

Abstract
The molecular photophysics and thermally activated delayed fluorescence (TADF) in spiro compounds are distinct because of the rigid orthogonal C–C bridging bond between donor and acceptor. The photophysics is found to be highly complex, with unprecedented multiple anti-Kasha emissions from three different singlet states, two of which are one-photon forbidden. The TADF mechanism is critically controlled by local acceptor nπ* states; the singlet nπ* state undergoes rapid intersystem crossing populating an energetically close acceptor ππ* triplet state. The acceptor triplet nπ* state couples nonadiabatically to a CT triplet state mediating reverse intersystem crossing. When the nπ* and CT states are energetically close, TADF is greatly enhanced with rISC rate reaching 107 s–1. We observe neither DF from the singlet nπ* state nor electron transfer (ET) to form the 1CT because there is no ET driving force; however, ET from the higher-energy donor singlet ππ* state readily occurs along with donor emission.
Organic light-emitting diodes (OLEDs) now underpin a large section of the display market because of their many appealing characteristics, including being high efficiency, flexible, and solution processable, and their ability to be fabricated on low-cost substrates.1−4 Development of suitable emitters for OLEDs is fundamental in increasing their output efficiency and optimizing device performance. One of the key requirements is an emitter that utilizes triplet excited states to maximize the achievable internal quantum efficiency close to 100%. Phosphorescent materials currently used in OLED displays have provided one route, using heavy-metal-containing organic complexes to facilitate spin–orbit coupling and efficient spin-mixing to give efficient phosphorescence.5,6 In addition, nonradiative decay of triplet excitons formed in the emission layer host material can be suppressed by ensuring that the triplet energy of the host is higher than that of the phosphorescent emitter.7
An alternative method for harvesting triplet states, thermally activated delayed fluorescence (TADF), has attracted tremendous interest for OLED applications by providing a means of harvesting triplet excitons without the need for rare and expensive heavy metals.8,9 Thermal activation of triplet excitons causes them to undergo reverse intersystem crossing (rISC) back to the singlet manifold, producing delayed fluorescence. To achieve efficient rISC, TADF molecules must meet several key criteria, starting with a small S1–T1 energy gap, ΔEST. One way to achieve this is through a lowest-energy excited state of charge-transfer (CT) character. Charge-transfer states achieve effective separation of electron and hole wave functions and small associated ΔEST values, either by possessing structurally orthogonal D and A moieties (decoupling their individual electronic systems) or through a large spatial separation of D and A fragments (e.g., in exciplexes10). Either approach results in minimal electron exchange interaction energy and a very small ΔEST, frequently <50 meV.11−13 However, when the electron exchange energy is so small, the singlet (1CT) and triplet (3CT) orbitals become degenerate and transition between the two is spin-forbidden as no change in orbital angular momentum can occur.14 To facilitate TADF a third triplet excited state, very close in energy to the 3CT state but having different orbital character, is required to mix (nonadiabatically) with 3CT to mediate a spin flip and couple the triplet back to the singlet manifold. This can be either a local triplet state (3LE) or a higher-lying triplet CT state.15
An orthogonal N–C donor–acceptor bridge is typically used in TADF materials to give the appropriate energy level ordering, but such bonding is not a prerequisite for TADF.8,9,16−18 While only a few examples have been reported, in spiro-linked D–A molecules the rigid and orthogonal donor–acceptor C–C bridge can yield efficient rISC.19−21 This is very important as a bridging C–N bond between D and A in a typical D–A TADF material is considered to be a very weak bond, prone to degradation. This is one of the suspected main causes of the short lifetimes achievable by TADF emitters in devices. Moving away from this architecture, to a spiro system for example, could greatly enhance emitter lifetime, and this provides a very compelling motivation for these studies to provide a better understanding of spiro TADF emitters.
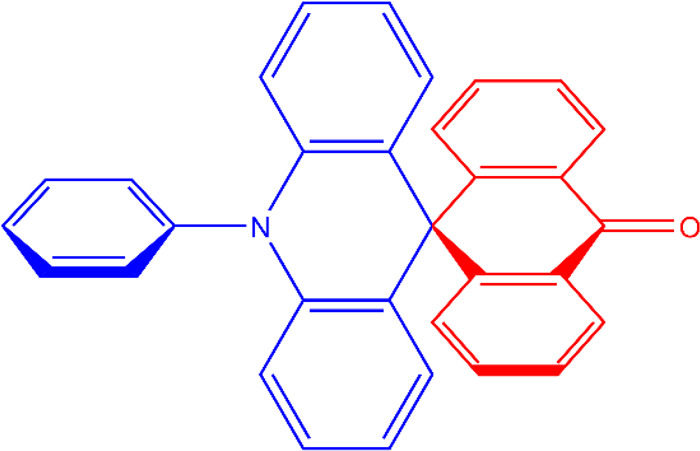
The spiro-linked acridine anthracenone derivative 10-phenyl-10H,10′H-spiro[acridine-9,9′-anthracen]-10′-one (ACRSA, Scheme 1) was one of the first reported spiro-linked TADF materials. It showed efficient solution-state photoluminescence (PL efficiency of 81%), with similar performance (∼75%) doped in the OLED host bis[2-(diphenylphosphino)phenyl] ether oxide (DPEPO) leading to moderately high device EQE of 16.5% in the same host.22,23 The generally moderate (but never outstanding) device performances reported for spiro-linked TADF materials (compared with up to 25% EQE in C–N linked D–A molecules) suggest a different photophysics and/or rISC mechanism may be active in the presence of the C–C spiro-bridge, warranting further investigation.
Scheme 1. Chemical Structure of ACRSA.
Lyskov and Marian have recently presented a very detailed quantum chemistry analysis of the ACRSA molecule. They highlight the complex interactions between charge transfer and local states, the role of state mixing (with high-lying excitonic transitions) that overcomes the forbidden nature of radiative decay from these states, and the potential for rapid nanosecond rISC mediated by n−π* to π–π* ISC transitions.24 The excited states (calculated in a toluene environment) and a composite energy state diagram from their data, using their state nomenclature, are given in Supporting Scheme 1.
Applying a range of spectroscopic measurements to ACRSA, we uncover highly complex photophysics, in which several general principles of molecular spectroscopy are contravened. From such a “simple” molecule, the complexity of its multiple decay channels reflects the critical roles that nonadiabatic vibronic coupling and the nπ* state of the acceptor have on molecular photophysics and also demonstrates the strongly decoupled nature of D and A units.
