

Abstract
Lymphocyte development requires ordered assembly and subsequent modifications of the antigen receptor genes through V(D)J recombination and Immunoglobulin class switch recombination (CSR), respectively. While the programmed DNA cleavage events are initiated by lymphocyte-specific factors, the resulting DNA double-strand break (DSB) intermediates activate the ATM kinase-mediated DNA damage response (DDR) and rely on the ubiquitously expressed classical non-homologous end-joining (cNHEJ) pathway including the DNA-dependent protein kinase (DNA-PK), and, in the case of CSR, also the alternative end-joining (Alt-EJ) pathway, for repair. Correspondingly, patients and animal models with cNHEJ or DDR defects develop distinct types of immunodeficiency reflecting their specific DNA repair deficiency. The unique end-structure, sequence context, and cell cycle regulation of V(D)J recombination and CSR also provide a valuable platform to study the mechanisms of, and the interplay between, cNHEJ and DDR. Here, we compare and contrast the genetic consequences of DNA repair defects in V(D)J recombination and CSR with a focus on the newly discovered cNHEJ factors and the kinase-dependent structural roles of ATM and DNA-PK in animal models. Throughout, we try to highlight the pending questions and emerging differences that will extend our understanding of cNHEJ and DDR in the context of primary immunodeficiency and lymphoid malignancies.
Keywords: Non-homologous end-joining, ATM, DNA-PK, V(D)J recombination, Class Switch Recombination
1. Introduction
Mammalian cells have two major DNA double-strand break (DSB) repair pathways – the classical non-homologous end-joining (cNHEJ) pathway that directly ligates two DNA ends together, and the homologous recombination (HR) pathway that repairs a DSB using the sister chromatid or a homologous chromosome as the template. In recent years, the alternative end-joining (Alt-EJ) pathway has been used to describe end-ligation in cells lacking core cNHEJ factors (e.g., KU, LIG4, XRCC4). Alt-EJ preferentially uses microhomology (MH) at the junctions, is mediated by single-strand DNA ligases, LIG1 and LIG3 with the help of POLQ, and partially overlaps with the microhomology (MH)-mediated end-joining (MMEJ) pathway originally described in yeast1–4. Since the cNHEJ pathway can also generate joining products with short MH (n<=4nt), we chose to use the term Alt-EJ here to describe end-ligations without core cNHEJ components regardless the exact number of MH usage. In addition to the specific repair pathways that work on the DNA, DSBs also activate ATM kinase and a cascade of post-translational modifications, collectively referred to as the DNA damage response (DDR), which promotes precise and efficient DSB repair in part by modifying the chromatin environments. There are excellent reviews on the detailed molecular mechanism of cNHEJ5,6, Alt-EJ1–4, and DDR7,8, here, we focus on the roles of DDR and cNHEJ on lymphocyte-specific gene rearrangements.
The mammalian adaptive immune system is renowned for its specificity, diversity, and memory. This is achieved at the DNA level through three programmed gene rearrangement events– V(D)J recombination, Immunoglobulin class switch recombination (CSR), and somatic hypermutation (SMH). SHM introduces mutations in the variable regions of Immunoglobulin (Ig) genes in B lymphocytes via the base excision repair and mismatch repair pathways without evidence for DNA DSBs9. Here we focus on the two events involving DNA DSB intermediates - V(D)J recombination and CSR. V(D)J recombination occurs in progenitor B and T lymphocytes, where the variable region exons encoding the antigen-binding domains are assembled from individual Variable (V), Diversity (D) and Joining (J) gene segments through a cut and paste mechanism (Fig. 1). V(D)J recombination occurs in all T cell receptor (TCR, α/δ, β, γ) and Ig (h, κ, λ) gene loci10, including the Ig Heavy Chain (IgH), depicted as an example in Figure 1. CSR occurs in naïve B cells upon antigen exposure and T cell contact, and replaces the initially expressed IgM constant region (Cμ) with another constant region (e.g., Cγ1 for IgG1), to generate an antibody with a different isotype, thus conferring a different effector function (Fig. 1). There are excellent reviews on the initiation and regulation of V(D)J recombination11–13 and CSR9,14,15, here we focus on the DSB repair phase. The DSB intermediates generated during V(D)J recombination and CSR activate the ATM kinase-mediated DDR and use the ubiquitous cNHEJ pathway, and, in the case of CSR, also the Alt-EJ pathway for completion16. Both V(D)J recombination and CSR are initiated in the G1 phase of the cell cycle when homologous templates are not readily available, explaining the lack of direct contribution from the HR pathway. That is been said, several HR factors contribute to lymphocyte development by supporting efficient proliferation and clonal expansion17,18. V(D)J recombination is completed within the G1 phase of the cell cycle and involves hairpin end intermediates19. In contrast, CSR occurs in proliferating cells and involves DSBs at highly repetitive and GC-rich switch regions15. These and other features explain the unique immunodeficient phenotypes associated with different DNA repair defects and also provide a valuable tool to understand the mechanisms of cNHEJ and DDR in general. Here, we compare and contrast the physiological roles of different cNHEJ and DDR factors during V(D)J recombination and CSR with an emphasis on newly discovered cNHEJ factors, the kinase-dependent structural functions of DNA-dependent protein kinase (DNA-PK), and the interplay between ATM-mediated DDR and cNHEJ (Table 1). Whenever possible, we try to highlight the challenges and new opportunities in understanding DNA repair during lymphocyte development using animal models and their implications in human diseases, especially immunodeficiency and lymphoid malignancies (Table 1). The clinical immunodeficiency associated with defects in cNHEJ and DDR are reviewed elsewhere20,21.
Figure 1. The overview of V(D)J recombination and class switch recombination.
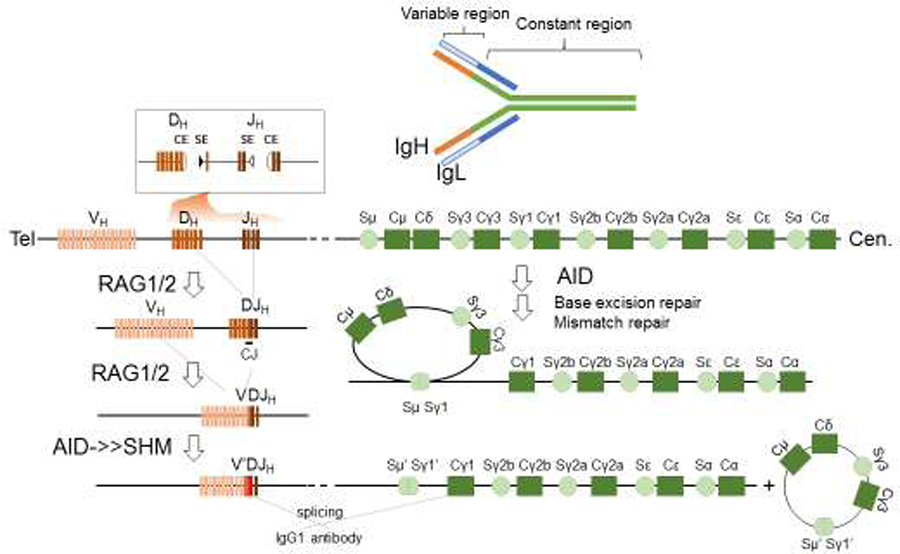
The Immunoglobulin (Ig) gene product, an antibody, is shown at the top. An antibody is formed by a pair of Ig Heavy chain gene products (IgH, in orange and green) and a pair of Ig light chain (IgL, in white and blue). The V(D)J recombination (on the lower left) assembles the variable gene exon that encodes the antigen-specific portion of the antibody (orange) from individual V, D or J gene segments in a two-step process, D to J first, then V to DJ. The reaction is initiated by RAG endonucleases, which introduces DNA double-strand breaks. An insert above shows the pair of RAG cleavage products at Dh and Jh with the hairpin coding ends and blunt signal ends adjacent to RSSs (triangles). Upon ligation by the cNHEJ pathway, the intermediate sequence is removed and the participating V, D, and J segments are fused to form the variable region exon that is spliced with downstream constant region exons (on the right) to form the IgH. While we only depict the IgH here, V(D)J recombination occurs in all 3 Ig (IgH, Igλ, and Igκ) and 4 T cell receptor (TCRσ/δ, TCRγ, TCRβ) gene loci. In naïve B cells, the IgH undergoes two additional modifications initiated by activating induced deaminase (AID). Somatic hypermutation (SHM) introduces point mutations in the variable region exon without creating DNA double-strand breaks. Class switch recombination (CSR) occurs in the constant region (on the right) and joins DNA double-strand breaks from two different switch (S) regions and effectively replaces the initially expressed IgM constant region (Cμ) with a downstream constant region, such as the Cγ1 for IgG1 depicted in the illustration. The constant region exons dictate the effector function of the antibody. CSR only occurs in the IgH locus of B cells, not in IgL or TCR loci. In contrast to RAG, which directly introduces DNA breaks, AID introduces U:G mismatches, which are processed by the base excision repair and mismatch repair pathways to generate mutations for somatic hypermutation (SHM) and DNA double-strand breaks for CSR.
Table 1.
Summary of V(D)J recombination and CSR phenotypes associated with major cNHEJ and DDR factors.
