

Abstract
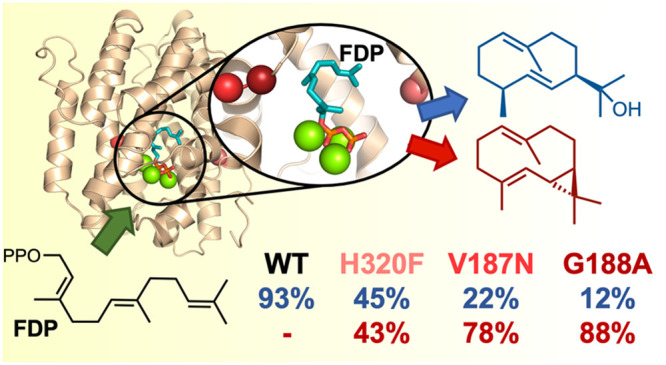
Natural sesquiterpene synthases have evolved to make complex terpenoids by quenching reactive carbocations either by proton transfer or by hydroxylation (water capture), depending on their active site. Germacradien-11-ol synthase (Gd11olS) from Streptomyces coelicolor catalyzes the cyclization of farnesyl diphosphate (FDP) into the hydroxylated sesquiterpene germacradien-11-ol. Here, we combine experiment and simulation to guide the redesign of its active site pocket to avoid hydroxylation of the product. Molecular dynamics simulations indicate two regions between which water molecules can flow that are responsible for hydroxylation. Point mutations of selected residues result in variants that predominantly form a complex nonhydroxylated product, which we identify as isolepidozene. Our results indicate how these mutations subtly change the molecular choreography in the Gd11olS active site and thereby pave the way for the engineering of terpene synthases to make complex terpenoid products.
Keywords: terpene synthase, terpenoid, molecular dynamics, mutagenesis, enzyme redesign, enzyme mechanism
Introduction
Terpenoids are the largest group of natural products universally present in all living systems with immense diversity in their structure and functions.1,2 To date, over 80000 naturally occurring isoprenoids have been reported, including more than 7000 sesquiterpenoids derived from ∼300 stereochemically distinct hydrocarbon skeletons.3,4 A highly complex and structurally diverse array of hydrocarbon or hydroxylated terpenes is synthesized by terpene synthase catalysis from acyclic isoprenyl diphosphates such as geranyl diphosphate (GDP), farnesyl diphosphate (FDP), and geranylgeranyl diphosphate (GGDP).4,5 The diversity of terpene skeletons arises not only from the number of terpene synthases but also from the ability of some terpene synthases to form multiple products from a single substrate.6−8 The product specificity and activity of terpene synthases are highly dependent on a small number of amino acids present inside or near the hydrophobic active site pocket. Previous studies have shown the profound effect that slight changes to these amino acids can have on the electronic distribution and the geometry of the active site pocket, which in turn affect the enzyme activity and specificity.9−16
In class I terpene synthases, a cluster of three Mg2+ ions (referred to as Mg2+A–C) is responsible for binding and triggering loss of the substrate diphosphate group to give a carbocation–inorganic diphosphate ion (PPi) pair. Two highly conserved metal-binding motifs on helices D (DDXXD/E motif) and H (NSE/DTE motif) face each other across the active site entrance. The motif on helix H binds a Mg2+ ion (Mg2+B) which allows substrate binding, whereupon Mg2+ binding to the motif on helix D triggers transition from an open to a closed active site conformation (including loop movement and changes in the G-helix).3,17,18 Once the active site is closed with all three Mg2+ ions bound, the carbocation–PPi pair is formed with the highly reactive carbocation intermediates sheltered from premature quenching by bulk solvent.3,17,18 The carbocations formed can undergo a series of intramolecular reactions involving cyclization, double-bond isomerization, hydride, methyl, and alkyl migrations, and finally proton loss to form hydrocarbons or hydroxylation to form terpene alcohols.3,19,20
Several class I terpene synthases have evolved to produce hydroxylated terpenes by mediating the capture of water in the reaction cascade.21−25 Although many terpene synthases likely harbor water molecules in the active site pocket (as observed in the crystal structures of several terpene synthases),26−30 the majority avoid water capture and do not produce hydroxylated compounds. Despite extensive experimental and theoretical investigations of the cyclization mechanism and structural elucidation of terpene synthases, there are still many questions regarding what determines water capture and what mechanism is involved. Some reports suggest that loop movements enable ingress of bulk water into the active site pocket to mediate water capture in germacradien-4-ol synthase31 and mutants of δ-cadinene synthase.32 Structural analysis of mutant Aspergillus terreus aristolochene synthase (ATAS) inhibitor complexes indicated that a Mg2+C-bound water molecule is most likely involved in the formation of (linear) non-native hydroxylated products.28 Isotope labeling studies on, for example, the epi-cedrol33 and epi-cubenol sesquiterpene synthases34 demonstrate that the oxygen atom present in these compounds is derived from bulk water. Mutation analysis of hedycaryol synthase showed the catalytic involvement of Asp82 of the DDXXD motif in the activation of water for the hydroxylation,35 whereas in bacterial cineole synthase, a specific active site Asn has been implicated in water activation.36 In the hydroxylating diterpene synthase CotB2, extensive experimental and computational work has highlighted key active site interactions and suggestions of how these are altered by mutations that lead to different main products.16,37 However, a complete picture of how (sesqui)terpene synthases mediate water capture remains elusive.