Because of its CT excited states, the photophysics of ACRSA is highly dependent on the solvent environment. Starting with the absorption spectrum of ACRSA in toluene (Supporting Figure S1), two strongly allowed transitions are observed to have onset at 3.64 and 3.83 eV, ascribed as ground state 11A1 → 11B2 and 11A1 → 11B1, respectively, allowed π–π* exciton transitions (Supporting Scheme 1).24
Following excitation at 330 nm (3.76 eV) into the 11B1 state, the resulting emission of both aerated and degassed toluene solutions (Figure 1a,d) shows a very large Stokes shift with emission onset at 2.7 eV and a structureless Gaussian line shape, implying pure CT character. This corresponds to the 11A2 → 11A1 transition (Supporting Scheme 1), experimentally confirming that the 11A2 is the lowest-energy singlet 1CT state. Addition of oxygen results in a large reduction in the emission intensity, consistent with an oxygen-quenched delayed fluorescence (DF) component that otherwise dominates the total emission. The reduced 1CT emission in aerated solution allows us to additionally detect the presence of a further high-energy emission band (onset, 3.7 eV). Upon comparing this emission to that previously observed in acridine25 it is clear that this emission emanates from the acridine 1LED ππ* state (11B1 in Supporting Scheme 1 and Figure S2). The fast radiative decay of this 1LED excitonic state competes effectively with internal conversion (IC), electron transfer, and intersystem crossing (ISC) nonradiative decay channels. The ππ* character is confirmed by the positive solvatochromic shift observed in this absorption band (Supporting Figure S3).
Figure 1.

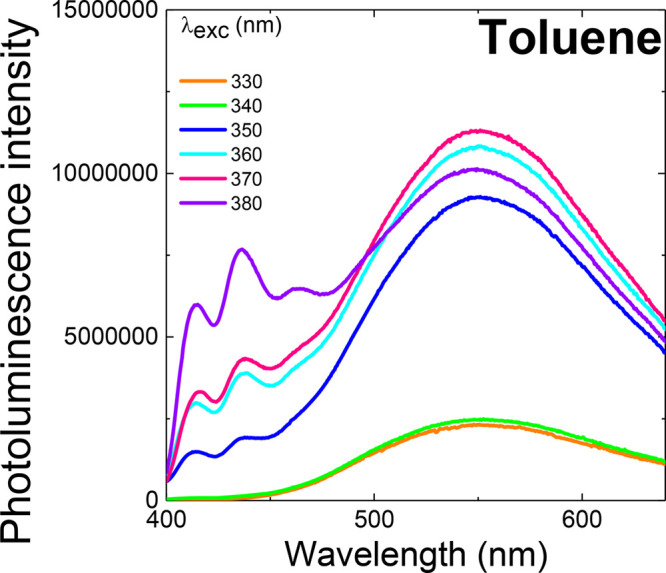
Steady-state emission from toluene solutions (50 μM) measured at three different excitation wavelengths: 330, 350, and 380 nm (non-normalized data). Top row spectra (a–c) are measured in aerated solution; bottom row (d–f) are measured in degassed solution showing the very strong delayed fluorescence contribution to emission, irrespective of excitation wavelength.
As both the emission of the 1LED ππ* state and a portion of the 1CT emission are not fully quenched by oxygen, this indicates that the exciton can undergo electron transfer yielding the 1CT state directly. High-energy triplet states formed by electron transfer and additionally by ISC from the 1LED ππ* state will cascade down to the lowest-energy triplet state—which can be harvested to give oxygen-sensitive TADF emission.
The relative yields of each emissive state for different excitation wavelengths are shown in Figure 2. Excitation of the solution below the onset of the 1LED transition (>350 nm, 3.54 eV, Figure 1b,c) reveals emission from another new structured band (onset at 3.1 eV as well as weak 1CT emission). As the 1LED transition has already been identified, we propose that this new localized emission arises from the symmetry-forbidden 1LEA nπ* state of the anthracenone acceptor unit, i.e., from the 21A2 → 11A1 transition. Transitions between states of A1 to A2 symmetry are one-photon, dipole-forbidden; however, coupling with the high-energy ππ* excitonic state can lend oscillator strength to these transitions.24 On degassing the solution we again observe a large increase in the intensity of the 1CT emission because of a DF contribution, such that the unaffected 1LEA nπ* emission becomes barely visible by comparison (Figure 1e,f). Dual emission through different channels at both high (330 nm) and low (350 nm) excitation energies establishes ACRSA as a doubly anti-Kasha emitter.
Figure 2.
Relative PL emission yields from aerated ACRSA toluene solution (1 mg/mL) normalized by the absorbance at each excitation wavelength, showing low quantum yield from excitation via the acridine excitonic state (330 nm excitation) compared to excitation into the direct low-energy 1CT and 1LEA transitions (370 nm excitation).
As we observe emission from both 1LEA and 1CT in aerated solution (380 nm excitation, well below the absorption tail of the 1LED excitonic state), we must conclude that both states are directly populated, yielding prompt emission. Calculation of oxygen-quenched delayed CT emission contribution (Supporting Figure S4) shows that direct 1CT absorption is more efficient at populating the 1CT state than ET from the 1LED exciton; 330 nm gives a PF/DF ratio of 6.8%, while at 380 nm PF/DF increases to 11.1% (lower triplet yield so lower DF). From the strong DF component we infer that the 1CT–3CT energy gap (11A2–13A2 gap, Supporting Scheme 1) must be small, indicating very low electron exchange energy from near degenerate orbitals. Consequently, ISC between 1CT and 3CT will be forbidden,14,26 and the strong DF CT emission observed when exciting at ≥350 nm can arise only through the photoexcited 1LEA state forming a large triplet population by ISC. Such efficient quenching by ISC—resulting in a large 3CT population and strong DF—fully supports the proposal made by Lyskov and Marian24 that the 1LEA nπ* state couples strongly to a 3LEA ππ* triplet state (13A1, Supporting Scheme 1) in competition with radiative decay.
These interpretations are further supported by excitation profiles measured at different emission collection wavelengths (Figure 3 and Supporting Figure S5). Monitoring emission from the 1CT state (at 515 and 600 nm), we observe emission arising from excitation of both the 1LED excitonic state (at <350 nm) as well as direct excitation of the 1CT (at ∼400 nm). Again, the CT emission is dominated by DF (degassed conditions), which is thoroughly quenched in aerated measurements. Monitoring at 412 nm (the peak of the 1LEA nπ* emission), excitation into the 1LED excitonic state gives weak emission, which indicates active IC from 1LED ππ* to 1LEA nπ*. Much stronger 1LEA nπ* emission is observed with excitation in the 350–400 nm range, resulting from direct 1LEA nπ* excitation. The 1LEA nπ* emission increases slightly on degassing, probably from enhancement of underlying delayed CT emission (with direct absorption from 350 to 415 nm). Excitation at wavelengths >425 nm yields no 1LEA nπ* emission, although we do observe weak 1CT emission (Supporting Figure S6), indicating that the onset of direct 1CT absorption is at lower energy than the 1LEA nπ* state. These excitation measurements demonstrate that excitation of the high-energy 1LED excitonic state gives rise to emission from three different singlet states simultaneously: 1LED exciton, 1LEA nπ*, and 1CT. While dual-emission TADF and room-temperature phosphorescent (RTP) materials are now frequently reported,27−30 extreme anti-Kasha behavior such that a molecule is capable of emitting from three singlet excited states (1LED exciton, 1LEA, and 1CT) simultaneously is exceedingly rare. We believe this is the first report of concurrent triple-pathway singlet emission from a single molecule.