| Gene | Type of Mutation | IR sensitivity | Mice | V(D)J recombination | Class switch recombination | Ref. | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Chromosomal | Episomal/Plasmid | IgG1 % of Ctrl | Resection | MH at junctions | ||||||||
| SJ (blunt) | CJ (hairpin) | SJ (blunt) | CJ (hairpin) | |||||||||
| non-homologous end-joining | KU70 | Null | ++++ | sick | ↓↓↓ | ↓↓↓ | ↓↓↓ | ↓↓↓ | ~30% | N.D. | ↑ | 45,167,196–198 |
| KU80/86 | Null | ++++ | sick | ↓↓↓ | ↓↓↓ | ↓↓↓ | ↓↓↓ | ~30% | N.D. | ↑ | 43,44,135,167,199,200 | |
| XRCC4 | Null | ++++ | lethal | ↓↓↓ | ↓↓↓ | ↓↓↓ | ↓↓↓ | ~30% | ↑↑ | ↑↑ | 157,201,202 | |
| LIG4 | Null | ++++ | lethal | ↓↓↓ | ↓↓↓ | ↓↓↓ | ↓↓↓ | ~30% | ↑↑ | ↑↑ | 46,159,166,203–206 | |
| XLF | Null | +++ | live | − | − | ↓↓ | ↓↓ | ~50% | N.D. | ↑↑ | 81,83,84,207 | |
| DNA-PKcs | Null | +++ | live | −1 | ↓↓↓ | ↓ | ↓↓↓ | 90% | ↑↑ | ↑↑ | 51,52,103–105,208 | |
| ARTEMIS | Null | ++ | live | − | ↓↓↓ | − | ↓↓↓ | >90% | N.D. | ↑↑ | 53,90,209–213 | |
| DNA-PKcs | KD | ++++ | lethal | ↓↓↓ | ↓↓↓ | ↓↓ | ↓↓↓ | 30% | ↑↑ | ↑↑ | 55,106 | |
| DNA-PKcs | T2609A | ++ | sick | − | − | ↓ | − | >90% | ↑ | ↑↑ | 70,72,73,76,78–80,89,214 | |
| DNA-PKcs | S2056A | + | live | − | − | ↓ | ↓ | 100% | − | − | 76,77,214 | |
| PAXX | Null | + | live | − | − | − | − | 100% | − | − | 132–136,215–217 | |
| MRI/CYREN | Null | + | live | − | − | N.D. | N.D. | ~90% | N.D. | N.D. | 139,140,218 | |
| DNA damage response | ATM | Null | ++++ | live | − | ↓2 | − | − | 50% | ↑ | ↑↑ | 85,123,148,190,219–224 |
| ATM | KD | ++++ | lethal | − | ↓2 | N.D. | N.D. | 50% | N.D. | N.D. | 123,151,152 | |
| ATM | S1987A | −4 | live | − | − | N.D. | N.D. | 100% | N.D. | N.D. | 125,126 | |
| H2AX | Null | +++ | live | − | − | N.D. | N.D. | ~30% | N.D. | ↑↑ | 178,179,190,225–228 | |
| MDC1 | Null | +++ | live | − | − | N.D. | N.D. | ~50% | N.D. | N.D. | 94,143,145,229 | |
| 53BP1 | Null | ++ | live | − | − | N.D. | N.D. | 5% | ↑↑ | ↑↑ | 146,188–190,230,231 | |
| RIF1 | Null | +++ | lethal | − | − | N.D. | N.D. | 10% | ↑↑ | ↑↑. | 187,188,190,232,233 | |
| REV7 | Null | +++ | sick | − | − | N.D. | N.D. | 10% | ↑↑ | N.D. | 183,234–236 | |
| SHLD1/2/3 | Null | ++ | N.D. | − | − | N.D. | N.D. | 10% | ↑↑ | N.D. | 183,185,186 | |
| PTIP | Null | ++ | lethal | N.D. | N.D. | N.D. | N.D. | ~50% | N.D. | N.D. | 181,237–241 | |
| Combinations | XLF&ATM | Null | ++++ | live | ↓↓↓ | ↓↓↓ | ↓↓ | ↓↓ | 30% | N.D. | N.D. | 85 |
| XLF&H2AX | Null | ++++ | lethal | ↓↓↓ | ↓↓↓ | N.D. | N.D. | N.D. | N.D. | N.D. | 85 | |
| XLF&MDC1 | Null | ++++ | lethal | ↓↓ | ↓↓ | N.D. | N.D. | N.D. | N.D. | N.D. | 94 | |
| XLF&53BP1 | Null | ++++ | live | ↓↓↓ | ↓↓↓ | N.D. | N.D. | N.D. | N.D. | N.D. | 91,92 | |
| XLF&ART | Null | ++++ | live | − | ↓↓↓ | N.D. | ↓↓↓ | N.D. | N.D. | N.D. | 90 | |
| XLF&DNA-PKcs | Null | ++++ | sick | ↓↓ | ↓↓↓ | N.D. | ↓↓↓ | ~50% | N.D. | N.D. | 90 | |
| XLF&PAXX | Null | ++++ | lethal | ↓↓↓ | ↓↓↓ | N.D. | N.D. | N.D. | N.D. | N.D. | 134–136,242,243 | |
| XLF&MRI | Null | ++++ | lethal | ↓↓↓ | ↓↓↓ | N.D. | N.D. | N.D. | N.D. | N.D. | 139 | |
| DNA-PKcs&ATM | Null | ++++ | lethal | ↓↓↓ | ↓↓↓ | ↓↓ | ↓↓↓ | ~20% | N.D. | N.D.3 | 70,87,88,93,244,245 | |
| DNA-PKcs&ATM | T2609A/Null | N.D. | N.D. | ↓↓↓ | ↓↓↓ | ↓ | ↓ | N.D. | N.D. | N.D. | 65,76,80 | |
| DNA-PKcs&ATM | S2056A/Null | N.D. | Live* | − | − | ↓ | ↓ | 50% | N.D. | N.D. | 65,76,77 | |
N.D., not determined; −, no change;
reduced fidelity;
Hybrid join formation;
increased insertion;
IR sensitive in human cells;
unpublished data.
2. DNA DSB repair during V(D)J Recombination
Recombination activating gene (RAG) - RAG1 and RAG2 - initiate V(D)J recombination in progenitor lymphocytes by recognizing a pair of compatible recombination signal sequences (RSSs) and introducing DSBs between the RSSs and the participating V, D, or J gene segments11 (Fig. 1). RAG form a Y-shape hetero-tetramer to synapse the involving V, D or J segments together and coordinately cleavage the two RSSs to generates two types of ends – a pair of covalently sealed hairpinned coding ends (CEs) and a pair of blunt and 5’ phosphorylated signal ends (SEs) (Fig. 2A). The top of the Y formed by RAG2 tilts toward one side to accommodate the two RSSs with different sizes of spacers (12bp and 23bp, respectively), which explain the 12–23 rule for RAG mediated recombination (Fig. 2B)22,23. After cleavage, the CEs are released and RAG holds the two SEs in a hairpin-forming complex (Fig. 2B) and promotes the ligation between the two SEs to generate a signal join. The two CEs undergo hairpin-opening and end-processing via the cNHEJ pathway before they are ligated to form a coding join (CJ) that eventually becomes part of the variable region exons (Fig. 2C). RAG expression is restricted to G1 arrested progenitor lymphocytes24 and RAG dictates the orientation-specific repair outcome of V(D)J recombination, explaining the exclusive dependence of V(D)J recombination on cNHEJ. RAG cleavage only occurs in transcriptionally active and epigenetically poised V, D, and J gene segments10,25, leading to the cell type (B vs T) and developmental stage (pro- vs pre-) specificity of V(D)J recombination.
Figure 2. The features of V(D)J recombination and the cNHEJ pathway.
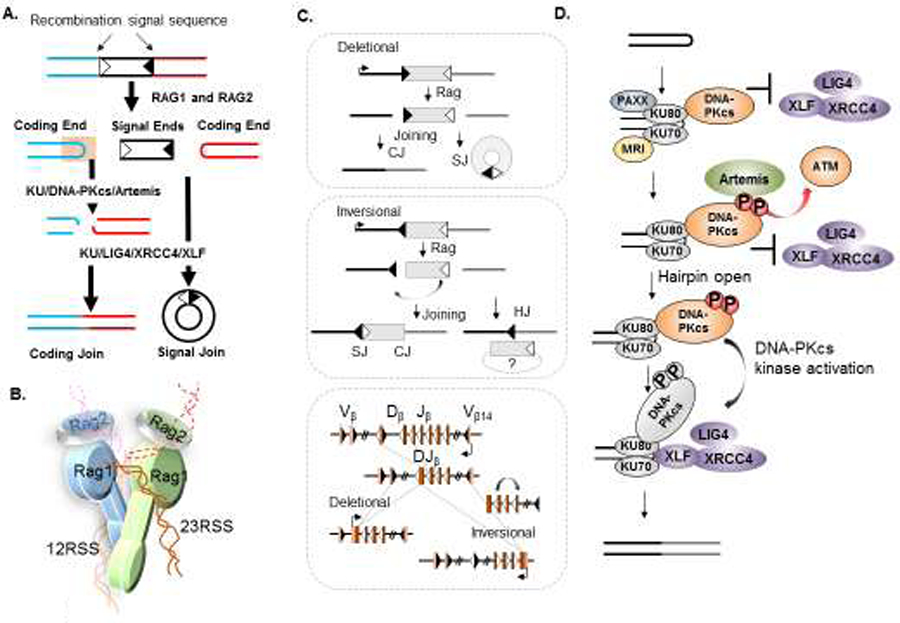
(A) RAG1/2 endonuclease cleavage occurs after synapsing two compatible RSSs (with different spacer length, 12 and 23bp, illustrated as white or black triangles) together and generates a pair of 5’ phosphorylated blunt signal ends (SE) and a pair of covalently sealed hairpinned coding ends (CE) (red and blue hairpins). The two SEs can be directly joined by KU together with the LIG4-XRCC4-XLF complex to form a signal join (SJ). The two CEs have to be opened by KU-DNA-PKcs and Artemis endonuclease before they can be ligated to form a coding join (CJ). (B) A cartoon of the RAG post-cleavage complex shows two RAG1 and two RAG2 proteins synapsing two compatible RSSs (with 12 bp, 12RSS or 23 bp, 23RSS spacers) together and also holding the resulting SEs (solid lines, open) and CEs (dash lines, hairpinned) in close proximity to facilitate ligation22,108,195. The precise position of the CEs is yet to be firmly determined. (C) V(D)J recombination can occur in a deletional (upper) or inversional (middle) configuration depending on the relative orientation of the participating RSSs (open or filled triangles). The vast majority of V(D)J recombination occurs in the deletional configuration (e.g., IgH in Figure 1) with the two RSSs facing each other, and the recombination removing the SJ-containing internal sequence as an excised circular DNA (top). In rare cases, such as the TCRβ locus diagrammed at the bottom, the V(D)J recombination occurs between a pair of RSSs in the same orientation, and, as such, the recombination leads to the inversion of the internal sequence (grey box) to form an SJ on the chromosome in addition to the CJ (bottom left of the middlebox). In the case of ATM deficiency151, the internal sequence is lost in ~50% of the cases, and, as such, the CE and SE on the genome join together to form a hybrid join (HJ) accompanied by the deletion of the internal sequence, indicated on the bottom right of the middlebox. A diagram of the murine TCRβ locus, including the Vβs with deletional rearrangements and the Vβ14 that undergoes inversional V(D)J recombination,12 is shown at the bottom. (D) A cartoon illustration of non-homologous end-joining at a CE. The KU heterodimer binds to the hairpin sealed CEs, and then recruits DNA-PKcs, which, upon phosphorylation by itself or by ATM kinase, serves as a platform to recruit and activate Artemis endonuclease. In the absence of kinase activation, DNA-PKcs at the ends precludes the DNA ligation complex from accessing the ends as well. The successful activation of DNA-PKcs triggers a conformational change that then allows end ligation by the LIG4-XRCC4-XLF complex.
2.1. The role of cNHEJ in V(D)J recombination
The cNHEJ pathway is exclusively required for V(D)J recombination26,27 and entails both an evolutionarily conserved end-ligation component and a largely vertebrate-specific end-processing component (Fig. 2D). Although putative orthologs of DNA-PKcs has been identified in other non-vertebrate species based on sequence homology, the biological function of DNA-PKcs has not been characterized. During V(D)J recombination, KU70 (gene name XRCC6) and KU86 (Ku80 in mouse, gene name XRCC5) form the KU heterodimer, which binds to hairpin CEs and blunt SEs and initiates cNHEJ. For end-ligation, KU recruits and stabilizes the ligation complex formed by XLF (gene name NHEJ1), XRCC4, and LIG4 to ligate the blunt SEs directly and precisely, forming a SJ28–30. For end-processing, KU, especially the C-terminus of KU8031, recruits and activates the catalytic subunit of DNA-PK (DNA-PKcs, gene name PRDKC), which activates Artemis endonuclease to open the CE hairpins32,33, thereby allowing the CEs to be ligated to form a CJ. Hairpin opening outside the apex introduces palindromic insertions (P elements)32–36 and terminal deoxynucleotidyl transferase (TDT) also adds non-templated insertions (N nucleotides)37,38 to further increase the diversity of the variable region exons. KU associated DNA polymerases (λ and μ)39,40 and WRN helicase41,42 further contribute to the V(D)J recombination. Correspondingly, mouse models deficient in end-ligation caused by loss of Ku70, Ku80, Lig4, or Xrcc4 - display severe neuronal apoptosis and accumulate both CEs and SEs43–47 (Table 1). In the case of Lig4, or Xrcc4, the neuronal apoptosis is accompanied by embryonic lethality that can be rescued by the co-deletion of KU48, which normally limits end-resection49 and Alt-EJ mediated repair. While Xlf-null mice do not develop overt neuronal apoptosis, patients with XLF/Cernunnos deficiency show microcephaly, consistent with a model in which end-ligation prevents neuron apoptosis30,50. Meanwhile, mice with complete loss of the largely vertebrate-specific end-processing factors DNA-PKcs or Artemis have no neuronal apoptosis, carry isolated immunodeficiency, and form SJs efficiently but with reduced fidelity51–53 (Table 1). Similarly, patients with complete loss of Artemis33 or DNA-PKcs mutation that only affects the hairpin opening and Artemis activation54 did not develop marked microcephaly. Notably, mouse models expressing a kinase-dead DNA-PKcs that blocks DNA end-ligation also have severe neuronal apoptosis and late embryonic lethality55, like Lig4−/− or Xrcc4−/− mice, highlight the importance of end-ligation in neurological development. In this regar, a patient with substantially decreased expression of catalytically inactive DNA-PKcs protein also suffers profound neurological abnormalities and show sign of end-ligation defects56. Together, the data supports an important role of cNHEJ mediated end-ligation in post-mitotic neurons in vivo. The cNHEJ factors discussed in this section contibutes to V(D)J recombination by directly working on the DNA itself (in contrast to chromatin) and have also been extensively characterized using plasmid-based substrates (Table 1).
