

Abstract
Objectives
To describe CSF‐defined neuronal intermediate filament (NIF) autoimmunity.
Methods
NIF‐IgG CSF‐positive patients (41, 0.03% of 118599 tested, 1996–2019) were included (serum was neither sensitive nor specific). Criteria‐based patient NIF‐IgG staining of brain and myenteric NIFs was detected by indirect immunofluorescence assay (IFA); NIF‐specificity was confirmed by cell‐based assays (CBAs, alpha internexin, neurofilament light [NF‐L]), heavy‐[NF‐H] chain).
Results
Sixty‐one percent of 41 patients were men, median age, 61 years (range, 21–88). Syndromes were encephalopathy predominant (23), cerebellar ataxia predominant (11), or myeloradiculoneuropathies (7). MRI abnormalities (T2 hyperintensities of brain, spinal cord white matter tracts. and peripheral nerve axons) and neurophysiologic testing (EEG, EMG, evoked potentials) co‐localized with clinical neurological phenotypes (multifocal in 29%). Thirty patients (73%) had ≥ 1 immunological perturbation: cancer (paraneoplastic), 22; systemic infection (parainfectious [including ehrlichosis, 3] or HIV), 7; checkpoint‐inhibitor cancer immunotherapy, 4; other, 5. Cancers were as follows: neuroendocrine‐lineage carcinomas, 12 (small cell, 6; Merkel cell, 5; pancreatic, 1 [11/12 had NF‐L‐IgG detected, versus 8/29 others, P = 0.0005]) and other, 11. Onset was predominantly subacute (92%) and accompanied by inflammatory CSF (75%), and immunotherapy response (77%). In contrast, CSF controls (15684 total) demonstrated NIF‐IgG negativity (100% of test validation controls), and low frequencies of autoimmune diagnoses (20% of consecutively referred clinical specimens) and neuroendocrine‐lineage carcinoma diagnosis (3.1% vs. 30% of NIF cases), P < 0.0001. Median NF‐L protein concentration was higher in 8 NF‐L‐IgG‐positive patients (median, 6718 ng/L) than 16 controls.
Interpretation
Neurological autoimmunity, defined by CSF‐detected NIF‐IgGs, represents a continuum of treatable axonopathies, sometimes paraneoplastic or parainfectious.
Introduction
Several proteins with prominent structural integrity roles in neurons and glia have been reported as substrates for neurological autoimmunity, including the axon‐abundant neuronal intermediate filaments (NIFs). 1 , 2 , 3 , 4 NIF‐IgGs from affected patients produce either of two criteria‐based patterns of IgG binding to neuronal filamentous elements on murine tissue‐based indirect immunofluorescence assay (IFA). 1 For neurological diagnosis, a testing algorithm that included alpha internexin (αIN, which is not a neurofilament), neurofilament light chain (NF‐L), and heavy chain (NF‐H)‐IgGs have proven sufficient for confirmation of tissue findings and clinical interpretation. An antibody profile that included NF‐L‐IgG positivity was associated with paraneoplastic CNS disorders accompanied by neuroendocrine‐lineage carcinomas, most commonly Merkel cell or small cell type. 1 Clinical significance remained uncertain in patients with other NF‐L‐IgG‐negative profiles, many of whom were historical cases with only serum and limited clinical data available. A parainfectious cause in some patients also appears plausible, as we have previously identified some cases of ehrlichosis preceding neurological symptom onset.
Herein, we describe in detail our entire clinical experience of 41 patients with NIF autoimmunity detected in CSF (1996–2020).
Methods
Patients
Patients were all those (both Mayo Clinic and external referrals) in whom NIF‐IgGs were detected in CSF in the Neuroimmunology Laboratory (NIL) of the Mayo Clinic in the course of clinical service evaluation for autoimmune neurological disorders. Testing was undertaken by a two‐step algorithm, as described previously. 1 First, all specimens were screened for NIF‐IgGs by IFA, and met criteria for either pattern 1 (typical of NF‐L, or αIN‐predominant IgG staining) or pattern 2 (typical of NF‐H‐predominant IgG staining), Figure 1 , left panels. Second, all positive specimens yielded by the first step were tested by three NIF‐specific CBAs (αIN, NF‐L, and NF‐H), and were positive in at least one of those assays, Figure 1 , right panels. Forty‐one such CSF‐positive cases were acquired consecutively between October 30th 1996 and June 30th 2020 (of 118599 patients who had CSF tested in the course of assessment for autoimmune neurological diagnoses, 0.03%). An additional 80 cases with serum NIF‐IgG positivity were also acquired over that time frame and were excluded from this study (77 had no available CSF to evaluate further; all 3 for whom CSF was available, but were NIF‐IgG negative, had alternative non‐autoimmune diagnoses [frontotemporal dementia, classical multiple sclerosis, or metabolic encephalopathy]).
Figure 1.
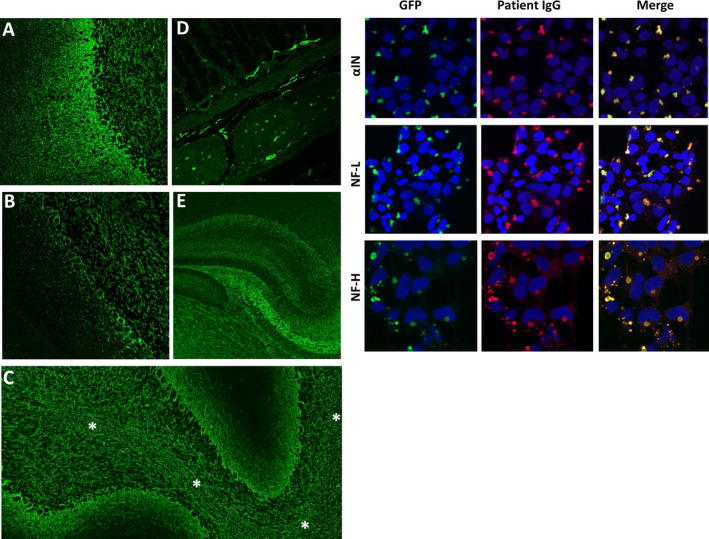
NIF‐IgG studies in CSF. Left panels, A‐E, patterns of NIF‐IgG staining of mouse tissue by indirect immunofluorescence. A, pattern 1 (αIN‐ or NF‐L‐predominant); staining throughout the cerebellum with prominent blush in the molecular layer (left). B, pattern 2 (NF‐H‐predominant); staining is mostly restricted to the granular layer (right) and Purkinje cell layer (middle), with sparse molecular layer staining (left). Staining of other elements, such as white matter (C, asterisks), myenteric plexus (D), and hippocampus (E), are identical for patterns 1 and 2. Right, HEK‐293 cell‐based assay confirming positivity for all three NIF‐IgG subtypes evaluated in a patient whose CSF produced pattern 1 NIF‐IgG staining by the tissue‐based assay.
Clinical and testing data were obtained from the Mayo Clinic electronic medical record (13) and outside neurologists (28) by CZ, SS, and AM. MRI images were reviewed and selected by AM, EF, and DD (CNS), and AAM (peripheral nervous system).
Ehrlichia chaffeensis testing
Each specimen was diluted (serum, 1:64; CSF 1:2) and placed in microscopic slide wells precoated with Ehrlichia chaffeenis‐infected cells. After incubation, the slides were washed and a fluorescein‐isothiocyanate conjugate was added to each well. The slides were then read using a fluorescence microscope. Fluorescent staining of intracellular organisms constituted a positive result.