Figure 3.

Excitation spectra from aerated and degassed ACRSA toluene solution (50 μM) showing the direct (≥350 nm excitation) and indirect population (from 1LED, <350 nm excitation) of the 1LEA nπ* and 1CT states.
Extending to solvents of different polarity, Figure 4 shows the emission spectra of ACRSA in MCH, toluene, and DCM (aerated and degassed) at different excitation wavelengths. Supporting Figure S7 directly compares emission between excitation at 330 and 380 nm in each solvent for additional clarity. In MCH and DCM, the same general trends are observed as in toluene. However, the structured emission from the local 1LEA nπ* state is clearly not affected by polarity (Supporting Figure S8). In MCH the 1CT emission is only very weakly stabilized, lies underneath the local 1LEA nπ* emission, but is readily distinguished by the strong CT emission observed with 330 nm excitation (into the 1LED state). The appearance of the structured local emission increasing on removal of oxygen is in fact due to this underlying CT emission increasing (Supporting Figure S9). This is confirmed by the apparent relative decrease in local emission on degassing in DCM, where the CT band is red-shifted out from underneath the local emission band (but still partially overlapping) such that the true behavior of the local transition can been seen clearly.
Figure 4.

Emission spectra of ACRSA (1 mg/mL) in aerated and degassed solvents (a and b) MCH (ε = 2.02), (c and d) toluene (ε = 2.38), and (e and f) DCM (ε = 8.93) at different excitation wavelengths.
It is interesting to compare this general emission behavior of ACRSA to its separate acridine donor and anthracenone acceptor units. Acridine emits at 337 nm (Supporting Figure S2), whereas anthracenone (or anthrone) is reported as nonemissive.31 However, Fujii et al. also report that the tautomeric form of anthrone, 9-anthrol, is emissive. In benzene the spectra are almost identical to the ACRSA emission in MCH: well structured with similar vibronic contributions. Moreover, 9-anthrol in strongly basic solvents (NaOH) emits with a broad Gaussian band, centered at ca. 550 nm, very similar to ACRSA in toluene. This shows that charge transfer is efficient in ACRSA and that even in the excited state, the D and A units are strongly decoupled. It may also highlight the mixed nature of the excited states (local and CT) and also possible similarities between the excited-state structure of the A unit in ACRSA and that of 9-anthrol. However, this cannot come from such a tautomeric form as proton transfer is impossible for the anthracenone unit that is spiro linked to the acridine in ACRSA.
We observe the largest 1CT DF emission contribution in toluene (Supporting Figures S10 and 11). The oxygen-dependent spectra in MCH and DCM (Supporting Figure S12) show that exciting into the 1LED exciton leads to an oxygen-dependent CT population (DF increasing by a factor 4 in MCH and 2 in DCM), whereas via direct excitation the increase in CT emission is only a factor of 2 in MCH and DCM. This clearly indicates that 1CT is populated significantly through the triplet manifold and most efficiently via the 1LED state.
Time-resolved emission was measured following excitation at both 337 and 355 nm (Figure 5), starting with (degassed) toluene solution. The 337 nm pulses excite the 1LED excitonic state and produce rapidly decaying emission from this state at 370 nm, within the time response of the laser/iCCD system (<1 ns). 1CT emission is also observed in the earliest spectrum (2 ns), indicating an ET rate (estimated at 108–109 s–1) that does not out-compete radiative decay and IC of the 1LED state but is faster than our time resolution. This relatively slow ET is consistent with the strongly decoupled D and A electronic systems through their near perfect orthogonality. A blue-edge shoulder on the CT emission band indicates a small contribution from 1LEA nπ* emission as well, confirming that the 1LED exciton couples to both lower-lying singlet states.
Figure 5.

Area-normalized time-resolved emission decay of ACRSA in degassed MCH (a), toluene (b), and DCM (c) solutions (50 μM) excited at 337 nm into the 11B1 exciton transition and at 355 nm into the direct mixed 21A2 and 11A2 transitions (d–f). Poor solubility in MCH leads to the observed emission from dimer/excimer states at long wavelengths. A corresponding set of peak normalized spectra are given in Supporting Figure S13.
In all solvents we observe little energy relaxation of the 1CT state with time, indicating a rigid dihedral angle with little dispersion of the 1CT state energy. From the kinetic decay traces in toluene (Figure 6), we observe that the lifetime of the 1LEA nπ* state is very short, of order 2 ns (beginning of region i). Because this emission is a nominally forbidden transition, the short lifetime implies a rapid parallel quenching mechanism, most likely ISC. The prompt CT emission (region ii) is much longer, 274 ± 5 ns.32 Comparing decays using 337 and 355 nm excitation, we observe stronger coupling of the high-energy excitonic state to the 1CT state giving relatively more 1CT prompt emission. Moreover, delayed CT emission from following 337 nm excitation starts to contribute at rather early times, from ca. 600 ns until beyond 10 μs (region iii) with a lifetime of 5.21 ± 0.02 μs, implying a rapid buildup of triplet population. In contrast, excitation at 355 nm directly into the 1LEA/ 1CT bands gives more intense local 1LEA emission at early times with lifetime ca. 5 ns (more pronounced region i). Long-lived prompt 1CT emission, 264 ± 5 ns, and DF lifetime of 5.23 ± 0.06 μs are observed. Kinetic modeling32 gives rISC rates of 5.29 × 105 and 6.2 × 105 s–1, respectively, with all kinetic data summarized in Supporting Table 1. Isoemissive points in the time-resolved area-normalized emission spectra (Figure 5) indicate that the 1LEA and 1CT states decay independently of each other, consistent with simultaneous photoexcitation but no IC, electron or energy transfer, or vibronic coupling between them. This all indicates very slow radiative decay rates from the CT state consistent with a forbidden transition. This slow decay also demonstrates that ISC to the isoenergetic 3CT is highly forbidden, as there are no close-lying local triplet states to mediate ISC to the 3CT. Nonradiative decay must also be strongly suppressed, indicating few coupling vibrational modes to mediate IC.
Figure 6.

Emission decay kinetics for ACRSA in MCH (a), toluene (b), and DCM (c) (50 μM) as a function of excitation wavelength. Three main kinetic decay regions can be defined for (i) fast 1LEA state decay, (ii) slow prompt 1CT decay, and (iii) delayed 1CT decay. Panels d, e, and f show the effect of oxygen quenching on the kinetic decays (in aerated solvents).
