Detect High Explosives RDX and PETN. J. Am. Chem. Soc 2007, 129, 7254–7255. [DOI] [PubMed] [Google Scholar]
- (133).Hanst PL; Spence JW; Miller M Atmospheric Chemistry of N-Nitroso Dimethylamine. Environ. Sci. Technol 1977, 11, 403–405. [Google Scholar]
- (134).Tuazon EC; Carter WPL; Atkinson R; Winer AM; Pitts JN Atmospheric Reactions of N-Nitrosodimethylamine and Dimethylnitramine. Environ. Sci. Technol 1984, 18, 49–54. [DOI] [PubMed] [Google Scholar]
- (135).Shah AD; Mitch WA Halonitroalkanes, Halonitriles, Haloamides, and N-Nitrosamines: A Critical Review of Nitrogenous Disinfection Byproduct Formation Pathways. Environ. Sci. Technol 2012, 46, 119–131. [DOI] [PubMed] [Google Scholar]
- (136).López-Rodríguez R; McManus JA; Murphy NS; Ott MA; Burns MJ Pathways for N-Nitroso Compound Formation: Secondary Amines and Beyond. Org. Process Res. Dev 2020, 24, 1558–1585. [Google Scholar]
- (137).Chen Z; Valentine RL Modeling the Formation of N-Nitrosodimethylamine (NDMA) from the Reaction of Natural Organic Matter (NOM) with Monochloramine. Environ. Sci. Technol 2006, 40, 7290–7297. [DOI] [PubMed] [Google Scholar]
- (138).Le Roux J; Gallard H; Croué JP Chloramination of Nitrogenous Contaminants (Pharmaceuticals and Pesticides): NDMA and Halogenated DBPs Formation. Water Res 2011, 45, 3164–3174. [DOI] [PubMed] [Google Scholar]
- (139).Spahr S; Cirpka OA; von Gunten U; Hofstetter TB Formation of N-Nitrosodimethylamine during Chloramination of Secondary and Tertiary Amines: Role of Molecular Oxygen and Radical Intermediates. Environ. Sci. Technol 2017, 51, 280–290. [DOI] [PubMed] [Google Scholar]
- (140).Schreiber IM; Mitch WA Nitrosamine Formation Pathway Revisited: The Importance of Chloramine Speciation and Dissolved Oxygen. Environ. Sci. Technol 2006, 40, 6007–6014. [DOI] [PubMed] [Google Scholar]
- (141).Huang ME; Huang S; McCurry DL Re-Examining the Role of Dichloramine in High-Yield N-Nitrosodimethylamine Formation from N,N-Dimethyl-α-Arylamines. Environ. Sci. Technol. Lett 2018, 5, 154–159. [Google Scholar]
- (142).McCurry DL; Ishida KP; Oelker GL; Mitch WA Reverse Osmosis Shifts Chloramine Speciation Causing Re-Formation of NDMA during Potable Reuse of Wastewater. Environ. Sci. Technol 2017, 51, 8589–8596. [DOI] [PubMed] [Google Scholar]
- (143).Furst KE; Pecson BM; Webber BD; Mitch WA Distributed Chlorine Injection To Minimize NDMA Formation during Chloramination of Wastewater. Environ. Sci. Technol. Lett 2018, 5, 462–466. [Google Scholar]
- (144).Bond T; Simperler A; Graham N; Ling L; Gan W; Yang X; Templeton MR Defining the Molecular Properties of N-Nitrosodimethylamine (NDMA) Precursors Using Computational Chemistry. Environ. Sci. Water Res. Technol 2017, 3, 502–512. [Google Scholar]
- (145).Shen R; Andrews SA Demonstration of 20 Pharmaceuticals and Personal Care Products (PPCPs) as Nitrosamine Precursors during Chloramine Disinfection. Water Res. 2011, 45, 944–952. [DOI] [PubMed] [Google Scholar]
- (146).Liu YD; Selbes M; Zeng C; Zhong R; Karanfil T Formation Mechanism of NDMA from Ranitidine, Trimethylamine, and Other Tertiary Amines during Chloramination: A Computational Study. Environ. Sci. Technol 2014, 48, 8653–8663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (147).Kemper JM; Walse SS; Mitch WA Quaternary Amines As Nitrosamine Precursors: A Role for Consumer Products? Environ. Sci. Technol 2010, 44, 1224–1231. [DOI] [PubMed] [Google Scholar]