Germacradien-11-ol synthase (Gd11olS), a class I sesquiterpene synthase, is the N-terminal domain of geosmin synthase from S. coelicolor which catalyzes the cyclization of FDP (1) into the hydroxylated sesquiterpene germacradien-11-ol (Gd11ol, 2) as the major product and germacrene D (3) as the minor product (Scheme 1).38−40 Formation of germacrene D involves a 1,3-hydride shift in the germacryl cation and final deprotonation at C15. However, formation of germacradien-11-ol putatively involves deprotonation of C1 of the germacryl cation and formation of the strained bicyclic intermediate isolepidozene (4), which then undergoes ring opening upon water attack to form Gd11ol (Scheme 1).38,41
Scheme 1. Reactions Catalyzed by Gd11olSWT and Mutants.

In the present study, a combination of a molecular dynamics (MD) simulation of the enzyme–substrate complex (Figure 1) and kinetic characterization of enzyme variants generated by site-directed mutagenesis is used to obtain insight into and modulate the water capture behavior of Gd11olS. A total of 15 mutants were created in regions that the simulations suggest are involved in guiding a reactive water molecule to quench the neutral intermediate isolepidozene (4) (Scheme 1). Mutation of these residues resulted in changes in the product profile and water capture behavior of Gd11olS. A Gd11olS mutant (G188A) was identified which (almost fully) avoids water capture and predominantly produces isolepidozene. The findings presented here will aid in the engineering of terpene synthases to channel the production of novel terpenoids by manipulating their water capture ability.
Figure 1.

Representative MD snapshot of the FDP·Gd11olSWT complex in binding mode I. Protein residues shown with sticks and transparent spheres are important for FDP binding. Hydrogens are omitted for clarity. In binding mode II, the farnesyl chain of FDP turns in the opposite direction (see Figure S2).
Results and Discussion
Characterization of Wild-Type Gd11olS and Identification of the FDP Binding Mode
First, we set out to confirm the product profile and kinetics for wild-type Gd11olS (hereafter Gd11olSWT). The gene for the N-terminal domain of geosmin synthase (coding for residues 1–366 only of the whole protein) was codon optimized for E. coli and synthesized in a pET28a vector (see the Supporting Information). After overexpression in E. coli, the recombinant protein was purified to homogeneity (see the Supporting Information for more details). Incubation of Gd11olSWT with FDP resulted in the formation of germacradien-11-ol as the major product (2, 92.4%) and germacrene D as the minor product (3, 7.6%) (Figure 2 and Table S2), and the kinetic parameters for turnover of FDP (KM = 0.5 ± 0.08 μM, kcat = (5.0 ± 0.02) × 10–4 s–1) were in good agreement with previous work.40
Figure 2.

Product distribution of Gd11olSWT and its mutants (obtained using GC-MS). The product yield here refers to the relative abundance expressed as a percentage of the total amount of products formed from FDP. Product yield % values are given in Table S2. Total ion chromatograms for all variants are given in Figures S9–S24, with mass spectra for all six compounds provided in Figures S28–S33.