In DCM excitation at both 337 and 355 nm photoexcites a much larger initial 1LEA population than in toluene, which is not well-structured (in line with the steady-state measurements). The 1LEA emission also persists for longer, 10 and 16 ns, respectively. This increase in lifetime reflects a potential blue shift of the 1LEA nπ* and red shift of 3LEA ππ* with increasing solvent polarity, reducing the ISC rate between them. The total emission intensity is however much lower in DCM than in either toluene or MCH, which may result from increased nonradiative quenching of the lower-energy 1CT and 3CT states (energy gap law). In comparson with the decay kinetics in toluene (Figure 6), at 355 nm excitation the prompt 1CT state decays much more rapidly than in toluene (91 ± 3 ns). The DF component in DCM dominates emission after 300 ns (lifetime 2.38 ± 0.06 μs) and has a slower ISC rate (1.18 × 106 s–1) than in toluene. The rISC rate is also slower at 4.83 × 105 s–1. The CT triplet state involved in rISC, which relaxes in energy with increasing polarity, must therefore open up the gap between itself and the TADF-mediating local triplet state, reducing the rate of rISC and thus DF contribution in polar DCM.26 In toluene the DF is much stronger but the rISC rate is similar, again indicative of the lower-energy DCM CT states experiencing increased nonradiative decay (energy gap law). The faster radiative decay rates calculated in DCM compared to toluene coupled with the lower overall emission intensity is also explained by an increased nonradiative decay rate; our kinetic model assumes no nonradiative decay (other than ISC) and the fitted “radiative” rate will include both radiative and nonradiative rates.
In MCH the picture is different again. As the 1CT and 1LEA nπ* states are much closer in energy, it is difficult to deconvolute the behavior of each. From the decay kinetics, the 1LEA nπ* decays very rapidly as in toluene, with lifetime of ca. 1–2 ns. The prompt CT states decay much faster; the prompt CT lifetime between 5 and 8 ns indicates rapid radiative decay (rates of 4 to 8 × 107 s–1) of the 1CT state because of its high LE character (structured emission, Figure 4a33). The DF also decays rapidly over only a few microseconds, with a lifetime of ca. 350 ns. With 337 nm excitation (Figure 6 and Supporting Figure S13), in the first 10 ns emission covers the whole spectral window between 360 and 650 nm. This we assume is simultaneous emission from multiple excited states, along with possible aggregate emission. Even at early times, the 1CT emission is much stronger than that from the local 1LEA nπ* state. All of these states decay rapidly leaving just the main 1CT emission, as seen in the steady-state emission.
Phosphorescence was collected from ACRSA in a zeonex polymer matrix at 80 K (Supporting Figure S14). From this we deduce that the lowest-energy triplet state is of local character, with energy 2.88 eV (0–0 vibronic peak) and well-resolved structure. From the spectral shape we conclude that the emission emanates from the acceptor anthracenone unit, i.e., 3LEA nπ* state. At very long delay times (80 ms) we observe the emergence of another phosphorescence band. With little structure and onset of 3.2 eV, this matches well with reported acridine phosphorescence.25 Such dual phosphorescence was previously reported by us in other TADF D–A systems arising from a thermal equilibrium between D and A triplet states which are vibrationally coupled.26,34 The observation of dual phosphorescence establishes ACRSA as an anti-Kasha material on the triplet manifold in addition to its profoundly anti-Kasha singlet behavior.
We also examine the effect of oxygen quenching on the emission dynamics (Figure 6 and Supporting Figure S18). In toluene and DCM the early time emission spectra and decays are unaffected by oxygen, as would be expected from prompt emitting states, but lifetimes are a little longer, which we have no clear idea about at this time. In toluene, the prompt CT emission is quenched after ca. 36 μs (presumably by oxygen), but we observe weak DF, lifetime of ca. 120–240 ns accounting for about 5% of the total intensity. It is therefore remarkable to observe DF CT emission for such a long time in air.
Figure 7 shows transient absorption spectra of ACRSA in oxygen-free toluene for the first 6 ns (343 nm excitation) and 400 ns after pump (355 nm excitation). A band centered at 620 nm is seen to grow in after an induction time of ∼25 ps. The induced band can be resolved into two peaks centered at 619 and at 675 nm. Both of these peaks have the same decay kinetics, with the entire band growing in for ∼8–10 ns and decaying after ∼200 ns. Estimation of the band edge energy of each peak (assuming a Gaussian peak shape) yields energies of 1.84 and 1.65 eV, giving an energy separation of 190 meV, corresponding precisely with the C=O stretch of anthracenone. This is also the mode that vibronically couples the LE and CT triplet states.35
Figure 7.

Nanosecond transient absorption spectra of ACRSA in toluene (1 mg/mL) from 0 to 5 ns following 343 nm excitation (left panel) and 0–400 ns following 355 nm excitation (right panel).
As the induced absorption has a lifetime of 200 ns it cannot correspond to transitions from the short-lived local singlet 1LED or 1LEA states. It also cannot be a transition from the long-lived (>3 μs) 1/3CT population, and the 1CT emission energy is too low to accommodate an induced absorption at the observed energies. By elimination, this induced absorption is assigned to a transition from a 3LE triplet state. Supporting this, measurements in aerated solution gave very poor signal, consistent with oxygen quenching of triplet states. The grow-in kinetics of this induced band matches well with the fast decay of the local 1LED state with 343 nm excitation. We thus conclude that the induced absorption comes from a transient 3LEA ππ* population, having lifetime of ∼200 ns as it undergoes subsequent IC to the lower energy 3LEAnπ* triplet state. With excitation at 355 nm we initially populate the 1LEA nπ* state which rapidly undergoes ISC to populate the 3LEA ππ*. In both cases we observe the same photoinduced absorption spectra, confirming the common 3LEA ππ* assignment. The induction time observed with 343 nm excitation corresponds to the slower ISC and subsequent IC steps required to reach the 3LEA ππ* state from the 1LED (ISC to 3LED then IC). At low temperatures when vibrational coupling and ISC/IC are inefficient, these 1LED/A states give rise to the observed dual phosphorescence.
The molecular photophysics of TADF in ACRSA is substantially different from that of established TADF molecules, with different ISC and rISC channels.36 Strong solvatochromic relaxation of the lowest-energy singlet 1CT state occurs, while a close-lying 1LEA nπ* state remains unaffected by polarity. Both of these A2 states are emissive despite being one-photon symmetry forbidden. The 1LEA nπ* is quenched to its 3LE ππ* triplet state through very fast ISC in accordance with El Sayed’s rule. In transient absorption a rapid grow-in of a 3LE ππ* induced absorption is in agreement with this explanation. This ISC step out competes any vibrational coupling to the 1CT state, as confirmed by the time-resolved emission spectra showing that these states decay independently. In MCH the 1CT state has strong LE character giving fast radiative decay rates and a short PF CT lifetime compared to toluene and DCM (Supporting Table 1). The PF CT emission has a long lifetime in toluene and DCM because the 1CT state is energetically distant from the 1LEA nπ* state, precluding second-order vibronic coupling to any local state (singlet or triplet). This, along with the forbidden nature of direct 1CT to 3CT SOC, is clearly observed through the extremely long 1CT lifetime. In DCM, the CT states shift so low in energy that coupling to the ground state increases nonradiative decay (energy gap law). However, the presence of TADF in all solvents confirms that isolation of the 1CT is not important in the formation of a long-lived 3CT population because of the role of the 1LEA nπ* state.
