

Abstract
Objective
To characterize the natural history of spinal muscular atrophy (SMA) over 24 months using innovative measures such as wearable devices, and to provide evidence for the sensitivity of these measures to determine their suitability as endpoints in clinical trials.
Methods
Patients with Type 2 and 3 SMA (N = 81) with varied functional abilities (sitters, nonsitters, nonambulant, and ambulant) who were not receiving disease‐modifying treatment were assessed over 24 months: motor function (Motor Function Measure [MFM]), upper limb strength (MyoGrip, MyoPinch), upper limb activity (ActiMyo®), quantitative magnetic resonance imaging (fat fraction [FFT2] mapping and contractile cross‐sectional area [C‐CSA]), pulmonary function (forced vital capacity [FVC], peak cough flow, maximum expiratory pressure, maximum inspiratory pressure, and sniff nasal inspiratory pressure), and survival of motor neuron (SMN) protein levels.
Results
MFM32 scores declined significantly over 24 months, but not 12 months. Changes in upper limb activity could be detected over 6 months and continued to decrease significantly over 12 months, but not 24 months. Upper limb strength decreased significantly over 12 and 24 months. FVC declined significantly over 12 months, but not 24 months. FFT2 increased over 12 and 24 months, although not with statistical significance. A significant increase in C‐CSA was observed at 12 but not 24 months. Blood SMN protein levels were stable over 12 and 24 months.
Interpretation
These data demonstrate that the MFM32, MyoGrip, MyoPinch, and ActiMyo® enable the detection of a significant decline in patients with Type 2 and 3 SMA over 12 or 24 months.
Introduction
Spinal muscular atrophy (SMA) is a severe, progressive neuromuscular disorder which leads to a progressive loss of motor function and reduced life expectancy. 1 , 2 , 3 , 4 SMA is caused by reduced levels of survival of motor neuron (SMN) protein due to deletions and/or mutations in the SMN1 gene. A second gene, SMN2, produces only low levels of functional SMN protein. 1 , 2 The pathophysiology resulting from reduced SMN protein levels is thought to affect multiple systems in the body. 3
SMA manifests as a broad spectrum of severities and is classified into clinical types based on the age of onset and the highest motor milestone achieved: 4 Type 1 (onset age < 6 months; patients are never able to sit independently), Type 2 (onset age < 18 months; patients acquire independent sitting but are unable to walk or stand independently), Type 3 (onset age > 18 months; patients achieve independent walking), and Type 4 (patients achieve motor milestones and symptoms develop in adulthood). However, while these classifications give some indication of expected survival and long‐term motor function, their predictive value is only moderate. 5 , 6 Therefore, the standard‐of‐care guidelines for SMA are now based on functional status (nonsitter, sitter, ambulant) instead of SMA type. 7
Three therapies are currently approved for the treatment of SMA: 8 nusinersen (SPINRAZA® [Ionis Pharmaceuticals/Biogen]), an intrathecally administered SMN2‐targeting antisense oligonucleotide; 9 onasemnogene abeparvovec‐xioi (ZOLGENSMA® [Novartis]), a gene‐transfer therapy that delivers a functional copy of an SMN1 gene by a single intravenous infusion in patients < 2 years old; 10 and risdiplam (EVRYSDI™ [Genentech, Inc.]), an orally administered SMN2 splicing modifier for adults and children ≥ 2 months old. 11 These therapies have demonstrated improvements in survival and motor function; 12 however, the long‐term efficacy of these treatments remains unknown, and access may be restricted by SMA type. 13 Therefore, there is a clear need to identify reliable outcome measures for different stages in SMA progression so that the full benefit or limitation of a potential therapy can be recognized, and patient burden reduced.
A key challenge with current clinical assessments relates to the slow progression of SMA. Unlike Type 1 SMA, in which rapid deterioration occurs, Type 2 and 3 SMA progress slowly, so decline can be challenging to detect over short periods using current measures such as the Revised Upper Limb Module (RULM), 6‐min walk test (6MWT), and Expanded Hammersmith Functional Motor Scale (HFMSE). 5 The inability of the HFMSE to capture some small functional changes in patients with Type 2 and 3 SMA was demonstrated in a natural history study which showed that pulmonary and motor function outcome measures remained relatively stable over 12 months, requiring longitudinal observational studies to extend beyond 12 months. 14 More recently, in two double‐blind, placebo‐controlled studies, the Motor Function Measure (MFM), 15 , 16 and RULM 16 captured a significant difference between treated and untreated patients, while the HFMSE failed to do this.
To reduce the number of patients included to adequately power clinical trials, the risk of false negatives, and the duration of clinical trials, more sensitive outcome measures are needed. This is true for all patients with Type 2 and 3 SMA, especially older patients with a longer disease duration, who have been shown to have a reduced response to disease‐modifying treatments. 17 Additionally, the increasing use of add‐on therapy in clinical trials, in which the effect of a second drug is smaller and more variable than a single drug compared with placebo, further highlights the need for more sensitive outcome measures to detect small functional changes.
Novel outcome measures aim to overcome some of the difficulties faced when using current clinical measures by capturing small functional changes over shorter periods. Recent studies have demonstrated the feasibility of wearable devices, such as ActiMyo®, which generate digital measurements and allow patients to be monitored remotely in their daily lives. This enables real‐world longitudinal data to be captured in a way that is currently not possible in a clinical setting. 18 Furthermore, such measures facilitate a deeper understanding of the natural history of SMA, which may lead to better‐informed treatment decisions, such as those related to continuing or modifying treatment.
Here, we present data of a 24‐month observational natural history study, which may be one of the last in SMA due to an increasing number of patients receiving SMA treatment as a result of the recent approval of disease‐modifying treatments. Data from the MFM are presented to characterize the disease course over 24 months in patients with Type 2 and 3 SMA. Further assessments include muscle strength (digital measures assessed by performance in grip and pinch strength tests through wearable devices), fat fraction (FFT2), and pulmonary function tests for the identification of (1) prognostic variables of SMA, (2) best‐outcome measures for future treatment studies, and (3) SMN biomarkers.
Methods
Study design and analysis population
The 2‐year NatHis‐SMA study was conducted between May 2015 and May 2018 in nine neuromuscular disease reference centers (located in Belgium, France, and Germany). Protocol approval was authorized in France by the regulatory health authority and the central Ethics committee and approved in Liège, Leuven, and Essen by the local Ethics Committees. Parental/legal guardian consent was obtained prior to inclusion in the study. The study is registered on ClinicalTrials.gov (NCT02391831).
Patient population
Baseline patient characteristics for this study have been published previously. 19 Patients included were aged 2–30 years, with Type 2 SMA (n = 53 [nonsitters, n = 19; sitters, n = 34]) or Type 3 SMA (n = 28 [nonambulant, n = 9; ambulant, n = 19]). Nonsitters are defined as patients with Type 2 SMA who scored 0 on Item 9 of the MFM, while sitters are patients with Type 2 SMA who scored ≥ 1 on Item 9 of the MFM. Ambulant is defined as patients able to walk 10 meters without assistance. Functional status was determined at baseline and was not changed regardless of whether a patient's status changed over time. Assessments were adjusted for each patient based on age (2–5 years old and 6–30 years old) and ambulant status. Exclusion criteria included patients exposed to disease‐modifying treatments within the 6 months prior to study recruitment, patients with comorbid conditions that could significantly interfere with disease outcome measures and pregnant or breastfeeding women.
