

Abstract
We describe the clinical, electrodiagnostic, and genetic findings of three homozygous FIG4‐c.122T>C patients suffering from Charcot‐Marie‐Tooth disease type 4J (AR‐CMT‐FIG4). This syndrome usually involves compound heterozygosity associating FIG4‐c.122T>C, a hypomorphic allele coding an unstable FIG4‐p.Ile41Thr protein, and a null allele. While the compound heterozygous patients presenting with early onset usually show rapid progression, the homozygous patients described here show the signs of relative clinical stability. As FIG4 activity is known to be dose dependent, these patients’ observations could suggest that the therapeutic perspective of increasing levels of the protein to improve the phenotype of AR‐CMT‐FIG4‐patients might be efficient.
Introduction
FIG4 is a phosphoinositide phosphatase, which along with kinase FAB1/PIKfyve and scaffold protein VAC14 act as a regulating complex for PI(3,5)P2, a phospholipid signal involved in endolysosomal trafficking. Dysregulation of this complex results in a cytoplasmic accumulation of vacuoles and ultimately tissue degeneration 1 , 2 which can involve nervous, osseous, and muscular tissue. 3 Mutations in the encoding gene FIG4 have been described to result in a diverse spectrum of syndromes, among which is Charcot‐Marie‐Tooth (CMT) disease type 4J (CMT4J or AR‐CMT‐FIG4), a rare type of recessively inherited peripheral neuropathy. 4 AR‐CMT‐FIG4 is a subtype of CMT accounting for a wide range of phenotypes, both in onset, presentation, and clinical severity, 5 yet most cases account for compound heterozygosity involving an Ile41 to Thr substitution (p.Ile41Thr) and a null mutation, leading to low levels of an unstable FIG4 protein. 1 , 4 , 5 , 6 Nicholson et al estimated the population frequency of this allele to be 0.001, 5 leading to a predicted frequency of homozygous subjects of one per million. We describe here three FIG4‐p.Ile41Thr homozygous patients from unrelated families.
Patients and Methods
Patient recruitment
Patients were referred for molecular screening by their physicians to the center for molecular diagnosis of peripheral neuropathies at Limoges University Hospital Center, France. Family history, clinical evaluation and nerve conduction studies were provided by the physicians. Clinical evaluation was assessed by the patients’ neurologists through a standardized form. Blood samples were collected in EDTA tubes after providing informed consent. The protocol was in accordance with the French ethics legislation.
Mutations detection
Genomic DNA was extracted using standard methods (Illustra DNA Extraction kit BACC3, GEHC). Next Generation Sequencing (NGS) was performed using a 92‐genes custom panel designed for the diagnosis of CMT and associated neuropathies 7 (Ampliseq Custom [Life Technologies, Carlsbad, CA, USA]). The amplified library, which corresponds to exonic and flanking (25bp) intronic regions, was prepared using the Ion P1‐HiQ‐Template‐OT2‐200 kit (Life Technologies), sequenced on a Proton sequencer (Life Technologies), and mapped to the human reference sequence hg19/GHCh37.
Confirmation of FIG4‐c.122T>C and additional variations (HSPB3‐c.67C>T and REEP1‐POLR1A breakpoint), familial screening as well as haplotype analysis (in search of the common ancestral North European FIG4‐I41T haplotype 5 ) were performed through Sanger sequencing (BigDye‐Terminator‐v1.1, capillary‐electrophoresis‐ABI3500xl DX, Sequence‐Navigator‐1.0.1‐Applied Biosystems) using forward and reverse custom primer pairs.
Databases, such as ExACGenome browser (http://exac.broadinstitute.org), dbSNP152 (National Center for Biotechnology Information [NCBI], Bethesda, MD, USA, http://www.ncbi.nlm.nih.gov/projects/SNP/), ClinVar (www.ncbi.nlm.nih.gov/clinvar), DECIPHER v9.29 (https://decipher.sanger.ac.uk/), and FrExAC (http://lysine.univ‐brest.fr/FrExAC/), were also screened.
A review of the literature was performed, based on PubMed (https://www.ncbi.nlm.nih.gov/pubmed), and all the published articles presenting mutations on FIG4, HSPB3, and REEP1‐POLR1A were collected.
Copy number variations (CNVs)
CNVs were assessed through CovCop‐software‐v.1.2.1. 8 The REEP1 duplication was confirmed through multiplexligation‐dependentprobeamplification (MLPA) (SALSA‐MLPA‐P213‐HSP mix‐2 probemix, MRC‐Holland). Further investigation was performed through CGH‐array on G3HumanCGHmicroarrays8x60K (Agilent Technologies).