Optically the DF emission intensity is low in MCH despite more appropriate energy level ordering. This we suggest is due to rISC from the 3CT state being in continuous competition with IC back to the strongly coupled 3LEA nπ* state. This is also why we can observe resolved phosphorescence from this local triplet state. In MCH we do still identify a fast DF signal quenched by oxygen. This confirms the strong enhancement of both radiative decay and rISC in MCH, achieved through near resonant 1CT and 3CT states (with high local character) and closely spaced 1LEA ππ* singlet, 3LEA ππ*, and 3LEA nπ* triplet states. This alignment of energy levels yields both small energy gaps for efficient nonadiabatic vibronic coupling and large SOC matrix elements, giving very fast rISC rate of ∼1 × 107 s–1. This fast rISC rate is required for it to compete with rapid IC to the well-coupled lowest-energy 3LEA nπ* state.
As the polarity of the solvent increases 1CT and 3CT energetically relax, with 3CT becoming the lowest-energy triplet state. A larger gap opens to the local states especially to the 3LEA nπ*, which increases in energy with increasing solvent polarity, as observed in other aromatic ketone systems.37,38 The measured rISC rates consequently fall by nearly 2 orders of magnitude with increasing polarity (Supporting Table 1), reflecting the increasing energy gap between 3CT and local states (but uniform energy gaps between the local singlet and triplet acceptor states).39 The highly structured phosphorescence from the anthracenone acceptor unit indicates that in nonpolar solvents the 3LEA nπ* triplet state must be below the TADF-active 3CT state.
In all solvents we never observe DF from the 1LEA nπ*, but we do observe dual 1LEA nπ* and 1CT emission. This clearly indicates the two states are decoupled. In MCH we see that the 1LEA nπ* and unrelaxed 1CT energies are very close, resulting in a very low driving force for ET from the 1LEA nπ* state insufficient to drive charge transfer, especially in competition with fast quenching by ISC.
From the time-resolved emission spectra (Figure 5) it is clear that in all solvents the CT state that gives prompt and DF emission does not red shift significantly with time. This emission is also relatively narrow, indicating solvent shell reorganization is very fast and there is no electronic relaxation of the CT state caused by slow molecular reorganization.40,41
ACRSA is unique in that it allows excitation into more than one singlet state. Below 350 nm, excitation of the 1LED ππ* exciton yields fast emission from this high-energy excitonic singlet state observed in aerated solutions, in competition with electron transfer to create the 1CT state. However, the main deactivation pathway of the 1LED ππ* exciton is via ISC to upper triplet states, which then decay down the triplet manifold to populate 3CT triplet states, yielding DF from subsequent rISC. This is confirmed by the strong oxygen dependence of the DF CT emission we observe and the results from photoinduced absorption measurements. Nonradiative decay can occur via other triplet states that do not couple to the rISC-active lowest-energy CT triplet state, leading to excited-state quenching. Excitation above 350 nm directly populates the 1CT and 1LEA nπ* excitonic states, yielding simultaneous prompt emission from both states.
These results confirm the very fast ISC channel from 1LEA nπ* to 3LEA ππ* in ACRSA, as predicted by Lyskov and Marian. rISC remains much slower than this in polar solvents, typically with 2–5 μs lifetime. However, in MCH far higher rISC rates are seen as the TADF-active 3CT state rises in energy toward the 3LE states.24
This leads us to a proposed “experimental” energy level scheme given in Figure 8, based on the calculations of Lyskov and Marian and our experimental observations.
Figure 8.
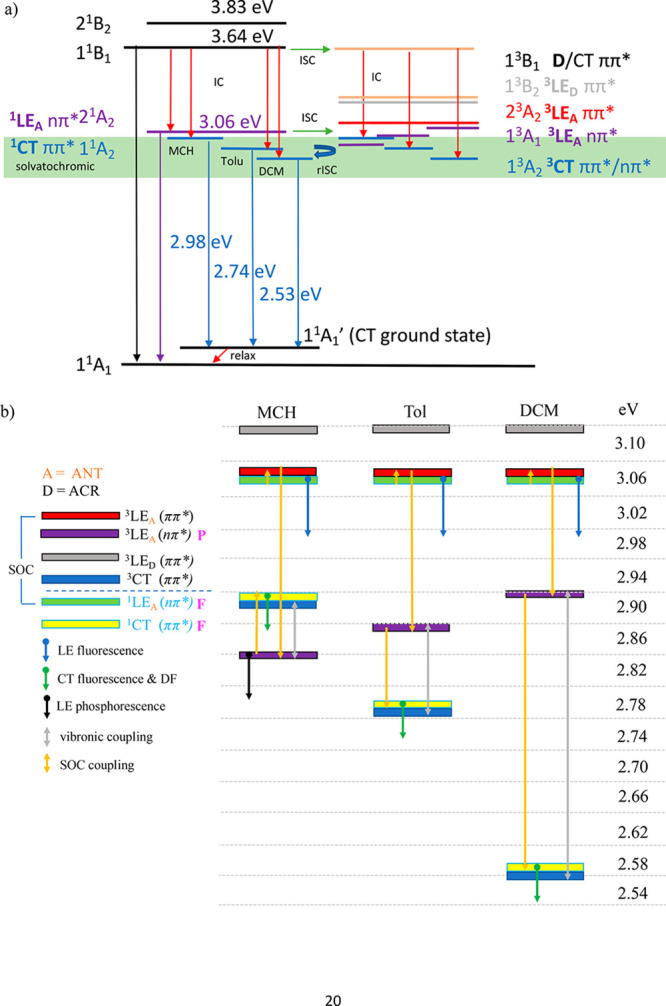
Energy level scheme for ACRSA. (a) Measured energy levels from spectral onsets of ACRSA in different polarity solvents, following the nomenclature of Lyskov and Marian.24 The green band represents the energy range over which we observe solvatochromic states. Red arrows represent nonradiative transitions. (b) The effect of solvent polarity on the states and the SOC (yellow arrows) and vibronic coupling (gray arrows) between states giving rise to ISC and rISC.
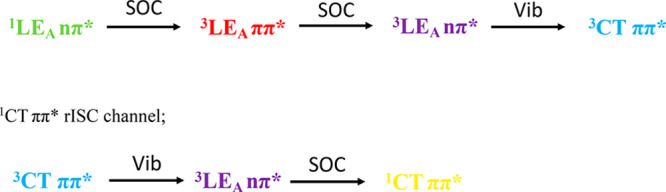
Combining all of our experimental observations we conclude the following ISC and rISC channels populate the CT states in ACRSA via direct optical excitation of the 1LEA nπ*.