Protocol
Outcomes assessed during the study include motor function (MFM), upper limb strength (MyoGrip, MyoPinch), upper limb activity (ActiMyo®), quantitative magnetic resonance imaging (MRI) (FFT2 mapping and muscle contractile cross‐sectional area [C‐CSA]), pulmonary tests (forced vital capacity [FVC], peak cough flow [PCF], maximum expiratory pressure [MEP], maximum inspiratory pressure [MIP], and sniff nasal inspiratory pressure [SNIP]), and measurement of blood SMN protein levels. These outcomes were evaluated at baseline and then every 6 months (±28 days) for the 24‐month duration of the study, except for the MRI assessment (performed annually in two French centers). The full protocol for this trial has been published previously. 19 One‐year ActiMyo® data are only available for a limited number of patients due to lack of availability of ActiMyo® devices at the time (Month 6, n = 25 [ambulant, n = 6; nonambulant, n = 19]; Month 12, n = 17 [ambulant, n = 4; nonambulant, n = 13]; Month 18, n = 7 [ambulant, n = 1; nonambulant, n = 6]; Month 24, n = 2 [ambulant, n = 1; nonambulant, n = 1]).
Other assessments measured during this study, including electrophysiological parameters, quality of life, and detailed MRI, are to be published separately.
Outcome measures
Motor function assessment
The MFM assesses motor function across three domains: standing position, ambulation, and transfers (D1), axial and proximal motor function (D2), and distal motor function (D3). A 4‐point Likert scale is used to rate the patient's maximal functional ability; higher scores indicate a higher level of function. MFM32, a 32‐item scale, was used for patients aged ≥ 6 years and the MFM20, a 20‐item scale, was used for children 2–5 years old. For patients 4–6 years old, the physiotherapist selected the most appropriate version based on the child's motor function abilities and overall development. The same scale was used throughout the study for each patient.
Upper limb strength assessment
Dominant and nondominant hand strength was assessed with the MyoGrip and MyoPinch devices (Ateliers Laumonier, Nesles‐la‐Vallée, France) in patients ≥ 6 years old, as previously described. 19 , 20 , 21 MyoGrip measures handgrip forces from 0 to 90 kg, and MyoPinch measures key pinch with a nominal scale of 10 kg. 22 Patients were given at least three attempts on both arms to demonstrate maximal isometric grip and key pinch strength. If improvements between attempts were observed, patients were given up to two additional attempts. Results are expressed as a percentage of predicted values based on age 23 with the maximal value used for analysis. Higher values reflect a higher level of upper arm strength.
Continuous movement recording
Upper arm activity was assessed with ActiMyo® in a subset of nonambulant patients ≥ 6 years old, as previously described. 19 ActiMyo® is a wearable device (Sysnav, Vernon, France) that measures patient movement in a real‐life setting. 20 , 24 The device was worn daily to longitudinally assess individualized changes in activity. One sensor was worn on the wrist, and the other was attached to the wheelchair. Five variables representing upper limb activity were generated: the wrist angular velocity (||Ω||), the wrist acceleration (||A||), the wrist vertical acceleration against gravity (vA), and the power and the percentage of activity time. Higher values reflect a higher level of upper limb activity.
In a limited number of ambulant patients, ActiMyo® was also used to measure stride length and velocity. 24 , 25 The 95th percentile of the stride‐velocity distribution has been recently qualified by the European Medicines Agency and has a valid secondary outcome in Duchenne Muscular Dystrophy (DMD). 25
MRI muscle assessment
FFT2 maps were derived from quantitative water‐fat imaging obtained using a 3‐point Dixon 3D gradient echo sequence; the Dixon‐based regions of interest were used in combination with the FFT2 mapping to provide the C‐CSA, as previously described. 19 These assessments were carried out in one study site with a limited number of patients. FFT2 and C‐CSA were calculated separately for arms, forearms, legs, and thighs based on outputs for different muscle groups (see supplemental material for more detailed information).
Pulmonary function tests
Patients ≥ 6 years performed several assessments while in the sitting position: FVC, PCF, MEP, MIP, and SNIP. FVC and PCF were measured using the Vitalograph spirometer and software, while MEP, MIP, and SNIP were evaluated using the MicroRPM device. Assessment results are expressed as percentages of predicted values based on age, weight, and height; 26 , 27 , 28 , 29 , 30 the best results of three measurements were selected for further analysis. (See supplemental material for more detailed information).
SMN protein levels
Blood SMN protein levels were measured by an optimized SMN protein assay (see supplemental material for detailed information). SMN1 and SMN2 copy numbers were determined, as previously described. 19
Statistical analyses
Four functional status groups were predefined for comparison analyses: 19 nonsitters with Type 2 SMA, sitters with Type 2 SMA, nonambulant patients with Type 3 SMA, and ambulant patients with Type 3 SMA. To determine whether outcome measures could differentiate between patients in different groups, proportions were compared using the Chi‐square test, and quantitative variables were compared using the Kruskal–Wallis one‐way analysis of variance; post hoc analyses based on the Dunn‐Bonferroni method were performed for significant results. The Wilcoxon signed‐rank test was used to assess changes over time. The relationships between variables were examined using Spearman's correlation coefficients. Standardized response means (SRM) were calculated for all assessments. All analyses were performed using SPSS Statistics 22 Statistical software (IBM Corp, Armonk, NY). The limit of statistical significance was set to 0.05.
Results
Participant disposition at Month 24
Eighty‐one patients were enrolled in this study (Fig. 1). A high number of these participants (n = 32) discontinued the study between Months 12 and 24 to pursue approved treatments or enroll in clinical trials. Baseline MFM scores increased with decreasing SMA severity; nonsitters with Type 2 SMA had the lowest scores, while ambulant patients with Type 3 SMA had the highest scores. Further baseline clinical and physical characteristics have been previously published. 19
Figure 1.
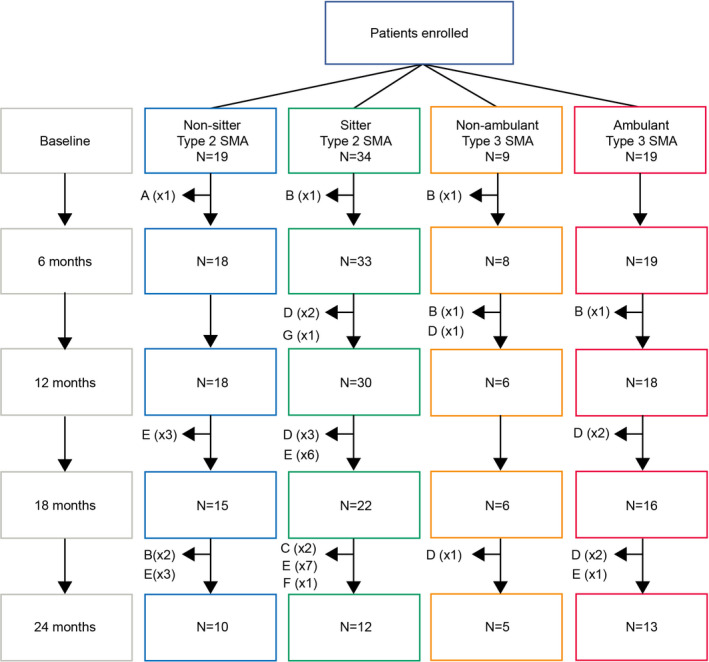
CONSORT diagram. Reasons for premature withdrawal include: (A) loss of follow‐up; (B) consent withdrawal; (C) physician decision; (D) enrollment into other clinical trials; (E) initiation of nusinersen treatment; (F) other; and (G) missed visit. SMA, spinal muscular atrophy.