Controls
The following CSF control cohorts were evaluated:
Laboratory analytical: from 70 patients without autoimmune CNS disease (50 adult; 20 pediatric) assayed in the course of test verification procedure to meet regulatory requirement for clinical use.
Clinical neurological: 120 consecutive Mayo Clinic Rochester patients (3:1 ratio with cases) evaluated in the Department of Neurology who had CSF tested in NIL during 2019.
Clinical cancer: All 15494 Mayo Clinic patients with CSF sent to NIL for neural autoantibody evaluation (1996–2018). Patient records were cross‐referenced for cancer diagnoses (historical or contemporary) pertinent to the patient results (neuroendocrine‐lineage carcinomas [small cell, Merkel cell, other] and lymphomas [Hodgkin and non‐Hodgkin type, not including those with CNS lymphoma).
Measurement of CSF NF‐L protein concentration: CSF specimens were from 8 of the NIF autoimmune cohort (all NF‐L‐IgG positive), 8 patients with multiple sclerosis (MS), and 8 patients with normal pressure hydrocephalus (NPH). Measurement of CSF NF‐L protein concentration was performed in duplicates by investigators blinded to clinical data using a commercially available enzyme‐linked immunosorbent assay (NF‐light; Uman Diagnostics, Umea, Sweden) as described by the manufacturer with the same batch of reagents.
Statistical analysis
Comparisons of categorical variables pertinent to NIF autoimmune patients and controls were undertaken utilizing Chi‐square test with Yates correction and Fisher exact test as appropriate.
Results
All 70 CSF analytical controls were negative by tissue IFA, and all 3 NIF CBAs (αIN, NF‐L, and NF‐H), testing undertaken in the course of test verification and validation for clinical use in our diagnostic laboratory. All 41 included patient CSF specimens that met diagnostic NIF‐IgG immunohistochemical criteria, had ≥ 1 NIF‐IgGs detected by CBAs: αIN (29); NF‐H (30); and NF‐L (20). CSF from 14 patients (9 had NF‐H‐IgG positivity alone by CBAs) produced pattern 2 (heavy‐chain typical) staining by IFA, and the remaining CSF from 27 patients produced pattern 1 staining (none had NF‐H‐IgG positivity alone), Figure 1. Companion serum was available in 27 of 41 patients, and was NIF‐IgG negative in 20 (74%).
Demographic and neurological phenotypes
Thirteen patients were evaluated neurologically at the Mayo Clinic. Twenty‐five patients were male (61%). Median age at CSF testing was 61 years (range, 21–88), Tables 1, 2, 3. Twenty‐one patients were previously reported, and are clinically described in further detail herein, along with the remaining 20 patients acquired 2018–2020. Neurological presentations were subacute over a course of 0‐3 months in 36 patients (90%), with a progressive course over 3–6 months in the other 4. Four patients were known to have a relapsing course.
Table 1.
Encephalopathy phenotypes: cancers, immunological findings, and paraclinical data.
| Pt no. Age/Gender | Syndrome | Symptoms, signs | Cancer, infection, autoimmune | NIF, CSF | MRI | CSF WCC/Pro/OCB/Ab | Neurophysiology, neuropsychology | |
|---|---|---|---|---|---|---|---|---|
| 1. M/77 | ENC | AMS, auditory halluci, dysarthria | URI | HL | Br: deep WM T2 abn + enhancement | 35/N/0/0 | EEG: triphasic waves | |
| 2. F/47* | ENC | Anxiety, agitation, AMS, confusion, dyskinesias, coma | SCLC | AL | Br: hazy deep WM ↑T2 FLAIR | N/42/4/NMDA‐R | EEG: gen delta slowing, seizures | |
| 3. M/52* | ENC | Depression, anxiety, AMS | ‐ | AHL | Br: limbic & anterior temporal T2 FLAIR Abns | 16/72/8 | EEG: N | |
| 4. F/74 | ENC | AMS, AMS, visual disturbance (ophthalmitis) | Merkel Cell ca, pembrolizumab | AHL | Br: N | 11/150/5/CRMP5 | ‐ | |
| 5. M/76 | ENC | Rapid‐onset amnesia, gait apraxia | Prostate adenoca | H | Br: N | N | ‐ | |
| 6. M/40 | ENC | Headache, meningism, blurred vision, seizures | NHL | H | Br: fronto‐parietal T2 WM changes | 8/64/N | EEG: seizures | |
| 7. M/41 | ENC | AMS, depressed mood | Enterovirus | H | Br: N | 14/68/N/0 | ‐ | |
| 8. F/66 | ENC | AMS, apraxia eyelid opening & gait, coma, | HIV CD4 > 200 | H | Br: N | N | ‐ | |
| 9. M/73 | ENC | AMS, cognitive decline | Lung adenoca | AH | Br: N | N/49/N/0 | ‐ | |
| 10. M/55 | ENC | Cognitive decline, motor apraxia | ‐ | A | Br: N | N | ‐ | |
| 11. M/47 | ENC | AMS, focal dyscognitive seizures | Symptoms post‐Ehrlicosis pneumonia; RA | A | Br: diffuse WM ↑ T2 | N/56/N | EEG: gen seizure discharges | |
| 12. M/63* | ENC & brach plexitis | HA, diplopia, nausea, meningism, AMS, R&L shoulder pain | Papillary renal cell ca; membranous glomerulonephritis | A | Br: L&R periventricular, hazy WM ↑T2.Thick , T2 signal abns L brach plexus | 11/41/4 | EMG: bilateral suprascapular neuropathies w/o re‐innervation | |
| 13. F/75 | ENC | AMS, amnesia,, diplopia, dysarthria, dysphagia, weakness | Merkel cell ca, pembrolizumab | AHL | Br: N | 12/58/N/0 | ‐ | |
| 14. F/60* | ENC | Visual disturbance, hemianopsia, delirium, confusion, VII CN palsy | Pernicious anemia | A | Br: hemispheric deep WM ↑T2; R parieto‐occipital, periventricular, & centrum semiovale regions, enhancement | N/79/N | EEG, N. NP: Global deficits, executive dysfunction, frontosubcortical. | |
| 15. M/79 | ENC | AMS, coma, dysarthria, dysphagia | Hepatocellular ca | A | ‐ | N/93/N/0 | EEG: generalized slowing | |