1LEA nπ* ISC channel:
 |
1 |
We observe that the 1LEA nπ* and ππ* singlet states are not affected by polarity, but from calculation and reports of other ketones, the triplet nπ* states are more affected, shifting up in energy, which we depict in Figure 7. Thus, the SOC step via3LEA ππ* states should be minimally affected by polarity changes. The rate-limiting step is likely the 3CT coupling to the 3LEA nπ* through vibronic coupling and so is not as impacted by the increasing energy gap in toluene and DCM. The overall rISC rate is controlled by this vibronic coupling, decreasing rISC rates in toluene and DCM compared to MCH from 107 s–1 to ca. 5 × 105 s–1. In air we observe 1CT quenching because of the very long natural lifetime in toluene and DCM. We propose this simpler rISC mechanism compared to that of Lyskov and Marian because we never observe delayed emission from the 1LEA nπ* state.
In this study, the “simple” spiro TADF molecule ACRSA is demonstrated to be anything but. We find both strong excitation energy and solvent polarity dependencies in its emission. Exciting the first allowed local ππ* excitonic state of the donor acridine gives rise to three different emissions: one from the excitonic state itself (that competes with both ISC and IC, and electron transfer to two lower lying, one-photon forbidden A2 symmetry states), one from the local nπ* state of the anthracenone acceptor, and also emission from a CT state. Dual emission from these symmetry forbidden (A2) states is indicative of mixing with the donor excitonic states in ACRSA. These lower energy states have weak direct absorption giving rise to prompt local 1LEA nπ* state emission and both prompt and delay CT emission. On the removal of oxygen, we observe very large increases in the emission from the CT state, indicating a large delayed emission contribution. We find no DF from the 1LEA nπ* state however. Both direct and indirect excitation (via the excitonic state) gives very large DF emission in degassed solutions.
In agreement with DFT/MRCI calculations, fast ISC between the lowest-energy 1LEA nπ* singlet state and 3LEA ππ* triplet state is established from the correspondence of 1LEA nπ* emission decay and grow-in of induced absorption from the 3LEA ππ* state, confirming strong SOC between these states. In MCH we observe very strong enhancement of both radiative decay and rISC, achieved through near-resonant 1CT and 3CT states (having high local character) as well as nearby 1LE nπ* singlet and 3LE ππ* triplet states. This alignment yields high radiative decay rates for the 1CT state, small energy gaps for nonadiabatic vibronic coupling, and high SOC rates. The net result is very high rISC rates of 107 s–1. However, rapid ISC from excitation to higher-energy singlet states yields fewer triplet states and lower DF. This indicates competing decay channels on the triplet manifold, with not all excitons reaching the lowest-energy TADF-active triplet state. In devices, similar ISC from high-energy excitons terminating in TADF-inactive triplet states may be a mechanism that explains the generally unexceptional OLED performance of ACRSA and other spiro-linked derivatives.
Throughout our optical measurements ACRSA gives radiative decay from up to three different excited states simultaneously (at early times) and two states even at tens of nanoseconds. Combined with observed dual phosphorescence this “simple” molecule therefore utterly disregards Kasha’s law. The perpendicular and rigid geometry of the molecule—which very effectively decouples A and D as required—is what allows us to see identifiable photophysical properties of the donor (1LED ππ* exciton emission, in competition with slow ET and nonradiative decay), alongside formally forbidden 1CT and 1LEA nπ* emission. ACRSA’s “simple” spiro structure therefore bestows it with unprecedented and previously unrecognized richness in its optical properties because of this very effective electronic decoupling of the donor and acceptor units, apart from the molecular CT state. This is not limited to ACRSA, nor spiro TADF molecules in general, but all TADF molecules because of the requirement to effectively decouple the D and A to yield CT states with vanishing singlet–triplet energy gaps, a necessary (but not sufficient) requirement for efficient TADF. This whole class of materials can be thought of as a combination of donor, acceptor, and molecular CT state giving their own photophysics, as is the case for an exciplex TADF systems10,42 and emerging through-space TADF materials, where the linkage and coupling between the D and A is even less well-defined. Thus, in its original form, does Kasha’s rule still apply to such a “system”?
Acknowledgments
We thank the EPSRC (EP/P012167/1) and acknowledge the TADFlife and HyperOLED projects funded by the European Union’s Horizon 2020-MCSA-ITN Research and Innovation Programme under Grant Agreements No. 812872 and No. 732013 under the action ICT-02-2016 for funding the research in Durham. We acknowledge fruitful conversations with Professor C. Marian.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpclett.0c03314.
Experimental details; additional photophysical data such as absorption spectra, time-resolved emission decays and steady-state spectra of ACRSA in different solvents, excitation, and concentrations; phosphorescence spectra of ACRSA in zeonex matrix; absorption and emission spectra of acridine; lifetime fittings (PDF)
Author Contributions
L.G.F., Y.L., A.D., and C.L. made the presented optical and time-resolved spectroscopy measurements. A.M. devised the research, undertook the data analysis, and supervised the work. A.M. wrote the manuscript with L.G.F. and A.D., with contributions from all authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Mizukami M.; Cho S. Il; Watanabe K.; Abiko M.; Suzuri Y.; Tokito S.; Kido J. Flexible Organic Light-Emitting Diode Displays Driven by Inkjet-Printed High-Mobility Organic Thin-Film Transistors. IEEE Electron Device Lett. 2018, 39, 39–42. 10.1109/LED.2017.2776296. [DOI] [Google Scholar]