Motor function
Overall, the MFM total score discriminated between the four functional groups (Fig. 2 and Fig. S1). At Month 12 (n = 40), there was a decrease in MFM32 median total score from baseline by 1.05 points (range: −14.58 to 5.21; [Wilcoxon test P = 0.073]; SRM: 0.37) in all patients with Type 2 and 3 SMA. This decrease was not significant. At Month 24 (n = 27), MFM32 median total score decreased significantly from baseline scores by 2.08 points (range: –17.71 to 4.17; [P = 0.009]; SRM: 0.55) in all patients (Table S1).
Figure 2.

MFM total scores from baseline to 24 months in individuals with SMA according to age and SMA type. MFM20 was used in patients < 6 years of age; MFM32 was used in patients ≥ 6 years of age. For patients aged 4–6 years old, the physiotherapist selected the most appropriate version based on the child's motor function abilities. Each line represents the trajectory of one patient over 24 months. Assessments were made at baseline, Month 6, 12, 18, and 24. MFM, motor function measure; MFM20, 20‐item MFM; MFM32, 32‐item MFM.
There was a significant decrease in MFM32 median total score of −3.13 points (range −8.33 to 4.17; P = 0.041; SRM: 0.80) over 24 months in nonsitters with Type 2 SMA (n = 11) (Fig. S1, Table S1); however, no significant decrease was found in patients in other functional groups (sitters with Type 2 SMA [−1.56 points; P = 0.285; SRM: 0.57]; nonambulant patients with Type 3 SMA [−1.04 points; P = 0.680; SRM: 0.39]; and ambulant patients with Type 3 SMA [0.00 points; P = 0.465; SRM: 0.36]) (Table S1).
There were no significant changes in MFM20 scores. At Month 12, median MFM20 scores increased by 1.67 points (n = 30; [range: −18.33 to −13.33]; P = 0.430) and decreased by 3.34 points (n = 10; [range: −11.66 to 10.00]; P = 0.313) at Month 24.
Upper limb strength
Overall, there was a significant decrease in median grip strength of 0.53% of predictive value at Month 12 (n = 38; [Wilcoxon test P = 0.003]; SRM: 0.42) and 0.95% at Month 24 (n = 27; [P = 0.002]; SRM: 0.49) independent of the SMA subtype (Fig. 3A, Table S2). Large interindividual variability in grip strength among participants with Type 3 SMA was observed, which decreased globally over time (Fig. 3B). There was also an overall significant decrease in median pinch strength of 0.79% of predictive value at Month 12 (n = 37; [P = 0.016]; SRM: 0.35) and a decrease of 1.21% at Month 24 (n = 27; [P = 0.037]; SRM: 0.38) (Fig. 4A, Table S3). There was no correlation between age and median pinch strength (Fig. 4B).
Figure 3.
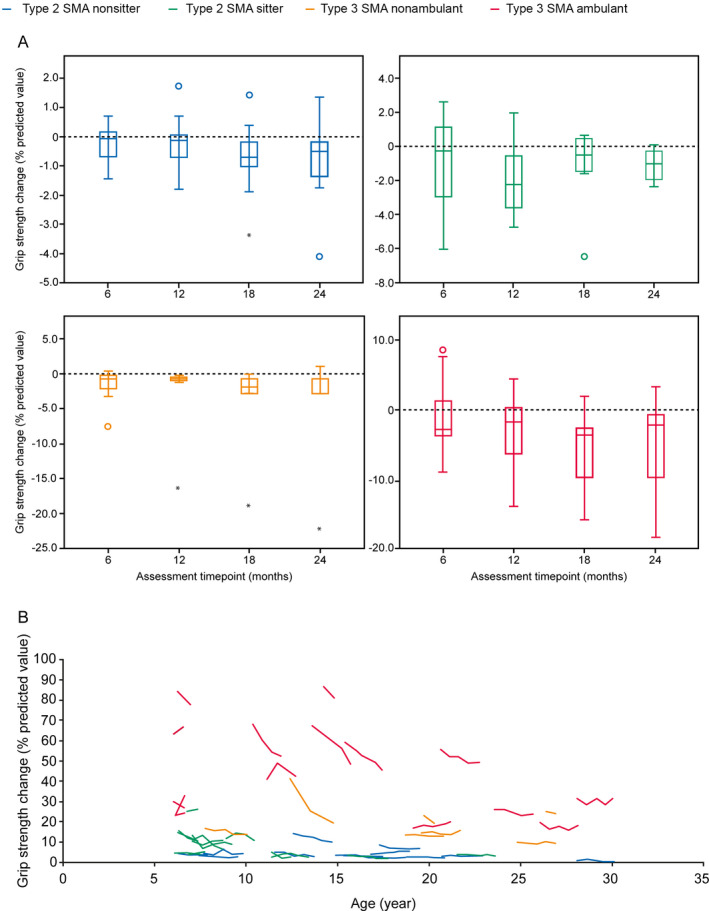
Change in grip strength over 24 months by (A) SMA type and (B) age. Grip strength was measured as a percentage of predictive values based on age using MyoGrip at baseline, Month 6, 12, 18, and 24 (Month 12, n = 38; Month 24, n = 27). Higher values reflect a higher level of upper arm strength. A) Closed circles represent outliers. B) Each line represents the trajectory of one patient over 24 months. SMA, spinal muscular atrophy.
Figure 4.
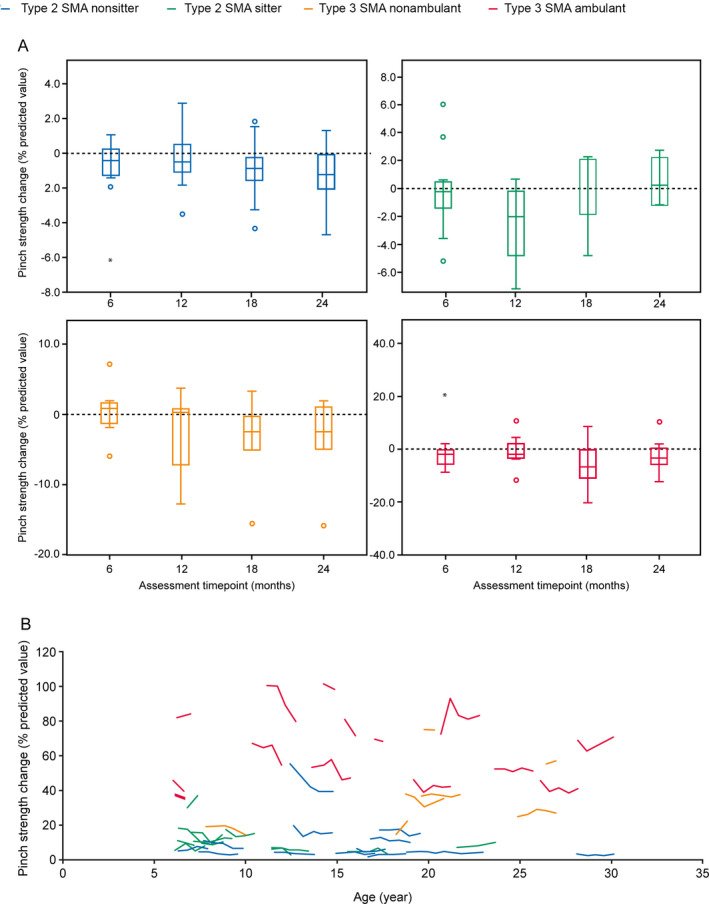
Change in mean pinch strength over 24 months by (A) SMA type and (B) age. Pinch strength was measured as a percentage of predictive values based on age using MyoPinch at baseline, Month 6, 12, 18, and 24. (Month 12, n = 37; Month 24, n = 27). Higher values reflect a higher level of upper arm strength. A) Closed circles represent outliers and B) Each line represents the trajectory of one patient over 24 months. SMA, spinal muscular atrophy.