Results
Clinical evaluation and Electrodiagnostic findings
Clinical data including nerve conduction studies are reported in Table 1. While probands A and C were adult patients with over 20 years of disease duration, patient B was only 15‐years‐old at the time of molecular diagnosis, 7 years after clinical onset. All three patients presented with early, childhood onset of neuropathy, at different developmental stages. While no clinical data could be obtained to narrow down the onset of symptoms for patient A, patient B developed clinical symptoms around the age of 10 and patient C had obvious gait abnormalities as soon as the age of 13 months. All patients presented with distal, bilateral lower limb weakness with severe amyotrophy of the calves. They all had a relatively preserved ambulation, although patients A and B were unable to run and jump. Patient C only suffered from cramps and muscular fatigue. All three patients were able to walk unaided. Patients A and B had diffuse areflexia, while patient C showed preserved reflexes. Sensory involvement was variable, with patient A showing no sensory loss, patient B presenting diffuse hyperesthesia, and patient C having superficial sensory loss in all four limbs, and deep sensory loss in lower limbs. Patients B and C presented with scoliosis while proband A did not show any sign of back deformation. In proband C’s case, severe kyphoscoliosis required a surgical intervention at age 15. Finally, patient A’s clinical history revealed a severe episode of Guillain–Barré syndrome at age 15 with respiratory failure. While the patient mostly recovered, persistent dyspnea on exertion was noted by the physician. Neurophysiological studies revealed severe demyelinating features with mild to severe degrees of axonal involvement, predominating in the lower limbs. While patients exhibited various degrees of upper limbs demyelination as shown through median nerve conduction velocities (Table 1), all presented with severely decreased velocities (<15m/s) in lower limbs with decreased amplitudes of compound motor action potentials.
Table 1.
Clinical features of probands
| Proband | Sex | Onset | Age at molecular diagnosis | Site of Weakness | Diffuse Areflexia | Sensory impairment | Median nerve conduction velocity (m.s‐1) left – right | Peroneal nerve conduction velocity (m.s‐1) left – right | Upper Limb Axonal Loss | Lower Limb Axonal Loss | Other features | Comments |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | M | Childhood | 37 | Distal | Yes | No | 12,7 – ND | ND | Mild | Severe |
Temporal dispersion after proximal stimulation No recordable SNAP |
Guillain–Barré syndrome (age 15) with residual respiratory symptoms. Pes cavus. Preserved ambulation (inability to run and jump). |
| B | M | Childhood (9‐10 years) | 15 | Distal | Yes | Hyperesthesia (UL and LL) | ND – 29 | 12.3 – 11.3 | Moderate | Severe |
Temporal dispersion after proximal stimulation Preserved SNAPs at the upper limbs |
Scoliosis. Pes cavus. Preserved ambulation (inability to run and jump). |
| C | M | Early childhood (13 months) | 44 | Distal | No | Superficial (UL and LL) Deep (LL) | 39 – 40 | 8.2 – 8.1 | No | Moderate | Preserved SNAPs at the upper limbs |
Severe kyphoscoliosis (arthrodesis). Pes cavus. Preserved ambulation (cramps and fatigability). |
UL: Upper Limbs; LL: Lower Limbs. Axonal loss severity criteria were estimated by reporting the measured amplitude of conduction over standard amplitude for each respective center. A ratio >70% indicates mild axonal loss, <70% and >30% indicates moderate axonal loss, and <30% accounts for severe axonal loss. ND: Not Done; L: Left; R: Right; SNAP: Sensory Nerve Action Potential.
Genetic testing
CMT genes and associated neuropathies were screened through our large custom NGS panel, which allowed the detection of a homozygous c.122T>C mutation (p.Ile41Thr) in FIG4 in all three probands, confirmed by Sanger sequencing. Hemizygosity was ruled out through Cov’Cop analysis software. In addition, molecular analysis of probands B and C’s parents showed that they all carried the heterozygous FIG4‐p.Ile41Thr mutation, confirming the homozygous state of p.Ile41Thr in these patients. Family history reported that the two probands’ families were of Western European origin. No family history was available for patient A since he was adopted. Pedigrees of the probands B and C are depicted in Figure 1. Heterozygous carriers were all asymptomatic. As the literature reported that FIG4‐pIle41Thr patients presented with a common ancestral North European haplotype, 4 , 5 we performed such an analysis to rule out the hypothesis of a French new founder effect: five of six haplotypes proved to correspond to the ancestral one and the other haplotype differed only at one distal SNP, indicating that our patients do carry the previously described ancestral variant (Figure 1).
Figure 1.