- Huang T.; Jiang W.; Duan L. Recent Progress in Solution Processable TADF Materials for Organic Light-Emitting Diodes. J. Mater. Chem. C 2018, 6, 5577–5596. 10.1039/C8TC01139G. [DOI] [Google Scholar]
- Forrest S. R. The Path to Ubiquitous and Low-Cost Organic Electronic Appliances on Plastic. Nature 2004, 428, 911–918. 10.1038/nature02498. [DOI] [PubMed] [Google Scholar]
- Kim J. J.; Han M. K.; Noh Y. Y. Flexible OLEDs and Organic Electronics. Semicond. Sci. Technol. 2011, 26, 030301. 10.1088/0268-1242/26/3/030301. [DOI] [Google Scholar]
- Baldo M. A.; O’Brien D. F.; You Y.; Shoustikov A.; Sibley S.; Thompson M. E.; Forrest S. R. Highly Efficient Phosphorescent Emission from Organic Electroluminescent Devices. Nature 1998, 395, 151–154. 10.1038/25954. [DOI] [Google Scholar]
- Adachi C.; Baldo M. A.; Thompson M. E.; Forrest S. R. Nearly 100% Internal Phosphorescence Efficiency in an Organic Light-Emitting Device. J. Appl. Phys. 2001, 90, 5048–5051. 10.1063/1.1409582. [DOI] [Google Scholar]
- Shizu K.; Uejima M.; Nomura H.; Sato T.; Tanaka K.; Kaji H.; Adachi C. Enhanced Electroluminescence from a Thermally Activated Delayed-Fluorescence Emitter by Suppressing Nonradiative Decay. Phys. Rev. Appl. 2015, 3, 014001. 10.1103/PhysRevApplied.3.014001. [DOI] [Google Scholar]
- Uoyama H.; Goushi K.; Shizu K.; Nomura H.; Adachi C. Highly Efficient Organic Light-Emitting Diodes from Delayed Fluorescence. Nature 2012, 492, 234–238. 10.1038/nature11687. [DOI] [PubMed] [Google Scholar]
- Dias F. B.; Bourdakos K. N.; Jankus V.; Moss K. C.; Kamtekar K. T.; Bhalla V.; Santos J.; Bryce M. R.; Monkman A. P. Triplet Harvesting with 100% Efficiency by Way of Thermally Activated Delayed Fluorescence in Charge Transfer OLED Emitters. Adv. Mater. 2013, 25, 3707–3714. 10.1002/adma.201300753. [DOI] [PubMed] [Google Scholar]
- Colella M.; Danos A.; Monkman A. P. Less Is More: Dilution Enhances Optical and Electrical Performance of a TADF Exciplex. J. Phys. Chem. Lett. 2019, 10, 793–798. 10.1021/acs.jpclett.8b03646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im Y.; Kim M.; Cho Y. J.; Seo J.-A.; Yook K. S.; Lee J. Y. Molecular Design Strategy of Organic Thermally Activated Delayed Fluorescence Emitters. Chem. Mater. 2017, 29, 1946–1963. 10.1021/acs.chemmater.6b05324. [DOI] [Google Scholar]
- Stachelek P.; Ward J. S.; Dos Santos P. L.; Danos A.; Colella M.; Haase N.; Raynes S. J.; Batsanov A. S.; Bryce M. R.; Monkman A. P. Molecular Design Strategies for Color Tuning of Blue TADF Emitters. ACS Appl. Mater. Interfaces 2019, 11, 27125. 10.1021/acsami.9b06364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang R.; Kukhta N. A.; Ward J. S.; Danos A.; Batsanov A. S.; Bryce M. R.; Dias F. B. Balancing Charge-Transfer Strength and Triplet States for Deep-Blue Thermally Activated Delayed Fluorescence with an Unconventional Electron Rich Dibenzothiophene Acceptor. J. Mater. Chem. C 2019, 7, 13224–13234. 10.1039/C9TC02175B. [DOI] [Google Scholar]
- Lim B. T.; Okajima S.; Chandra A. K.; Lim E. C. Radiationless Transitions in Electron Donor-Acceptor Complexes: Selection Rules for S1 → T Intersystem Crossing and Efficiency of S1 → S0 Internal Conversion. Chem. Phys. Lett. 1981, 79, 22–27. 10.1016/0009-2614(81)85280-3. [DOI] [Google Scholar]
- Gibson J.; Monkman A. P.; Penfold T. J. The Importance of Vibronic Coupling for Efficient Reverse Intersystem Crossing in Thermally Activated Delayed Fluorescence Molecules. ChemPhysChem 2016, 17, 2956–2961. 10.1002/cphc.201600662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatakeyama T.; Shiren K.; Nakajima K.; Nomura S.; Nakatsuka S.; Kinoshita K.; Ni J.; Ono Y.; Ikuta T. Ultrapure Blue Thermally Activated Delayed Fluorescence Molecules: Efficient HOMO-LUMO Separation by the Multiple Resonance Effect. Adv. Mater. 2016, 28, 2777–2781. 10.1002/adma.201505491. [DOI] [PubMed] [Google Scholar]
- Romanov A. S.; Jones S. T. E.; Gu Q.; Conaghan P. J.; Drummond B. H.; Feng J.; Chotard F.; Buizza L.; Foley M.; Linnolahti M.; Credgington D.; Bochmann M. Carbene Metal Amide Photoemitters: Tailoring Conformationally Flexible Amides for Full Color Range Emissions Including White-Emitting OLED. Chem. Sci. 2020, 11, 435–446. 10.1039/C9SC04589A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall D.; Suresh S. M.; dos Santos P. L.; Duda E.; Bagnich S.; Pershin A.; Rajamalli P.; Cordes D. B.; Slawin A. M. Z.; Beljonne D.; Köhler A.; Samuel I. D. W.; Olivier Y.; Zysman-Colman E. Improving Processability and Efficiency of Resonant TADF Emitters: A Design Strategy. Adv. Opt. Mater. 2020, 8, 1901627. 10.1002/adom.201901627. [DOI] [Google Scholar]
- Méhes G.; Nomura H.; Zhang Q.; Nakagawa T.; Adachi C. Enhanced Electroluminescence Efficiency in a Spiro-Acridine Derivative through Thermally Activated Delayed Fluorescence. Angew. Chem., Int. Ed. 2012, 51, 11311–11315. 10.1002/anie.201206289. [DOI] [PubMed] [Google Scholar]
- Nakagawa T.; Ku S. Y.; Wong K. T.; Adachi C. Electroluminescence Based on Thermally Activated Delayed Fluorescence Generated by a Spirobifluorene Donor–Acceptor Structure. Chem. Commun. 2012, 48, 9580–9582. 10.1039/c2cc31468a. [DOI] [PubMed] [Google Scholar]
- Ohkuma H.; Nakagawa T.; Shizu K.; Yasuda T.; Adachi C. Thermally Activated Delayed Fluorescence from a Spiro-Diazafluorene Derivative. Chem. Lett. 2014, 43, 1017–1019. 10.1246/cl.140360. [DOI] [Google Scholar]
- Wang Y. K.; Huang C. C.; Ye H.; Zhong C.; Khan A.; Yang S. Y.; Fung M. K.; Jiang Z. Q.; Adachi C.; Liao L. S. Through Space Charge Transfer for Efficient Sky-Blue Thermally Activated Delayed Fluorescence (TADF) Emitter with Unconjugated Connection. Adv. Opt. Mater. 2020, 8, 1901150. 10.1002/adom.201901150. [DOI] [Google Scholar]