Continuous movement recording
Upper limb activity was assessed by ActiMyo® in nonsitters and sitters with Type 2 SMA and nonambulant patients with Type 3 SMA. The overall median 95th percentile of ||Ω||, ||A||, vA, and power significantly decreased over 6 and 12 months of observation: at Month 6 (n = 19), ||Ω|| decreased by 9.12°/s (Wilcoxon test P = 0.018, SRM: 0.64); ||A|| decreased by 0.019 m/s2 (P = 0.014; SRM: 0.63); vA increased by 0.003 m/s2 (P = 0.04; SRM: 0.60); and power decreased by 0.037 W/kg (P = 0.03; SRM: 0.55). At Month 12 (n = 13), ||Ω|| decreased by 5.28°/s (P = 0.011; SRM: 0.74); ||A|| decreased by 0.015 m/s2 (P = 0.016; SRM: 0.75); vA increased by 0.0004 m/s2 (P = 0.345; SRM: 0.40); and power decreased by 0.049 W/kg (P = 0.064; SRM: 0.55). (Table 1, Fig. S2). Due to the small number of patients recorded for follow‐up at Month 24 (n = 1), decline over a longer period could not be calculated (data not shown).
Table 1.
Changes in ActiMyo® upper limb activity over 6 and 12 months.
| ||Ω|| mean (°/s) | ||Ω|| 95th percentile (°/s) | ||A|| mean (m/s2) | ||A|| 95th percentile (m/s2) | vA mean (m/s2) | vA 95th percentile (m/s2) | Power mean (W/kg) | Power 95th percentile (W/kg) | |
|---|---|---|---|---|---|---|---|---|
| Changes from baseline at Month 6 | ||||||||
| N | 19 | 19 | 19 | 19 | 19 | 19 | 19 | 19 |
| Mean | −2.66 | −9.62 | −0.01 | −0.02 | 0.01 | 0.00 | −0.01 | −0.05 |
| SD | 4.33 | 15.11 | −0.01 | 0.02 | 0.02 | 0.01 | 0.02 | 0.08 |
| Median | −2.68 | −9.12 | −0.01 | −0.02 | 0.01 | 0.00 | −0.01 | −0.04 |
| Min | −8.18 | −32.49 | −0.02 | −0.05 | −0.03 | 0.00 | −0.05 | −0.22 |
| Max | 8.04 | 24.71 | 0.01 | 0.04 | 0.03 | 0.02 | 0.03 | 0.12 |
| P‐value* | 0.02 | 0.02 | 0.01 | 0.01 | 0.07 | 0.04 | 0.01 | 0.03 |
| SRM | 0.61 | 0.64 | 0.71 | 0.63 | 0.47 | 0.60 | 0.63 | 0.55 |
| Changes from baseline at Month 12 | ||||||||
| N | 13 | 13 | 13 | 13 | 13 | 13 | 13 | 13 |
| Mean | −2.63 | −9.12 | −0.01 | −0.02 | 0.02 | 0.00 | −0.01 | −0.05 |
| SD | 3.51 | 12.40 | 0.01 | 0.02 | 0.03 | 0.01 | 0.02 | 0.10 |
| Median | −1.37 | −5.28 | 0.00 | −0.02 | 0.01 | 0.00 | −0.01 | −0.05 |
| Min | −9.53 | −38.59 | −0.02 | −0.05 | −0.02 | −0.01 | −0.06 | −0.26 |
| Max | 3.55 | 11.21 | 0.01 | 0.04 | 0.10 | 0.03 | 0.02 | 0.12 |
| P‐value* | 0.01 | 0.01 | 0.02 | 0.02 | 0.13 | 0.35 | 0.02 | 0.06 |
| SRM | 0.75 | 0.74 | 0.71 | 0.75 | 0.50 | 0.40 | 0.67 | 0.55 |
Statistical significance was reached at Months 6 and 12 but was not reached at Month 18 (data not shown). Only one patient performed a follow‐up at Month 24 (data not shown).
||Ω||, wrist angular velocity; ||A||, wrist acceleration; SD, standard deviation; SRM, standardized response means; vA, wrist vertical acceleration.
P‐value from Wilcoxon test.
No statistically significant changes in stride velocity were observed in ambulant participants throughout the study period at Month 6 (n = 6) or Month 12 (n = 4). (Table S4, Fig. S3).
MRI muscle assessment
Overall, there was an increase in FFT2 at Months 12 and 24 that was not statistically significant: median arm FFT2 increased by 0.0060% at Month 12 (n = 16; [Wilcoxon test P = 0.501]; SRM: 0.23) and decreased by 0.0051% at Month 24 (n = 9; [P = 0.767]; SRM: 0.18); median forearm FFT2 increased by 0.0087% (n = 13; [P = 0.422]; SRM: 0.35) at Month 12 and by 0.0134% at Month 24 (n = 10; [P = 0.203]; SRM: 0.39); median thigh FFT2 increased by 0.0211% at Month 12 (n = 7; [P = 0.398]; SRM: 0.36) and by 0.0449% at Month 24 (n = 3; [P = 0.109]; SRM: 10.68); median leg FFT2 increased by 0.0291% at Month 12 (n = 7; [P = 0.063]; SRM: 0.87) and by 0.0258% at Month 24 (n = 3; [P = 0.109]; SRM: 1.40) (Table 2, Figs. S4 and S5).
Table 2.
FFT2 changes over 12 and 24 months.
| FF T2 changes (%) | Arm | Forearm | Thigh | Leg |
|---|---|---|---|---|
| Changes from baseline at Month 12 | ||||
| N | 16 | 13 | 7 | 7 |
| Mean | 0.0090 | 0.0115 | 0.0093 | 0.0194 |
| SD | 0.0383 | 0.0326 | 0.0255 | 0.0223 |
| Median | 0.0060 | 0.0087 | 0.0211 | 0.0291 |
| Min | −0.0433 | −0.0246 | −0.0356 | −0.0217 |
| Max | 0.1284 | 0.0909 | 0.0385 | 0.0397 |
| P‐value* | 0.501 | 0.422 | 0.398 | 0.063 |
| SRM | 0.23 | 0.35 | 0.36 | 0.87 |
| Changes from baseline at Month 24 | ||||
| N | 9 | 10 | 3 | 3 |
| Mean | 0.0085 | 0.0161 | 0.0438 | 0.0300 |
| SD | 0.0470 | 0.0411 | 0.0041 | 0.0215 |
| Median | −0.0551 | 0.0134 | 0.0449 | 0.0258 |
| Min | −0.0556 | −0.0471 | 0.0393 | 0.0109 |
| Max | 0.0703 | 0.0903 | 0.0473 | 0.0532 |
| P‐value* | 0.767 | 0.203 | 0.109 | 0.109 |
| SRM | 0.18 | 0.39 | 10.68 | 1.40 |
FF T2, fat fraction; SD, standard deviation; SRM, standardized response means.
P‐value from Wilcoxon test.