2.2. The role of DNA-PK and its kinase activity in cNHEJ and V(D)J recombination
The KU heterodimer and DNA-PKcs together form the DNA-PK holoenzyme57 that is related to the ATM kinase58. V(D)J recombination has been a valuable system to study the function of DNA-PK. DNA-PKcs is the best-characterized substrate of DNA-PK. To understand its regulation in vivo, we generated a mouse model expressing kinase-dead (KD) DNA-PKcs (D3922A, DNA-PKcsKD)55,59. In contrast to the normal development of DNA-PKcs null mice51,52,60, DNA-PKcsKD/KD mice die in utero with severe neuronal apoptosis like in Lig4- or Xrcc4- deficient mice55, and accumulate both CEs and SEs, indicative of end-ligation defects55 (Table 1). Nevertheless, the deletion of KU or the Ku80 C-terminal domain that recruits DNA-PKcs to the DNA ends rescues the embryonic lethality and SJ formation defects in DNA-PKcsKD/KD mice55, indicating that, once loaded to DNA ends, DNA-PKcs requires its kinase activity to “license” end-ligation. Together with the observation that purified DNA-PKcs blocks DNA ligation by a T4 DNA ligase in the absence of hydrolyzable ATP61, these findings support a cap function of DNA-PKcs that is regulated by its own kinase activity. A recent study using ectopically expressed DNA-PKcs KD protein in human cells did not detect additional end-ligation defects
ATP hydrolysis is also essential for Artemis and DNA-PK mediated hairpin opening in vitro32. To identify the phosphorylation targets of DNA-PKcs, alanine substitutions were introduced to either Artemis or DNA-PKcs, the results suggest that DNA-PKcs phosphorylation at the T2609 cluster facilitates Artemis activation62. Denature gel electrophoresis shows that the hair pinned CEs are fully opened in DNA-PKcsKD/KD cells and that hairpin opening occurs efficiently in DNA-PKcsKD/KD cells and can be blocked by ATM kinase inhibitor55, suggesting that Artemis activation requires DNA-PKcs protein and the kinase activity from either DNA-PK or ATM. A patient carrying R3062L mutation in DNA-PKcs retains full kinase activity, yet loss the ability to activate Artemis20,54,56, also consistent with a structural rather kinase role of DNA-PKcs in Artemis activation. Given the strict requirement for DNA-PK’s kinase activity for end-ligation and the redundancy between ATM and DNA-PK activity for end-processing, we initially speculated that these differential requirements might reflect specific roles for DNA-PKcs autophosphorylation vs DNA-PKcs transphosphorylation by ATM.
Human DNA-PKcs has two well-characterized phosphorylation clusters63. Upon DSBs (e.g., radiation), the S2056 cluster is mainly inter-molecularly auto-phosphorylated by DNA-PK64–66 and the T2609 cluster is phosphorylated by ATM64,67–69 and DNA-PK65,70. The relative contribution of ATM vs DNA-PK to the T2609 cluster phosphorylation varies by relative abundance and activities of ATM vs DNA-PK. Upon UV irradiation, ATR kinase can also phosphorylate DNA-PKcs at the T2609 cluster71. Although not essential for cNHEJ in human cell extracts in vitro72, expression of human DNA-PKcs with an alanine substitution at the S2056 and/or T2609 cluster in DNA-PKcs-deficient CHO cells fails to restore IR resistance63,73–75 and Artemis mediated end-processing62,76. Moreover, episomal analyses of the DNA-PKcs with alanine substitutions at T2609 or S2056 clusters show reduced end-ligation that is comparable to the loss of DNA-PKcs65,73 and loss of both S2056 and T2609 cluster phosphorylation has a synergistic effect on end-ligation and IR sensitivity64,65,73. Yet, despite mild IR sensitivity, mouse models with knock-in alanine substitutions at either the S2056 cluster77 or the T2609 cluster78 support chromosomal V(D)J recombination (Table 1)77–80. Given DNA-PKcs is not essential for SJ formation during chromosomal V(D)J recombination or end-joining, the results support a model in which DNA-PKcs phosphorylation at the T2609 and S2056 cluster promotes end-ligation, but are not essential for either end-processing and end-ligation72.
That is being said, there is several notably difference among results acquired from different experimental systems. While additional experiments are necessary to resolve the difference, we offer some thoughts for consideration. First, chromosomal V(D)J recombination vs episomal reporters. In general, loss of DNA-PKcs and its phosphorylation causes more severe defects on plasmid-based assays than on chromosomal substrates or in vivo. One possibility is that the chromatin and chromatin bounded DNA damage response factors promote end-ligation and mask the subtle end-ligation defects. Plasmid-based DNA repair substrates are not able to access these beneficial chromatin effects and thus more sensitive to mild cNHEJ defects. Consistent with this hypothesis, Xlf-deficiency substantially reduced plasmids based V(D)J recombination30,81,82, while largely dispensable for chromosomal V(D)J recombination83–85. Similarly, loss of DNA-PKcs or the T2609 cluster phosphorylation of DNA-PKcs have a significant impact on the end-ligation of plasmid substrates65,70,86, but not chromosomal substrates87,88 or in vivo51,77,78,89. Strikingly, in both Xlf-deficient or DNA-PKcs-deficient cells, loss of ATM kinase activity or chromatin bounded ATM substrates – H2AX, MDC1, and 53BP1 abolish the residual end-ligation on chromosomal susbstrate85,87,88,90–94, supporting a critical role of chromatin and DNA damage response in promoting end-ligation. ATM kinase inhibition or deletion also abolishes SJ formation in murine cells carrying DNA-PKcs with alanine substitutions at the T2609A cluster80, but not those with alanine substitutions at the S2056 cluster77. Furthermore, RAG has extensive C-terminal chromatin interaction domains that are required for chromatin V(D)J recombination, but not for episomal assay26,95–99, suggesting RAG might also facilitate end-ligation through chromatin interaction. In addition to the availability of chromatin bounded DDR response and RAG interaction, the cell cycle could be another factor. While most episomal assays were performed in cycling cells when RAG and other nuclease were ectopically expressed throughout the cell cycle, chromosomal V(D)J recombination and V(D)J recombination in vivo occurs only in G1 arrested cells due to the transcription and post-translation regulation of RAG24,100. One notably difference is at the hairpin-opening. While Artemis is strictly required for the hairpin-opening during V(D)J recombination, several other endonucleases including MRE11 in complex with CtIP can open hairpin, including RAG generated hairpin in S or G2 phase of the cell cycle18,101,102. S and G2 phase cells also express other DNA repair factors involved in end-resection, and HR that are not available in G1 cells, plus the availability of sister chromatin nearby could introduce additional complexity and possibilities for DNA repair.
Second, IR sensitivity vs defects in V(D)J recombination. From a DNA repair point of view, RAG cleavage generates clean DNA ends that might be less vulnerable to end-ligation defects than complex DNA lesions including oxidized bases generated during IR89,103–106. Consistent with this hypothesis, Artemis is not required for SJ formation during chromosomal V(D)J recombination, but clear responsible for ~10% of complex ends generated by IR33,53,107. RAG also holds the SEs after cleavage and has extensive C-terminal chromatin interaction domains uniquely important for chromatin V(D)J recombination22,23,26,108–112, which can also facilitate end-ligation. Moreover, IR sensitivity in proliferating cells reflects a complex response to DNA damage beyond simple DNA repair. For example, loss of TP53 or its regulators and targets (e.g., ATM, ARF, CHK1, MDM2, P21, PUMA, and others) can all affect IR sensitivity in a cell-type and context-specific manner without directly affect DNA repair. This is particularly true for human cancer cell lines, which often carry complex genetic alterations. In this context, DNA-PKcs has been implicated in numbers of functions outside cNHEJ, including telomere biology113,114, Golgi stability115, DNA damage checkpoints116, and others117. In certain human cells including HCT116 and 293T cells, loss of DNA-PKcs also leads to adaptive loss of ATM protein expression, which further modulates IR sensitivity and DNA repair70,86. Notably, ATM activity, measured by the phosphorylation of KAP1, seems intact if not hyperactive in DNA-PKcs null, DNA-PKcs-T2609A, or DNA-PKcsKD/KD murine cells80,118 (Crowe and Zha unpublished observations). Biochemical analyses have shown that DNA-PKcs can phosphorylate ATM kinase to inhibit ATM kinase activity118. A uniformed hypothesis regarding how DNA-PKcs protein or activity loss might lead to an adaptive decrease of ATM protein levels remain pending, would hold the key to understand the cross-talk between ATM and DNA-PKcs. Moreover, we and others recently showed that mouse models with an alanine substitution at the T2609 cluster succumb to bone marrow failure79 with macrocytic anemia associated with ribosome biogenesis defects78. These and other cNHEJ independent functions of DNA-PKcs might be particularly important in understanding complex responses to radiation or other environmental insults beyond simple DSB repair.
Third, the role of DNA-PKcs in human vs in mice. It is noted early on, the abundancy of KU and DNA-PKcs, but not other cNHEJ factors, is nearly 50-fold higher in human cells than in mouse cells77, suggests that KU and DNA-PK might have cNHEJ-independent functions in human cells. The high concentration of KU and DNA-PKcs could potentiate relative low-affinity interaction with structured RNA, including the telomerase RNA template, which might contribute to the prominent telomere abnormalities119,120 in human cells lost KU or DNA-PKcs. Accordingly, while DNA-PKcs or KU null mice are viable, human cells, even cancer cells, cannot tolerate the complete and persistent loss of KU120 or DNA-PKcs without adaptive changes of other genes, including reducing ATM protein levels70,121. Several human patients derived cell lines contain spontaneous mutations of DNA-PKcs are viable, but whether the residual DNA-PKcs protein or activity or adaptive changes are essential for their long-term culture is yet to be determined54,56,122.
Finally, the difference between the kinase-dead and the phosphorylation site mutant proteins is not unique to DNA-PKcs. The expression of kinase-dead ATM also leads to different biological consequences than the alanine substitutions of its proposed auto-phosphorylation sites123–127. These genetic models demonstrate a structural role of DNA-PKcs that is modulated by its kinase activity, resulting in different phenotypes for loss of DNA-PKcs, expression of kinase-dead, and expression of an alanine substitution at the T2609 and/or S2056 clusters. The recent cryogenic electron microscopy structure of the DNA-bound and DNA-free forms of the DNA-PK holoenzyme uncovered an extended interface between KU and DNA-PK128–131. Several questions are pending, including the contribution of the proposed phosphorylation sites to the allosteric activation of DNA-PKcs, the exact mechanism by which DNA binding triggers KU-dependent activation of DNA-PKcs, the exact roles of the KU70 SAP domain and the KU80 C-terminal domain in cNHEJ and V(D)J recombination, and the interaction surface and the regulation between DNA-PKcs and Artemis. Collectively, this information would help us understand the dynamic movements that lead to DNA-PK activation and how DNA-PKcs caps the DNA ends and activates Artemis endonuclease.