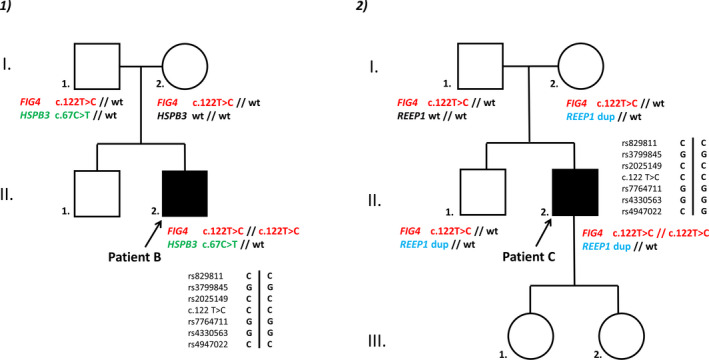
Pedigrees of the two probands and haplotype analysis. 1: patient B, 2: patient C.
Our NGS strategy associated with CNVs detection using Cov’Cop allowed to detect additional variations in probands B and C. Proband B presented with a heterozygote HSPB3‐c.67C>T variation (rs755999042) leading to a premature stop codon (p.Arg23*). Parental screening showed inheritance of this allele from his father (c.67C/c.67T) who is completely asymptomatic. In proband C, Cov’Cop analysis allowed to detect a large multi‐exonic duplication of REEP1, starting from exon 2 onwards to exon 7 included (ENST00000538924.1, NM001164730), confirmed by MLPA. CGH‐Array showed a gain of genetic material extending beyond REEP1 3’ UTR, involving neighboring gene POLR1A, resulting in a direct tandem repeat fusing the 3’ end of REEP1 with the 5’ end of POLR1A. Parental screening (Figure 1) revealed a familial pattern of inheritance, with both the unaffected patient’s mother and brother presenting with this duplication. No other suspect variation has been detected in these three patients.
Discussion
We report here three patients harboring the FIG4‐p.Ile41Thr mutation at the homozygous state, among which two are the first confirmed cases. 6 Out of 764 patients screened for CMT‐related mutations in our laboratory, 12 (1.57%) presented with a FIG4‐p.Ile41Thr mutation. 3/12 were homozygous for this mutation.
The first surprising feature of this report is the presence of a high proportion of homozygotes in our cohort of FIG4‐p.Ile41Thr patients. Indeed, as the prevalence of this mutation is estimated to be of 1.10‐3 in the general population, 9 homozygous cases are expected to be relatively rare. The presence of three cases in a priori unrelated families in the French population could have suggested a possible increase of FIG4‐p.Ile41Thr heterozygosity in this population. However, screening of the FrExAC database showed an allele frequency in accordance with the estimates in the literature. 10 Moreover, the involvement of a new founder effect in the French population seems unlikely insofar as we observed no new haplotype common to our three patients.
In addition, two variations in genes known for their implication in neurological disorders have been detected in two of our three patients. Although our bibliographic study did not suggest any clinical relevance of this particular HSPB3 variant, several articles and databases have related REEP1 duplications, involving or not POLR1A. 11 , 12 Exposed cases show different phenotypes, from spastic paraplegia to autism spectrum disorders. In the case of proband C, none of these symptoms were reported by the physician, neither in the patient nor in his mother and brother suggesting the noncausative effect of this REEP1‐CNV. In the same fashion, pedigree analysis for proband B seems to exclude any causative effect of his HSPB3 variant.
An interesting feature of AR‐CMT‐FIG4 lies in its recessive nature, usually associating a hypomorphic to a null allele. In recent years, it has been demonstrated that the p.Ile41Thr mutation impairs the association between FIG4 and its scaffold protein VAC14, resulting in a catalytically active yet unstable protein. 1 , 13 Further research in the pale tremor mouse showed that overexpression of a mutant p.Ile41Thr transcript on a null background could rescue lethality in a dose‐dependent fashion. They also predicted that the expression of the FIG4‐p.Ile41Thr protein at 10% of the normal level might be sufficient for long‐term survival. 1 The hypomorphic nature of this allele can thus be compensated or not by the second allele: heterozygous carriers show no symptoms, whereas association with a null allele can result in AR‐CMT‐FIG4, since those patients would associate low levels of an unstable yet active FIG4 protein to an inactive one. Lastly, association of two null alleles results in Yunis–Varón syndrome, which associates skeletal defects and severe, often lethal neurologic involvement. 3
Somewhere in‐between, homozygous FIG4‐pIle41Thr patients were expected to be severely affected, as they would express two hypomorphic alleles. Indeed, the three patients reported here presented with an initial background of severe clinical symptoms associating early onset with major distal axon loss, scoliosis, amyotrophy, and gait disturbance. However, they also showed preserved ambulation over time and no signs of clinical evolution, at least in the two cases that lasted for over two decades, while early onset compound heterozygous patients seem to undergo rapid evolution 5 . This observation might be consistent with the murine model, as homozygous patients would express twice the levels of p.Ile41Thr protein than compound heterozygous patients, therefore leading to less severe forms of the disease. Such an observation might thus support the prediction that efforts to increase the levels of this protein in AR‐CMT‐FIG4 patients could prove therapeutic 1 , 2 . However, it is of note that this hypothesis has been tested in vitro, through a Western Blot of protein lysates extracted from fibroblastcells in a cohort of CMT4J patients including compound heterozygous patients and a potential homozygous 9‐year‐old boy 6 . The results did not display any significant FIG4 protein increase in this patient, which comes as a surprise considering the dose‐dependent activity shown in the murine model. Unfortunately, the case’s parents were not demonstrated to be carriers for the mutation, and it remains possible that this patient could be a potential compound heterozygous with a deletion of this area that could have been missed.