There was an overall statistically significant increase in muscle C‐CSA at Month 12, with no significant increase at Month 24: median arm C‐CSA increased by 42.62 cm2 at Month 12 (n = 16; [Wilcoxon test P = 0.017]; SRM: 0.69) and by 22.87 cm2 at Month 24 (n = 9; [P = 0.110]; SRM: 0.45); median forearm C‐CSA increased by 27.31 cm2 at Month 12 (n = 13; [P = 0.039]; SRM: 0.72) and by 26.77 cm2 at Month 24 (n = 10; [P = 0.059]; SRM: 0.73); median thigh C‐CSA increased by 169.88 cm2 at Month 12 (n = 7; [P = 0.043]; SRM: 1.01) and by 178.36 cm2 at Month 24 (n = 3; [P = 0.285]; SRM: 0.66); median leg C‐CSA increased by 91.14 cm2 at Month 12 (n = 7; [P = 0.018]; SRM: 0.82) and by 308.18 cm2 at Month 24 (n = 3; [P = 0.285]; SRM: 0.87) (Table S5, Fig. S5).
Pulmonary function tests
In all functional groups, the median percentage of predicted FVC decreased significantly at Month 12 by 2.63% (n = 40; [Wilcoxon test P = 0.012]; SRM: 0.45), but the change in FVC of 0.02% from baseline was not significant at Month 24 (n = 27; [P = 0.885]; SRM: 0.10) (Fig. 5A and B, Table S6). Overall, MEP, PCF, and SNIP decreased at Month 12 and increased at Month 24, while MIP increased at Months 12 and 24; however, these changes were not significant: median MIP increased by 6.90% at Month 12 (n = 40; [P = 0.90]; SRM: 0.28) and increased by 6.30% at Month 24 (n = 27; [P = 0.203]; SRM: 0.30); median MEP decreased by 0.55% at Month 12 (n = 40; [P = 0.778]; SRM: 0.09) and increased by 2.30% at Month 24 (n = 27; [P = 0.199]; SRM: 0.30); median PCF decreased by 0.50% at Month 12 (n = 40; [P = 0.697]; SRM: 0.10) and increased by 1.80% at Month 24 (n = 26; [P = 0.657]; SRM: 0.15); median SNIP decreased by 0.60% at Month 12 (n = 40; [P = 0.888]; SRM: 0.01) and increased by 6.60% at Month 24 (n = 27; [P = 0.230]; SRM: 0.19) (Fig. 5C–E).
Figure 5.
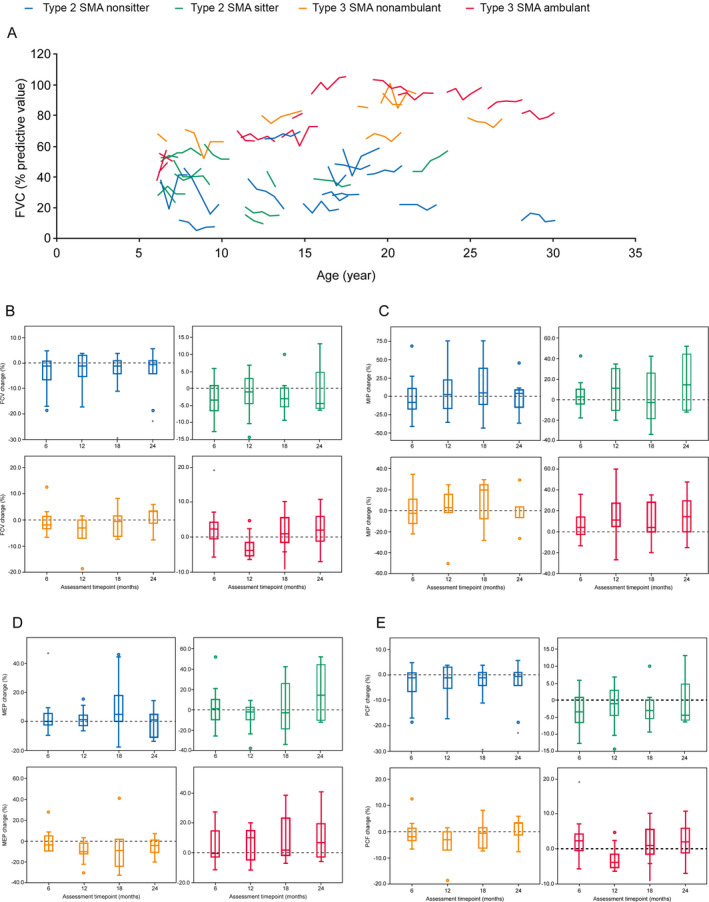
(A) Change in FVC over 24 months according to age. B–E) Change in FVC, MIP, MEP, and PCF over 24 months by SMA type. (A/B) FVC change was measured using the Vitalograph spirometer at baseline, Month 6, 12, 18, and 24 (Month 12, n = 40; Month 24, n = 27). Each line represents the trajectory of one patient over 24 months. (C) MIP was evaluated using the MicroRPM device at baseline, Month 6, 12, 18, and 24 (Month 12, n = 40; Month 24, n = 27). (D) MEP was evaluated using the MicroRPM device at baseline, Month 6, 12, 18, and 24 (Month 12, n = 40; Month 24, n = 27). (E) PCF was measured using the Vitalograph spirometer at baseline, Month 6, 12, 18, and 24 (Month 12, n = 40; Month 24, n = 26). Closed circles represent outliers. FVC, forced vital capacity; MEP, minimum expiratory pressure; MIP, minimum inspiratory pressure; PCF, peak cough flow; SMA, spinal muscular atrophy.
SMN protein levels
The 2‐year longitudinal measurements of blood SMN protein levels revealed that there were no significant changes in the overall levels of SMN protein (median range 3.24–3.44 ng/mL) (Fig. 6). Comparison with baseline values for each group showed no significant changes over 12 months (n = 26; Wilcoxon test P = 0.216) and 24 months (n = 35; Wilcoxon test P = 0.902). This was true for all groups (Table S7).
Figure 6.
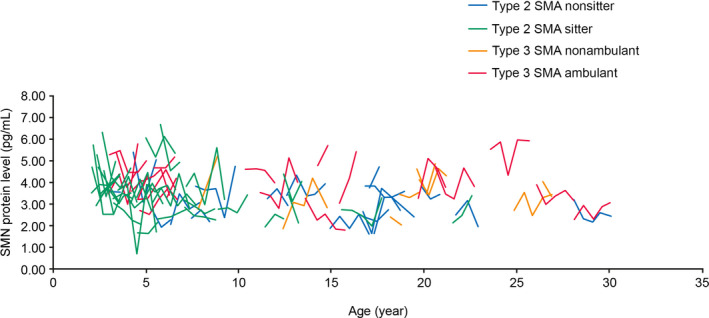
SMN protein levels over 24 months in individuals with SMA according to age and SMA type. Each line represents an individual with SMA. Assessments were made at baseline, Month 6, 12, 18, and 24 (Month 12, n = 69; Month 24, n = 35). SMA, spinal muscular atrophy; SMN, survival of motor neuron.
Correlations between clinical outcome measures
The relationship between MFM total score with MyoGrip, MyoPinch, and ActiMyo® measurements was evaluated in all functional groups (data not shown). High correlations were observed between MFM and grip strength (Spearman = 0.917; P < 0.001) and MFM and pinch strength (Spearman = 0.911; P < 0.001). Of the ActiMyo® assessments, MFM32 was most highly correlated with ||A|| (Spearman = 0.824; P < 0.001). A high correlation was also found between ||A|| with grip strength (Spearman = 0.823; P < 0.001) and pinch strength (Spearman = 0.718; P < 0.001).