2.3. The new cNHEJ factors, and their roles in cNHEJ and V(D)J recombination
In the past few years, several new cNHEJ factors have been described - PAXX and MRI - based on their interaction with KU and their essential role in V(D)J recombination in mice or cells lacking XLF, a non-essential cNHEJ factor that is associated with mild lymphocyte development defects in mouse models41,81,84. PAXX binds to the core region of the KU heterodimer132,133. Paxx−/− mice develop normally with normal levels of mature lymphocytes, but Xlf/Paxx double deficient cells accumulate both CEs and SEs, suggesting a critical role of Paxx in end-ligation that is masked by Xlf134–136. Several models have been proposed to explain the role of XLF in cNHEJ, including the formation of XLF-XRCC4 filament that brings the two DNA ends together5,137 and facilitating Lig4 re-adenylation138. We envision that the presence of XLF increases ligation frequency, thus indirectly compensates for the compromised cNHEJ in Paxx-deficient cells. Notably, Paxx shares some structural features with XLF132,133. Similarly, Mri also binds to KU and is essential for chromosomal V(D)J recombination in Xlf−/− cells139. While Mri promotes cNHEJ and V(D)J recombination in G1 arrested B cells, it is also thought to suppress cNHEJ mediated telomere fusion in S or G2 phase cells140. Exactly how MRI shifts between these two seemingly opposing functions remains unknown. In addition to KU, MRI also binds to ATM and the MRE11-RAD50-NBS1 (MRN) complex. MRI might mediate different functions by associating with different molecular complexes139. On this note, whether cell cycle or cyclin-associated kinases might play a role in regulating MRI association and function also needs to be tested. Notably, unlike LIG4, XRCC4, XLF, DNA-PKcs, and ARTEMIS, the mutations of which have been linked to human primary immunodeficiency20,30,33,50,54,56,141, PAXX, and MRI be not mutated in immunodeficient patients and their connection to human immunodeficiency remain unknown.
2.4. The role of the DNA damage response in V(D)J recombination
DSBs generated during V(D)J recombination activate the DDR. Briefly, the MRN complex recruits and activates ATM kinase, which phosphorylates many substrates, including the histones H2AX flanking the DNA, break at S139 to form γH2AX142. MDC1 directly binds to γH2AX143–145 and recruits the E3 ubiquitin ligases RNF8 and RNF168, which further modify H2A and eventually leads to the recruitment of 53BP17. ATM and the downstream factors H2AX and 53BP1 are not required for lymphocyte development, but in mouse models, their absence causes a mild reduction in surface TCRβ/CD3ε levels in CD4+CD8+ double-positive T cells146–148 and the accumulation of unrepaired RAG-dependent DSBs in ~4% of Atm-deficient B cells149. The need for sequential V(D)J recombination at the TCRα/δ150 likely contributes to its hypersensitivity to DNA repair defects (Table 1). Correspondingly, loss of ATM, H2AX, or 53BP1 does cause major development blockade associated with V(D)J recombination in the majority of the Ig and TCR loci(Fig. 1 and 2C). Using an inversional chromosomal V(D)J recombination substrate, Sleckman and colleagues showed that MRN and ATM prevent hybrid joins – the aberrant ligation of a CE with a SE during inversional V(D)J recombination151,152 - providing a mechanism for the mild lymphocyte development defects associated with ATM-deficiency149(Fig. 2C). Interestingly, ATM and ATM kinase activities are required for SJ formation in DNA-PKcs null cells87,88, supporting overlapping roles between ATM and DNA-PKcs kinase activity. Moreover, in cells and mice lacking XLF, a non-essential cNHEJ factor, ATM kinase activity, and its substrates H2AX and 53BP1 are required for SJ formation and end-ligation85,91,92 also, providing genetic evidence for an important role of the DDR in V(D)J recombination. In this context, whether other DDR factors are also required for V(D)J recombination in the sensitized XLF-deficient background and whether DDR factors have an additional role in V(D)J recombination beyond ligation (such as end-protection) remain to be tested. How the chromatin-based DDR network interacts with the epigenetic environment at the TCR and Ig loci required for V(D)J recombination remains unclear. It is possible that the epigenetically active TCR or Ig loci are poised to enhance repair and prevent mis-repair through higher order topologically associated domains (TADs)153 to promote effective V(D)J recombination.
3. DNA repair during Immunoglobulin Class Switch Recombination
Ig CSR generates antibodies with different isotypes and thus different effector functions. B cell-specific activation-induced cytidine deaminase (AID) initiates CSR by converting cytosine to uracil in the transcribed S regions preceding each set of constant region coding exons154. These mismatches are processed by both base excision repair and mismatch repair machinery and are eventually converted to DSBs in proliferating B cells. In the second phase, DSBs in the upstream IgM switch region (Sμ) are joined with DSBs in the downstream switch region (e.g., Sγ1 for IgG1) to complete IgH isotype switching (Fig. 1) and generate antibodies with different isotypes. The DSB repair that completes CSR is mediated by the cNHEJ and the Alt-EJ pathway with the help of ATM kinase and its chromatin substrates H2AX, 53BP1, etc.
3.1. The role of cNHEJ in CSR
To study the role of cNHEJ in CSR, B cells can be generated in cNHEJ-deficient backgrounds via the introduction of germ-line knock-in alleles harboring productive V(D)J rearrangements at both the IgH155 and Igκ156 loci. Unexpectedly, Ku70-, Xrcc4- or Lig4-deficient mature B cells generated using this approach157–159 and germline Xlf-deficient B cells82,84 all have significant residual CSR (25–50% of the WT levels) that is mediated by an Alt-EJ pathway biased towards MH (Table 1). Expression of kinase-dead DNA-PKcs causes severe CSR defects, phenocopies Xrcc4- or Lig4-deficiency106 (Table 1). In contrast, DNA-PKcs or Artemis null B cells have at most moderate defects in CSR efficiency that is detectable only by sensitive IgH FISH analysis160,161, consistent with the limited impacts of DNA-PKcs or Artemis deletion in end-ligation (Table 1). The recently developed high throughput genome-wide translocation sequencing (HTGTS)162 can isolate thousands of CSR junctions. CSR junctions recovered from Xrcc4−/− and DNA-PKcsKD/KD B cells are enriched for MHs, with nearly 50% of junctions containing 2–3 nt MHs, in contrast to 20–25% in WT cells106. Perhaps most surprising is that the CSR junctions from DNA-PKcs−/− B cells with nearly 90% of WT levels CSR are also equally enriched for MHs106, suggesting MHs facilitate CSR. But whether this MH-mediated CSR in DNA-PKcs−/− B cells depends on Alt-EJ or LIG4-mediated cNHEJ remain to be determined. Two features of CSR might contribute to the robust residual CSR in cNHEJ deficient cells that prefer the MH-mediated end-joining – the highly repetitive and GC rich switch region that is ideal for MH-mediated repair163 (Fig. 3A) and the S/G2 phase of the cell cycle that allows resection and promotes end-annealing. The loss of the newly discovered cNHEJ factor PAXX does not affect CSR efficiency136 and whether PAXX or even Artemis affects CSR junctions remains to be determined.
Figure 3. The features of class switch recombination and the DNA damage response.

(A) The switch region sequence is highly repetitive and GC rich. A collection of representative murine Sμ regions, (chr12 113424221–113424368, mm10) is shown. The genomic coordinates of the first bp of each sequence segment are listed in the first column and the GC% within the segment is noted in the last column. The overall GC% is 61%. (B) A cartoon of cNHEJ vs Alt-EJ during CSR. The cNHEJ mediated by KU heterodimers and the LIG4-XRCC4-XLF ligation complex directly ligate both strands of a double-strand break. Alternatively, annealing between complementary single-strand overhangs converts one double-strand break into two single-strand nicks, which can then be ligated by LIG3 or LIG1 through Alt-EJ. (C) A simplified cartoon illustrating the main DDR players that have been implicated in CSR. Upon DNA damage, ATM kinase phosphorylates histone H2AX to form γH2AX, which recruits MDC1. MDC1 subsequently recruits E3 ubiquitin ligases RNF8 and RNF168 to modify adjacent H2A. Histone Ub modifications, together with H4K20me2, recruit 53BP1 and leads to hyperphosphorylation of 53BP1. This phosphorylated 53BP1 recruits RIF1 and the SHIELDIN (SHLD1–3 and REV7) complex. Among the SHIELDIN complex, SHLD2 serves as a scaffold. The N-terminus of SHLD2 binds to SHLD3 and REV7, while the C-terminus of SHLD2 interacts with SHLD1 and single-strand DNA. These factors work together to prevent excessive end-resection and promote productive CSR.
3.2. The role of Alt-EJ in Class switch recombination
The robustness of Alt-EJ mediated CSR provides a unique tool to study the Alt-EJ pathway. In contrast to cNHEJ that ligates both strands of the DNA double helix simultaneously, the Alt-EJ pathway uses MH-mediated annealing to convert one DSB into two single-strand nicks (Fig. 3B). Mammalian cells have three DNA ligases, LIG1, LIG3, and LIG4, which repair the phosphodiester backbone in each DNA strand in an adenylation-dependent manner. Each strand ligation requires re-adenylation of the ligases138. LIG4 can potentially form higher-order oligomers through its interaction with the XRCC4-XLF filament137,164, which might explain the unique ability for LIG4 to mediate double-strand break repair. The conversion of a DSB to two single-strand nicks bypasses the requirement for LIG4 and allows the single-strand DNA ligases - LIG3 in complex with XRCC1 or LIG1 – to complete the Alt-EJ mediated CSR165. End-resection, which is required for MH-mediated repair to expose flanking MHs, is also evident in Xrcc4−/−, DNA-PKcs−/− and DNA-PKcsKD/KD B cells106. However, it remains unknown whether end-resection is required for Alt-EJ mediated CSR in the highly repetitive switch region. Nevertheless, the similar CSR efficiency and the MH and end-resection patterns in Xrcc4−/− and DNA-PKcsKD/KD cells suggest that the presence of kinase-dead DNA-PKcs does not block the Alt-EJ pathway106. Moreover, the deletion of Ku70 in Lig4−/− B cells reduces the bias toward MHs166,167, suggesting there might be more than one Alt-EJ pathway – the one requires long MH and the one that does not. Also, pol θ expression increases in activating B cells168 and might contributes to Alt-EJ through templated nucleotide insertions. Although Polq−/− B cells have normal CSR efficiency169, the percentage of recovered junctions with insertions is significantly lowered170. Interestingly, Polq−/− B cells also show increased IgH-Myc chromosomal translocations170, suggesting a role of pol θ in suppressing MH mediated translocations. The genetic requirement for Alt-EJ mediated CSR is still being investigated. Loss of Alt-EJ factors, including XRCC1, an obligatory partner of Lig3 in the nucleus171, PARP1, or POLQ, all did not compromise CSR efficiency alone165,169,172,173. Loss of DNA-PKcs103–105,161,174 or DNA-PKcs phosphorylation at the T2609 cluster89 has a mild effect on IgG1 CSR efficiency in vitro. But the CSR junctions recovered from the DNA-PKcs−/− or DNA-PKcs5A/5A mice are highly enriched for MH89,106. The results suggest that MH-mediated end-joining could be quite robust during CSR. Given the much more severe CSR defects in Lig4−/− B cells, the end-ligation that generates these small MH are likely including contributions from both cNHEJ pathway and the Alt-EJ pathway. The exact contribution of Alt-EJ to physiological CSR in cNHEJ-proficient cells also remains to be determined.
3.3. The role of the DNA damage response in CSR.
In contrast to V(D)J recombination that is only mildly affected by DDR-deficiency or in sensitized backgrounds, CSR highly depends on the DDR. CSR is significantly (>50%) impaired in cells deficient in ATM175–177 or its downstream targets H2AX178,179, MDC1143,180, RNF8, RNF168, or 53BP175, or even PTIP181,182, REV7(gene name MD2L2)183,184, SHIELDIN complex183,185,186, or RIF1187,188. which prevents end-resection downstream of 53BP1(Fig. 3C). ATM kinase inhibitors or expression of kinase-dead ATM have similar impacts on CSR as Atm loss123, suggesting that ATM contributes to CSR mainly through its kinase activity. Notably, although 53BP1 has a similar, if not a weaker, role in general DSB repair and irradiation resistance than ATM or H2AX, loss of 53BP1 leads to a 95% reduction of CSR that is much more prominent than all other repair factors179,180,189. It has been proposed that overreaction in the 53BP1-deficient cells exaggerates the CSR defects189. Moreover, 53BP1 is also thought to have a role in CSR upstream of DSB repair that is mediated by its downstream effector protein PTIP, which regulates the histone acetylation complex MLL2/3181,182. More recently, it has also been proposed that 53BP1 ensures that the orientation of CSR junctions to favor productive CSR beyond end-ligation181. Recent HTGTS sequence analyses of CSR junctions recovered from ATM-deficient or H2AX-deficient cells also show increased MHs190, suggesting the repetitive sequence in the switch region might poise CSR for MH-mediated joining independent of specific repair pathways. In the future, systematic analyses of CSR junctions with HTGTS might uncover additional regulators of Alt-EJ at the chromosomal levels.