Having established the product profile of Gd11olSWT, we turned to a MD simulation to obtain detailed structural information to guide in the engineering of its active site to change the product profile. For Gd11olSWT, a crystal structure in a complex with three Mg2+ ions and alendronic acid (PDB code 5DZ2) mimics the active “closed” form of the enzyme (with alendronic acid indicating the location of the diphosphate);42 however, the positioning of the farnesyl moiety of the FDP substrate is unknown. We thus modeled the farnesyl chain in four distinct possible orientations that could lead to cyclization. The first was based on the orientation of the substrate analogue in a complex with aristolochene synthase (PDB code 4KUX),43 with the others obtained by changing the rotation of single bonds accompanied by geometry optimization (Figure S2). For each of these FDP·Gd11olSWT complexes, 10 independent 30 ns MD simulations were performed in explicit solvent (see Supporting Information for details).
Only two of the four orientations of the farnesyl chain were stable in the MD simulations: binding modes I and II (Figure 1 and Figure S2). The other two starting orientations changed into either binding mode I or II during the MD simulation; thus, they were discarded from further analysis. Although all simulations of binding modes I and II indicate the same overall orientation, the farnesyl chain is flexible and thus shows a wide ensemble of conformations during simulation (Figure S3), as shown, for example, by the wide distribution sampled of the interatomic distance between the atoms C1 and C10 (Figure 3a,b; see Scheme 1 for atom numbering). The C1–C10 distance reaches values of ≤5 Å in both binding modes, suggesting that the farnesyl chain could, in principle, cyclize into a germacryl cation (upon ionization of FDP) regardless of the binding mode. However, while binding mode I shows a marked preference for the formation of conformations of FDP that can lead to the expected R enantiomer of the germacryl cation, mode II almost exclusively samples conformations in line with the formation of the S enantiomer (Figures 3c,d). Thus, mode I is identified to be the most likely binding mode of FDP. This binding mode is equivalent to that observed in the crystal structure of other sesquiterpene synthases in complexes with FDP analogues (e.g., aristolochene synthase with FSDP and selinadiene synthase with (3S,6E)-3,7,11-trimethyldodeca-6,10-dien-1-yl trihydrogen diphosphate; see Figure S4).30,43
Figure 3.

Histograms of the C1–C10 distance (a, b) and percentage of (total, pro-R, and pro-S) FDP conformations with C1–C10 ≤ 5 Å (c, d) for the FDP·Gd11olS (WT and mutant variants) complexes in both binding modes I and II, calculated over the 10 MD runs for each (200 ns). Histograms are in 0.1 Å bins. A C1–C10 distance of ≤5 Å is used here as a qualitative measure of the ability of FDP to undergo cyclization and form a germacryl cation. To define the prochirality of the C10 atom, apart from the C1–C10 distance, a proper re or si orientation of the C1 atom with respect to the C10=C11 double bond was considered (to determine an FDP conformation as pro-R or pro-S; Figure S5). The trends remain the same when a smaller cutoff value of the C1–C10 distance is used.
To further confirm the FDP binding mode in Gd11olS obtained from simulation, we turned to mutagenesis. The bulky side chains of residues Y79, F83, W312, and H320 line the farnesyl moiety in binding mode I (Figure 1 and Figure S3), with W312 and H320 likely enforcing the final part of the farnesyl chain to bend back toward the C1 atom, facilitating C1–C10 cyclization. We thus generated and assayed the W312A and H320A mutants of Gd11olS experimentally, to corroborate this prediction. As expected, both mutations lead to significant amounts of the “early exit” noncyclic product nerolidol (6) and reduce production of 2 (Figure 2 and Table S2), which further supports binding mode I to be the catalytically relevant substrate complex (Michaelis complex, MC) in