MFM32 was also highly correlated with muscle imaging measurements. MFM32 was most positively correlated with thigh C‐CSA (Spearman = 0.813, P = 0.026). High negative correlations were found between MFM32 and forearm FFT2 (Spearman = −0.868, P < 0.001) and MFM32 and leg FFT2 (Spearman = −0.815, P = 0.025).
Discussion
We demonstrate that a significant decline over a 12‐ or 24‐month period can be reliably measured in a broad range of SMA patients using the MFM32, MyoGrip, MyoPinch, and ActiMyo®, and when using magneto‐inertial technology to continuously measure movements, changes can even be measured over a 6‐month period.
The SRM represents the sensitivity of these measures, which indicates that clinical studies may be conducted in this population using a smaller number of patients over a 12‐month study duration. In SMA natural history studies, decline can be captured within a 12‐month period in patients with Type 1 SMA, the most severe form of SMA, which occurs in nonsitters < 6 months old who experience rapid deterioration. 31 In Type 2 and 3 SMA, however, studies up to 3 years long or sub‐group analyses are typically needed to detect a significant decline in patients using the HFMSE. 14 , 32 Current clinical measures have a low sensitivity to change over 12 months: the SRM of the RULM over 12 months is 0.14 33 and the mean change in the 6MWT over 12 months is low (−7.8 m/year) and age dependent. 34 A recent study has also illustrated that the SRM of HFMSE in patients with Type 3 SMA is low and very variable according to age and ambulant status. 35 In this study, we aim to identify additional ways to detect a change in SMA severity over shorter periods. This will enable more sensitive and reliable outcome measures to be identified for the development of SMA treatments in clinical trials.
The reliability and validity of the MFM have been previously demonstrated in a group of patients with SMA. 36 Our previously reported MFM baseline results confirmed findings that the MFM can discriminate between SMA type successfully, as well as sitter and ambulant status, with no significant overlap between SMA type. 19 In the current study, we report the MFM scores of the same participants over 24 months at regular 6‐month intervals. Overall, there was a significant decline in MFM32 scores at Month 24, with no significant decline at Month 12. Post hoc analyses demonstrated that differences between MFM32 scores at baseline and Month 24 in patients with Type 3 SMA did not reach statistical significance; this could be interpreted as a slower disease progression in individuals with Type 3 SMA, or this may be related to the smaller number of patients in this group. A more extensive study in a cohort of patients with Type 3 SMA over 24 months may be required to confirm this hypothesis. Similar findings of difficulties in identifying significant decline in patients with Type 3 SMA have been observed using the RULM. 33
Upper limb muscle strength (i.e., grip and pinch strength assessed by high‐precision dynamometry) decreased over the 12‐ and 24‐month periods in nonambulant patients. Grip strength decline appeared to be consistent across all ages in patients with Type 2 SMA. In participants with Type 3 SMA, grip strength appeared to have the greatest increase between 10 and 15 years old, compared with predicted values based on age, before declining. Studies with a larger sample size and further analyses are needed to confirm whether this demonstrates a peak in grip strength as shown in previous findings, where upper limb strength (expressed in absolute values) increased before the age of 14 and decreased after. 21 Similarly, a recent, retrospective study in patients with Type 3 SMA demonstrated increased motor function, as measured by the HFMSE, up to the age of 7 and a decrease beyond this age. 35 A similar time course in motor function has been observed in DMD, where the distance covered in a 6MWT continuously increased before the age of 7 and decreased thereafter. 37 This is also true for upper limb strength in DMD when expressed in absolute values, 38 but not in relative values for age. 23 The increase in grip strength in patients with Type 3 SMA may correspond to puberty; however, data on the pubertal status of patients were not collected in the current study, therefore it is not possible to correlate pubertal status with upper limb strength. Together these data demonstrate that patients with Type 3 SMA, as in DMD, may typically experience a short period of increasing motor function in early childhood and adolescence, followed by decline.
Upper limb activity, assessed by ActiMyo®, was shown to decrease significantly over 6 and 12 months, as measured in patients with Type 2 SMA and nonambulant patients with Type 3 SMA. Longer‐term assessment of upper limb activity could not be measured due to the small number of patients assessed at 24 months (n = 1). The SRM of ActiMyo® demonstrates its high sensitivity, in line with findings previously described in DMD. 24 , 25 This may be due to the ability of the assessment to overcome daily fluctuations in patient conditions that may impact performance in clinical assessments, through the longitudinal monitoring and averaging of recorded data. Further to this, the use of wearable sensors at home will potentially allow more patient‐relevant tasks to be captured in future clinical trials.
Pulmonary function, as measured by FVC, decreased significantly over 12 months, in contrast to previous findings. 14 There was no significant change in FVC over 24 months, possibly due to a smaller sample size as a result of many participants discontinuing the study between Months 12 and 24 (n = 32). All other pulmonary measurements were stable over the 24 months. This supports FVC as a more suitable assessment of the pulmonary function in patients with SMA, potentially reducing the need to perform multiple time‐consuming and tiring pulmonary tests in clinical trials.
Although an increase in the FFT2 data was observed at Months 12 and 24 for all SMA types, it was not considered statistically significant. This may be due to the low number of participants available at Month 24 (arm FFT2, n = 9; forearm FFT2, n = 10; thigh FFT2, n = 3; leg FFT2, n = 3). An unexpected significant increase in muscle C‐CSA was observed at Month 12, possibly due to patient growth; however, no significant increase was observed at Month 24. This is in line with previous findings, where the data also revealed a significant correlation between muscle‐imaging assessments such as FFT2 and muscle C‐CSA and motor function assessments such as the MFM. 39 Taken together, this suggests that although muscle imaging alone may not serve as a suitable marker of disease progression in SMA, it has the potential to serve as a valuable outcome measure in clinical trials in the context of other assessments of motor function.
SMN protein levels were relatively stable over the 24‐month observation period throughout the whole SMA patient population and across different functional groups, even though individual fluctuations were detected. In addition, as expected, SMN levels were lower over 12 and 24 months compared with healthy volunteers. This is in good concordance to a previously conducted observational cross‐sectional study in individuals with Type 3 SMA, 39 where no changes in the SMN protein were observed over 52 weeks. Recent data support the hypothesis that increased SMN protein levels are restricted to early development (until 3 months of age), where the levels show a substantial decrease then remain stable (>3 months through 14 years). 40 Overall, this indicates that SMN protein expression seems to be stable over time in SMA patients. While SMN protein levels may, therefore, not be a suitable biomarker for disease progression; however, they are an essential biomarker where interventional trials are required to demonstrate increases in SMN protein levels in response to SMA treatment.
Study limitations
As anticipated, several participants (particularly those within the Type 2 cohort) discontinued this 24‐month study to pursue an approved therapy or to join clinical trials particularly between Month 12 and Month 24 (n = 32). Although the number of patients included in this international prospective natural history study is high relative to similar studies, statistical significance was difficult to reach due to small group sizes when accounting for age and physical conditions in subgroup analyses. This was especially true for muscle imaging and for ActiMyo® assessments, in which participant numbers were small. Finally, differences in standard of care between multiple centers and countries may have been a confounding factor in this study.
CONCLUSIONS
The data presented here demonstrate that the MFM32, MyoGrip, Myopinch, and ActiMyo® allow for the detection of significant decline over a 12‐ or 24‐month period. The ActiMyo® assessment was also able to detect changes over 6 months. Moreover, the SRM that represents the sensitivity of change in these measures has the potential to reduce the necessary sample size and study duration of future clinical trials. The collective use of these measures in a therapeutic trial protocol could provide a more holistic evaluation of disease progression in individuals with SMA, potentially improving treatment options for patients.