| 16. F/73* | ENC | Neck & body pain, AMS, delusions, & auditory hallucinations (mild asymptomatic neuropathy at F/U | Ehrlichosis: URTI, ↓Plts, ↑ LFTs. Tick bite 2 weeks prior. | HL | Br C & T SC, N; LS spine: enhancing anterior thecal sac & LS nerve roots | 55/150/N/0, ↑IgG index | EEG: focal L temporal slowing; EMG mild polyrad | |
| 17. M/69 | ENC & ataxia | AMS, gait apraxia, dysarthria, cerebellar signs | ‐ | AHL | Br: diffuse atrophy | N/N/4/0 | ‐ | |
| 18. F/43 | ENC & ataxia | AMS, dysarthria, dysphagia, tremor, cerebellar ataxia | HIV, medication noncompliant | A | Br: hazy ↑T2 WM, R frontal, deep WM, BS, cerebellar hemispheres/ peduncles, (frontal enhancement) | 199/N/P/0 | ‐ | |
| 19. M/88* | ENC & ataxia | Delirium, pan‐cerebellar ataxia, large amplitude rubral tremor, dysphagia, dysarthria | Pancreas neuroendocrine ca | AHL | Br: hazy ↑T2 brain & cerebellar WM | N/46/N/0 | ‐ | |
| 20. F/74* | ENC | Headache, vertigo, nausea, vomiting, deep body aching, spasticity | SCLC | AHL | Br: Hazy ↑T2 (deep WM, brainstem, SC corticospinal tracts), enhancing L VII CN | 11/77/9/0 | ‐ | |
| 21. F/48 | Encephalomyelopathy | Vision disturbance, multimodal cognitive decline, seizures weakness | RA, hypothyroid history | AHL | Br: R&L hemispheric WM & full SC T2 changes with enhancement (brain & conus) | 11/94/P/GFAP | ‐ | |
| 22. M/57 | Encephalomyelopathy | Headache, nausea, visual disturbance, AMS, hemianopsia, weakness | Lung rhabdoid adenoca, ipilimumab & novolumab | AL | Br: hypophysitis | 20/93/NA | ‐ | |
| 23. M/21* | Encephalomyelopathy | Visual disturbance, AMS; relapse with myelopathy | ‐ | A | Br: R&L hemispheric hazy WM T2 FLAIR changes. Spine: N | 36/NA/9 | ‐ | |
A, alpha‐internexin; Ab, antibody; Abn, anormal; AMS, altered mental status; brach, brachial; Ca, carcinoma; CRMP5, collapsin response‐mediator protein‐5 EEG, electroencephalogram; EMG, electromyography; ENC, encephalopathy; F, female; FLAIR, fluid‐attenuated inversion recovery; F/U, follow‐up; gen, generalized; GFAP, glial fibrillary acidic protein; H, heavy chain; Halluci, hallucinations; HIV, human immunodeficiency virus; L, left; Li, light chain; LS, lumbosacral; M, male; Mo, months; N, normal; NA, Not available; NMDA‐R, N‐methyl‐D‐aspartate receptor; NP, neuropsychometric test; OCBs, oligoclonal bands (CSF‐exclusive); Polyrad, polyradiculoneuropathy; Pro, protein; Pt no., patient number; R, right; RA, rheumatoid arthritis; SCLC, small cell lung cancer; URI, upper respiratory tract infection; WCC, white cell count; WM, white matter.
Patient evaluated in person at Mayo Clinic; ↑ = increased/hyperintense; ↓ = reduced.
Table 2.
Cerebellar ataxia‐predominant phenotypes: accompanying cancers, immunological findings, and paraclinical data.
| Pt no. Age/Gender | Neurological syndrome | Symptoms, signs | Cancer, potential immunologic contributors | NIF CBA Profile, CSF | MRI | CSF WCC/ Pro/ OCB/ Co‐existing Ab | Neurophysiology | |
|---|---|---|---|---|---|---|---|---|
| 24. F/81 | Ataxia | Pan‐cerebellar ataxia | Hodgkin lymphoma | H | Brain: leptomeningeal enhancement | N | ‐ | |
| 25. M/57 | Ataxia | Pan‐cerebellar ataxia | Hepatocellular carcinoma | AH | ‐ | N | ‐ | |
| 26. F/74 | Ataxia | Pan‐cerebellar ataxia | Merkel cell carcinoma, pembrolizumab | AH | Brain: normal | N/N/N/Zic4 | ‐ | |
| 27. M/64 | Ataxia | Pan‐cerebellar ataxia | SCLC | AHL | ‐ | N | ‐ | |
| 28. F/74* | Ataxia | Pan‐cerebellar ataxia vertigo, diplopia |
Merkel cell carcinoma; post‐URI |
AHL | Brain: initially normal, marked cerebellar atrophy at follow‐up | 32/71/8 | ‐ | |
| 29. M/65 | Ataxia | Pan‐cerebellar ataxia, diplopia, nausea, vomiting | Merkel cell carcinoma | AHL | Brain: Cerebellum T2 changes | 15/N/P/0 | ‐ | |
| 30. M/64 | Ataxia | Pan‐cerebellar ataxia, vertigo, nausea, vomiting | SCLC, RA history | AHL | Brain: focal FLAIR changes in the left cerebellar hemisphere suggestive of cerebellitis | 29/97/N/ANNA‐3 | ‐ | |
| 31. M/74 | Ataxia | Encephalopathy, vertigo, diplopia, dysarthria, cerebellar ataxia | ‐ | H | Brain: bilateral T2 hyperintensities in cerebellum | 31/93/N/0 | ‐ | |
| 32. F/64 | Spinocerebellar | Cerebellar ataxia, leg paresthesia, ascending weakness | Leiomyosarcoma | AHL | ‐ | N/N/N/GAD65 | EMG: length‐dependent axonal polyneuropathy | |
| 33. M/56 | Spinocerebellar | Cerebellar ataxia, leg paresthesias | SCLC | AHL | Brain: N | 13/128/P | ‐ | |
| 34. F/61* | Bulbospinal | Ptosis, diplopia, altered taste, pseudobulbar palsy, progressive pain, hyperpathia, weakness in arms | ‐ | AHL | Brain: diffuse T2 FLAIR changes hemispheric white matter & pons, and cervical cord (hazy) | 26/88/N | Normal EMG; slow somatosensory‐evoked potentials, localizing to cervical spinal cord. | |
A, alpha‐internexin; Ab, antibody; ANNA‐3, antineuronal nuclear anitbody type‐3; EEG, electroencephalogram; EMG = electromyography; F, female; FLAIR = fluid‐attenuated inversion recovery; GAD65, glutamic acid decarboxylase 65; H, heavy chain; HIV, human immunodeficiency virus; L, light chain; M, male; N, normal; OCBs, oligoclonal bands (CSF‐exclusive); P, positive; Pro, protein; Pt no., patient number; RA, rheumatoid arthritis; SCLC, small cell lung cancer; WCC, white cell count.
Patient evaluated in person at Mayo Clinic.
Table 3.
Myelopathy‐ and neuropathy‐predominant phenotypes: accompanying cancers, immunological findings, and paraclinical data.