Gd11olSWT. Note that H320A still forms approximately the same amount of germacrene D, indicating that His320 is unlikely to be the base required for its formation (leaving PPi and His226 as the possible bases involved). The formation of nerolidol was confirmed by coeluting the assay sample of W312A with an authentic standard (Figure S25). As a complement, using the same computational approach used with Gd11olSWT, we modeled and simulated the W312A and H320A mutants with both FDP binding modes I and II. For binding mode I, there is a significant decrease (by >70%) in the number of FDP conformations with C1–C10 ≤ 5 Å (hereafter referred to as PCCs, productive conformations for cyclization) for both mutants in comparison to Gd11olSWT, whereas the number of extended conformations (C1–C10 > 7 Å) increases from 14% to 42% for W312A and to 25% for H320A (Figure 3a,c). This agrees well with the reduction in the total yield of cyclic products and the formation of nerolidol observed experimentally. The higher proportion of more extended conformations of FDP in W312A Gd11olS also correlates well with the higher yield of nerolidol (41.6% vs 22.5%) that it produces (Figure 2). Meanwhile, due to the larger space available in the active site of the mutants for the distal end of the farnesyl chain, there is no clear preference for pro-R or pro-S PCCs (Figure 3c). This indicates that the active site cavity contour of Gd11olSWT guides the stereoselectivity of germacryl cation formation in mode I, while for the W312A and H320A mutants the formation of the observed (R)-germacryl cation products are enforced by the reaction energetics.44 In binding mode II, pro-S PCCs are highly favored over pro-R PCCs for both mutants (as for Gd11olSWT). Further, a decrease in PCCs in comparison to Gd11olSWT is only observed for H320A (33%), which also shows a higher increase in extended FDP configurations in comparison to W312A (Figure 3b,d). This is in contrast with the higher reduction in cyclization and higher yield of nerolidol observed with the latter (Figure 2). Thus, the simulations with the mutants also support binding mode I to be the MC. The mutant W312A has a similar KM (0.4 ± 0.07 μM) and the H320A variant has an ∼3-fold higher KM (1.12 ± 0.10 μM) in comparison to Gd11olSWT, whereas an ∼10-fold reduction is observed in kcat for both mutants (Table 1), indicating that such alterations of the active site cavity lead to less efficient turnover. Replacement of W312 with phenylalanine leads to the formation of fully cyclized products germacrene A (5, 71.5%) and 2 (28.5%). The formation of 5 was confirmed by coinjection with an authentic standard generated from FDP upon incubation with aristolochene synthase from Penicillium roquefortii (which also makes 5; Figure S26). The kinetic efficiency of this mutant was found to be similar to that of Gd11olSWT (Table 1). These results show that Phe312 can still provide sufficient stabilization for C1–C10 closure but that Trp312 is required to selectively produce 2 (e.g., by stabilizing conversion of the germacryl cation to 4). The stabilization of carbocation intermediates by cation−π interactions with aromatic residues is frequently indicated in terpene synthase catalysis.3,30,45,46
Table 1. Kinetic Parameters for Gd11olSWT and Mutants.
| KM (μM) | kcat (10–5 s–1) | kcat/KM (10–5 μM–1 s–1) | |
|---|---|---|---|
| Gd11olSWT | 0.5 ± 0.08 | 52.4 ± 0.02 | 116.4 |
| R228V | 0.8 ± 0.10 | 18.14 ± 0.04 | 22.7 |
| Q313A | 1.10 ± 0.14 | 6.9 ± 0.02 | 6.3 |
| Q313N | 0.5 ± 0.07 | 5.02 ± 0.01 | 10.04 |
| H320A | 1.12 ± 0.10 | 3.8 ± 0.01 | 3.4 |
| H320F | 1.8 ± 0.5 | 2.3 ± 0.02 | 1.3 |
| V187D | N/Aa | ||
| V187N | NDb | ||
| V187A | 0.3 ± 0.06 | 19.4 ± 0.01 | 64.7 |
| G188V | N/Aa | ||
| G188A | 3.8 ± 1.5 | 3.7 ± 0.5 | 1.0 |
| G188S | 5.7 ± 0.9 | 4.08 ± 0.2 | 0.7 |
| A190V | 4.6 ± 2.2 | 1.3 ± 0.3 | 0.3 |
| A190L | N/Aa | ||
| W312F | 0.9 ± 0.09 | 25.5 ± 0.06 | 28.3 |
| W312A | 0.4 ± 0.07 | 4.02 ± 0.01 | 10.05 |
Not applicable (inactive).
Not determined due to low activity.