- (148).Park S-H; Wei S; Mizaikoff B; Taylor AE; Favero C; Huang C-H Degradation of Amine-Based Water Treatment Polymers during Chloramination as N-Nitrosodimethylamine (NDMA) Precursors. Environ. Sci. Technol 2009, 43, 1360–1366. [DOI] [PubMed] [Google Scholar]
- (149).An D; Chen Y; Gu B; Westerhoff P; Hanigan D; Herckes P; Fischer N; Donovan S; Croue JP; Atkinson A Lower Molecular Weight Fractions of PolyDADMAC Coagulants Disproportionately Contribute to N-Nitrosodimethylamine Formation during Water Treatment. Water Res. 2019, 150, 466–472. [DOI] [PubMed] [Google Scholar]
- (150).Choi J; Valentine RL N-Nitrosodimethylamine Formation by Free-Chlorine-Enhanced Nitrosation of Dimethylamine. Environ. Sci. Technol 2003, 37, 4871–4876. [DOI] [PubMed] [Google Scholar]
- (151).Mitch WA; Schreiber IM Degradation of Tertiary Alkylamines during Chlorination/Chloramination: Implications for Formation of Aldehydes, Nitriles, Halonitroalkanes, and Nitrosamines. Environ. Sci. Technol 2008, 42, 4811–4817. [DOI] [PubMed] [Google Scholar]
- (152).Zhang A; Li Y; Song Y; Lv J; Yang J Characterization of Pharmaceuticals and Personal Care Products as N-Nitrosodimethylamine Precursors during Disinfection Processes Using Free Chlorine and Chlorine Dioxide. J. Hazard. Mater 2014, 276, 499–509. [DOI] [PubMed] [Google Scholar]
- (153).Walse SS; Mitch WA Nitrosamine Carcinogens Also Swim in Chlorinated Pools. Environ. Sci. Technol 2008, 42, 1032–1037. [DOI] [PubMed] [Google Scholar]
- (154).Breider F; Salihu I; von Gunten U Formation of N-Nitrosamines by Micelle-Catalysed Nitrosation of Aliphatic Secondary Amines. Environ. Sci. Process. Impacts 2018, 20, 1479–1487. [DOI] [PubMed] [Google Scholar]
- (155).Sgroi M; Roccaro P; Oelker GL; Snyder SA N-Nitrosodimethylamine Formation upon Ozonation and Identification of Precursors Source in a Municipal Wastewater Treatment Plant. Environ. Sci. Technol 2014, 48, 10308–10315. [DOI] [PubMed] [Google Scholar]
- (156).Lim S; Lee W; Na S; Shin J; Lee Y N-Nitrosodimethylamine (NDMA) Formation during Ozonation of N,N-Dimethylhydrazine Compounds: Reaction Kinetics, Mechanisms, and Implications for NDMA Formation Control. Water Res. 2016, 105, 119–128. [DOI] [PubMed] [Google Scholar]
- (157).von Gunten U; Salhi E; Schmidt CK; Arnold WA Kinetics and Mechanisms of N-Nitrosodimethylamine Formation upon Ozonation of N,N-Dimethylsulfamide-Containing Waters: Bromide Catalysis. Environ. Sci. Technol 2010, 44, 5762–5768. [DOI] [PubMed] [Google Scholar]
- (158).Zeng T; Glover CM; Marti EJ; Woods-Chabane GC; Karanfil T; Mitch WA; Dickenson ERV Relative Importance of Different Water Categories as Sources of N-Nitrosamine Precursors. Environ. Sci. Technol 2016, 50, 13239–13248. [DOI] [PubMed] [Google Scholar]
- (159).Gan W; Bond T; Yang X; Westerhoff P Role of Chlorine Dioxide in N-Nitrosodimethylamine Formation from Oxidation of Model Amines. Environ. Sci. Technol 2015, 49, 11429–11437. [DOI] [PubMed] [Google Scholar]
- (160).Velo-Gala I; Farré MJ; Radjenovic J; Gernjak W N-Nitrosodimethylamine (NDMA) Degradation by the Ultraviolet/Peroxodisulfate Process. Environ. Sci. Technol. Lett 2019, 6, 106–111. [Google Scholar]
