

Abstract
Focal cortical dysplasia (FCD) and hemimegalencephaly (HME) are related malformations with shared etiologies. We report three patients with a spectrum of cortical malformations associated with pathogenic brain‐specific somatic Ras homolog enriched in brain (RHEB) variants. The somatic variant load directly correlated with the size of the malformation, with upregulated mTOR activity confirmed in dysplastic tissues. Laser capture microdissection showed enrichment of RHEB variants in dysmorphic neurons and balloon cells. Our findings support the role of RHEB in a spectrum of cortical malformations confirming that FCD and HME represent a disease continuum, with the extent of dysplastic brain directly correlated with the somatic variant load.
Introduction
Focal cortical dysplasia (FCD) and hemimegalencephaly (HME) can cause drug‐resistant epilepsy, with histopathological features of FCDII including cortical dyslamination and dysmorphic neurons (DNs) with (FCDIIB) or without (FCDIIA) balloon cells (BCs). 1 Pathogenic somatic variants restricted to brain tissue in PI3K–AKT–mTOR pathway genes are implicated in a range of cortical malformations. 2 We and others have hypothesized that the lesion size may correlate with the degree of mosaicism (the “variant load”) within dysplastic tissue. 3 , 4 , 5
Recently, somatic variants in the gene encoding Ras homolog enriched in brain (RHEB) were reported in cortical malformations; a RHEB p.E40V substitution in one HME patient (21% variant load) 6 and a p.Y35L substitution in one FCDIIA patient (6% variant load). 7 In concordance with these findings, we previously reported genetic data in one FCDIIB patient with RHEB p.E40V substitution (8.9% variant load) and one HME patient with RHEB p.Y35L substitution (17.6% variant load). 3
Here, we characterize a new patient with a transmantle FCD due to a brain‐specific somatic variant in RHEB with a low variant load. In combination with newly presented genetic and imaging data from our two previously described patients with somatic RHEB variants, 3 we determined that variant load correlates with the variable severity observed across the spectrum of FCD. We investigated the enrichment of RHEB variants in abnormal cell types using laser capture microdissection (LCM) as an extension of our previous work in the related mTOR pathway genes DEPDC5, MTOR, and PIK3CA. 3 , 4
Methods
Standard protocol approvals, registrations and patient consents
This study was approved by the Human Research Ethics Committee at the Royal Children’s Hospital (ID 29077) and ethical committee CPP Ile de France II (N° ID‐RCB/EUDRACT‐2015‐A00671‐48). Patient 2 and Patient 3 were registered on ClinicalTrials.gov (N° NCT02890641). Clinical details were obtained from medical records. Written informed consent was obtained from participants or their parents.
Molecular analyses
Brain tissues were collected at the time of surgery and stored as frozen and formalin‐fixed paraffin‐embedded (FFPE) tissue, taken from the left postcentral gyrus in Patient 1, from the opercular and insular region in Patient 2 (first surgery), and the left medial frontal lobe from Patient 3. Blood and FCD‐derived DNA were sequenced using deep targeted sequencing, and variants were validated by droplet digital PCR (ddPCR), site‐specific amplicon sequencing, or Sanger sequencing as previously described. 3 , 4 For immunostaining, FFPE tissues were processed and incubated with anti‐Phospho‐S6 Ribosomal Protein (Ser235/236) antibody (Cell‐Signalling, #2211, #4856), a routinely used antibody to assess mTOR activation. LCM was performed as previously described. 3 Pools of 500 cells (DNs from Patient 2 and DNs and BCs from Patient 3) were isolated. There was insufficient tissue available from Patient 1 for LCM studies.
Results
Clinical descriptions
Patient 1
This 21‐year‐old female presented at age 7 years with sensorimotor seizures manifest by ascending right numbness then weakness. Seizures occurred in clusters of multiple daily events over a few weeks with seizure free periods of up to 12 months in between. Video EEG showed low amplitude left centroparietal slowing and sharp activity and seizures associated with low voltage left centroparietal periodic rhythms. Brain MRI showed a transmantle FCD involving left pre‐ and postcentral gyri (Fig. 1A). Leading up to surgery, seizures had been present for 6 months occurring on a multiple weekly to multiple daily frequency and accompanied by altered responsiveness. Partial resection was performed at age 16 years while awake with resection limited by right arm weakness, which did not persist. Brief right hand sensory seizures continued postoperatively and gradually reduced in frequency until ceasing 15 months postsurgery. During this time, triple anticonvulsant therapy with topiramate, lamotrigine, and clobazam was slowly weaned to low dose topiramate monotherapy. Her brief sensory seizures returned after a 14‐month period of seizure freedom and she has had ongoing incomplete seizure control since then despite trials of additional anticonvulsant medications, varying from a multiple daily to multiple weekly frequency.
Figure 1.