Conflicts of Interest
MA is an employee of Sysnav; AD received personal fees from AveXis for Scientific Advisory Boards (SAB) and is Principal Investigator for SMA trials for Hoffmann‐La Roche and AveXis; LDW attended SAB of F. Hoffmann‐La Roche Ltd, Biogen and AveXis and received consultancy fees from Biogen and AveXis; VL received consulting fees for Biogen, Roche, Avexis, PTC therapeutics, Sarepta; US received honoraria for participation in SAB and industry symposia from Biogen, Roche, and Avexis; CL attended SAB of Biogen and received consultancy fees from Biogen, Sysnav, and F. Hoffmann‐La Roche Ltd; JYH received consulting fees for Biogen and Sarepta. He is co‐inventor of Myogrip and Myopinch; KG and NH are employees of, and holds shares in F. Hoffmann‐La Roche Ltd; MLL is an employee of Institut Roche; TS, CC, and RH are former employees and hold shares in F. Hoffmann‐La Roche Ltd; LS is a principal investigator of SMA studies for F. Hoffmann‐La Roche Ltd, Biogen, and AveXis; he has attended SAB of F. Hoffmann‐La Roche Ltd, Biogen, and AveXis and received consultancy fees from Biogen; he serves on the board for Cytokinetics. He is co‐inventor in the patent 20190029605 (Method for estimating physical activity of the upper limb) from which he has not perceived any financial interest; CC, EF, YP, AMS, PC, CV, LL, and TG report no conflict of interest.
Author Contributions
All authors contributed to the study conception and design. Analysis and interpretation were performed by all authors. All authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Supporting information
Figure S1. Change in MFM over 24 months stratified by function.
Figure S2. Individual mean and 95th percentile ActiMyo® plots according to age and SMA type.
Figure S3. Individual mean and 95th percentile stride velocity and length in Type 3 ambulant patients.
Figure S4. Individual arm and forearm and fat fraction changes according to age.
Figure S5. FFT2 and C‐CSA changes in thigh and leg according to age.
Table S1. Change in MFM32 score from baseline over 12 and 24 months.
Table S2. Changes in grip strength from baseline at Months 12 and 24.
Table S3. Mean and median pinch strength changes from baseline at Months 12 and 24.
Table S4. Change from baseline stride velocity and stride length in ambulant patients with Type 3 SMA.
Table S5. Muscle C‐CSA changes from baseline over 12 and 24 months.
Table S6. Change in FVC from baseline at Months 12 and 24.
Table S7. Change in SMN protein level from baseline at Months 12 and 24.
Appendix S1. Methods: Further information.
Acknowledgments
The authors would like to thank all the individuals enrolled in the natural history study and the site staff involved. The authors thank the NatHis‐SMA study group for data collection and protocol organization (numbers in bracket refer to authors affiliations): Silvana de Lucia1, Stéphanie Gilabert1, Marianne Hezode1, Valérie Decostre1, Aurélie Chabanon1, Aurélie Phelep1, Mélanie Villeret1, Arnaud Jollet1, Rose‐Diana Ho1, Isabelle Ledoux1, Aurélie Canal1, Severine Denis3, Stéphanie Delstanche3, Fabian Dal Farra3, Armelle Magot4, Lucie Le Goff4, Christelle Peseux4, Raphaële Chasserieau4, Pascal Cintas5, Valérie Bellio5, Françoise Auriol6, Stéphanie Fontaine7, Véronique Manel7, Aurelie Barrière7, Dominique Vincent Genod7, Manel Saidi7, Nathalie Goemans9, Marleen Van den Hauwe9, Annelies Van Impe9, Annelies Vanden Eynden9, Olivier Blouet11, Sihame Messai11, Heike Kölbel12, Andrea Gangfuβ12, Corinna Seifert12, Barbara Andres12, Eli Toledano16*, Omar Khwaja17*, Anne Marquet17, Anna Bayfield20, and Laura Vandenbrande1. The lead author for this group is Laurent Servais.1,13,19 Finally, the authors thank Nosheen Hussain of MediTech Media, UK, for providing medical writing support, which was funded by F. Hoffmann‐La Roche Ltd, Basel, Switzerland, in accordance with Good Publication Practice (GPP3) guidelines (http://www.ismpp.org/gpp3). This study is co‐funded by Institut Roche and Association Institut de Myologie.
*No longer an employee of Roche at the time of publication.
20CPS R&D Early Development & Reagent Design, Roche Diagnostics GmbH, Penzberg, Germany.
Data are available upon request. Data contain potentially identifying and sensitive patient information, and the participants did not consent to have their full transcripts made publicly available. Interested and qualified researchers can contact the sponsor quality manager, Fanny Moraux, Association Institut de Myologie, at f.moraux@institut-myologie.org in order to request data access.
Funding Information
This study is co‐funded by Institut Roche and Association Institut de Myologie. The authors thank Nosheen Hussain of MediTech Media, UK, for providing medical writing support, which was funded by F. Hoffmann‐La Roche Ltd, Basel, Switzerland, in accordance with Good Publication Practice (GPP3) guidelines (http://www.ismpp.org/gpp3).
Funding Statement
This work was funded by Association Institut de Myologie grant ; Institut Roche grant ; F. Hoffmann‐La Roche Ltd grant .
References
- 1. Lefebvre S, Burglen L, Reboullet S, et al. Identification and characterization of a spinal muscular atrophy‐determining gene. Cell 1995;80:155–165. [DOI] [PubMed] [Google Scholar]
- 2. Lorson CL, Hahnen E, Androphy EJ, Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. PNAS USA 1999;96:6307–6311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hamilton G, Gillingwater TH. Spinal muscular atrophy: going beyond the motor neuron. Trends Mol Med 2013;19:40–50. [DOI] [PubMed] [Google Scholar]
- 4. Mercuri E, Bertini E, Iannaccone ST. Childhood spinal muscular atrophy: controversies and challenges. Lancet Neurol 2012;11:443–452. [DOI] [PubMed] [Google Scholar]
- 5. Tizzano EF, Finkel RS. Spinal muscular atrophy: a changing phenotype beyond the clinical trials. Neuromusc Disord 2017;27:883–889. [DOI] [PubMed] [Google Scholar]
- 6. Rudnik‐Schoneborn S, Hausmanowa‐Petrusewicz I, Borkowska J, Zerres K. The predictive value of achieved motor milestones assessed in 441 patients with infantile spinal muscular atrophy Types II and III. Eur Neurol 2001;45:174–181. [DOI] [PubMed] [Google Scholar]
- 7. Mercuri E, Finkel RS, Muntoni F, et al. Diagnosis and management of spinal muscular atrophy: part 1: recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromusc Disord 2018;28:103–115. [DOI] [PubMed] [Google Scholar]
- 8. Schorling DC, Pechmann A, Kirschner J. Advances in treatment of spinal muscular atrophy ‐ new phenotypes, new challenges, new implications for care. J Neuromuscul Dis 2020;7:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Biogen Inc . SPINRAZA® (nusinersen) US prescribing information. [Web Page] 2016. [updated Dec 2016; cited 2020 29 June]; Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/209531lbl.pdf.
- 10. A veXis Inc . ZOLGENSMA® (onasemnogene abeparvovec‐xioi) US prescribing information. [Web Page] [updated May 2019; cited 2020 29 June]; Available from: https://www.fda.gov/media/126109/download.