4. Closing remarks
Efficient DNA repair during V(D)J recombination and CSR is not only important for lymphocyte development but also plays a critical role in suppressing oncogenic translocation. Indeed, the majority of recurrent chromosomal translocations in human lymphoid malignancies involve the Ig or TCR loci and arise from aberrant DSB repairs during V(D)J recombination or CSR191. While defects in the cNHEJ pathway often cause severe combined immunodeficiency (SCID)192, defects in the DDR, especially in ATM kinase which also has a checkpoint function, often lead to early-onset lymphoma and leukemia, providing a model to study the mechanism of chromosomal translocations147,193,194.
In summary, V(D)J recombination highly depends on core cNHEJ factors, while the DDR has more prominent roles in CSR. Two factors might contribute to this difference; the postcleavage complex formed by RAG and the strict G1 phase repair during V(D)J recombination. Moreover, the repetitive switch region sequences and the proliferative nature of B cells undergoing CSR render Alt-EJ very robust during CSR and provide unique opportunities to study the genetic requirement for Alt-EJ at the physiological level. On the other hand, the hairpin structure and exclusive G1 phase repair of V(D)J recombination provide an opportunity to study end-processing and the regulation of DNA-PKcs during end-joining. With the development of high-throughput sequencing methods and chromosomal V(D)J recombination reporters, significant strides have been made in understanding the regulation and function of cNHEJ and the DDR and their interaction in vivo. With the development of CRISPR genetic screens, we expect that additional DDR factors can be identified based on their role in V(D)J recombination and CSR, and further expand our knowledge of DNA double-strand break repair networks.
Acknowledgment
We thank all members of the Zha lab for helpful discussions. We apologize to colleagues whose valuable original publications were not cited here due to space limitations. This work was support by the National Institutes of Health grants R01CA158073, R01CA215067, R01CA184187, and R01CA226852 to S.Z. X.W is supported by NIH P01 CA174653 to S.Z..
Abbreviations
- Alt-EJ
Alternative end-joining
- CE
coding end
- CJ
coding join
- cNHEJ
classical non-homologous end-joining
- CSR
Class Switch Recombination
- DDR
DNA damage response
- DSB
DNA double-strand break
- HR
homologous recombination
- HTGTS
High throughput genome-wide translocation sequencing
- IgH
Immunoglobulin Heavy Chain
- MH
microhomology
- MMEJ
microhomology-mediated end-joining
- RSS
recombination signal sequences
- SE
signal end
- SJ
signal join
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference
- 1.Symington LS & Gautier J Double-strand break end resection and repair pathway choice. Annu Rev Genet 45, 247–71 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Sfeir A & Symington LS Microhomology-Mediated End Joining: A Back-up Survival Mechanism or Dedicated Pathway? Trends Biochem Sci 40, 701–714 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McVey M & Lee SE MMEJ repair of double-strand breaks (director’s cut): deleted sequences and alternative endings. Trends Genet 24, 529–538 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wood RD & Doublie S DNA polymerase theta (POLQ), double-strand break repair, and cancer. DNA Repair (Amst) 44, 22–32 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reid DA et al. Organization and dynamics of the nonhomologous end-joining machinery during DNA double-strand break repair. Proc Natl Acad Sci U S A 112, E2575–84 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ochi T, Wu Q & Blundell TL The spatial organization of non-homologous end joining: from bridging to end joining. DNA Repair (Amst) 17, 98–109 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ciccia A & Elledge SJ The DNA damage response: making it safe to play with knives. Mol Cell 40, 179–204 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Setiaputra D & Durocher D Shieldin - the protector of DNA ends. EMBO Rep 20(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Methot SP & Di Noia JM Molecular Mechanisms of Somatic Hypermutation and Class Switch Recombination. Adv Immunol 133, 37–87 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Jung D, Giallourakis C, Mostoslavsky R & Alt FW Mechanism and Control of V(D)J Recombination at the Immunoglobulin Heavy Chain Locus. Annu.Rev.Immunol (2006). [DOI] [PubMed] [Google Scholar]
- 11.Teng G & Schatz DG Regulation and Evolution of the RAG Recombinase. Adv Immunol 128, 1–39 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Jung D & Alt FW Unraveling V(D)J recombination; insights into gene regulation. Cell 116, 299–311 (2004). [DOI] [PubMed] [Google Scholar]
- 13.Dudley DD, Chaudhuri J, Bassing CH & Alt FW Mechanism and control of V(D)J recombination versus class switch recombination: similarities and differences. Adv.Immunol 86, 43–112 (2005). [DOI] [PubMed] [Google Scholar]
- 14.Casellas R et al. Mutations, kataegis and translocations in B cells: understanding AID promiscuous activity. Nat Rev Immunol 16, 164–76 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu K & Lieber MR Current insights into the mechanism of mammalian immunoglobulin class switch recombination. Crit Rev Biochem Mol Biol 54, 333–351 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alt FW, Zhang Y, Meng FL, Guo C & Schwer B Mechanisms of programmed DNA lesions and genomic instability in the immune system. Cell 152, 417–29 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mak TW et al. Brca1 required for T cell lineage development but not TCR loci rearrangement. Nat Immunol 1, 77–82 (2000). [DOI] [PubMed] [Google Scholar]
- 18.Liu X et al. CtIP is essential for early B cell proliferation and development in mice. J Exp Med 216, 1648–1663 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schatz DG & Swanson PC V(D)J recombination: mechanisms of initiation. Annu Rev Genet 45, 167–202 (2011). [DOI] [PubMed] [Google Scholar]
- 20.Woodbine L, Gennery AR & Jeggo PA The clinical impact of deficiency in DNA non-homologous end-joining. DNA Repair (Amst) 16, 84–96 (2014). [DOI] [PubMed] [Google Scholar]
- 21.de Villartay JP Congenital defects in V(D)J recombination. Br Med Bull 114, 157–67 (2015). [DOI] [PubMed] [Google Scholar]
- 22.Kim MS, Lapkouski M, Yang W & Gellert M Crystal structure of the V(D)J recombinase RAG1-RAG2. Nature 518, 507–11 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ru H et al. DNA melting initiates the RAG catalytic pathway. Nat Struct Mol Biol 25, 732–742 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiang H et al. Ubiquitylation of RAG-2 by Skp2-SCF links destruction of the V(D)J recombinase to the cell cycle. Mol Cell 18, 699–709 (2005). [DOI] [PubMed] [Google Scholar]
- 25.Liu Y, Zhang L & Desiderio S Temporal and spatial regulation of V(D)J recombination: interactions of extrinsic factors with the RAG complex. Adv Exp Med Biol 650, 157–65 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deriano L et al. The RAG2 C terminus suppresses genomic instability and lymphomagenesis. Nature 471, 119–23 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taccioli GE et al. Impairment of V(D)J recombination in double-strand break repair mutants. Science 260, 207–210 (1993). [DOI] [PubMed] [Google Scholar]
- 28.Lieber MR The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem 79, 181–211 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ahnesorg P, Smith P & Jackson SP XLF interacts with the XRCC4-DNA ligase IV complex to promote DNA nonhomologous end-joining. Cell 124, 301–313 (2006). [DOI] [PubMed] [Google Scholar]
- 30.Buck D et al. Cernunnos, a novel nonhomologous end-joining factor, is mutated in human immunodeficiency with microcephaly. Cell 124, 287–299 (2006). [DOI] [PubMed] [Google Scholar]
- 31.Singleton BK, Torres-Arzayus MI, Rottinghaus ST, Taccioli GE & Jeggo PA The C terminus of Ku80 activates the DNA-dependent protein kinase catalytic subunit. Mol.Cell Biol 19, 3267–3277 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ma Y, Pannicke U, Schwarz K & Lieber MR Hairpin opening and overhang processing by an Artemis/DNA-dependent protein kinase complex in nonhomologous end joining and V(D)J recombination. Cell 108, 781–794 (2002). [DOI] [PubMed] [Google Scholar]
- 33.Moshous D et al. Artemis, a novel DNA double-strand break repair/V(D)J recombination protein, is mutated in human severe combined immune deficiency. Cell 105, 177–186 (2001). [DOI] [PubMed] [Google Scholar]
- 34.Wood C & Tonegawa S Diversity and joining segments of mouse immunoglobulin heavy chain genes are closely linked and in the same orientation: implications for the joining mechanism. Proc Natl Acad Sci U S A 80, 3030–4 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tonegawa S Somatic generation of antibody diversity. Nature 302, 575–81 (1983). [DOI] [PubMed] [Google Scholar]
- 36.Lieber MR et al. The defect in murine severe combined immune deficiency: joining of signal sequences but not coding segments in V(D)J recombination. Cell 55, 7–16 (1988). [DOI] [PubMed] [Google Scholar]
- 37.Gilfillan S, Dierich A, Lemeur M, Benoist C & Mathis D Mice lacking TdT: mature animals with an immature lymphocyte repertoire. Science 261, 1175–1178 (1993). [DOI] [PubMed] [Google Scholar]
- 38.Komori T, Okada A, Stewart V & Alt FW Lack of N regions in antigen receptor variable region genes of TdT-deficient lymphocytes. Science 261, 1171–1175 (1993). [DOI] [PubMed] [Google Scholar]
- 39.Bertocci B, De SA, Weill JC & Reynaud CA Nonoverlapping functions of DNA polymerases mu, lambda, and terminal deoxynucleotidyltransferase during immunoglobulin V(D)J recombination in vivo. Immunity 25, 31–41 (2006). [DOI] [PubMed] [Google Scholar]
- 40.Bogue MA, Wang C, Zhu C & Roth DB V(D)J recombination in Ku86-deficient mice: distinct effects on coding, signal, and hybrid joint formation. Immunity 7, 37–47 (1997). [DOI] [PubMed] [Google Scholar]
- 41.Grundy GJ et al. The Ku-binding motif is a conserved module for recruitment and stimulation of non-homologous end-joining proteins. Nat Commun 7, 11242 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen L et al. WRN, the protein deficient in Werner syndrome, plays a critical structural role in optimizing DNA repair. Aging Cell 2, 191–9 (2003). [DOI] [PubMed] [Google Scholar]
- 43.Nussenzweig A et al. Requirement for Ku80 in growth and immunoglobulin V(D)J recombination. Nature 382, 551–555 (1996). [DOI] [PubMed] [Google Scholar]
- 44.Zhu C, Bogue MA, Lim DS, Hasty P & Roth DB Ku86-deficient mice exhibit severe combined immunodeficiency and defective processing of V(D)J recombination intermediates. Cell 86, 379–389 (1996). [DOI] [PubMed] [Google Scholar]
- 45.Gu Y, Jin S, Gao Y, Weaver DT & Alt FW Ku70-deficient embryonic stem cells have increased ionizing radiosensitivity, defective DNA end-binding activity, and inability to support V(D)J recombination. Proc.Natl.Acad.Sci.U.S.A 94, 8076–8081 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Frank KM et al. DNA ligase IV deficiency in mice leads to defective neurogenesis and embryonic lethality via the p53 pathway. Mol.Cell 5, 993–1002 (2000). [DOI] [PubMed] [Google Scholar]
- 47.Gao Y et al. Interplay of p53 and DNA-repair protein XRCC4 in tumorigenesis, genomic stability and development. Nature 404, 897–900 (2000). [DOI] [PubMed] [Google Scholar]
- 48.Karanjawala ZE et al. The embryonic lethality in DNA ligase IV-deficient mice is rescued by deletion of Ku: implications for unifying the heterogeneous phenotypes of NHEJ mutants. DNA Repair (Amst) 1, 1017–26 (2002). [DOI] [PubMed] [Google Scholar]
- 49.Mimitou EP & Symington LS Ku prevents Exo1 and Sgs1-dependent resection of DNA ends in the absence of a functional MRX complex or Sae2. EMBO J 29, 3358–69 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Callebaut I et al. Cernunnos interacts with the XRCC4 x DNA-ligase IV complex and is homologous to the yeast nonhomologous end-joining factor Nej1. J.Biol.Chem 281, 13857–13860 (2006). [DOI] [PubMed] [Google Scholar]
- 51.Gao Y et al. A targeted DNA-PKcs-null mutation reveals DNA-PK-independent functions for KU in V(D)J recombination. Immunity 9, 367–376 (1998). [DOI] [PubMed] [Google Scholar]
- 52.Taccioli GE et al. Targeted disruption of the catalytic subunit of the DNA-PK gene in mice confers severe combined immunodeficiency and radiosensitivity. Immunity 9, 355–366 (1998). [DOI] [PubMed] [Google Scholar]
