

Abstract
The decarboxylation reaction of phenylglyoxylic acid with hydrogen peroxide is studied by real-time hyperpolarized carbon-13 nuclear magnetic resonance (13C NMR) spectroscopy at room temperature. A non-observable reaction intermediate is identified using blind selective saturation pulses in the expected chemical shift range, thereby revealing information on the reaction mechanism.
Graphical Abstract
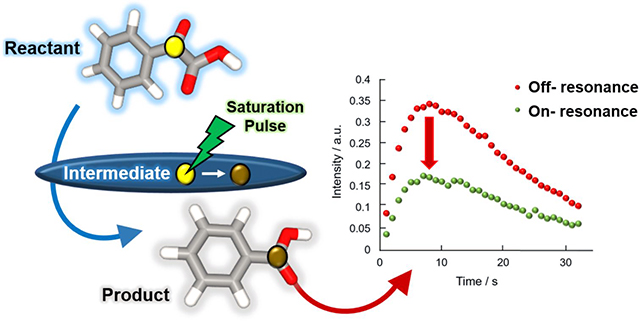
Nuclear spin hyperpolarization is used to enhance NMR signals for observation of chemical reactions in real-time. Here, we introduce a blind saturation technique, which enables the indirect detection of otherwise unobservable reaction intermediates.
Nuclear magnetic resonance (NMR) spectroscopy has long played a pivotal role in the study of reaction mechanisms in the fields of chemistry and biology, as the technique reveals information on molecular structure and the interactions between reaction substrates with atomic resolution. However, due to the intrinsically low sensitivity of NMR, the method is mainly applied for analysing reaction products without yielding detailed transient information for a reaction. A potentially more powerful approach to characterizing reaction mechanisms is to monitor the transient species that arise as a reaction occurs. The advent of dissolution-dynamic nuclear polarization (D-DNP) technology has further increased the applicability of NMR spectroscopy since the hyperpolarization technique can enhance NMR signals by several orders of magnitude1, allowing for an investigation of reaction mechanisms and kinetics in real time.2,3
In the D-DNP method, hyperpolarization takes place at cryogenic temperatures (typically ~ 1 K) in a moderate magnetic field of a few Tesla. The sample incorporates target molecules and free radicals in a matrix that forms a glass at low temperatures. Under these conditions, high electron spin polarization from the stable radical is transferred to the nuclear spins of target molecules. Hyperpolarization of the target molecules can be preserved by rapidly dissolving the sample in a preheated solvent, and NMR data is subsequently obtained at ambient temperature, allowing utilization of strongly enhanced NMR signals. With this method, direct observations of transient or low-population species in ongoing reactions can be achieved with high sensitivity.4,5 In this regard, the D-DNP technique is an emerging analytical tool to investigate reaction mechanisms, as an alternative to traditional stopped-flow and rapid injection NMR methods, and low temperature NMR including with chemical exchange saturation transfer (CEST) detection.6–9
The application of the D-DNP technique has been detailed in diverse areas, ranging from complex biochemical processes such as in-cell metabolic conversion, and enzyme-catalyzed reactions to organic reactions including polymerization.10 Low-population reaction intermediates can be identified and quantified in real-time by hyperpolarized 13C NMR spectroscopy, thus yielding direct chemical information on reaction mechanisms.11,12 Moreover, the spin states in a chemical reaction under non-equilibrium conditions contain information about events that occurred at earlier points in time. Based on this principle, the use of D-DNP can be extended to mechanistic studies by encoding the reactant spin states at the beginning of a reaction, and observing the signals transferred to intermediates and/or products.13–15 In the aforementioned mechanistic studies, reaction intermediates with a sufficiently high stability and population during the NMR measurement time were observable. Isotope labelling of the entire molecule or of specific molecular positions can further be performed to amplify the signal-to-noise ratio of acquired spectra.16,17 Although resonance signals of interest are significantly enhanced via hyperpolarization, the ability to observe reaction intermediates remains contingent on favorable reaction conditions as described above. The main focus of this work is to present a way to successfully detect information on invisible intermediates via a pulse scheme of blind selective saturations of nuclear spins. By eliminating the requirement for direct observation, the application range of NMR is substantially expanded. For a proof of concept, the decarboxylation reaction of an alpha-keto acid with hydrogen peroxide is characterized. Alpha-keto acids are significant in biological processes such as glycolysis and the tricarboxylic acid cycle. Phenylglyoxylic acid (PhGA) is also used as a non-toxic antioxidant that chemically protects the stability of pharmaceuticals by undergoing a decarboxylation reaction.18