- 11. Genentech . Evrysdi (risdiplam) US prescribing information. 2020. [cited 2020 August]; Available from: https://www.gene.com/download/pdf/evrysdi_prescribing.pdf
- 12. Ramdas S, Servais L. New treatments in spinal muscular atrophy: an overview of currently available data. Expert Opin Pharmacother 2020;21:307–315. [DOI] [PubMed] [Google Scholar]
- 13. Mendell JR. Single‐dose gene‐replacement therapy for spinal muscular atrophy. N Engl J Med 2017;377:1713–1722. [DOI] [PubMed] [Google Scholar]
- 14. Kaufmann P, McDermott MP, Darras BT, et al. Prospective cohort study of spinal muscular atrophy types 2 and 3. Neurology 2012;79:1889–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bertini E, Dessaud E, Mercuri E, et al. Safety and efficacy of olesoxime in patients with Type 2 or non‐ambulatory Type 3 spinal muscular atrophy: a randomized, double‐blind, placebo‐controlled Phase 2 trial. Lancet Neurol 2017;16:513–522. [DOI] [PubMed] [Google Scholar]
- 16. Mercuri E, Barisic N, Boespflug‐Tanguy O, et al. SUNFISH Part 2: efficacy and safety of risdiplam (RG7916) in patients with Type 2 or non‐ambulant Type 3 spinal muscular atrophy (SMA). Presented at SMA. Europe 2020. 2020.
- 17. Dangouloff T, Servais L. Clinical evidence supporting early treatment of patients with spinal muscular atrophy: current perspectives. Ther Clin Risk Manag 2019;15:1153–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Le Moing AG, Seferian AM, Moraux A, et al. A movement monitor based on magneto‐inertial sensors for non‐ambulant patients with Duchenne muscular dystrophy: a pilot study in controlled environment. PLoS One 2016;11:e0156696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chabanon A, Seferian AM, Daron A, et al. Prospective and longitudinal natural history study of patients with Type 2 and 3 spinal muscular atrophy: baseline data NatHis‐SMA study. PLoS One 2018;13:e0201004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Annoussamy M, Lilien C, Giadaro T. X‐linked myotubular myopathy: a prospective international natural history study. Neurology 2019;92:e1852–e1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Seferian A, Moraux A, Canal A, et al. Upper limb evaluation and one‐year follow up of non‐ambulant patients with spinal muscular atrophy: an observational multicenter trial. PLoS One 2015;10:e0121799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Servais L, Deconinck N, Moraux A, et al. Innovative methods to assess upper limb strength and function in non‐ambulant Duchenne patients. Neuromusc Disord 2013;23:139–148. [DOI] [PubMed] [Google Scholar]
- 23. Hogrel JY, Decostre V, Ledoux I, et al. Normalized grip strength is a sensitive outcome measure through all stages of Duchenne muscular dystrophy. J Neurol 2020;267:2022–2028. [DOI] [PubMed] [Google Scholar]
- 24. Lilien C, Gasnier E, Gidaro T, et al. Home‐based monitor for gait and activity analysis. JoVE 2019. 10.3791/59668 [DOI] [PubMed] [Google Scholar]
- 25. Haberkamp M, Moseley J, Athanasiou D, et al. European regulators' views on a wearable‐derived performance measurement of ambulation for Duchenne muscular dystrophy regulatory trials. Neuromusc Disord 2019;29:514–516. [DOI] [PubMed] [Google Scholar]
- 26. Crapo RO, Morris AH, Gardner RM. Reference spirometric values using techniques and equipment that meet ATS recommendations. Am Rev Respir Dis 1981;123:659–664. [DOI] [PubMed] [Google Scholar]
- 27. Bianchi C, Baiardi P. Cough peak flows: standard values for children and adolescents. Am J Phys Med Rehabil 2008;87:461–467. [DOI] [PubMed] [Google Scholar]
- 28. Wilson SH, Cooke NT, Edwards RH, Spiro SG. Predicted normal values for maximal respiratory pressures in Caucasian adults and children. Thorax;39:535–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Stefanutti D, Fitting JW. Sniff nasal inspiratory pressure. Reference values in Caucasian children. Am J Respir Crit Care Med 1999;159:107–111. [DOI] [PubMed] [Google Scholar]
- 30. Uldry C, Fitting JW. Maximal values of sniff nasal inspiratory pressure in healthy subjects. Thorax 1995;50:371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mercuri E, Lucibello S, Perulli M, et al. Longitudinal natural history of type I spinal muscular atrophy: a critical review. Orphanet J Rare Dis 2020;15:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mercuri E, Darras BT, Chiriboga CA, et al. Nusinersen versus Sham control in later‐onset spinal muscular atrophy. N Engl J Med 2018;378:625–635. [DOI] [PubMed] [Google Scholar]
- 33. Pera MC, Coratti G, Mazzone ES, et al. Revised upper limb module for spinal muscular atrophy: 12 month changes. Muscle Nerve. 2019;59:426–430. [DOI] [PubMed] [Google Scholar]
- 34. Montes J, McDermott MP, Mirek E, et al. Ambulatory function in spinal muscular atrophy: age‐related patterns of progression. PLoS One 2018;13:e0199657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Coratti G, Messina S, Lucibello S, et al. Clinical variability in spinal muscular atrophy type III. Ann Neurol 2020;2020:14. [DOI] [PubMed] [Google Scholar]
- 36. Trundell D, Scouiller S, Gorni K, et al. Validity and reliability of the 32‐item Motor function measure across age groups in spinal muscular atrophy in 2–5 year olds with neuromuscular disorders and 2–25 year olds with spinal muscular atrophy. Neurol Ther 2020;9:575–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. McDonald CM, Henricson EK, Abresch RT, et al. The 6‐minute walk test and other clinical endpoints in duchenne muscular dystrophy: reliability, concurrent validity, and minimal clinically important differences from a multicenter study. Muscle Nerve 2013;48(3):357–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hogrel JY, Wary C, Moraux A. Longitudinal functional and NMR assessment of upper limbs in Duchenne muscular dystrophy. Neurology 2016;86:1022–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bonati U, Holiga S, Hellbach N, et al. Longitudinal characterization of biomarkers for spinal muscular atrophy. Ann Clin Transl Neurol 2017;4:292–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ramos D, d'Ydewalle C, Gabbeta V, et al. Age‐dependent SMN expression in disease‐relevant tissue and implications for SMA treatment. J Clin Investig 2019;129:4817–4831. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Change in MFM over 24 months stratified by function.
Figure S2. Individual mean and 95th percentile ActiMyo® plots according to age and SMA type.
Figure S3. Individual mean and 95th percentile stride velocity and length in Type 3 ambulant patients.
Figure S4. Individual arm and forearm and fat fraction changes according to age.
Figure S5. FFT2 and C‐CSA changes in thigh and leg according to age.
Table S1. Change in MFM32 score from baseline over 12 and 24 months.
Table S2. Changes in grip strength from baseline at Months 12 and 24.
Table S3. Mean and median pinch strength changes from baseline at Months 12 and 24.
Table S4. Change from baseline stride velocity and stride length in ambulant patients with Type 3 SMA.
Table S5. Muscle C‐CSA changes from baseline over 12 and 24 months.
Table S6. Change in FVC from baseline at Months 12 and 24.
Table S7. Change in SMN protein level from baseline at Months 12 and 24.
Appendix S1. Methods: Further information.