| Pt no. Age/Gender | Neurological syndrome | Symptoms, signs | Cancer, potential immunologic contributors | NIF CBA Profile, CSF | MRI | CSF WCC/Pro/OCB/Co‐existing Ab | Neurophysiology & biopsy data |
|---|---|---|---|---|---|---|---|
| 35. M/64*, * | Myelopathy | Right‐sided weakness, bowel and bladder dysfunction. Wheel‐chair bound | Ehrlichosis preceding | H | Spinal cord: longitudinally extensive C2‐5 T2 changes; central enhancement. | 6//N/N/N | Cord biopsy revealed histiocyte‐rich lesion with myelin loss, scattered T lymphocytes |
| 36. M/59 | Myelopathy | Progressive weakness | ‐ | A | Brain/spine: diffuse T2 changes brain & spinal cord; enhancement | NA/NA/P | ‐ |
| 37. F/60 | Myeloneuropathy | Progressive painful paresthesias with upper limb and trigeminal distribution, leg spasticity | SCLC | AHL | ‐ | N | ‐ |
| 38. M/59*, * | Myeloneuropathy | Progressive painful neuropathy with trigeminal, trunk, & 4 limb distribution. Hearing loss. | ‐ | H | Brain: N; Spine:T2 hyperintensity T3 level; thickened lumbosacral nerve roots; T2 hyperintense femoral nerves R > Le | N/67/N/0 | Right sensorineural hearing loss, slow right blink response, slow somatosensory‐evoked potentials localizing to thoracic spinal cord, & axonal sensorimotor polyneuropathy |
| 39. F/43 | Lumbosacral polyradiculoneuropathy | Severe diffuse limb pain & weakness | ‐ | HL | Spine: N | 35/53/5 | ‐ |
| 40. M/65*, * | Lumbosacral polyradiculoneuropathy | Progressive limb weakness | Non‐Hodgkin lymphoma | H | Spine: enhancing anterior thecal sac & lumbosacral nerve roots | N/83/0/0 (cytology normal) | EMG lumbosacral radiculopathies |
| 41. M/45 | Multifocal motor neuropathy | Deep aching & progressive upper extremity weakness, neuropathy | ‐ | A |
Brain & spine: N |
N/50/N/0 | EMG: 50% partial conduction block right ulnar nerve; F‐wave prolonged |
A, alpha‐internexin; Ab, antibody; EMG, electromyography; F, female; H, heavy chain; HIV, human immunodeficiency virus; L, light chain; Le, left; M, male; N, normal; NA, not available; OCBs, oligoclonal bands (CSF‐exclusive); Pro, protein; P, positive; Pt no., patient number; R, right; RA, rheumatoid arthritis; SCLC, small cell lung cancer; WCC, white cell count.
Evaluated at Mayo Clinic.
Neurological phenotypes among 120 neurological controls consecutively referred to the Neuroimmunology Laboratory for CSF antibody testing (all NIF‐IgG negative) were diverse. Twenty‐four (20%) had final clinical autoimmune or other inflammatory diagnoses: encephalitis, 14; neuropathy, 3 (all large fiber [axonal, 2; demyelinating, 1]); ataxia, 2; MS, 2; neuromyelitis optica, 1; myelitis, 1; stiff‐person syndrome, 1. The remaining 96 controls (80%) had: psychiatric disorders, 22; unspecified neurological disorders, 17 (cognitive, 12; ataxia, 1; peripheral nervous/autonomic, 4); degenerative dementias, 17; primary headache disorders, 15; metabolic encephalopathies, 7; stroke, 5; primary seizure disorders, 5; other non‐neurologic, 5; brain neoplasms, 2; and hereditary, 1.
In contrast, NIF autoimmune phenotypes were restricted to three groups (Tables 1, 2, 3): encephalopathy predominant, 23 (56%); ataxia predominant, 11 (29%); and myelopolyradiculoneuropathy, 7 (15%). Twelve patients had multifocal neurological disease (29%). In contrast with controls, 31 of 41 NIF‐IgG cases (77%, including all 13 Mayo Clinic cases) had documentation supportive of nervous system inflammation: ≥1 of increased post‐gadolinium enhancement on MRI, increased white cells or oligoclonal bands in CSF, or previously characterized neural IgG detected in serum or CSF, Tables 1, 2, 3, P < 0.0001, Fisher Exact Test.
The 23 patients with the most common phenotype, encephalopathy predominant, had rapid onset and progression of cognitive impairment, typically multimodal. All had rapid onset delirium and memory problems, and one or more additional features: vision disturbance (6: 5 had blurred vision without ophthalmologic findings, including 2 with hemianopsia; 1 had ophthalmitis and coexisting CRMP‐5‐IgG), motor apraxia (4, [of gait, 4, of eyelids, 1]), headache (5, 3 had meningism), mood change (4), seizures (3, [generalized, 2; focal dyscognitive, 1]), or language disorder and dyskinesias (1, and coexisting NMDA‐R antibody). Eleven had ≥ 1 accompanying signs of brainstem dysfunction (8: dysarthria, 6; dysphagia, 4; diplopia, 2; nausea, 1; facial palsy, 1; tremor, 1; ptosis and bulbar palsy, 1), myelopathy (4), cerebellar ataxia (3), and polyradiculoneuropathy (1). Detailed neuropsychometric testing in Patient 14 revealed deep white matter localization (moderate global cognitive deficits, with executive dysfunction particularly notable, localizing to frontal and subcortical pathways).
A cerebellar or brainstem predominant neurological phenotype was encountered in 11 patients (30%), 4 of whom also had myelopathic findings (Table 2). Additional brainstem findings included: diplopia, 4; vertigo, 2; vomiting, 2; and dysarthria, 2.
The remaining 7 patients (18%) had, as the sole neurological manifestation: inflammatory lumbosacral polyradiculopathies (3), myelitis (2), myeloneuropathy (1), and multifocal motor neuropathy with conduction block (GM‐1 antibody negative, 1), Table 3. Among 15 total patients with myelopathic or neuropathic findings, 8 had considerable limb or generalized body pain at onset (Tables 1, 2, 3).
MRI findings
Data were available in 36 patients (88%). Twelve of 36 patients (35%) were reported to have normal imaging, all evaluated outside Mayo Clinic. A further four had normal brain imaging initially, which subsequently demonstrated deep white matter T2 hyperintensities. Among the 16 non‐Mayo cases with abnormal imaging, T2 abnormalities were reported in ≥ 1 of hemispheric deep white matter (6, with accompanying enhancement in 3, including 1 with radial periventricular enhancement, and coexisting GFAP‐IgG), brainstem (4), cerebellum, (3) and spinal cord (3, with accompanying enhancement in 2).
MR images were reviewed in detail for 13 Mayo Clinic patients; all had abnormalities detected (Figures 2 and 3, Tables 1, 2, 3). Observed brain abnormalities were confluent, hazy T2 change in white matter of ≥ 1 of cerebral periventricular deep white matter (7), brainstem (2), temporal lobes (1), and cerebellum (1). Although individually nonspecific in appearance (and generally reported as such by radiology), those changes were neurological localization‐relevant throughout the group (Tables 1 and 2). Post‐gadolinium enhancement was also observed in Patient 14 (periventricular radial and leptomeningeal radial [GFAP‐IgG negative]) and patient 20 (left facial nerve). One patient with severe pancerebellar ataxia (27) had initial normal imaging, but diffuse cerebellar atrophy at follow‐up. Patient 12, with shoulder pain after encephalitis onset, also had thickening and increased T2 signal of brachial plexus, without enhancement (Figure 3). Three patients with myelopathy (1) or encephalomyelopathy (2) had longitudinally extensive T2 signal change in the cord (dorsal cord and white matter tract predominant, hazy in 2, with accompanying dorsal cord‐predominant postgadolinium enhancement in 1), Figure 2. Patient 38 with myeloneuropathy had short segment dorsal thoracic T2 signal change, thickened and T2 hyperintense lumbosacral roots, and T2 hyperintensity of bilateral femoral nerves. Both patients 16 (encephalopathy and asymptomatic polyradiculoneuropathy detected on EMG) and 40 (symptomatic polyradiculoneuropathy) had enhancement of the anterior thecal sac and nerve roots.
Figure 2.