Fig. 1a shows the proposed mechanism for decarboxylation of PhGA based on a general decarboxylation mechanism of alpha-keto acids in the presence of hydrogen peroxide.19,20 Here, the PhGA was hyperpolarized by DNP, and then rapidly dissolved in a heated buffer solution. All 13C signals in this substrate were substantially enhanced, with the most intense signals from C2-PhGA showing >5,000-fold enhancements relative to a conventional NMR spectrum (see Supplementary Information). For the decarboxylation reaction, 25μL of hydrogen peroxide (30%) was preloaded into the NMR tube and mixed with the hyperpolarized PhGA solution after dissolution. As the reaction proceeds, the high spin polarization from the PhGA is transferred to the reaction products. As a result, the appearance of signals from the products benzoate and carbon dioxide/bicarbonate signals was observed in real time (Fig. 1b). However, any additional signals that would be attributed to the intermediate were not identified. The absence of these signals could be due to a relatively low population or short lifetime of the reaction intermediate. In order to examine the decarboxylation mechanism, and especially proving the presence of the invisible intermediate during the reaction time, a blind saturation pulse scheme (see Supplementary Information) was implemented. In this experiment, selective saturation pulses were applied in the expected chemical shift range of the intermediate to alter spin polarization of the intermediate if it resonates at a frequency within the saturation band. As the transfer of hyperpolarization from the intermediate to the product is reduced, a decrease of the product signals arising from the saturated spins results. This decrease enables to visualize the reaction progress or connectivity between reaction species without affecting the reaction itself by the saturation pulses. To demonstrate this concept, prior to conducting blind saturation with PhGA, a reaction with 1,2-13C-pyruvic acid was employed. In this reaction, the presence of the intermediate 2-hydroperoxy-2-hydroxypropanoate was previously observed by low-temperature NMR, as well as by D-DNP NMR.21,22
Fig. 1.

(a) Proposed mechanism of decarboxylation reaction of PhGA with H2O2. (b) Time‐resolved 13C NMR spectra of hyperpolarized PhGA mixed with H2O2. The spectra were acquired by a series of small flip angle pulses with 1 s time resolution. Final concentrations of PhGA and H2O2 after dissolution were 25 mM and 544 mM, respectively (pseudo-1st-order reaction condition). All of the resonance signals from the hyperpolarized reactant (C1-PhGA: 173.1 ppm, C2-PhGA: 197.1 ppm) and product (benzoate: 175.8 ppm, CO2: 124.8 ppm, bicarbonate: 160.4 ppm) were detected. Aromatic carbons in the reactant and product (Ar-PhGA, Ar-Benzoate) were observed in the range of 136.3 ppm ~ 128.8 ppm.
This reaction, depicted in Fig. 2a, was performed using hyperpolarized 1,2-13C-pyruvic acid. In this case, it was possible to detect 13C signals from the intermediate as well as from the products during the reaction (see Supplementary Information). As can be seen from the top spectrum in Fig. 2b, the C1 and C2 intermediate peaks were observed as doublets due to 13C-13C J-coupling, at chemical shifts of approximately 101.8 ppm and 175.5 ppm, respectively. To test the blind saturation pulse scheme (100 Hz bandwidth), two experiments were performed. The centre frequency of the selective saturation pulse was set to (i) the frequency of the C2-intermediate (101.8 ppm), and (ii) off-resonance (82 ppm) for a reference. Corrections for differences in the polarization levels were implemented by scaling of the signals, such that extrapolated signal intensities became unit intensity for C2-PY at t=0. The scaling factor was determined from single exponential curve fitting of the measured C2-PY signal intensities. A marked decrease in the signal intensity of acetate (181.3 ppm) can be seen in the spectrum with on-resonance saturation. This result can be explained by a loss of hyperpolarization on the C2-intermediate, caused by the saturation pulses, which is subsequently included in the reaction product acetate. The signal decrease demonstrates the successful observation of the intermediate generated in the reaction, and indicates the utility of the selective saturation scheme for investigating reaction mechanisms.