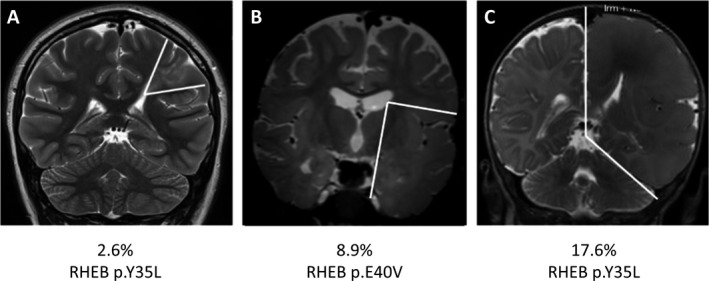
Correlation between somatic variant load in RHEB and the extent of lesion. Coronal T2 MRI scans showing transmantle FCD in Patient 1 (A) lobar FCD in Patient 2 (B), and HME in Patient 3 (C). The boundaries of the malformations are illustrated by white lines. Somatic variants in RHEB were identified in all three patients, with positive correlation between the variant load and the size of the brain lesion. The corresponding genetic variant and somatic variant load are indicated for each patient.
Patient 2
This 6‐year‐old male presented at age 6 months with focal seizures manifest by right arm tonic posturing and head deviation to the left. Video EEG showed left fronto‐temporal epileptic discharges. Stereo EEG showed a left operculo‐insular and lateral temporal seizure onset zone. Brain MRI showed a left temporo‐opercular FCD (Fig. 1B). First surgery at age 2 consisted of resection of the opercular and insular region but did not control seizures (Engel outcome score 4). Due to ongoing seizures, a hemispherotomy was subsequently performed at age 4. The patient has been seizure free for 4 years without AED (Engel outcome score 1).
Patient 3
This 3‐year‐old male presented at age 3 days with multiple daily focal seizures manifest by right‐sided clonic jerking, eyelid blinking, and cyanosis. EEG revealed multiple left hemisphere foci and 3T MRI showed left HME (Fig. 1C). A syndromic diagnosis of epidermal naevus syndrome was made. Hemispherotomy was performed at age 5 months. This patient has been seizure free for 2.5 years without AED (Engel outcome score 1).
Molecular findings
We identified a brain‐specific somatic RHEB variant (NM_005614.4:c.104_105delACinsTG; p.Y35L) in Patient 1. Brain‐specific somatic RHEB variants were previously identified in Patient 2 and Patient 3 3 (Table 1). Variants were not detected in the matched blood samples.
Table 1.
Somatic RHEB variants detected in the patients with cortical malformations.
| Patient ID | Histopathology | cDNA Change | Protein Change | Brain VAF (targeted sequencing) | Validation |
|---|---|---|---|---|---|
| Patient 1 | FCDIIB | c.104_105delACinsTG | p.Y35L | 2.6% | Droplet Digital PCR (2.6%) |
| Patient 2 1 | FCDIIB | c.119A > T | p.E40V | 8.9% | Site‐specific amplicon sequencing (8.9%) |
| Patient 3 1 | FCDIIB (HME) | c.104_105delACinsTA | p.Y35L | 17.6% | Sanger sequencing |
VAF, variant allele frequency.
Patient 2 and Patient 3 have previously been reported.
Analysis of imaging showed that the extent of the malformation correlated with somatic variant load. In Patient 1 (2.6% variant load), the small FCD was restricted to a transmantle dysplasia affecting left pre‐ and postcentral gyri (Fig. 1A). In Patient 2 (8.9% variant load), a lobar FCD affected the entire left temporal lobe (Fig. 1B) and in Patient 3 (17.6% variant load), left HME was present (Fig. 1C).
We performed immunostaining against p‐S6 (canonical hallmark of mTOR kinase activity) and identified abundant DNs and BCs in brain specimens of all patients with strong immunoreactivity (Fig. 2A–C), indicating upregulated mTOR activity. In contrast, immunoreactivity against p‐S6 was not observed in non‐mTOR‐related FCDI and postmortem control specimens (Fig. 2D–E).
Figure 2.