Dynamics of Water in the Gd11olS Active Site Pocket
With the (catalytically relevant) FDP-Gd11olS complex confirmed to be binding mode I, we focused on analyzing the behavior of water surrounding FDP, with the aim of identifying water molecules that could react with the FDP:C11 atom at the final stages of the reaction to form 2 (Scheme 1). There are two major regions of the protein close to FDP where water molecules are often present during the MD simulations: (i) the region composed of the residues R228, Q313, and H320, adjacent to FDP:C11 (hereafter referred to as the RQH site) and (ii) the region composed of the residues E161, R184, H226, and D230, next to the G-helix (hereafter referred to as the G-helix site) (Figure 4a). Water molecules are observed to flow between the G-helix site and the RQH site (Figure 4b), suggesting that even though FDP:C11 is closer to the latter, both sites can play a role in the availability of a reactive water molecule and changes in either could affect the hydroxylating activity of the enzyme. Notably, exchange of waters between the G-helix site and bulk water is observed regularly in our simulations, whereas direct exchange between the RQH site and bulk water is not. The highly conserved kink of the G-helix is an important catalytic feature of terpene synthases that has been previously shown to have a profound effect on product specificity.7,11,12,47 For example, TPS4 and TPS5 (sesquiterpene synthases from Zea mays that share 98% sequence identity) produce the same mixture of compounds but with different proportions of certain stereoisomers, an amino acid substitution (G by A) in the kink of the G-helix being the major factor determining the stereoselectivity.11 Another example is the conversion of δ-cadinene synthase (DCS) into a germacradien-4-ol synthase (Gd4olS) by mutation of the hinge points of the kink (DCS-N403P/L405H).47 Moreover, in bacterial class I terpene synthases, two residues (G188 and V187 in Gd11olS) of the so-called effector triad, which is involved in substrate binding, substrate ionization, and active site closure, are located at the kink.30 Thus, the RQH site and G-helix kink (187VGGA190) were selected as mutation targets to modulate the water capture behavior of Gd11olS.
Figure 4.

(a) Representative MD snapshot of the FDP·Gd11olSWT complex in binding mode I showing the two regions of the active site where water molecules are observed during the MD simulations: the G-helix and R228Q313H320 sites. Non-water hydrogen atoms are omitted for clarity. (b) Superimposed structures of the FDP·Gd11olSWT complex in binding mode I (at intervals of 4 ps for water molecules and 200 ps for the rest of the system) during one of the 10 MD runs. Hydrogens are omitted for clarity; the distribution of red spheres thus indicates the location of water along the trajectory. Water can flow between the G-helix and RQH sites.
Mutation of the RQH Site
Each residue of the RQH site was mutated (R228 to V, Q313 to N and A, and H320 to F), aiming to reduce the polarity and thus the likely availability of water in this site. H320 was mutated to F to keep a similar side-chain size, considering that the H320A mutant displayed a shift to linear product formation (see above and Figure 2). R228 was changed to V and Q313 to N because, on the basis of the substrate–analogue complex structure of the (nonhydroxylating) ATAS (PDB code 4KUX),43 residues V212, N299, and S303 of ATAS are positioned equivalently to R228, Q313, and H320 of Gd11olSWT (Figure 4a), respectively.42,43 Note that, despite the absence of hydroxylating activity in ATAS, a water molecule is found at its active site hydrogen-bonded to N299 and S303.31,43 To avoid any possible water-activating role of the residue at 313, Q313 was also changed to the nonpolar A; previously, a water-activating role for an active-site Asn residue was indicated (N305 in the hydroxylating bacterial 1,8-cineole synthase, bCinS).36
Overall, modifications of the RQH site residues to nonpolar residues led to an increase in the total yield of nonhydroxylated compounds (i.e., isolepidozene and germacrene D, and also germacrene A for H320F; Figure 2 and Table S2) with a concomitant decrease in the total yield for hydroxylated compounds (i.e., Gd11ol). However, a significant shift away from hydroxylation occurred only with the H320F mutant, which produced isolepidozene as the major nonhydroxylated compound, with a 43.2% yield. On consideration that the modification of H320 by F is almost isosteric, it is reasonable to expect that FDP still binds in binding mode I (Figure 1). Since the H320F variant still produces 2 (45.3%), H320 is not a required general base for hydroxylation at C11. A likely explanation for the large shift away from hydroxylation with F320 is that FDP:C11 is now less accessible to water, possibly due to the disruption of the G-helix–RQH water flow observed in Gd11olSWT (for which H320 is in a key position; Figure 4). Since R228 is exposed to bulk solvent, the small perturbation of hydroxylation of the R228V mutation indicates that a flux of water directly from the solvent to the RQH site (only observed once in one of the Gd11olSWT MD simulations) is not a major factor determining water availability for the reaction. While the Q313A mutation also has a slight effect on hydroxylation, the product profile remains the same with Q313N. The kinetic constants for these two mutants are very similar to wild type (Table 1). This suggests that Q313 mediates in the availability of a reactive water molecule but does not participate in the reaction as a (transient) proton acceptor. The results with the mutants thus show that the three residues of the RQH site work in synergy to favor hydroxylation of the isolepidozene intermediate in Gd11olSWT catalysis, with H320 being the most important.