Fig. 2.

(a) The decarboxylation reaction of 1,2-13C-pyruvate with H2O2. A selective saturation pulse applied to the C2-intermediate to interrupt the signal flow from the reactant to product is illustrated with a red bolt symbol. (b) Selective saturation with the pulse applied off-resonance and on-resonance with respect to the 2-13C-pyruvate-intermediate. The 5th scan of a time-resolved 13C data set is shown. For the off-resonance experiment, the offset of the saturation pulse was set −20 ppm away from the intermediate signal. For data normalization, signal intensity of C2-PY was fit to a single exponential decay function, and the fitted intensity at t = 0 was scaled to unit value. The selected region for the saturation in each case is indicated in the spectrum with an orange trapezoid.
The reaction intermediate of 1,2-13C-pyruvate and hydrogen peroxide was detectable due to a high initial concentration (14 M), and an available labelled isotope compound. Unlike 1,2-13C-pyruvate, both limited solubility and the natural 13C abundance of PhGA make it less likely to detect the reaction intermediate in real time. However, it was anticipated that PhGA would potentially follow a decarboxylation mechanism similar to that of 1,2-13C-pyruvate. Therefore, we applied blind selective saturation pulses in a range close to the C2-intermediate in the reaction with 1,2-13C-pyruvate. These experiments aimed at supporting the reaction mechanism and provide chemical shift information for the invisible PhGA intermediate, 2-hydroperoxy-2-hydroxy-2-phenylacetate.
First, the presence of the proposed intermediate was examined by conducting the blind saturation experiment with the bandwidth of the saturation pulse expanded to 600 Hz (Fig. 3). The saturation pulse was applied at 102 ppm, affecting a frequency range from 99 to 105 ppm (on-resonance). A series of selective saturation pulses were applied for a total of 720 ms before each acquisition with a scan delay of 1 s. As a control, the same experiments were conducted with off-resonance saturation at −20 ppm away from the expected intermediate regions (off-resonance) and with the saturation pulse power turned off (PW-off). If the intermediate signal is within the excitation range, the signal intensity of the product sharing the same carbon with the intermediate is expected to decrease in the blind saturation experiment. A correction for different polarization levels was applied as described in the figure caption. As displayed in Fig 3c, a substantial drop in the signal of benzoate was observed in the saturation experiment, indicating that the C2 resonance frequency of the intermediate is within the chemical shift range affected by the saturation pulse. In contrast, the signal intensities of bicarbonate (Fig. 3d) and carbon dioxide (see Supplementary information) generated from C1 of PhGA were almost identical in all of the data sets. In addition, when compared to the off-resonance experiment, the apparent conversion rate in the on-resonance experiment was found to be slightly higher when the invisible intermediate signal was attenuated by saturation pulses (Fig. 3a). This effect can be seen in both the PhGA and pyruvate reactions (Fig. 2b and Fig. 3a). It may support the presence of a dynamic equilibrium between the reactant and intermediate, whereby a portion of the saturation is carried back to the reactant signal.
Fig. 3.

Intensities of the reactant signals (a) C2-PhGA and (b) C1-PhGA, as well as of the product signals (c) benzoate, (d) bicarbonate from the blind saturation and two control experiments. Signal intensities of the reactant are fit to a single exponential decay function and fitted intensity with C2-PhGA at t = 0 is scaled to unit value in order to compensate for different polarization levels. The scaling factor is determined from the measured C2-PhGA signal intensities with a single exponential decay function. Hyperpolarized 13C NMR spectra of (e) benzoate and (f) bicarbonate at the maximum signal intensity (8th scan).