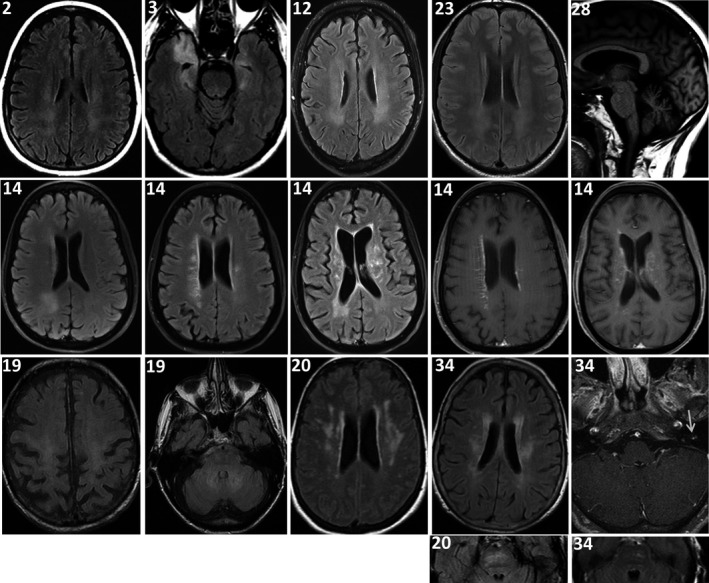
MRI brain findings. Images are all axial T2 FLAIR except: last panel right, top row (T1 sagittal); second‐to‐last and last right panels, 2nd row, and last panel, right, 3rd row (T1 axial postgadolinium). Clinical localization‐related hazy‐appearing T2 white matter hyperintensities were observed in deep white matter of cerebral hemispheres in patients 2, 3, 12, 14, 19, 20, 23, and 34 (Tables 1 & 3), and pontine white matter in patients 20 and 34. Patient 14 also had postgadolinium enhancement (linear radial, left; and leptomeningeal, right [GFAP‐IgG negative]). Patient 28 had cerebellar atrophy at follow‐up. Patient 34 also had enhancement of the left facial nerve (arrow).
Figure 3.
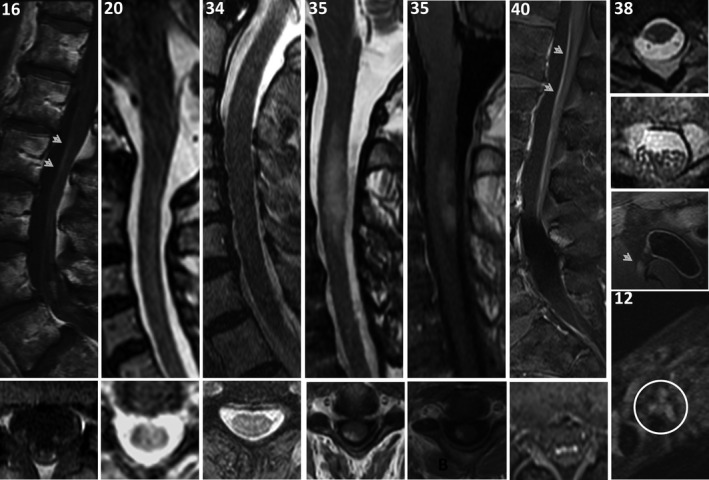
MRI spine and nerve findings. MRI spine, sagittal, top row; axial, bottom row, except right‐most column, which are all axial T2 images. Lumbosacral spine, T1 postgadolinium images in patients 16 (asymptomatic EMG‐documented lumbosacral polyradiculoneuropathy and encephalopathy [Figure 2]) and 40 (symptomatic lumbosacral polyradiculoneuropathy) reveal enhancement of anterior thecal sac (top, arrow heads) and nerve roots (bottom). Patients 20, 34, and 35 with myelopathy had dorsal cord white matter predominant longitudinally extensive T2 hyperintensities, hazy‐appearing in patients 20 and 34, with cord expansion and enhancement in patient 35. Axial cord images reveal T2 hyperintensities and T1 postgadolinium to be white matter tract predominant. Patient 38 with myeloneuropathy had T2 hyperintensity of right‐sided white matter tract in the thoracic cord (top), T2 signal change and enlargement of lumbosacral nerve roots (middle top), and T2 hyperintensity of bilateral femoral nerves (right‐side demonstrated, middle bottom, arrow head). Patient 12 with encephalopathy (Figure 2) and bilateral shoulder pain had T2 hyperintensity of the brachial plexus (bottom, circled).
Neurophysiology
EEG was abnormal in 4 of 6 patients with encephalopathy (generalized diffuse delta slowing in all, with superimposed electrographic seizures in 2), Table 1. EMG revealed lumbosacral polyradiculoneuropathy (2), axonal sensorimotor neuropathy (2), and multifocal motor neuropathy with conduction block (1). Two patients evaluated at Mayo Clinic had spinal cord localizing slowing of central proprioceptive pathways on somatosensory‐evoked potentials (both had MRI T2 cord lesions). Electrophysiological testing in Patient 38 (Table 3, with hearing loss, right face pain, and diffuse truncal and limb pain) revealed right sensorineural hearing loss by audiogram, absent right blink response, slowing of central proprioceptive, and sensorimotor axonal neuropathy on EMG. All 13 patients evaluated at Mayo Clinic had normal metabolic laboratory test results including vitamin B12, methylmalonic acid, and folate.
Oncological and immunological findings
In total, 30 patients (73%) had ≥ 1 documented risk factor for neurological autoimmunity: contemporaneous cancer (paraneoplastic), 22; contemporaneous preceding respiratory infection (post‐infectious), 5; checkpoint inhibitor cancer immunotherapy, 4; coexisting autoimmune disease, 5; and long‐standing HIV, 2, (Tables 1, 2, 3).
Contemporaneous cancer diagnoses (within 24 months of neurological symptom onset) were made in 22 patients (54%), and 13 after onset of neurological symptoms. These cancers were as follows: carcinoma of neuroendocrine lineage (12: SCLC, 6; Merkel cell, 5; pancreatic, 1), adenocarcinomas, (3: lung, 2; metastatic prostate, 1), systemic lymphoma (3: non‐Hodgkin, 2; Hodgkin, 1), hepatocellular carcinoma, 2; papillary cell renal carcinoma, 1; and leiomyosarcoma, 1.
NF‐L‐IgG was detected in 11 of 12 patients with neuroendocrine‐lineage carcinomas, and in 8 of 29 other patients (P = 0.0005), Fisher Exact Test, Tables 1, 2, 3. Individual NIF‐IgGs served to confirm the overall diagnosis, but were not otherwise predictive of neurological phenotype or cancer (P > 0.05). All three patients with lymphoma had NF‐H‐IgG positivity only. The cancer control cohort of 15494 patients revealed 478 had small cell carcinoma or other neuroendocrine‐lineage carcinoma (3.1%), and 432 had lymphoma (2.8%). Neuroendocrine lineage carcinomas were more common among the NIF‐IgG cohort (12/40, 30%) than the control group (chi‐square, 86.4, P < 0.0001); no statistical difference was observed for lymphoma (chi‐squared, 1.52, P = 0.185).
Five further patients developed neurological symptoms during recovery from systemic infections; ehrlichosis in 3 and undocumented pathogen in 2. Patient 16, evaluated in detail at Mayo Clinic (Table 1) developed diffuse body pain and neck ache, followed by encephalopathy and inflammatory CSF, 2 weeks after a tick bite. She had laboratory findings typical of systemic ehrlichosis, without evidence of CNS disease. She had elevated ALT (236 U/L, normal value, 7‐45) and thrombocytopenia (58/μL; normal value, 157‐371) on hospital admission; both values normalized over the course of her 10‐day hospitalization. Ehlichia chaffeensis testing revealed: PCR, serum positive, CSF negative; IgG, serum positive (1:2048; normal < 1:64), and CSF positive (1:32; normal, <1:2) with no cross‐reactivity detected for other ehrlichia forms, anaplasmosis, or Lyme disease. She was treated with doxycycline for 3 weeks. Her encephalopathy gradually resolved to near normal over the course of 8 weeks, without further intervention. Ehrlichia chaffeensis IgG testing was undertaken in a further 10 patients with available serum (6 with cancer, 4 without), all were negative.