- 53.Rooney S et al. Leaky Scid phenotype associated with defective V(D)J coding end processing in Artemis-deficient mice. Mol.Cell 10, 1379–1390 (2002). [DOI] [PubMed] [Google Scholar]
- 54.van der Burg M et al. A DNA-PKcs mutation in a radiosensitive T-B- SCID patient inhibits Artemis activation and nonhomologous end-joining. J Clin Invest 119, 91–8 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jiang W et al. Differential phosphorylation of DNA-PKcs regulates the interplay between end-processing and end-ligation during nonhomologous end-joining. Mol Cell 58, 172–85 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Woodbine L et al. PRKDC mutations in a SCID patient with profound neurological abnormalities. J Clin Invest 123, 2969–80 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gottlieb TM & Jackson SP The DNA-dependent protein kinase: requirement for DNA ends and association with Ku antigen. Cell 72, 131–42 (1993). [DOI] [PubMed] [Google Scholar]
- 58.Hartley KO et al. DNA-dependent protein kinase catalytic subunit: a relative of phosphatidylinositol 3-kinase and the ataxia telangiectasia gene product. Cell 82, 849–56 (1995). [DOI] [PubMed] [Google Scholar]
- 59.Menolfi D & Zha S ATM, ATR and DNA-PKcs kinases-the lessons from the mouse models: inhibition not equal deletion. Cell Biosci 10, 8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bosma M, Schuler W & Bosma G The scid mouse mutant. Curr.Top.Microbiol.Immunol 137, 197–202 (1988). [DOI] [PubMed] [Google Scholar]
- 61.Block WD et al. Autophosphorylation-dependent remodeling of the DNA-dependent protein kinase catalytic subunit regulates ligation of DNA ends. Nucleic Acids Res 32, 4351–7 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Goodarzi AA et al. DNA-PK autophosphorylation facilitates Artemis endonuclease activity. EMBO J 25, 3880–9 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Meek K, Dang V & Lees-Miller SP DNA-PK: the means to justify the ends? Adv Immunol 99, 33–58 (2008). [DOI] [PubMed] [Google Scholar]
- 64.Nagasawa H et al. Differential role of DNA-PKcs phosphorylations and kinase activity in radiosensitivity and chromosomal instability. Radiat Res 175, 83–9 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Meek K, Douglas P, Cui X, Ding Q & Lees-Miller SP trans Autophosphorylation at DNA-dependent protein kinase’s two major autophosphorylation site clusters facilitates end processing but not end joining. Mol Cell Biol 27, 3881–90 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen BP et al. Cell cycle dependence of DNA-dependent protein kinase phosphorylation in response to DNA double strand breaks. J Biol Chem 280, 14709–15 (2005). [DOI] [PubMed] [Google Scholar]
- 67.Shrivastav M et al. DNA-PKcs and ATM co-regulate DNA double-strand break repair. DNA Repair (Amst) 8, 920–9 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yajima H, Lee KJ, Zhang S, Kobayashi J & Chen BP DNA double-strand break formation upon UV-induced replication stress activates ATM and DNA-PKcs kinases. J Mol Biol 385, 800–10 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen BP et al. Ataxia telangiectasia mutated (ATM) is essential for DNA-PKcs phosphorylations at the Thr-2609 cluster upon DNA double strand break. J Biol Chem 282, 6582–7 (2007). [DOI] [PubMed] [Google Scholar]
- 70.Neal JA, Xu Y, Abe M, Hendrickson E & Meek K Restoration of ATM Expression in DNA-PKcs-Deficient Cells Inhibits Signal End Joining. J Immunol 196, 3032–42 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yajima H, Lee KJ & Chen BP ATR-dependent phosphorylation of DNA-dependent protein kinase catalytic subunit in response to UV-induced replication stress. Mol Cell Biol 26, 7520–8 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Povirk LF, Zhou RZ, Ramsden DA, Lees-Miller SP & Valerie K Phosphorylation in the serine/threonine 2609–2647 cluster promotes but is not essential for DNA-dependent protein kinase-mediated nonhomologous end joining in human whole-cell extracts. Nucleic Acids Res 35, 3869–78 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ding Q et al. Autophosphorylation of the catalytic subunit of the DNA-dependent protein kinase is required for efficient end processing during DNA double-strand break repair. Mol Cell Biol 23, 5836–48 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Meek K, Gupta S, Ramsden DA & Lees-Miller SP The DNA-dependent protein kinase: the director at the end. Immunol Rev 200, 132–41 (2004). [DOI] [PubMed] [Google Scholar]
- 75.Chan DW et al. Autophosphorylation of the DNA-dependent protein kinase catalytic subunit is required for rejoining of DNA double-strand breaks. Genes Dev 16, 2333–8 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cui X et al. Autophosphorylation of DNA-dependent protein kinase regulates DNA end processing and may also alter double-strand break repair pathway choice. Mol Cell Biol 25, 10842–52 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jiang W et al. Phosphorylation at S2053 in Murine (S2056 in Human) DNA-PKcs Is Dispensable for Lymphocyte Development and Class Switch Recombination. J Immunol 203, 178–187 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shao Z et al. DNA-PKcs has KU-dependent function in rRNA processing and haematopoiesis. Nature 579, 291–296 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhang S et al. Congenital bone marrow failure in DNA-PKcs mutant mice associated with deficiencies in DNA repair. J Cell Biol 193, 295–305 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lee BS et al. Functional Intersection of ATM and DNA-PKcs in Coding End Joining During V(D)J Recombination. Mol Cell Biol (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Roch B, Abramowski V, Chaumeil J & de Villartay JP Cernunnos/Xlf Deficiency Results in Suboptimal V(D)J Recombination and Impaired Lymphoid Development in Mice. Front Immunol 10, 443 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zha S, Alt FW, Cheng HL, Brush JW & Li G Defective DNA repair and increased genomic instability in Cernunnos-XLF-deficient murine ES cells. Proc Natl Acad Sci U S A 104, 4518–23 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lescale C et al. RAG2 and XLF/Cernunnos interplay reveals a novel role for the RAG complex in DNA repair. Nat Commun 7, 10529 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Li G et al. Lymphocyte-Specific Compensation for XLF/Cernunnos End-Joining Functions in V(D)J Recombination. Mol.Cell 31, 631–640 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zha S et al. ATM damage response and XLF repair factor are functionally redundant in joining DNA breaks. Nature 469, 250–4 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Neal JA & Meek K Deciphering phenotypic variance in different models of DNA-PKcs deficiency. DNA Repair (Amst) 73, 7–16 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zha S et al. Ataxia telangiectasia-mutated protein and DNA-dependent protein kinase have complementary V(D)J recombination functions. Proc Natl Acad Sci U S A 108, 2028–33 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gapud EJ et al. Ataxia telangiectasia mutated (Atm) and DNA-PKcs kinases have overlapping activities during chromosomal signal joint formation. Proc Natl Acad Sci U S A 108, 2022–7 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Crowe JL et al. DNA-PKcs phosphorylation at the T2609 cluster alters the repair pathway choice during immunoglobulin class switch recombination. BioRxiv (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Oksenych V et al. Functional redundancy between the XLF and DNA-PKcs DNA repair factors in V(D)J recombination and nonhomologous DNA end joining. Proc Natl Acad Sci U S A 110, 2234–9 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Oksenych V et al. Functional redundancy between repair factor XLF and damage response mediator 53BP1 in V(D)J recombination and DNA repair. Proc Natl Acad Sci U S A 109, 2455–60 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Liu X et al. Overlapping functions between XLF repair protein and 53BP1 DNA damage response factor in end joining and lymphocyte development. Proc Natl Acad Sci U S A 109, 3903–8 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Callen E et al. Essential role for DNA-PKcs in DNA double-strand break repair and apoptosis in ATM-deficient lymphocytes. Mol Cell 34, 285–97 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Beck C, Castaneda-Zegarra S, Huse C, Xing M & Oksenych V Mediator of DNA Damage Checkpoint Protein 1 Facilitates V(D)J Recombination in Cells Lacking DNA Repair Factor XLF. Biomolecules 10(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Liu Y, Subrahmanyam R, Chakraborty T, Sen R & Desiderio S A plant homeodomain in RAG-2 that binds Hypermethylated lysine 4 of histone H3 is necessary for efficient antigen-receptor-gene rearrangement. Immunity 27, 561–571 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.West KL et al. A direct interaction between the RAG2 C terminus and the core histones is required for efficient V(D)J recombination. Immunity 23, 203–212 (2005). [DOI] [PubMed] [Google Scholar]
- 97.Elkin SK et al. A PHD finger motif in the C terminus of RAG2 modulates recombination activity. J.Biol.Chem 280, 28701–28710 (2005). [DOI] [PubMed] [Google Scholar]
- 98.Akamatsu Y et al. Deletion of the RAG2 C terminus leads to impaired lymphoid development in mice. Proc.Natl.Acad.Sci.U.S.A 100, 1209–1214 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sadofsky MJ, Hesse JE & Gellert M Definition of a core region of RAG-2 that is functional in V(D)J recombination. Nucleic Acids Res 22, 1805–1809 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jiang H, Ross AE & Desiderio S Cell cycle-dependent accumulation in vivo of transposition-competent complexes between recombination signal ends and full-length RAG proteins. J Biol Chem 279, 8478–86 (2004). [DOI] [PubMed] [Google Scholar]
- 101.Makharashvili N et al. Catalytic and noncatalytic roles of the CtIP endonuclease in double-strand break end resection. Mol Cell 54, 1022–33 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wang H et al. CtIP maintains stability at common fragile sites and inverted repeats by end resection-independent endonuclease activity. Mol Cell 54, 1012–21 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bosma GC et al. DNA-dependent protein kinase activity is not required for immunoglobulin class switching. J.Exp.Med 196, 1483–1495 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Manis JP, Dudley D, Kaylor L & Alt FW IgH class switch recombination to IgG1 in DNA-PKcs-deficient B cells. Immunity 16, 607–617 (2002). [DOI] [PubMed] [Google Scholar]
- 105.Kiefer K et al. The catalytic subunit of DNA-protein kinase (DNA-PKcs) is not required for Ig class-switch recombination. Proc.Natl.Acad.Sci.U.S.A. %20;104, 2843–2848 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Crowe JL et al. Kinase-dependent structural role of DNA-PKcs during immunoglobulin class switch recombination. Proc Natl Acad Sci U S A (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Riballo E et al. A pathway of double-strand break rejoining dependent upon ATM, Artemis, and proteins locating to gamma-H2AX foci. Mol.Cell 16, 715–724 (2004). [DOI] [PubMed] [Google Scholar]
- 108.Ru H, Zhang P & Wu H Structural gymnastics of RAG-mediated DNA cleavage in V(D)J recombination. Curr Opin Struct Biol 53, 178–186 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Giblin W et al. Critical in vivo roles for the postcleavage activities of the RAG proteins (2008). [Google Scholar]
- 110.Ramon-Maiques S et al. The plant homeodomain finger of RAG2 recognizes histone H3 methylated at both lysine-4 and arginine-2. Proc.Natl.Acad.Sci.U.S.A 104, 18993–18998 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Matthews AG et al. RAG2 PHD finger couples histone H3 lysine 4 trimethylation with V(D)J recombination. Nature 450, 1106–1110 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Corneo B et al. Rag mutations reveal robust alternative end joining. Nature 449, 483–486 (2007). [DOI] [PubMed] [Google Scholar]
- 113.Williams ES et al. Telomere dysfunction and DNA-PKcs deficiency: characterization and consequence. Cancer Res 69, 2100–7 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Beattie TL & Lees-Miller SP Unraveling the roles of WRN and DNA-PKcs at telomeres. Aging (Albany NY) 2, 257–8 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Farber-Katz SE et al. DNA damage triggers Golgi dispersal via DNA-PK and GOLPH3. Cell 156, 413–27 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Woo RA, McLure KG, Lees-Miller SP, Rancourt DE & Lee PW DNA-dependent protein kinase acts upstream of p53 in response to DNA damage. Nature 394, 700–4 (1998). [DOI] [PubMed] [Google Scholar]