- (161).Lee C; Yoon J; von Gunten U Oxidative Degradation of N-Nitrosodimethylamine by Conventional Ozonation and the Advanced Oxidation Process Ozone/Hydrogen Peroxide. Water Res. 2007, 41, 581–590. [DOI] [PubMed] [Google Scholar]
- (162).Chaplin BP; Schrader G; Farrell J Electrochemical Oxidation of N-Nitrosodimethylamine with Boron-Doped Diamond Film Electrodes. Environ. Sci. Technol 2009, 43, 8302–8307. [DOI] [PubMed] [Google Scholar]
- (163).Almassi S; Li Z; Xu W; Pu C; Zeng T; Chaplin BP Simultaneous Adsorption and Electrochemical Reduction of N-Nitrosodimethylamine Using Carbon-Ti4O7 Composite Reactive Electrochemical Membranes. Environ. Sci. Technol 2019, 53, 928–937. [DOI] [PubMed] [Google Scholar]
- (164).Davie MG; Reinhard M; Shapley JR Metal-Catalyzed Reduction of N-Nitrosodimethylamine with Hydrogen in Water. Environ. Sci. Technol 2006, 40, 7329–7335. [DOI] [PubMed] [Google Scholar]
- (165).Huo X; Liu J; Strathmann TJ Ruthenium Catalysts for the Reduction of N-Nitrosamine Water Contaminants. Environ. Sci. Technol 2018, 52, 4235–4243. [DOI] [PubMed] [Google Scholar]
- (166).Ma H; Li S; Wang H; Schneider WF Water-Mediated Reduction of Aqueous N-Nitrosodimethylamine with Pd. Environ. Sci. Technol 2019, 53, 7551–7563. [DOI] [PubMed] [Google Scholar]
- (167).Hatzinger PB; Lippincott DR Field Demonstration of N-Nitrosodimethylamine (NDMA) Treatment in Groundwater Using Propane Biosparging. Water Res. 2019, 164, 114923. [DOI] [PubMed] [Google Scholar]
- (168).Webster TS; Condee C; Hatzinger PB Ex Situ Treatment of N-Nitrosodimethylamine (NDMA) in Groundwater Using a Fluidized Bed Reactor. Water Res. 2013, 47, 811–820. [DOI] [PubMed] [Google Scholar]
- (169).Ho L; Grasset C; Hoefel D; Dixon MB; Leusch FDL; Newcombe G; Saint CP; Brookes JD Assessing Granular Media Filtration for the Removal of Chemical Contaminants from Wastewater. Water Res. 2011, 45, 3461–3472. [DOI] [PubMed] [Google Scholar]
- (170).Fujioka T; Khan SJ; Poussade Y; Drewes JE; Nghiem LD N-Nitrosamine Removal by Reverse Osmosis for Indirect Potable Water Reuse - A Critical Review Based on Observations from Laboratory-, Pilot- and Full-Scale Studies. Sep. Purif. Technol 2012, 98, 503–515. [Google Scholar]
- (171).Fujioka T; Khan SJ; McDonald JA; Roux A; Poussade Y; Drewes JE; Nghiem LD N-Nitrosamine Rejection by Reverse Osmosis Membranes: A Full-Scale Study. Water Res 2013, 47, 6141–6148. [DOI] [PubMed] [Google Scholar]
- (172).Fujioka T; Kodamatani H; Nghiem LD; Shintani T Transport of N-Nitrosamines through a Reverse Osmosis Membrane: Role of Molecular Size and Nitrogen Atoms. Environ. Sci. Technol. Lett 2019, 6, 44–48. [Google Scholar]
- (173).Fujioka T; Ishida KP; Shintani T; Kodamatani H High Rejection Reverse Osmosis Membrane for Removal of N-Nitrosamines and Their Precursors. Water Res. 2018, 131, 45–51. [DOI] [PubMed] [Google Scholar]
- (174).Croll H; Soroush A; Pillsbury ME; Romero-Vargas Castrillón S Graphene Oxide Surface Modification of Polyamide Reverse Osmosis Membranes for Improved N-Nitrosodimethylamine (NDMA) Removal. Sep. Purif. Technol 2019, 210, 973–980. [Google Scholar]
- (175).Hanigan D; Zhang J; Herckes P; Krasner SW; Chen C; Westerhoff P Adsorption of N-Nitrosodimethylamine Precursors by Powdered and Granular Activated Carbon. Environ. Sci. Technol 2012, 46, 12630–12639. [DOI] [PubMed] [Google Scholar]