Mutation of the G-Helix Kink Region
Residues V187 and G188A of the G-helix kink (187VGGA190) of Gd11olS correspond to the so-called linker and effector residues of the conserved effector triad of class I terpene synthases. The former plays a key role in rearrangement of the G-helix in active-site closure, and the latter may assist in substrate ionization.30 In bacterial terpene synthases, the most common residues present at the linker position are V, D, T, and N, and those at the effector position are G, A, V, and S.30 This was used as a reference to generate several Gd11olS mutants of the G-helix kink region (G188A, G188S, G188V, V187D, and V187N) to be tested. Three additional mutants of the G-helix kink (V187A, A190V, and A190L) were included for further comparison. The corresponding product profiles for all of these mutants are depicted in Figure 2 and Table S2. Replacement of V187 by the smaller hydrophobic residue alanine had no effect on the product profile apart from producing small quantities of 4 (4.1%). However, V187N and (mainly) G188A led to a pronounced shift of the product profile in favor of the formation of 4 (77.9% and 88.4%, respectively), with reduced production of 2. Meanwhile, replacing G188 by the small polar serine reduced the formation of 2 to 59.4% and yielded 37.4% of 4. The mutant A190V displayed the widest product diversity and generated four products in total: 2 (25.3%), 4 (62.5%), 5 (5.7%), and 7 (6.5%). The remaining mutants V187D, G188V, and A190L led to a complete loss of activity, which may be attributed to the inability of the protein to form the corresponding reactive closed enzyme–substrate complex (MC) due to a significant perturbation of G-helix conformational behavior. Interestingly, mutation of the equivalent residue of G188 in selinadiene synthase (G182) with valine also led to a complete loss of activity.30 The importance of the G-helix for the formation of the relevant Michaelis complex is further indicated by the >8-fold increase in the KM value obtained for the mutants G188A, G188S, and A190V in comparison to Gd11olSWT (Table 1).
These results show that the G-helix kink region has a major influence on hydroxylation (as well as MC formation). To shed more light on this, we modeled the MC of the mutants G188S and G188A using MD simulations. Again, we performed simulations for both binding modes I and II. A preference for pro-R PCCs was again observed for both mutants in mode I, with no pro-R PCCs sampled in G188A and no pro-R/S preference for G188S in mode II (Figure 3). It is thus likely that mode I is again the relevant MC for these variants, and we focused our analysis on this binding mode.
As with Gd11olSWT, water molecules were observed in the RQH and G-helix sites for both mutants, in similar quantities. At least one water is present in the RQH site for the majority of the time in Gd11olSWT and the G188A and G188S variants (>85%, Figure S6). Further, an evaluation of the interatomic distance between FDP:C11 and the closest RQH site water molecule when pro-R PCCs are formed does not reveal a significant difference either (Figure S7), with water molecules able to come close to FDP:C11 similarly. Thus, simulations of the MC complexes do not indicate water availability and its proximity to FDP:C11 to be a factor in the reduction of product hydroxylation in the G188S and G188A mutants.
The MD simulations do indicate that both the G188A and G188S mutations affect the conformation of the G-helix. As expected due to their preferred backbone orientations, the kink is more helical in the mutant enzymes than in Gd11olSWT; hydrogen bonds between the amide of residue 188 and the carbonyl oxygens of R185 and R184 are formed more frequently (Figure 5 and Figure S8). In the G188S mutant, the hydroxyl group of S188 can additionally form hydrogen bonds with surrounding polar residues (e.g., H226 and S222) and the backbone of A190 and R185, which also changes the helix kink conformation with respect to Gd11olSWT. For example, the hydrogen bond between the residues M183 and V187 is less stable in the G188S mutant (Figure 5 and Figure S8). The changes in hydrogen bonds subtly affect the hydration pattern around the G-helix site. Further, the shape of the active site varies among these enzyme variants, which will have an influence on the reaction energetics and therefore on product specificity, by subtly changing substrate–enzyme–water interactions along the reaction.