To determine the C2 chemical shift for the intermediate in the PhGA reaction more accurately, the bandwidth of the saturation pulse was reduced to 100 Hz, and the centre frequency of the saturation pulse was varied between 99 and 105 ppm in increments of 1 ppm. The resulting kinetic profiles of C1 signals of benzoate are shown in Fig. 4a. A comparison of the time dependent curves shows that signal intensities were significantly attenuated when the saturation frequency was fsat = 103 ppm. Since both the initial concentrations of the reactants and reaction conditions were nominally the same in the experiments, relatively consistent signal intensities of the C2-PhGA were observed (Fig. 4b). The significant change of benzoate signal at only fsat = 103 ppm is expected to be caused by successful targeting of the invisible intermediate signal by the selective saturation (Fig. 4c). Two different control experiments, (i) decreasing the saturation time by half and (ii) turning off the saturation, supported the direct targeting of the invisible intermediate by the selective saturation (see Supplementary information). Furthermore, the apparent conversion rate of PhGA to Benzoate (kPhGA→Benzoate) in the 7 datasets was investigated using a single-step, irreversible kinetic model that includes additional signal depletion pathways for the application of fixed small flip angle pulses and T1 relaxation. In each scan, a hard pulse with a fixed small flip angle α is used to convert sin(α) of the total longitudinal magnetization to the observable transverse magnetization, while cos(α) of the longitudinal magnetization is preserved for the following scans. Thus, this fixed flip angle scheme causes signal decay with a factor of λ=(-ln(cosα))/Δt where α is the flip angle and Δt is the time delay between scans (see Supplementary information). The experimental data suggests a kinetic model that involves a dynamic equilibrium between the reactant and intermediate. However, due to the lack of information about the intermediate, the simplest kinetic model is used. With experimentally determined T1 relaxation rates of the reactant and product from separate measurements, kPhGA→Benzoate values were obtained from the decaying C2-PhGA signals (see Supplementary information). The heat maps included in Fig. 4a represent the kinetic profiles generated for C2-PhGA and benzoate when assuming kPhGA→Benzoate = 0.10 – 0.20 s-1. From a series of blind saturation experiments, the decrease in the intensity of the product signal observed for fsat = 103 ppm is clearly significant even when a large variation in the reaction rate is considered. Overall, these findings confirm a successful indirect detection of the invisible intermediate signal and the corresponding chemical shift range. Therefore, blind saturation provides information on the expected intermediate. These experiments observe the temporal correlation between a reaction product and the invisible intermediate while the reaction is occurring. While the underlying experimental concepts of this method may be similar to the principles of chemical exchange saturation transfer (CEST)9 at low temperature ranges, the blind saturation approach can be applied under non-equilibrium conditions at ambient temperatures without stabilizing reagents, rather than those mostly involving chemical exchange. In general, the results of this study may be applied for monitoring chemical reactions involving single or multi-step conversions. In summary, the presence of a reaction intermediate in the decarboxylation reaction of PhGA with H2O2 was identified by real-time D-DNP NMR with a method relying on blind selective saturations. Blind saturation pulses applied in the expected chemical shift ranges of the intermediate confirmed that the hyperpolarized PhGA signal is transmitted to benzoate through the invisible intermediate, thereby revealing information on the decarboxylation reaction pathway of PhGA. In real-time mechanistic studies, blind saturation pulses can potentially be applied to complex reactions such as metabolic processes, where multiple intermediates exist, and where transient molecules are not detectable due to their short lifetime and low signal-to-noise ratio.
Fig. 4.

(a) Normalized C1 signal intensities of benzoate in selective saturation experiments with a reduced bandwidth of the saturation pulse of 100 Hz. The center frequency of the saturation pulse (fsat) was varied from 99 to 105 ppm, in steps of 1 ppm. (b) Normalized 13C spectra of C2-PhGA as a function of saturation frequency. (c) Normalized 13C spectra of benzoate as a function of saturation frequency. In (b) and (c), peaks are shown from the spectra of the 1st, 4th, and 8th scans in the time-resolved data set. Lines are drawn to connect the peak maxima. The same normalization method utilized in Fig. 3 was applied for (b) and (c).
Supplementary Material
Acknowledgements
This research was supported by the National Research Foundation of Korea (NRF-2019R1C1C1005412, NRF-2020R1A4A4079870), the Korea Health Industry Development Institute (HI17C0830), Korea Research Institute of Standards and Science (KRISS–2020–GP2020–0004), the Ministry of SMEs and Startups (S2603827), and the National Institutes of Health (R01-GM132655).