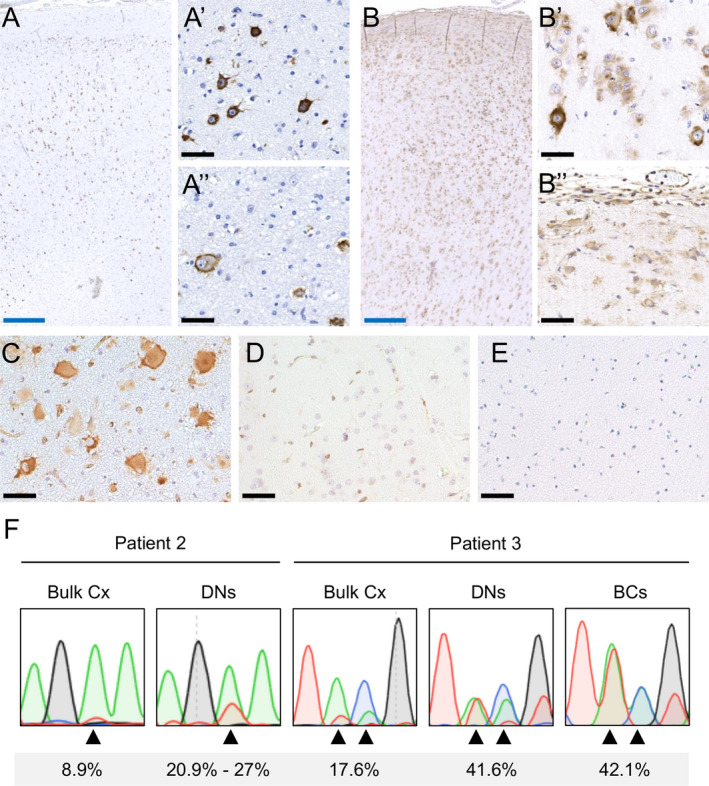
RHEB‐mutated DNs and BCs show p‐S6 immunoreactivity. Immunostaining of patient brain sections with an antibody directed against p‐S6 shows abnormal cells with strong immunoreactivity in Patient 2 (A), Patient 3 (B), and Patient 1 (C). In contrast, immunostaining of p‐S6 in an FCDI resection specimen (D) and a normal postmortem specimen (E) did not show strong immunoreactivity. In Patient 2, p‐S6‐positive cells are distributed in all cortical layers (A), a predominance of DNs is observed (A’), while scattered BCs are found at the gray–white matter borders (A’’). In Patient 3 cortical layering is completely lost (B), with high density of BCs all over the specimen, including layer 1 (B), and often organized in clusters (B’’), while DNs are more interspersed (B’). Black scale bar = 50 μm, blue scale bar = 500 μm. Sequencing of LCM‐isolated cells shows variant enrichment in DNs and BCs (F). There were insufficient BCs from Patient 2 for LCM. Arrowheads indicate the altered bases in Patient 2 (A > T) and Patient 3 (AC > TA). Variant allele frequencies quantified by site‐specific amplicon sequencing are shown. DN: Dysmorphic neuron; BC: Balloon cell.
Density of pS6‐positive cells in cortical layers could not be well appreciated in Patient 1 due to limited tissue. Density of pS6‐positive cells was lower in Patient 2 compared to Patient 3, consistent with the lower variant load. In Patient 2, pS6‐positive cells were distributed in all cortical layers (except layer 1), with a higher proportion and distribution of DNs compared to scattered BCs at the gray–white junction (Fig. 2A). In Patient 3, cortical layering was completely absent, with high density of BCs throughout the specimen, including layer 1, often organized in small clusters, while DNs were rarer (Fig. 2B).
To investigate which cell types harbor the somatic variants, we used LCM to isolate pools of DNs from Patient 2, and pools of DNs and BCs from Patient 3. There was insufficient material to perform LCM from Patient 1 and insufficient number of BCs from frozen tissue of Patient 2. Sanger and site‐specific amplicon sequencing of each pool revealed clear enrichment of the variants in DNs and BCs compared to bulk tissue, suggesting enrichment in these cell types (Fig. 2F).
Discussion
Here, we report clinical, neuropathological and genetic data of three patients with brain mosaic RHEB variants leading to a spectrum of FCD, the extent of which correlates with the somatic variant load. RHEB is a member of the Ras superfamily GTPases that can activate mTOR signaling pathway. 8 Immunostaining of dysplastic tissues showed that RHEB p.Y35L and p.E40V substitutions result in hyperactivation of mTOR. LCM findings show enrichment of somatic RHEB variants in DNs and BCs, as we previously reported in other mTOR pathway genes DEPDC5, MTOR, and PIK3CA, 3 , 4 confirming that these variants account for the abnormal cell types. The presence of the variants, at similar allele frequencies in both DNs and BCs suggests that both cell types share a common developmental lineage.
Patient 1 and Patient 3 both carried the p.Y35L substitution, with lower variant load causing small FCD and higher variant load causing HME. This supports the hypothesis that the timing of the postzygotic somatic mutation is crucial in determining lesion size. In accordance with this notion, germline de novo RHEB mutations have been previously reported in three patients with macrocephaly. 9 Furthermore, a mouse model expressing a constitutively active form of RHEB (RHEBCA) showed a dose‐dependent phenotype, characterized by no seizures at low RHEBCA levels and spontaneous recurrent seizures increasing proportionally with higher doses in the mutant. 10 The findings in human patients and mouse models further strengthen the correlation between the lesion size and variant load.