- Nasu K.; Nakagawa T.; Nomura H.; Lin C.-J.; Cheng C.-H.; Tseng M.-R.; Yasuda T.; Adachi C. A Highly Luminescent Spiro-Anthracenone-Based Organic Light-Emitting Diode Exhibiting Thermally Activated Delayed Fluorescence. Chem. Commun. 2013, 49, 10385. 10.1039/c3cc44179b. [DOI] [PubMed] [Google Scholar]
- Lyskov I.; Marian C. M. Climbing up the Ladder: Intermediate Triplet States Promote the Reverse Intersystem Crossing in the Efficient TADF Emitter ACRSA. J. Phys. Chem. C 2017, 121, 21145–21153. 10.1021/acs.jpcc.7b06187. [DOI] [Google Scholar]
- dos Santos P. L.; Ward J. S.; Bryce M. R.; Monkman A. P. Using Guest–Host Interactions to Optimize the Efficiency of TADF OLEDs. J. Phys. Chem. Lett. 2016, 7, 3341–3346. 10.1021/acs.jpclett.6b01542. [DOI] [PubMed] [Google Scholar]
- Dias F. B.; Santos J.; Graves D. R.; Data P.; Nobuyasu R. S.; Fox M. A.; Batsanov A. S.; Palmeira T.; Berberan-Santos M. N.; Bryce M. R.; et al. The Role of Local Triplet Excited States and D-A Relative Orientation in Thermally Activated Delayed Fluorescence: Photophysics and Devices. Adv. Sci. 2016, 3, 1600080. 10.1002/advs.201600080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang R.; Ward J. S.; Kukhta N. A.; Avó J.; Gibson J.; Penfold T.; Lima J. C.; Batsanov A. S.; Berberan-Santos M. N.; Bryce M. R.; et al. The Influence of Molecular Conformation on the Photophysics of Organic Room Temperature Phosphorescent Luminophores. J. Mater. Chem. C 2018, 6, 9238–9247. 10.1039/C8TC02987C. [DOI] [Google Scholar]
- Ward J. S.; Nobuyasu R. S.; Fox M. A.; Batsanov A. S.; Santos J.; Dias F. B.; Bryce M. R. Bond Rotations and Heteroatom Effects in Donor–Acceptor–Donor Molecules: Implications for Thermally Activated Delayed Fluorescence and Room Temperature Phosphorescence. J. Org. Chem. 2018, 83, 14431–14442. 10.1021/acs.joc.8b02187. [DOI] [PubMed] [Google Scholar]
- Nobuyasu R. S.; Ward J. S.; Gibson J.; Laidlaw B. A.; Ren Z.; Data P.; Batsanov A. S.; Penfold T. J.; Bryce M. R.; Dias F. B. The Influence of Molecular Geometry on the Efficiency of Thermally Activated Delayed Fluorescence. J. Mater. Chem. C 2019, 7, 6672–6684. 10.1039/C9TC00720B. [DOI] [Google Scholar]
- Ward J. S.; Nobuyasu R. S.; Fox M. A.; Aguilar J. A.; Hall D.; Batsanov A. S.; Ren Z.; Dias F. B.; Bryce M. R. Impact of Methoxy Substituents on Thermally Activated Delayed Fluorescence and Room-Temperature Phosphorescence in All-Organic Donor-Acceptor Systems. J. Org. Chem. 2019, 84, 3801–3816. 10.1021/acs.joc.8b02848. [DOI] [PubMed] [Google Scholar]
- Fujii T.; Mishima S.; Tanaka N.; Kawauchi O.; Kodaira K.; Nishikiori H.; Kawai Y. Absorption and Fluorescence Spectra of 9-Anthrol and Its Chemical Species in Solution. Res. Chem. Intermed. 1997, 23, 829–839. 10.1163/156856797X00105. [DOI] [Google Scholar]
- Haase N.; Danos A.; Pflumm C.; Morherr A.; Stachelek P.; Mekic A.; Brütting W.; Monkman A. P. Kinetic Modeling of Transient Photoluminescence from Thermally Activated Delayed Fluorescence. J. Phys. Chem. C 2018, 122, 29173–29179. 10.1021/acs.jpcc.8b11020. [DOI] [Google Scholar]
- De Sa Pereira D.; Menelaou C.; Danos A.; Marian C.; Monkman A. P. Electroabsorption Spectroscopy as a Tool for Probing Charge Transfer and State Mixing in Thermally Activated Delayed Fluorescence Emitters. J. Phys. Chem. Lett. 2019, 10, 3205–3211. 10.1021/acs.jpclett.9b00999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobuyasu R. S.; Ren Z.; Griffiths G. C.; Batsanov A. S.; Data P.; Yan S.; Monkman A. P.; Bryce M. R.; Dias F. B. Rational Design of TADF Polymers Using a Donor-Acceptor Monomer with Enhanced TADF Efficiency Induced by the Energy Alignment of Charge Transfer and Local Triplet Excited States. Adv. Opt. Mater. 2016, 4, 597–607. 10.1002/adom.201500689. [DOI] [Google Scholar]
- Marian C. M. Mechanism of the Triplet-to-Singlet Upconversion in the Assistant Dopant ACRXTN. J. Phys. Chem. C 2016, 120, 3715–3721. 10.1021/acs.jpcc.6b00060. [DOI] [Google Scholar]
- Etherington M. K.; Gibson J.; Higginbotham H. F.; Penfold T. J.; Monkman A. P. Revealing the Spin–Vibronic Coupling Mechanism of Thermally Activated Delayed Fluorescence. Nat. Commun. 2016, 7, 13680. 10.1038/ncomms13680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundt R.; Villnow T.; Ziegenbein C. T.; Gilch P.; Marian C.; Rai-Constapel V. Thioxanthone in Apolar Solvents: Ultrafast Internal Conversion Precedes Fast Intersystem Crossing. Phys. Chem. Chem. Phys. 2016, 18, 6637–6647. 10.1039/C5CP06849E. [DOI] [PubMed] [Google Scholar]
- Rai-Constapel V.; Villnow T.; Ryseck G.; Gilch P.; Marian C. M. Chimeric Behavior of Excited Thioxanthone in Protic Solvents: II. Theory. J. Phys. Chem. A 2014, 118, 11708–11717. 10.1021/jp5099415. [DOI] [PubMed] [Google Scholar]
- Dias F. B.; Penfold T. J.; Monkman A. P. Photophysics of Thermally Activated Delayed Fluorescence Molecules. Methods Appl. Fluoresc. 2017, 5, 012001. 10.1088/2050-6120/aa537e. [DOI] [PubMed] [Google Scholar]
- Ward J. S.; Nobuyasu R. S.; Batsanov A. S.; Data P.; Monkman A. P.; Dias F. B.; Bryce M. R. The Interplay of Thermally Activated Delayed Fluorescence (TADF) and Room Temperature Organic Phosphorescence in Sterically-Constrained Donor–Acceptor Charge-Transfer Molecules. Chem. Commun. 2016, 52, 2612. 10.1039/C5CC09645F. [DOI] [PubMed] [Google Scholar]
- Santos P. L.; Ward J. S.; Data P.; Batsanov A. S.; Bryce M. R.; Dias F. B.; Monkman A. P. Engineering the Singlet–Triplet Energy Splitting in a TADF Molecule. J. Mater. Chem. C 2016, 4, 3815–3824. 10.1039/C5TC03849A. [DOI] [Google Scholar]
- dos Santos P. L.; Dias F. B.; Monkman A. P. Investigation of the Mechanisms Giving Rise to TADF in Exciplex States. J. Phys. Chem. C 2016, 120, 18259–18267. 10.1021/acs.jpcc.6b05198. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.