- (176).Beita-Sandí W; Ersan MS; Uzun H; Karanfil T Removal of N-Nitrosodimethylamine Precursors with Powdered Activated Carbon Adsorption. Water Res. 2016, 88, 711–718. [DOI] [PubMed] [Google Scholar]
- (177).Chen Z; Valentine RL The Influence of the Pre-Oxidation of Natural Organic Matter on the Formation of N-Nitrosodimethylamine (NDMA). Environ. Sci. Technol 2008, 42, 5062–5067. [DOI] [PubMed] [Google Scholar]
- (178).Shah AD; Krasner SW; Lee CFT; von Gunten U; Mitch WA Trade-Offs in Disinfection Byproduct Formation Associated with Precursor Preoxidation for Control of N-Nitrosodimethylamine Formation. Environ. Sci. Technol 2012, 46, 4809–4818. [DOI] [PubMed] [Google Scholar]
- (179).Atkinson AJ; Fischer N; Donovan S; Bartlett J; Alrehaili O; Sinha S; Kommineni S; Herckes P; Westerhoff P Purification and Removal of the Low Molecular Weight Fraction of PolyDADMAC Reduces N-Nitrosodimethylamine Formation during Water Treatment. Environ. Sci. Water Res. Technol 2020, 6, 2492–2498. [Google Scholar]
- (180).Lijinsky W; Taylor HW Carcinogenicity of Methylated Derivatives of N-Nitrosodiethylamine and Related Compounds in Sprague-Dawley Rats. J. Natl. Cancer Inst 1979, 62, 407–410. [PubMed] [Google Scholar]
- (181).Dai N; Mitch WA Relative Importance of N-Nitrosodimethylamine Compared to Total N-Nitrosamines in Drinking Waters. Environ. Sci. Technol 2013, 47, 3648–3656. [DOI] [PubMed] [Google Scholar]
- (182).United States Environmental Protection Agency. Regional Screening Levels for Chemical Contaminants at Superfund Sites https://www.epa.gov/risk/regional-screening-levels-rsls-generic-tables (accessed Aug 4, 2020).
- (183).Massachusetts Department of Environmental Protection. Drinking Water Standards and Guidelines https://www.mass.gov/guides/drinking-water-standards-and-guidelines (accessed 2020-08-05).
- (184).Massachusetts Department of Environmental Protection. Supporting Documentation for Drinking Water Standards and Guidelines; 2004. [Google Scholar]
- (185).Boyd JM; Hrudey SE; Li XF; Richardson SD Solid-Phase Extraction and High-Performance Liquid Chromatography Mass Spectrometry Analysis of Nitrosamines in Treated Drinking Water and Wastewater. TrAC, Trends Anal. Chem 2011, 30, 1410–1421. [Google Scholar]
- (186).Munch JW; Bassett MV Method 521: Determination of Nitrosamines in Drinking Water by Solid Phase Extraction and Capillary Column Gas Chromatography with Large Vol. Injection and Chemical Ionization Tandem Mass Spectrometry (MS/MS); U.S. Environmental Protection Agency, 2004. [Google Scholar]
- (187).Grebel JE; Young CC; Suffet IHM Solid-Phase Microextraction of N-Nitrosamines. J. Chromatogr. A 2006, 1117, 11–18. [DOI] [PubMed] [Google Scholar]
- (188).Cha W; Fox P; Nalinakumari B High-Performance Liquid Chromatography with Fluorescence Detection for Aqueous Analysis of Nanogram-Level N-Nitrosodimethylamine. Anal. Chim. Acta 2006, 566, 109–116. [Google Scholar]
- (189).Noffsinger JB; Danielson ND Generation of Chemiluminescence upon Reaction of Aliphatic Amines with Tris(2,2′-Bipyridine)Ruthenium(III). Anal. Chem 1987, 59, 865–868. [Google Scholar]
- (190).Pérez-Ruiz T; Martínez-Lozano C; Tomés V; Martín J Automated Solid-Phase Extraction and High-Performance Liquid Chromatographic Determination of Nitrosamines Using Post-Column Photolysis and Tris(2,2′-Bipyridyl) Ruthenium(III) Chemiluminescence. J. Chromatogr. A 2005, 1077, 49–56. [DOI] [PubMed] [Google Scholar]