Among the 9 patients who developed cancer prior to neurological symptom onset, 4 had been treated with immune checkpoint inhibitors (pembrolizumab, 3; ipilimumab and nivolumab, 1). These four patients were being treated for Merkel cell carcinoma (3) or lung adenocarcinoma (1). Coexisting medical disorders indicative of background immune dysregulation prior to neurological symptom onset were present in 7 patients (rheumatoid arthritis, 3; HIV, 2 [1 with CD4 count > 400; 1 noncompliant with antiretroviral medication]; pernicious anemia, 1; autoimmune thyroiditis, 1; membranous glomerulonephritis, 1).
Additional CSF findings
Routine CSF data were available in 28 patients (74%, Tables 1, 2, 3), and was inflammatory (at least one of white cell count or oligoclonal bands elevated) in 21 of those (75%). CSF protein was elevated in all patients tested (median value, 71 mg/dL [range, 41–150 mg/dL; normal, ≤35]). White cell count was elevated in 20 patients (71%: median value, 16/μL [range, 8‐199; normal, ≤ 5]), all lymphocyte predominant. CSF‐exclusive supernumerary oligoclonal band numbers were elevated in 14 patients (50%: median value, 5 [range, 4–9; normal, <4]). Two of those 14 patients had elevated IgG index in addition.
Coexisting IgG antibodies
Six patients, including five with cancer (neuroendocrine‐lineage, 4), had coexisting IgGs detected; 2 in CSF, and 4 in serum (15%), Tables 1 and 2. In CSF, one patient each had either NMDA‐R‐IgG (a woman in her 40s with typical anti‐NMDA‐R encephalitis phenotype, and accompanying small cell carcinoma) or GFAP‐IgG (typical autoimmune GFAP astrocytopathy phenotype without cancer). In serum, one patient with Merkel cell carcinoma had coexisting CRMP‐5‐IgG (1:15360; normal, ≤240; typical CRMP‐5 ophthalmitis, in addition to encephalopathy). The remaining three patients had ataxia in the context of high‐titer GAD65‐IgG (397 nmol/L; normal, ≤0.02), ANNA‐3 (1:3840; normal, ≤240), or Zic4 antibody; all had cancer.
NF‐L protein values
CSF NF‐L protein values were higher among patients with NF‐L‐IgG (median, 6718 ng/L; range, 165‐10000) in comparison to age‐matched controls with MS at onset (median, 940 ng/L; range, 426‐8166) and NPH (median, 959 ng/L; range, 734–9929), Figure 4.
Figure 4.
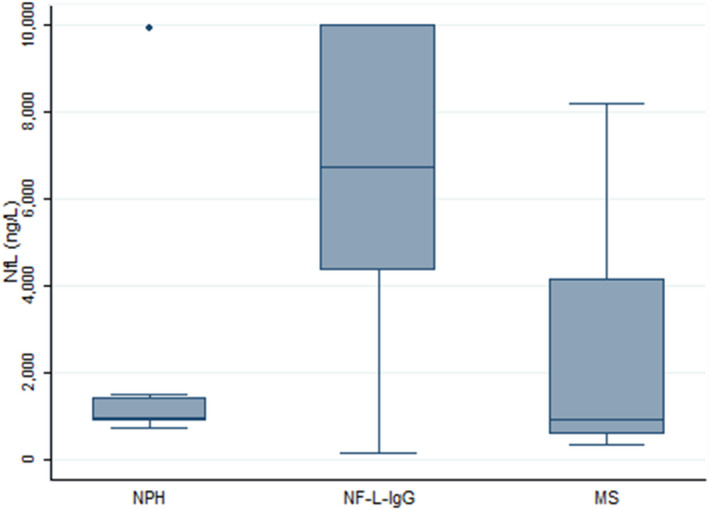
CSF NF‐L protein concentration comparison between patients with NF‐L‐IgG and controls. Higher NF‐L values indicative of more pronounced axonal damage were detected in the CSF of patients with NF‐L‐IgG in comparison with age‐matched MS cases and patients with NPH included as controls.
Treatment and outcome
Median duration of follow‐up was 9 months (range, 1–36). Treatment or outcome data were available for 31 patients (Table 4). Nineteen patients received ≥ 1 neurological symptom‐directed immune therapy in diverse combinations (≥1 of corticosteroids [15], rituximab [7], IVIg [4], plasma exchange [3], cyclophosphamide [2], cancer chemotherapy [2], and azathioprine [1]). Of those 19 patients, 13 (77%) responded neurologically (improvement, 8 or complete remission 5), 1 did not improve, and 4 worsened. Of 12 patients who had no neurological attack‐related treatment (including Patient 23 who had a subsequent attack treated), 6 had spontaneous improvement, and 6 worsened. Seven patients were reported to have died, including 3 who died from cancer and 4 who died from progression of neurological disease (3 of those 4 were untreated). Of those 11 who worsened or died, 4 had cancer, 3 had a background of infection (none with ehrlichosis), and 4 had no etiology established (Table 4). Three of four who developed neurological symptoms in the context of checkpoint inhibitor cancer treatment responded to immunotherapy, and one died.
Table 4.
Treatments and outcomes
| Pt no. Age/Gender | Neurological syndrome, cancer, or other potential trigger | Treatment (time to treatment, months) | Outcome |
|---|---|---|---|
| 1. M/77 | Encephalopathy, URI | None | Died |
| 2. F/47* | Encephalopathy, SCLC | Steroids, rituximab, azathioprine (1) | Resolved |
| 3. M/52* | Encephalopathy | Steroids, IVIg (3) | Worsened |
| 4. F/74* | Encephalopathy, Merkel cell | Steroids, IVIG, Rituximab, cyclophosphamide (6) | Improved |
| 7. M/41 | Encephalopathy, enterovirus | None | Died |
| 8. F/66 | Encephalopathy, HIV | None | Died |
| 10. M/55 | Encephalopathy | Steroids | Improved |
| 11. M/47 | Encephalopathy, ehrlichosis | None | Spontaneous improvement |
| 12. M/63* | Encephalopathy, papillary renal cell carcinoma | Rituximab, steroids (3) | Resolved |
| 13. F/75 | Encephalopathy & ataxia, Merkel cell | Steroids | Improved |
| 14. F/60* | Encephalopathy | IVIg, Steroids, Rituximab (4) | Worsened, died |
| 15. M/79 | Encephalopathy, hepatocellular carcinoma | None | Died** |
| 16. F/73* | Encephalopathy, ehrlichosis | None | Spontaneous resolution |
| 17. M/69 | Encephalopathy | PLEX, steroids, and rituximab | Worsened |
| 18. F/43 | Encephalopathy & ataxia, HIV | None | Spontaneous resolution |
| 19. M/88* | Encephalopathy & ataxia, pancreas neuroendocrine | None | Died** |
| 21. F/48* | Encephalomyelopathy | Steroids | Resolved |
| 22. M/57 | Encephalomyelopathy, lung rhabdoid adenocarcinoma | Steroids | Resolved |
| 23. M/21 | Encephalomyelopathy |
Attack 1: None; Attack 2: steroids (1) |
Resolved on both occasions |
| 24. F/81 | Ataxia, Hodgkin lymphoma | Chemotherapy | Improved |
| 26. F/74 | Ataxia, Merkel cell | Rituximab | Worsened, died** |
| 27. M/64 | Ataxia, SCLC | IVIg | Improved |
| 28. F/74* | Ataxia, Merkel cell | Steroids, Rituximab, cyclophosphamide (1) | Improved, but profound disability (walker or wheelchair) |
| 29. M/65 | Ataxia, Merkel cell carcinoma | Steroids, Chemotherapy | Improved |
| 30. M/64 | Ataxia, SCLC | Steroids | Improved |
| 33. M/56 | Spinocerebellar, SCLC | None | Spontaneous improvement |
| 34. F/61* | Bulbospinal | PLEX (5) | No response to PLEX. Fluctuating course, PEG dependent |
| 35. M/64* | Myelitis, ehrlichia | Steroids, PLEX (1) | Resolved |
| 38. M/59* | Myeloneuropathy | IVIg, rituximab (36) | Worsened. Progression of symptoms from lower to upper extremities |
| 39. F/43 | Lumbosacral polyradiculoneuropathy | None | Spontaneous improvement |
| 40. M/65* | Lumbosacral polyradiculoneuropathy, NHL | None | Worsened |
F, female; IVIg, intravenous immunoglobulin; M, male; NHL, non‐Hodgkin lymphoma; PLEX, plasma exchange; Pt no., patient number; URI, upper respiratory tract infection; SCLC, small cell carcinoma.