Our findings support the role of RHEB somatic variants in the etiology of FCD and HME, confirming that both entities are a continuum of mTOR‐related cortical malformations. The detection of somatic variants in lesional tissue relies on deep targeted sequencing to identify mosaic variants at low (<5%) allele frequencies. The finding of recurrent RHEB variants in FCD and HME emphasizes that RHEB should be included with other mTOR pathway genes in sequencing panels for cortical malformations. Precision therapeutic strategies targeting the mTOR pathway are currently under investigation for the treatment of seizures and neurodevelopmental symptoms associated with FCD. For example, an ongoing clinical trial uses the mTOR inhibitor everolimus to treat patients with FCDII who have ongoing seizures after more than two antiepileptic drugs and surgery (NCT03198949).
Conflict of Interest
The authors report no disclosures.
Acknowledgments
We thank the families involved in this research and acknowledge the assistance of Kate Pope and Greta Gillies in manuscript preparation.
Funding information
This study was funded by the National Health and Medical Research Council (GNT1128933 to RJL and PJL and GNT1161549 to PJL and RJL), European Research Council (N°682345 to SB), "Investissements d'avenir" (ANR‐10‐IAIHU‐06 and ANR‐18‐RHUS‐0005 to SB) and the Brain Foundation (to RJL). Additional funding was provided by the Independent Research Institute Infrastructure Support Scheme and the Victorian State Government Operational Infrastructure Program. RJL is supported by a Melbourne Children’s Clinician Scientist Fellowship. PJL is supported by the Vincent Chiodo Foundation. WSL is supported by the Melbourne Research Scholarship and the Research Training Program Scholarship. This study was not industry sponsored.
Funding Statement
This work was funded by National Health and Medical Research Council grants GNT1128933 and GNT1161549; European Research Council grant N°682345; Investissements d'avenir grants ANR‐10‐IAIHU‐06 and ANR‐18‐RHUS‐0005; Brain Foundation grant ; Independent Research Institute Infrastructure Support Scheme grant ; Victorian State Government Operational Infrastructure Program grant ; Melbourne Children’s Clinician Scientist Fellowship grant ; Vincent Chiodo Foundation grant ; Melbourne Research Scholarship grant ; Research Training Program Scholarship grant .
Contributor Information
Stéphanie Baulac, Email: stephanie.baulac@icm-institute.org.
Richard J. Leventer, Email: richard.leventer@rch.org.au.
References
- 1. Blumcke I, Thom M, Aronica E, et al. The clinicopathologic spectrum of focal cortical dysplasias: a consensus classification proposed by an ad hoc Task Force of the ILAE Diagnostic Methods Commission. Epilepsia 2011;52:158–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Marsan E, Baulac S. Review: mechanistic target of rapamycin (mTOR) pathway, focal cortical dysplasia and epilepsy. Neuropathol Appl Neurobiol 2018;44:6–17. [DOI] [PubMed] [Google Scholar]
- 3. Baldassari S, Ribierre T, Marsan E, et al. Dissecting the genetic basis of focal cortical dysplasia: a large cohort study. Acta Neuropathol 2019;138:885–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lee WS, Stephenson SEM, Howell KB, et al. Second‐hit DEPDC5 mutation is limited to dysmorphic neurons in cortical dysplasia type IIA. Ann Clin Transl Neurol 2019;6:1338–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. D'Gama AM, Woodworth MB, Hossain AA, et al. Somatic mutations activating the mTOR pathway in dorsal telencephalic progenitors cause a continuum of cortical dysplasias. Cell Rep 2017;21:3754–3766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Salinas V, Vega P, Piccirilli MV, et al. Identification of a somatic mutation in the RHEB gene through high depth and ultra‐high depth next generation sequencing in a patient with Hemimegalencephaly and drug resistant Epilepsy. Eur J Med Genet 2019;62:103571. [DOI] [PubMed] [Google Scholar]
- 7. Zhao S, Li Z, Zhang M, et al. A brain somatic RHEB doublet mutation causes focal cortical dysplasia type II. Exp Mol Med 2019;51:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Goitre L, Trapani E, Trabalzini L, Retta SF. The Ras superfamily of small GTPases: the unlocked secrets. Methods Mol Biol 2014;1120:1–18. [DOI] [PubMed] [Google Scholar]
- 9. Reijnders MRF, Kousi M, van Woerden GM, et al. Variation in a range of mTOR‐related genes associates with intracranial volume and intellectual disability. Nat Commun 2017;8:1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nguyen LH, Mahadeo T, Bordey A. mTOR hyperactivity levels influence the severity of epilepsy and associated neuropathology in an experimental model of tuberous sclerosis complex and focal cortical dysplasia. J Neurosci 2019;39:2762–2773. [DOI] [PMC free article] [PubMed] [Google Scholar]