- (191).Bellec G; Cauvin JM; Salaun MC; Le Calvé K; Dréano Y; Gouérou H; Ménez JF; Berthou F Analysis of N-Nitrosamines by High-Performance Liquid Chromatography with Post-Column Photohydrolysis and Colorimetric Detection. J. Chromatogr. A 1996, 727, 83–92. [DOI] [PubMed] [Google Scholar]
- (192).Lee M; Lee Y; Soltermann F; von Gunten U Analysis of N-Nitrosamines and Other Nitro(so) Compounds in Water by High-Performance Liquid Chromatography with Post-Column UV Photolysis/Griess Reaction. Water Res. 2013, 47, 4893–4903. [DOI] [PubMed] [Google Scholar]
- (193).Padhye L; Wang P; Karanfil T; Huang C-H Unexpected Role of Activated Carbon in Promoting Transformation of Secondary Amines to N-Nitrosamines. Environ. Sci. Technol 2010, 44, 4161–4168. [DOI] [PubMed] [Google Scholar]
- (194).Huang C-H; Padhye LP; Wang Y-L Catalytic Impact of Activated Carbon on the Formation of Nitrosamines from Different Amine Precursors. ACS Symp. Ser 2013, 1150, 79–100. [Google Scholar]
- (195).Chuang YH; Shabani F; Munoz J; Aflaki R; Hammond SD; Mitch WA Formation of N-Nitrosamines during the Analysis of Municipal Secondary Biological Nutrient Removal Process Effluents by US EPA Method 521. Chemosphere 2019, 221, 597–605. [DOI] [PubMed] [Google Scholar]
- (196).Kodamatani H; Yamazaki S; Saito K; Amponsaa-Karikari A; Kishikawa N; Kuroda N; Tomiyasu T; Komatsu Y Highly Sensitive Method for Determination of N-Nitrosamines Using High-Performance Liquid Chromatography with Online UV Irradiation and Luminol Chemiluminescence Detection. J. Chromatogr. A 2009, 1216, 92–98. [DOI] [PubMed] [Google Scholar]
- (197).Fujioka T; Takeuchi H; Tanaka H; Nghiem LD; Ishida KP; Kodamatani H A Rapid and Reliable Technique for N-Nitrosodimethylamine Analysis in Reclaimed Water by HPLC-Photochemical Reaction-Chemiluminescence. Chemosphere 2016, 161, 104–111. [DOI] [PubMed] [Google Scholar]
- (198).Fujioka T; Kodamatani H; Takeuchi H; Tanaka H; Nghiem LD Online Monitoring of N-Nitrosodimethylamine for the Removal Assurance of 1,4-Dioxane and Other Trace Organic Compounds by Reverse Osmosis. Environ. Sci. Water Res. Technol 2018, 4, 2021–2028. [Google Scholar]
- (199).Roback SL; Kodamatani H; Fujioka T; Plumlee MH Validation of a Novel Direct-Injection Chemiluminescence-Based Method for N-Nitrosamine Analysis in Advanced-Treated Recycled Water, Drinking Water, and Wastewater. Environ. Sci. Water Res. Technol 2020, 6, 1106–1115. [Google Scholar]
- (200).Fiddler W The Occurrence and Determination of N-Nitroso Compounds. Toxicol. Appl. Pharmacol 1975, 31, 352–360. [DOI] [PubMed] [Google Scholar]
- (201).Daiber D; Preussmann R Quantitative Colorimetrische Bestimmung Organischer N-Nitroso-Verbindungen Durch Photo-chemische Spaltung Der Nitrosaminbindung. Fresenius’ Z. Anal. Chem 1964, 206, 344–352. [Google Scholar]
- (202).Preussmann R; Neurath G; Wulf-Lorentzen G; Daiber D; Hengy H Anfärbemethoden Und Dünnschicht-Chromatographie von Organischen N-Nitrosoverbindungen. Fresenius’ Z. Anal. Chem 1964, 202, 187–192. [Google Scholar]
- (203).Eisenbrand G; Preussmann R Eine Neue Methode Zur Kolorimetrischen Bestimmung von Nitrosaminen Nach Spaltung Der N-Nitrosogruppe Mit Bromwasserstoff in Eissessig. Arzneim.-Forsch 1970, 20, 1512–1517. [PubMed] [Google Scholar]