- 117.Jette N & Lees-Miller SP The DNA-dependent protein kinase: A multifunctional protein kinase with roles in DNA double strand break repair and mitosis. Prog Biophys Mol Biol 117, 194–205 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhou Y et al. Regulation of the DNA Damage Response by DNA-PKcs Inhibitory Phosphorylation of ATM. Mol Cell 65, 91–104 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ruis BL, Fattah KR & Hendrickson EA The catalytic subunit of DNA-dependent protein kinase regulates proliferation, telomere length, and genomic stability in human somatic cells. Mol Cell Biol 28, 6182–95 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Li G, Nelsen C & Hendrickson EA Ku86 is essential in human somatic cells. Proc.Natl.Acad.Sci.U.S.A 99, 832–837 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hendrickson CL, Purkayastha S, Pastwa E, Neumann RD & Winters TA Coincident In Vitro Analysis of DNA-PK-Dependent and -Independent Nonhomologous End Joining. J Nucleic Acids 2010, 823917 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lu H, Saha J, Beckmann PJ, Hendrickson EA & Davis AJ DNA-PKcs promotes chromatin decondensation to facilitate initiation of the DNA damage response. Nucleic Acids Res 47, 9467–9479 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Yamamoto K et al. Kinase-dead ATM protein causes genomic instability and early embryonic lethality in mice. J Cell Biol 198, 305–13 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Daniel JA et al. Loss of ATM kinase activity leads to embryonic lethality in mice. J Cell Biol 198, 295–304 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Pellegrini M et al. Autophosphorylation at serine 1987 is dispensable for murine Atm activation in vivo. Nature (2006). [DOI] [PubMed] [Google Scholar]
- 126.Daniel JA et al. Multiple autophosphorylation sites are dispensable for murine ATM activation in vivo. J.Cell Biol 183, 777–783 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Bakkenist CJ & Kastan MB DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421, 499–506 (2003). [DOI] [PubMed] [Google Scholar]
- 128.Sibanda BL, Chirgadze DY, Ascher DB & Blundell TL DNA-PKcs structure suggests an allosteric mechanism modulating DNA double-strand break repair. Science 355, 520–524 (2017). [DOI] [PubMed] [Google Scholar]
- 129.Sharif H et al. Cryo-EM structure of the DNA-PK holoenzyme. Proc Natl Acad Sci U S A 114, 7367–7372 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Yin X, Liu M, Tian Y, Wang J & Xu Y Cryo-EM structure of human DNA-PK holoenzyme. Cell Res 27, 1341–1350 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Sibanda BL, Chirgadze DY & Blundell TL Crystal structure of DNA-PKcs reveals a large open-ring cradle comprised of HEAT repeats. Nature 463, 118–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ochi T et al. DNA repair. PAXX, a paralog of XRCC4 and XLF, interacts with Ku to promote DNA double-strand break repair. Science 347, 185–8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Xing M et al. Interactome analysis identifies a new paralogue of XRCC4 in non-homologous end joining DNA repair pathway. Nat Commun 6, 6233 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kumar V, Alt FW & Frock RL PAXX and XLF DNA repair factors are functionally redundant in joining DNA breaks in a G1-arrested progenitor B-cell line. Proc Natl Acad Sci U S A 113, 10619–24 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lescale C et al. Specific Roles of XRCC4 Paralogs PAXX and XLF during V(D)J Recombination. Cell Rep (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Liu X, Shao Z, Jiang W, Lee BJ & Zha S PAXX promotes KU accumulation at DNA breaks and is essential for end-joining in XLF-deficient mice. Nat Commun 8, 13816 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Mahaney BL, Hammel M, Meek K, Tainer JA & Lees-Miller SP XRCC4 and XLF form long helical protein filaments suitable for DNA end protection and alignment to facilitate DNA double strand break repair. Biochem Cell Biol 91, 31–41 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Riballo E et al. XLF-Cernunnos promotes DNA ligase IVXRCC4 re-adenylation following ligation. Nucleic Acids Research 37, 482–492 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Hung PJ et al. MRI Is a DNA Damage Response Adaptor during Classical Non-homologous End Joining. Mol Cell 71, 332–342 e8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Arnoult N et al. Regulation of DNA repair pathway choice in S and G2 phases by the NHEJ inhibitor CYREN. Nature 549, 548–552 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.de Villartay JP et al. Human and animal models of V(D)J recombination deficiency. Curr.Opin.Immunol 15, 592–598 (2003). [DOI] [PubMed] [Google Scholar]
- 142.Rogakou EP, Pilch DR, Orr AH, Ivanova VS & Bonner WM DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J.Biol.Chem 273, 5858–5868 (1998). [DOI] [PubMed] [Google Scholar]
- 143.Lou Z et al. MDC1 maintains genomic stability by participating in the amplification of ATM-dependent DNA damage signals. Mol.Cell 21, 187–200 (2006). [DOI] [PubMed] [Google Scholar]
- 144.Stucki M et al. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell 123, 1213–1226 (2005). [DOI] [PubMed] [Google Scholar]
- 145.Stewart GS, Wang B, Bignell CR, Taylor AM & Elledge SJ MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature 421, 961–966 (2003). [DOI] [PubMed] [Google Scholar]
- 146.Difilippantonio S et al. 53BP1 facilitates long-range DNA end-joining during V(D)J recombination. Nature 456, 529–33 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Zha S et al. ATM-deficient thymic lymphoma is associated with aberrant tcrd rearrangement and gene amplification. Journal of Experimental Medicine 207, 1369–1380 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Borghesani PR et al. Abnormal development of Purkinje cells and lymphocytes in Atm mutant mice. Proc.Natl.Acad.Sci.U.S.A 97, 3336–3341 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Callen E et al. ATM prevents the persistence and propagation of chromosome breaks in lymphocytes. Cell 130, 63–75 (2007). [DOI] [PubMed] [Google Scholar]
- 150.Carico Z & Krangel MS Chromatin Dynamics and the Development of the TCRalpha and TCRdelta Repertoires. Adv Immunol 128, 307–61 (2015). [DOI] [PubMed] [Google Scholar]
- 151.Bredemeyer AL et al. ATM stabilizes DNA double-strand-break complexes during V(D)J recombination. Nature 442, 466–470 (2006). [DOI] [PubMed] [Google Scholar]
- 152.Helmink BA et al. MRN complex function in the repair of chromosomal Rag-mediated DNA double-strand breaks. J.Exp.Med 206, 669–679 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Zhang Y et al. The fundamental role of chromatin loop extrusion in physiological V(D)J recombination. Nature 573, 600–604 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Franco S, Alt FW & Manis JP Pathways that suppress programmed DNA breaks from progressing to chromosomal breaks and translocations. DNA Repair (Amst) 5, 1030–1041 (2006). [DOI] [PubMed] [Google Scholar]
- 155.Sonoda E et al. B cell development under the condition of allelic inclusion. Immunity 6, 225–33 (1997). [DOI] [PubMed] [Google Scholar]
- 156.Pelanda R, Schaal S, Torres RM & Rajewsky K A prematurely expressed Ig(kappa) transgene, but not V(kappa)J(kappa) gene segment targeted into the Ig(kappa) locus, can rescue B cell development in lambda5-deficient mice. Immunity 5, 229–39 (1996). [DOI] [PubMed] [Google Scholar]
- 157.Yan CT et al. IgH class switching and translocations use a robust non-classical end-joining pathway. Nature 449, 478–482 (2007). [DOI] [PubMed] [Google Scholar]
- 158.Pan-Hammarstrom Q et al. Impact of DNA ligase IV on nonhomologous end joining pathways during class switch recombination in human cells. J.Exp.Med 201, 189–194 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Rucci F et al. Homozygous DNA ligase IV R278H mutation in mice leads to leaky SCID and represents a model for human LIG4 syndrome. Proc Natl Acad Sci U S A 107, 3024–9 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Rooney S, Alt FW, Sekiguchi J & Manis JP Artemis-independent functions of DNA-dependent protein kinase in Ig heavy chain class switch recombination and development. Proc.Natl.Acad.Sci.U.S.A 102, 2471–2475 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Franco S et al. DNA-PKcs and Artemis function in the end-joining phase of immunoglobulin heavy chain class switch recombination. J.Exp.Med 205, 557–564 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Hu J et al. Detecting DNA double-stranded breaks in mammalian genomes by linear amplification-mediated high-throughput genome-wide translocation sequencing. Nat Protoc 11, 853–71 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Dunnick W, Hertz GZ, Scappino L & Gritzmacher C DNA sequences at immunoglobulin switch region recombination sites. Nucleic Acids Res 21, 365–72 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Ropars V et al. Structural characterization of filaments formed by human Xrcc4-Cernunnos/XLF complex involved in nonhomologous DNA end-joining. Proc Natl Acad Sci U S A 108, 12663–8 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Boboila C et al. Robust chromosomal DNA repair via alternative end-joining in the absence of X-ray repair cross-complementing protein 1 (XRCC1). Proc Natl Acad Sci U S A 109, 2473–8 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Boboila C et al. Alternative end-joining catalyzes robust IgH locus deletions and translocations in the combined absence of ligase 4 and Ku70. Proc Natl Acad Sci U S A 107, 3034–9 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Boboila C et al. Alternative end-joining catalyzes class switch recombination in the absence of both Ku70 and DNA ligase 4. J Exp Med 207, 417–27 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Kawamura K et al. DNA polymerase theta is preferentially expressed in lymphoid tissues and upregulated in human cancers. Int J Cancer 109, 9–16 (2004). [DOI] [PubMed] [Google Scholar]
- 169.Li Y, Gao X & Wang JY Comparison of two POLQ mutants reveals that a polymerase-inactive POLQ retains significant function in tolerance to etoposide and gamma-irradiation in mouse B cells. Genes Cells 16, 973–83 (2011). [DOI] [PubMed] [Google Scholar]
- 170.Zhang X et al. Fundamental roles of chromatin loop extrusion in antibody class switching. Nature 575, 385–389 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Simsek D et al. Crucial role for DNA ligase III in mitochondria but not in Xrcc1-dependent repair. Nature 471, 245–8 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Ghosh R, Roy S, Kamyab J, Danzter F & Franco S Common and unique genetic interactions of the poly(ADP-ribose) polymerases PARP1 and PARP2 with DNA double-strand break repair pathways. DNA Repair (Amst) 45, 56–62 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Rybanska I et al. PARP1 and DNA-PKcs synergize to suppress p53 mutation and telomere fusions during T-lineage lymphomagenesis. Oncogene 32, 1761–71 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Bjorkman A et al. DNA-PKcs Is Involved in Ig Class Switch Recombination in Human B Cells. J Immunol 195, 5608–15 (2015). [DOI] [PubMed] [Google Scholar]
- 175.Lumsden JM et al. Immunoglobulin class switch recombination is impaired in Atm-deficient mice. J.Exp.Med 200, 1111–1121 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Reina-San-Martin B, Chen HT, Nussenzweig A & Nussenzweig MC ATM is required for efficient recombination between immunoglobulin switch regions. J.Exp.Med 200, 1103–1110 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Pan-Hammarstrom Q et al. Disparate roles of ATR and ATM in immunoglobulin class switch recombination and somatic hypermutation. J.Exp.Med 203, 99–110 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Reina-San-Martin B et al. H2AX is required for recombination between immunoglobulin switch regions but not for intra-switch region recombination or somatic hypermutation. J.Exp.Med 197, 1767–1778 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Franco S et al. H2AX prevents DNA breaks from progressing to chromosome breaks and translocations. Mol.Cell 21, 201–214 (2006). [DOI] [PubMed] [Google Scholar]
- 180.Manis JP et al. 53BP1 links DNA damage-response pathways to immunoglobulin heavy chain class-switch recombination. Nat Immunol 5, 481–7 (2004). [DOI] [PubMed] [Google Scholar]