Evaluated at Mayo Clinic.
Died from cancer.
Discussion
CSF‐detected NIF autoimmunity unifies patients with subacute onset, treatable inflammatory neurological disorders, usually affecting the CNS, with a paraneoplastic cause in over half, and other documented contemporary immunological perturbation in another fifth. Autoimmune encephalopathy and ataxia‐predominant brainstem disorders were most common, and the remainder had myelopathy or inflammatory polyradiculoneuropathy (including 1 patient with the established autoimmune phenotype, multifocal motor neuropathy). This is distinct from the broader profile of phenotypes encountered in patients referred to our laboratory, and have autoimmune diagnoses, exemplified by the control group (which also included neuromyelitis optica, transverse myelitis, and stiff‐person syndrome). The NIF‐IgG profiles by cell‐based assay (one or more of αIN‐, NF‐L‐, NF‐H‐IgGs) provided confirmation of NIF specificity and prediction of cancer type (small cell or Merkel cell when NF‐L‐IgG was detected), although not specific neurological phenotypes. NIFs are cytoplasmic proteins, and thus, the antibodies are likely nonpathogenic biomarkers of NIF‐directed T‐cell effectors.
In keeping with the diffuse localization of NIF antigens, one quarter had overlapping neurological phenotypes, previously observed in patients with autoimmunity targeting GFAP, the astrocyte abundant intermediate filament. 5 White matter‐predominant radiological findings and electrophysiological abnormalities (EEG, audiology blink reflexes, SSEPs, EMG) from 13 patients studied in detail at Mayo Clinic support the concept of a continuum of dysfunction, sometimes spanning both central and peripheral nervous systems. Some findings, such as abnormal white matter on MRI and reduced nerve conduction amplitudes, support the concept of axonal dysfunction in the context of NIF‐directed autoimmunity. Although the NIF autoimmune neurological presentations were not uniform, they were distinct from the profile of cases generally referred to our laboratory for CSF evaluation, 80% of whom had neurological autoimmunity as initial differential diagnostic consideration, but ultimately proved to have other diagnoses.
Radiological abnormalities, on a case‐by‐case basis, were often hazy appearing mostly without postgadolinium enhancement. These abnormalities of white matter in cerebral hemispheres, pons, cerebellum, and spinal cord, and nerve roots and peripheral nerves, correlated with the neurological localization, although did not have specifying features. The expression of all three proteins studied (NF‐H, NF‐L, and αIN) is abundant in the pons and brainstem as well as the cerebral white matter, cerebellum, and to various degrees in the hippocampus, midbrain, spinal cord (https://www.proteinatlas.org/), and the peripheral nervous system. There are inter‐protein differences in expression outside the nervous system (https://www.proteinatlas.org/). 6
A paraneoplastic cause was found in just over half of our patients, which is within the typical 50‐80% cancer detection frequency in classical paraneoplastic neurological disorders. 7 Small cell carcinoma and other related neuroendocrine‐lineage carcinomas, including Merkel cell, were most common, and had a strong association in general with NIF autoimmunity, in particular with NF‐L‐IgG, a protein known to be abundantly expressed in these tumors. 1 , 8 , 9 , 10 NF‐H‐IgG may also have some biological significance in lymphoma, although numbers were small and any paraneoplastic association was not borne out by statistical analysis, and neoplastic tissue was not available for further study. All three lymphomas and the one prostate adenocarcinoma detected were among nine patients (7 men) positive for NF‐H‐IgG only. NF‐H protein is abundant in male prostate, female ovary, colon, adipose and soft tissue, monocytes and T cells, and is also known to be expressed in different cancers, including some identified in this cohort (neuroendocrine carcinoma [Merkel, pancreatic, SCLC], and prostate adenocarcinoma) (https://www.proteinatlas.org/). 1 , 8 , 9 , 11
We previously reported a patient with paraneoplastic cerebellar ataxia in the setting of Merkel cell carcinoma, whose NIF‐IgG profile (αIN, NF‐L, NF‐M, and NF‐H, but not peripherin) matched the NIF protein expression profile in her biopsied neoplasm. 1 These observations support an onconeural antigen‐driven immune response in the paraneoplastic NIF cases. While the etiology (in some) and the pathophysiology (in all) of NIF autoimmunity remain to be elucidated, the rarity of detection of NIF‐IgGs in our reference laboratory (41 cases in 24 years) argues against them being epiphenomenal byproducts of neurodegeneration. The concept of NIFs as specific immunological targets is also supported by the higher levels of NF‐L protein in CSF of affected patients than NPH or MS controls.
The paraneoplastic patients and several others had other underlying immunologic perturbations that may have predisposed patients to developing neurological autoimmunity (viz., infection [acute ehrlichosis or chronic HIV], T‐cell regulation‐inhibiting monoclonal cancer therapies, and preexisting autoimmune diseases). HIV infection is a predisposition for autoimmunity to develop. The pathophysiology of parainfectious NIF autoimmunity is otherwise unknown. In the cases of cancer immunotherapy, the neurological syndrome was recognizable as it arose after cancer diagnosis, and was time‐locked to treatment. Neurological symptoms in the context of these therapies should raise suspicion for an autoimmune cause and broad cancer‐specific neural autoantibody profiles may assist diagnosis. 12
CSF positivity was required for both sensitivity and specificity of NIF‐IgG testing. Cases were carefully ascertained, utilizing strict immunohistochemical criteria and confirmation by a trio of cell‐based assays in CSF only. CSF analytical control specimens, as required by U.S. federal regulations of clinical laboratories, were also assessed and were universally negative by both assays, confirming our previous findings of the high specificity of these biomarkers in CSF for neurological autoimmunity. In contrast, serum positivity was only detected in 26% of our CSF‐positive cases (insensitive), and was always accompanied by a nonautoimmune diagnosis in those who were CSF negative (nonspecific), as also previously reported for GFAP‐IgG and NMDAR‐IgG. 5 , 13 , 14 , 15 We also previously demonstrated the even lower clinical specificity of NIF‐IgG testing (93%) when undertaken in serum using a single molecular protein assay alone, such as western blot or cell‐based assay. 1
Various immune therapies, administered either alone or in diverse combinations, were utilized. Three quarters of those treated were reported to have had neurological improvements. Among those untreated, some improved spontaneously, while others died from cancer complications. However, others again without cancer who remained untreated died from neurological disease, highlighting the importance of early immune therapy initiation in autoimmune neurological disorders generally. It was notable that all three patients with encephalopathy or myelopathy had full recoveries, either spontaneously or after immune therapies.