- (204).Neurath G; Pirmann B; Dünger M Identifizierung von N-Nitroso-Verbindungen Und Asymmetrischen Hydrazinen Als 5-Nitro-2-Hydroxy-Benzal-Derivate Und Anwendung Im Mikromaßstab. Chem. Ber 1964, 97, 1631–1638. [Google Scholar]
- (205).Serfontein WJ; Smit JH Evidence for the Occurrence of N-Nitrosamines in Tobacco. Nature 1967, 214, 169–170. [Google Scholar]
- (206).Yang KW; Brown EV A Sensitive Analysis for Nitrosamines. Anal. Lett 1972, 5, 293–304. [Google Scholar]
- (207).Kulshrestha P; McKinstry KC; Fernandez BO; Feelisch M; Mitch WA Application of an Optimized Total N-Nitrosamine (TONO) Assay to Pools: Placing N-Nitrosodimethylamine (NDMA) Determinations into Perspective. Environ. Sci. Technol 2010, 44, 3369–3375. [DOI] [PubMed] [Google Scholar]
- (208).Breider F; von Gunten U Quantification of Total N-Nitrosamine Concentrations in Aqueous Samples via UV-Photolysis and Chemiluminescence Detection of Nitric Oxide. Anal. Chem 2017, 89, 1574–1582. [DOI] [PubMed] [Google Scholar]
- (209).Wang J; Chan WG; Haut SA; Krauss MR; Izac RR; Hempfling WP Determination of Total N-Nitroso Compounds by Chemical Denitrosation Using CuCl. J. Agric. Food Chem 2005, 53, 4686–4691. [DOI] [PubMed] [Google Scholar]
- (210).Hausladen A; Rafikov R; Angelo M; Singel DJ; Nudler E; Stamler JS Assessment of Nitric Oxide Signals by Triiodide Chemiluminescence. Proc. Natl. Acad. Sci. U. S. A 2007, 104, 2157–2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (211).Cetó X; Saint CP; Chow CWK; Voelcker NH; Prieto-Simón B Electrochemical Detection of N-Nitrosodimethylamine Using a Molecular Imprinted Polymer. Sens. Actuators, B 2016, 237, 613–620. [Google Scholar]
- (212).Lin CH; Wang PH; Wang TH; Yang LJ; Wen TC The Surface-Enhanced Raman Scattering Detection of N-Nitro-sodimethylamine and N-Nitrosodiethylamine via Gold Nanorod Arrays with a Chemical Linkage of Zwitterionic Copolymer. Nanoscale 2020, 12, 1075–1082. [DOI] [PubMed] [Google Scholar]
- (213).Pilot R SERS Detection of Food Contaminants by Means of Portable Raman Instruments. J. Raman Spectrosc 2018, 49, 954–981. [Google Scholar]
- (214).Choi J; Kim JH; Oh JW; Nam JM Surface-Enhanced Raman Scattering-Based Detection of Hazardous Chemicals in Various Phases and Matrices with Plasmonic Nanostructures. Nanoscale 2019, 11, 20379–20391. [DOI] [PubMed] [Google Scholar]
- (215).Minami T; Esipenko NA; Zhang B; Kozelkova ME; Isaacs L; Nishiyabu R; Kubo Y; Anzenbacher P Supramolecular Sensor for Cancer-Associated Nitrosamines. J. Am. Chem. Soc 2012, 134, 20021–20024. [DOI] [PubMed] [Google Scholar]
- (216).United States Food and Drug Administration. Tobacco Product Standard for N-Nitrosonornicotine Level in Finished Smokeless Tobacco Products (Proposed Rule) https://www.fda.gov/about-fda/economic-impact-analyses-fda-regulations/tobacco-product-standard-n-nitrosonornicotine-level-finished-smokeless-tobacco-products-proposed (accessed 2020-09-18).
- (217).He M; Croy RG; Essigmann JM; Swager TM Chemiresistive Carbon Nanotube Sensors for N-Nitrosodialkylamines. ACS Sens. 2019, 4, 2819–2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (218).OSHA. Hazard Information Bulletins: N-Nitroso Compounds in Industry; 1990. [Google Scholar]