Figure 5.
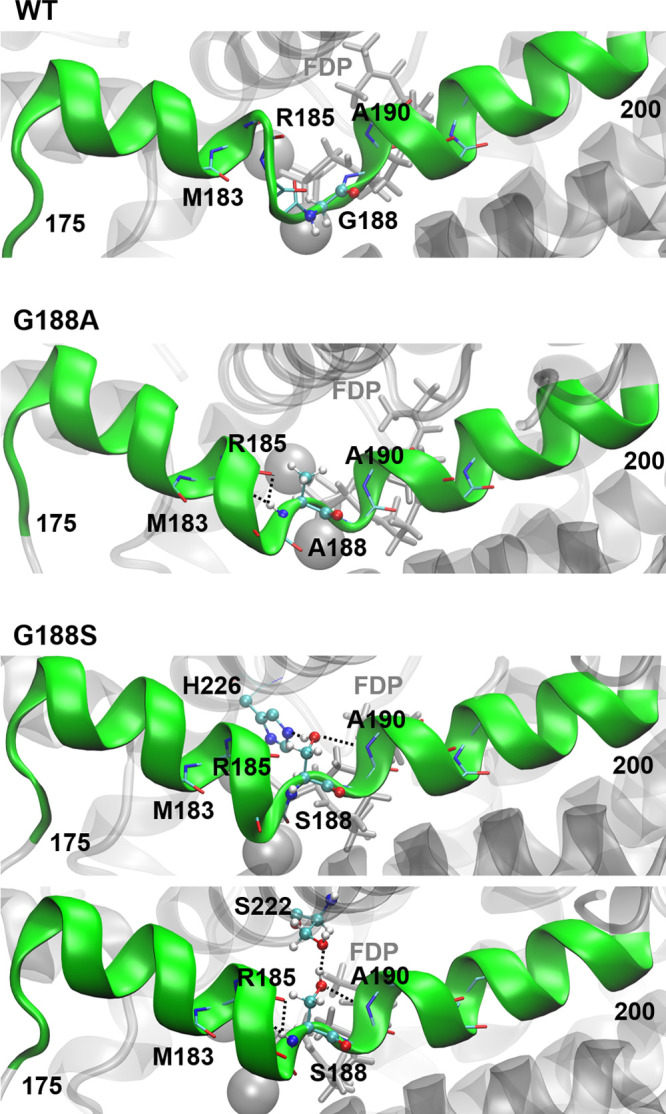
Dominant configurations of the G-helix (green, mutation target) in the MD simulations of the MCs of Gd11olSWT and the mutants G188A and G188S. Relevant hydrogen bond interactions of residue 188 are shown as dashed lines.
Confirmation of Isolepidozene as the Major Product in Gd11olS G188A
To determine the structure of the major sesquiterpene produced by the G188A mutant, preparative-scale incubation with FDP (see the Supporting Information for more details) was performed, resulting in 12 mg of product. After chromatographic purification using deactivated silica (neutralized with 1% triethylamine) with n-pentane as eluent, a total of 7.5 mg of a colorless oil was isolated with >99% purity, as judged by GCMS (Figure S27). The purified compound was analyzed by NMR spectroscopy and found to have a fused dimethylcyclopropane–germacrene type skeleton (spectroscopic assignment of all the 1H and 13C NMR spectra is given in Figures S46–S52 in the Supporting Information). The 13C and 13C DEPT 135 spectra revealed the presence of a distinctive peak for a fused cyclopropane ring with signals at δC 24.3, 36.2, and 41.1 ppm corresponding to C11, C1, and C10 respectively. Hence, the major product could be either isolepidozene or its cis-fused isomer bicyclogermacrene (Figure 6).48,49 To determine the relative stereochemistry of the C1 and C10 atoms and to confirm the product, the coupling constant of the H1 and H10 protons (3JHH) was measured by 1H homonuclear decoupling NMR spectroscopy and predicted computationally (see the Supporting Information for more details). The experimentally determined coupling constant was 5.20 Hz (Figure S53). Spin–spin coupling constant calculations50,51 were performed at the ωB97X-D52/6-311G(d,p)53 level on QM optimized structures (using Gaussian 09;54 see the Supporting Information for details), which predicted the coupling constant between the H1 and H10 protons to be 4.84 Hz for isolepidozene and 8.82 Hz for bicyclogermacrene. This confirms that the major product formed by Gd11olS G188A and also produced by other variants, is indeed isolepidozene (4). This supports the proposal that 4 is the neutral intermediate that undergoes ring opening and water attack to form germacradien-11-ol.55
Figure 6.