Limitations of this study include some missing data from a few of the non‐Mayo Clinic cases, and the retrospective nature of data collection. NIF autoimmune cases appear to be rare, thus far, although we encountered almost half of our 25‐year experience since 2018, possibly attributable to a general increase in CSF testing request numbers, and improved NIF‐IgG IFA pattern recognition by technologists at assay screening. At present, we encounter approximately two cases per month in our laboratory, as compared to 4 cases per week of anti‐NMDA‐R encephalitis. Because all three patients we had encountered with serum positivity where CSF was negative had non‐autoimmune diagnoses, we also excluded another 77 historical patients for whom only serum was available to study.
NIF autoimmunity should be included in the differential diagnosis for neurological autoimmunity. Imaging and neurophysiology confirms an array of unifocal or multifocal neurological dysfunction spanning both central and peripheral nervous systems. Cancer search should be broad, although type may be predicted by IgG profile (NF‐L‐IgG small cell carcinoma). A possible NF‐H‐IgG association with other cancer types needs further study. Other immunological perturbations should be considered as potential precipitating factors (post‐acute infection, HIV, or checkpoint‐inhibiting cancer therapeutics). Although some patients die from cancer, the neurological disorders are treatable, more often than not. Diagnosis requires detection of 1 of 2 distinct patterns of IgG staining of neuronal intermediate filaments by indirect immunofluorescence assay of patient CSF, and confirmation by a trio of NIF‐specific cell‐based assays, also in CSF. Serum testing lacks sensitivity and specificity.
Author Contributions
Conception and design of the study: A.M. Acquisition and analysis of data: all authors. Drafting the manuscript or figures: AM, AAM, and SM. Critical revision of manuscript: all authors.
Conflicts of Interest
None.
Acknowledgments
Mayo Clinic Center for Individualized Medicine and Department of Laboratory Medicine and Pathology, Mayo Clinic, for funding. Dr Elitza Theel, PhD, Division of Microbiology, Department of Laboratory Medicine and Pathology, Mayo Clinic for performing ehrlichosis testing. Neuroimmunology Laboratory staff for excellent logistical and technical assistance.
Funding Information
None.
References
- 1. Basal E, Zalewski N, Kryzer TJ, et al. Paraneoplastic neuronal intermediate filament autoimmunity. Neurology 2018;91:e1677–e1689. 10.1212/WNL.0000000000006435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Honorat JA, Lopez‐Chiriboga AS, Kryzer TJ, et al. Autoimmune septin‐5 cerebellar ataxia. Neurol Neuroimmunol Neuroinflamm 2018;5:e474 10.1212/NXI.0000000000000474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fang B, McKeon A, Hinson SR, et al. Autoimmune glial fibrillary acidic protein astrocytopathy: a novel meningoencephalomyelitis. JAMA Neurol 2016;73:1297–1307. 10.1001/jamaneurol.2016.2549. [DOI] [PubMed] [Google Scholar]
- 4. Gadoth A, Kryzer TJ, Fryer J, et al. Microtubule‐associated protein 1B: Novel paraneoplastic biomarker. Ann Neurol 2017;81:266–277. 10.1002/ana.24872[published Online First: 2017/01/12]. [DOI] [PubMed] [Google Scholar]
- 5. Flanagan EP, Hinson SR, Lennon VA, et al. Glial fibrillary acidic protein immunoglobulin G as biomarker of autoimmune astrocytopathy: Analysis of 102 patients. Ann Neurol 2017;81:298–309. 10.1002/ana.24881. [DOI] [PubMed] [Google Scholar]
- 6. Xu Z, Dong DL, Cleveland DW. Neuronal intermediate filaments: new progress on an old subject. Curr Opin Neurobiol 1994;4:655–661. 10.1016/0959-4388(94)90006-x [published Online First: 1994/10/01]. [DOI] [PubMed] [Google Scholar]
- 7. Pittock SJ, Kryzer TJ, Lennon VA. Paraneoplastic antibodies coexist and predict cancer, not neurological syndrome. Ann Neurol 2004;56:715–719. 10.1002/ana.20269 [published Online First: 2004/10/07]. [DOI] [PubMed] [Google Scholar]
- 8. Lehto VP, Stenman S, Miettinen M, et al. Expression of a neural type of intermediate filament as a distinguishing feature between oat cell carcinoma and other lung cancers. Am J Pathol 1983;110(2):113–118. [published Online First: 1983/02/01]. [PMC free article] [PubMed] [Google Scholar]
- 9. Broers JL, Rot MK, Oostendorp T, et al. Spontaneous changes in intermediate filament protein expression patterns in lung cancer cell lines. J Cell Sci 1988;91 (Pt 1):91–108. [published Online First: 1988/09/01]. [DOI] [PubMed] [Google Scholar]
- 10. Virtanen I, Heikinheimo K, Hormia M, et al. Expression of intermediate filaments (IF) in tissues and cultured cells. Int J Dev Biol 1989;33(1):55–61. [published Online First: 1989/03/01]. [PubMed] [Google Scholar]
- 11. Schleicher RL, Hunter SB, Zhang M, et al. Neurofilament heavy chain‐like messenger RNA and protein are present in benign prostate and down‐regulated in prostatic carcinoma. Cancer Res 1997;57(16):3532–3536. [published Online First: 1997/08/15]. [PubMed] [Google Scholar]
- 12. Zekeridou A, Lennon VA. Neurologic autoimmunity in the era of checkpoint inhibitor cancer immunotherapy. Mayo Clin Proc 2019;94(9):1865–1878. 10.1016/j.mayocp.2019.02.003. [DOI] [PubMed] [Google Scholar]
- 13. Dubey D, Hinson SR, Jolliffe EA, et al. Autoimmune GFAP astrocytopathy: Prospective evaluation of 90 patients in 1year. J Neuroimmunol 2018;321:157–163. 10.1016/j.jneuroim.2018.04.016 [published Online First: 2018/05/26]. [DOI] [PubMed] [Google Scholar]
- 14. Zandi MS, Paterson RW, Ellul MA, et al. Clinical relevance of serum antibodies to extracellular N‐methyl‐D‐aspartate receptor epitopes. J Neurol Neurosurg Psychiatry 2015;86(7):708–713. 10.1136/jnnp-2014-308736 [published Online First: 2014/09/24]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McCracken L, Zhang J, Greene M, et al. Improving the antibody‐based evaluation of autoimmune encephalitis. Neurol Neuroimmunol Neuroinflamm 2017;4(6):e404 10.1212/NXI.0000000000000404 [published Online First: 2017/10/28]. [DOI] [PMC free article] [PubMed] [Google Scholar]