- 181.Daniel JA et al. PTIP promotes chromatin changes critical for immunoglobulin class switch recombination. Science 329, 917–23 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Starnes LM et al. A PTIP-PA1 subcomplex promotes transcription for IgH class switching independently from the associated MLL3/MLL4 methyltransferase complex. Genes Dev 30, 149–63 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Ghezraoui H et al. 53BP1 cooperation with the REV7-shieldin complex underpins DNA structure-specific NHEJ. Nature 560, 122–127 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Xu G et al. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature 521, 541–544 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Findlay S et al. SHLD2/FAM35A co-operates with REV7 to coordinate DNA double-strand break repair pathway choice. EMBO J 37(2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Dev H et al. Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat Cell Biol 20, 954–965 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Di Virgilio M et al. Rif1 prevents resection of DNA breaks and promotes immunoglobulin class switching. Science 339, 711–5 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Chapman JR et al. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol Cell 49, 858–71 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Bothmer A et al. 53BP1 regulates DNA resection and the choice between classical and alternative end joining during class switch recombination. J Exp Med 207, 855–65 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Panchakshari RA et al. DNA double-strand break response factors influence end-joining features of IgH class switch and general translocation junctions. Proc Natl Acad Sci U S A 115, 762–767 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Nussenzweig A & Nussenzweig MC Origin of chromosomal translocations in lymphoid cancer. Cell 141, 27–38 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Buckley RH The multiple causes of human SCID. J.Clin.Invest 114, 1409–1411 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Jiang W et al. Aberrant TCRdelta rearrangement underlies the T-cell lymphocytopenia and t(12;14) translocation associated with ATM deficiency. Blood 125, 2665–8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Zhang Y et al. The role of mechanistic factors in promoting chromosomal translocations found in lymphoid and other cancers. Adv Immunol 106, 93–133 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Lapkouski M, Chuenchor W, Kim MS, Gellert M & Yang W Assembly Pathway and Characterization of the RAG1/2-DNA Paired and Signal-end Complexes. J Biol Chem 290, 14618–25 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Fell VL & Schild-Poulter C Ku regulates signaling to DNA damage response pathways through the Ku70 von Willebrand A domain. Mol Cell Biol 32, 76–87 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Gu Y et al. Growth retardation and leaky SCID phenotype of Ku70-deficient mice. Immunity 7, 653–65 (1997). [DOI] [PubMed] [Google Scholar]
- 198.Ouyang H et al. Ku70 is required for DNA repair but not for T cell antigen receptor gene recombination In vivo. J Exp Med 186, 921–9 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Difilippantonio MJ et al. DNA repair protein Ku80 suppresses chromosomal aberrations and malignant transformation. Nature 404, 510–514 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Nussenzweig A, Sokol K, Burgman P, Li L & Li GC Hypersensitivity of Ku80-deficient cell lines and mice to DNA damage: the effects of ionizing radiation on growth, survival, and development. Proc.Natl.Acad.Sci.U.S.A 94, 13588–13593 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Gao Y et al. A critical role for DNA end-joining proteins in both lymphogenesis and neurogenesis. Cell 95, 891–902 (1998). [DOI] [PubMed] [Google Scholar]
- 202.Yan CT et al. XRCC4 suppresses medulloblastomas with recurrent translocations in p53-deficient mice. Proc Natl Acad Sci U S A 103, 7378–83 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Frank KM et al. Late embryonic lethality and impaired V(D)J recombination in mice lacking DNA ligase IV. Nature 396, 173–177 (1998). [DOI] [PubMed] [Google Scholar]
- 204.Han L & Yu K Altered kinetics of nonhomologous end joining and class switch recombination in ligase IV-deficient B cells. J Exp Med 205, 2745–53 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Barnes DE, Stamp G, Rosewell I, Denzel A & Lindahl T Targeted disruption of the gene encoding DNA ligase IV leads to lethality in embryonic mice. Curr Biol 8, 1395–8 (1998). [DOI] [PubMed] [Google Scholar]
- 206.Shull ER et al. Differential DNA damage signaling accounts for distinct neural apoptotic responses in ATLD and NBS. Genes Dev 23, 171–80 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Vera G et al. Cernunnos deficiency reduces thymocyte life span and alters the T cell repertoire in mice and humans. Mol Cell Biol 33, 701–11 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Kurimasa A et al. Catalytic subunit of DNA-dependent protein kinase: impact on lymphocyte development and tumorigenesis. Proc Natl Acad Sci U S A 96, 1403–8 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Loser DA et al. Sensitization to radiation and alkylating agents by inhibitors of poly(ADP-ribose) polymerase is enhanced in cells deficient in DNA double-strand break repair. Mol Cancer Ther 9, 1775–87 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Rivera-Munoz P et al. Reduced immunoglobulin class switch recombination in the absence of Artemis. Blood 114, 3601–9 (2009). [DOI] [PubMed] [Google Scholar]
- 211.Touvrey C et al. Distinct effects of DNA-PKcs and Artemis inactivation on signal joint formation in vivo. Mol Immunol 45, 3383–91 (2008). [DOI] [PubMed] [Google Scholar]
- 212.Huang Y et al. Impact of a hypomorphic Artemis disease allele on lymphocyte development, DNA end processing, and genome stability. J Exp Med 206, 893–908 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Li L et al. Targeted disruption of the Artemis murine counterpart results in SCID and defective V(D)J recombination that is partially corrected with bone marrow transplantation. J Immunol 174, 2420–8 (2005). [DOI] [PubMed] [Google Scholar]
- 214.Nagasawa H et al. Coordination of the Ser2056 and Thr2609 Clusters of DNA-PKcs in Regulating Gamma Rays and Extremely Low Fluencies of Alpha-Particle Irradiation to G0/G1 Phase Cells. Radiat Res 187, 259–267 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Dewan A et al. Robust DNA repair in PAXX-deficient mammalian cells. FEBS Open Bio 8, 442–448 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Gago-Fuentes R et al. Normal development of mice lacking PAXX, the paralogue of XRCC4 and XLF. FEBS Open Bio 8, 426–434 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Tadi SK et al. PAXX Is an Accessory c-NHEJ Factor that Associates with Ku70 and Has Overlapping Functions with XLF. Cell Rep 17, 541–555 (2016). [DOI] [PubMed] [Google Scholar]
- 218.Castaneda-Zegarra S et al. Generation of a Mouse Model Lacking the Non-Homologous End-Joining Factor Mri/Cyren. Biomolecules 9(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Herzog KH, Chong MJ, Kapsetaki M, Morgan JI & McKinnon PJ Requirement for Atm in ionizing radiation-induced cell death in the developing central nervous system. Science 280, 1089–91 (1998). [DOI] [PubMed] [Google Scholar]
- 220.Spring K et al. Atm knock-in mice harboring an in-frame deletion corresponding to the human ATM 7636del9 common mutation exhibit a variant phenotype. Cancer Res 61, 4561–8 (2001). [PubMed] [Google Scholar]
- 221.Elson A et al. Pleiotropic defects in ataxia-telangiectasia protein-deficient mice. Proc Natl Acad Sci U S A 93, 13084–9 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Xu Y & Baltimore D Dual roles of ATM in the cellular response to radiation and in cell growth control. Genes Dev 10, 2401–10 (1996). [DOI] [PubMed] [Google Scholar]
- 223.Barlow C et al. Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell 86, 159–71 (1996). [DOI] [PubMed] [Google Scholar]
- 224.Campbell A et al. A novel mouse model for ataxia-telangiectasia with a N-terminal mutation displays a behavioral defect and a low incidence of lymphoma but no increased oxidative burden. Hum Mol Genet 24, 6331–49 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Bassing CH et al. Increased ionizing radiation sensitivity and genomic instability in the absence of histone H2AX. Proc Natl Acad Sci U S A 99, 8173–8 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Bassing CH et al. Histone H2AX: a dosage-dependent suppressor of oncogenic translocations and tumors. Cell 114, 359–70 (2003). [DOI] [PubMed] [Google Scholar]
- 227.Petersen S et al. AID is required to initiate Nbs1/gamma-H2AX focus formation and mutations at sites of class switching. Nature 414, 660–665 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Economopoulou M et al. Histone H2AX is integral to hypoxia-driven neovascularization. Nat Med 15, 553–8 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Ermakova O, Salimova E, Piszczek L & Gross C Construction and phenotypic analysis of mice carrying a duplication of the major histocompatibility class I (MHC-I) locus. Mamm Genome 23, 443–53 (2012). [DOI] [PubMed] [Google Scholar]
- 230.Bothmer A et al. Regulation of DNA End Joining, Resection, and Immunoglobulin Class Switch Recombination by 53BP1. Mol Cell 42, 319–29 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Ward IM, Minn K, van Deursen J & Chen J p53 Binding protein 53BP1 is required for DNA damage responses and tumor suppression in mice. Mol Cell Biol 23, 2556–63 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Cornacchia D et al. Mouse Rif1 is a key regulator of the replication-timing programme in mammalian cells. EMBO J 31, 3678–90 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Buonomo SB, Wu Y, Ferguson D & de Lange T Mammalian Rif1 contributes to replication stress survival and homology-directed repair. J Cell Biol 187, 385–98 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Barazas M et al. Radiosensitivity Is an Acquired Vulnerability of PARPi-Resistant BRCA1-Deficient Tumors. Cancer Res 79, 452–460 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Watanabe N et al. The REV7 subunit of DNA polymerase zeta is essential for primordial germ cell maintenance in the mouse. J Biol Chem 288, 10459–71 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Pirouz M, Pilarski S & Kessel M A critical function of Mad2l2 in primordial germ cell development of mice. PLoS Genet 9, e1003712 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Su D et al. PTIP chromatin regulator controls development and activation of B cell subsets to license humoral immunity in mice. Proc Natl Acad Sci U S A 114, E9328–E9337 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Gong Z, Cho YW, Kim JE, Ge K & Chen J Accumulation of Pax2 transactivation domain interaction protein (PTIP) at sites of DNA breaks via RNF8-dependent pathway is required for cell survival after DNA damage. J Biol Chem 284, 7284–93 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Wang J et al. PTIP associates with Artemis to dictate DNA repair pathway choice. Genes Dev 28, 2693–8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Cho EA, Prindle MJ & Dressler GR BRCT domain-containing protein PTIP is essential for progression through mitosis. Mol Cell Biol 23, 1666–73 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Patel SR, Kim D, Levitan I & Dressler GR The BRCT-domain containing protein PTIP links PAX2 to a histone H3, lysine 4 methyltransferase complex. Dev Cell 13, 580–92 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Musilli S, Abramowski V, Roch B & de Villartay JP An in vivo study of the impact of deficiency in the DNA repair proteins PAXX and XLF on development and maturation of the hemolymphoid system. J Biol Chem 295, 2398–2406 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Hung PJ et al. Deficiency of XLF and PAXX prevents DNA double-strand break repair by non-homologous end joining in lymphocytes. Cell Cycle 16, 286–295 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Sekiguchi J et al. Genetic interactions between ATM and the nonhomologous end-joining factors in genomic stability and development. Proc.Natl.Acad.Sci.U.S.A 98, 3243–3248 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Gurley KE & Kemp CJ Synthetic lethality between mutation in Atm and DNA-PK(cs) during murine embryogenesis. Curr Biol 11, 191–4 (2001). [DOI] [PubMed] [Google Scholar]