It would be of interest to gather more follow‐up data of other confirmed FIG4‐p.Ile41Thr homozygous patients, to define whether those patients really do display a tendency to clinical stability over time, and an overall milder, more homogenous phenotype compared to the diversity of compound heterozygous patients’ phenotypes.
Authors Contribution
ML, CM, and ASL contributed to the study concept and design and the drafting and revising of the manuscript for content and acquisition of data. LM contributed to the drafting and revising of the manuscript for content and interpretation of data. SB, HBD, PD, MCAB, CS, FL, and AM contributed to the revising of the manuscript for content, acquisition, and interpretation of data.
Conflict of Interest
The authors have no conflict of interest to disclose.
References
- 1. Lenk GM, Ferguson CJ, Chow CY, et al. Pathogenic mechanism of the FIG4 mutation responsible for charcot‐marie‐tooth disease CMT4J. PLoS Genet 2011;7(6):e1002104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vaccari I, Carbone A, Previtali SC, et al. Loss of Fig4 in both Schwann cells and motor neurons contributes to CMT4J neuropathy. Hum Mol Genet 2015;24(2):383–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Campeau PM, Lenk GM, Lu JT, et al. Yunis‐Varón syndrome is caused by mutations in FIG4, encoding a phosphoinositide phosphatase. Am J Hum Genet 2013;92(5):781–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chow CY, Zhang Y, Dowling JJ, et al. Mutation of FIG4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J. Nature 2007;448(7149):68–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nicholson G, Lenk GM, Reddel SW, et al. Distinctive genetic and clinical features of CMT4J: a severe neuropathy caused by mutations in the PI(3,5)P2 phosphatase FIG4. Brain 2011;134(7):1959–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hu B, McCollum M, Ravi V, et al. Myelin abnormality in Charcot–Marie–Tooth type 4J recapitulates features of acquired demyelination. Ann Neurol 2018;83(4):756–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lerat J, Cintas P, Beauvais‐Dzugan H, et al. A complex homozygous mutation in ABHD12 responsible for PHARC syndrome discovered with NGS and review of the literature. J Peripher Nerv Syst JPNS 2017;22(2):77–84. [DOI] [PubMed] [Google Scholar]
- 8. Derouault P, Parfait B, Moulinas R, et al. “COV’COP” allows to detect CNVs responsible for inherited diseases among amplicons sequencing data. Bioinforma Oxf Engl. 2017;33(10):1586–1588. [DOI] [PubMed] [Google Scholar]
- 9. VCV000001721.3 ‐ ClinVar ‐ NCBI [Internet]. [cited 2019 Jul 17 ] Available from: https://www.ncbi.nlm.nih.gov/clinvar/variation/1721/#id_first.
- 10. FrExAc database [Internet]. [cited 2020 Jul 30 ] Available from: http://lysine.univ‐brest.fr/FrExAC/showAnnotations?chr=6&position=110036336.
- 11. Beetz C, Schüle R, Deconinck T, et al. REEP1 mutation spectrum and genotype/phenotype correlation in hereditary spastic paraplegia type 31. Brain J Neurol 2008;131(Pt 4):1078–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Holt R, Sykes NH, Conceição IC, et al. CNVs leading to fusion transcripts in individuals with autism spectrum disorder. Eur J Hum Genet 2012;20(11):1141–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ikonomov OC, Sbrissa D, Fligger J, et al. ArPIKfyve regulates Sac3 protein abundance and turnover: disruption of the mechanism by Sac3I41T mutation causing Charcot‐Marie‐Tooth 4J disorder. J Biol Chem 2010;285(35):26760–26764. [DOI] [PMC free article] [PubMed] [Google Scholar]