Footnotes
Conflicts of interest
There are no conflicts to declare.
Electronic Supplementary Information (ESI) available: dynamic nuclear polarization, NMR spectroscopy and data analysis, Single-step conversion kinetic model. See DOI: 10.1039/x0xx00000x
Notes and references
- 1.Ardenkjaer-Larsen JH, Fridlund B, Gram A, Hansson G, Hansson L, Lerche MH, Servin R, Thaning M and Golman K, PNAS, 2003, 100, 10158–10163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bowen S and Hilty C, Angew. Chem. Int. Ed, 2008, 47, 5235–5237. [DOI] [PubMed] [Google Scholar]
- 3.Lee Y, Heo GS, Zeng H, Wooley KL and Hilty C, J. Am. Chem. Soc, 2013, 135, 4636–4639. [DOI] [PubMed] [Google Scholar]
- 4.Jensen PR, Meier S, Ardenkjaer-Larsen JH, Duus JO, Karlsson M and Lerche MH, Chem. Commun, 2009, 34, 5168–5170. [DOI] [PubMed] [Google Scholar]
- 5.Lee Y, Zacharias NM, Piwnica-Worms D and Bhattacharya PK, Chem. Commun, 2014, 50, 13030–13033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Christianson MD, Tan EHP and Landis CR, J. Am. Chem. Soc, 2010, 132, 11461–11463. [DOI] [PubMed] [Google Scholar]
- 7.Bertz SH, Hardin RA, Murphy MD, Ogle CA, Richter JD and Thomas AA, J. Am. Chem. Soc, 2012, 134, 9557–9560. [DOI] [PubMed] [Google Scholar]
- 8.Ramanujam V, Charlier C and Bax A, Angew. Chem. Int. Ed, 2019, 58, 15309–15312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lokesh N, Seegerer A, Hioe J and Gschwind RM, J. Am. Chem. Soc, 2018, 140, 1855–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ardenkjaer-Larsen J-H, Boebinger GS, Comment A, Duckett S, Edison AS, Engelke F, Griesinger C, Griffin RG, Hilty C, Maeda H, Parigi G, Prisner T, Ravera E, van Bentum J, Vega S, Webb A, Luchinat C, Schwalbe H and Frydman L, Angew. Chem. Int. Ed, 2015, 54, 9162–9185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jensen PR, Meier S, Ardenkjaer-Larsen JH, Duus JO, Karlsson M and Lerche MH, Chem. Commun, 2009, 34, 5168–5170. [DOI] [PubMed] [Google Scholar]
- 12.Jensen PR and Meier S, Chem. Commun, 2020, 56, 6245–6248. [DOI] [PubMed] [Google Scholar]
- 13.Kim Y, Chen CH and Hilty C, Chem. Commun, 2018, 54, 4333–4336. [DOI] [PubMed] [Google Scholar]
- 14.Chen CH, Shih WC and Hilty C, J. Am. Chem. Soc, 2015, 137, 6965–6971. [DOI] [PubMed] [Google Scholar]
- 15.Bowen S and Hilty C, Anal. Chem, 2009, 81, 4543–4547. [DOI] [PubMed] [Google Scholar]
- 16.Jannin S, Dumez JN, Giraudeau P and Kurzbach D, J. Magn. Reson, 2019, 305, 41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lerche MH, Yigit D, Frahm AB, Ardenkjaer-Larsen JH, Malinowski RM and Jensen PR, Anal. Chem, 2018, 90, 674–678. [DOI] [PubMed] [Google Scholar]
- 18.Lopalco A and Stella VJ, J. Pharm. Sci, 2016, 105, 2879–2885. [DOI] [PubMed] [Google Scholar]
- 19.Holleman AF, Proc. K. Ned. Akad. Wet, 1904, 6, 715. [Google Scholar]
- 20.Aleksankin MM, Vysotskaya NA and Brodskii AE, Kernenergie, 1962, 5, 362–365. [Google Scholar]
- 21.Asmus C, Mozziconacci O and Schoneich C, J. Phys. Chem. A, 2015, 119, 966–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Drachman N, Kadlecek S, Duncan I and Rizi R, Phys. Chem. Chem. Phys, 2017, 19, 19316–19325. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.