QM (DFT) optimized geometries of (a) isolepidozene and (b) bicyclogermacrene, with H1 and H10 protons highlighted.
Conclusions
We have used a combination of computational simulation and site-directed mutagenesis to gain insight into the water activation and hydroxylation catalyzed by the sesquiterpene Gd11ol synthase. The FDP binding mode in the active site was confirmed by a combination of MD simulation and mutagenesis to be similar to that observed in other (nonhydroxylating) sesquiterpene synthases. On the basis of this binding mode, we identified two active site regions, namely the G-helix and RQH sites, that are directly or indirectly involved in managing water access to FDP:C11 and its hydroxylation at the right stage of the reaction. Water can flow freely between these sites in Gd11olSWT and investigated mutants, with the RQH site able to hold a water molecule close to FDP:C11 for nucleophilic attack on isolepidozene. Mutagenesis indicates, however, that none of the residues in this site are essential for hydroxylation: e.g., as a general base for water activation. Mutations in the G-helix kink region confirm its involvement in forming a reactive enzyme–substrate complex, as previously observed for other sesquiterpene synthases. Remarkably, certain mutations in this region resulted in a drastic change in the product profile, with G188A in particular leading to formation of the complex nonhydroxylated isolepidozene as the major product. Our work thereby shows that alteration of the G-helix kink motif reduces hydroxylation more efficiently than alteration of the RQH site. This indicates that subtle changes in the water binding regions of terpene synthases can profoundly affect their hydroxylation activities, and understanding the precise mechanism of such changes is a major challenge. Future work, including simulation of key intermediates and reactions alongside an experimental determination of mutant structures, may provide further insight into the exact origin of the reduction in hydroxylation caused by G-helix kink mutations. Our work highlights the intricate molecular choreography involved in water management by terpene synthases. A detailed understanding of how terpene synthases control substrate hydroxylation will pave the way for targeted enzyme engineering to allow efficient production of specific and novel terpenoids.
Acknowledgments
This work was supported by the BBSRC (grant numbers BB/R001596/1 and BB/R001332/1). M.W.v.d.K. is a BBSRC David Phillips Fellow (grant BB/M026280/1). Simulations were performed using the computational facilities of the Advanced Computing Research Centre, University of Bristol. We thank Dr. Robert Jenkins (Cardiff University) for his help with NMR spectroscopy. We also thank to Prof. John Pickett CBE, DSc, FRS (Cardiff University) for a critical reading of the manuscript and valuable suggestions.
Glossary
Abbreviations
- Gd11olS
germacradien-11-ol synthase
- MC
Michaelis complex
- PCCs
productive conformations for cyclization
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.0c04647.
Cloning, mutagenesis, expression and purification of protein variants, GC chromatograms, kinetics and product analysis (including NMR), and MD simulation and QM calculation methods (PDF)
Author Contributions
§ P.L.S. and A.M.E. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Tholl D. Terpene Synthases and the Regulation, Diversity and Biological Roles of Terpene Metabolism. Curr. Opin. Plant Biol. 2006, 9, 297–304. 10.1016/j.pbi.2006.03.014. [DOI] [PubMed] [Google Scholar]
- Kirby J.; Keasling J. D. Biosynthesis of Plant Isoprenoids: Perspectives for Microbial Engineering. Annu. Rev. Plant Biol. 2009, 60, 335–355. 10.1146/annurev.arplant.043008.091955. [DOI] [PubMed] [Google Scholar]
- Christianson D. W. Structural and Chemical Biology of Terpenoid Cyclases. Chem. Rev. 2017, 117, 11570–11648. 10.1021/acs.chemrev.7b00287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava P. L.; Daramwar P. P.; Krithika R.; Pandreka A.; Shankar S. S.; Thulasiram H. V. Functional Characterization of Novel Sesquiterpene Synthases from Indian Sandalwood, Santalum album. Sci. Rep. 2015, 5, 10095. 10.1038/srep10095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada Y.; Kuzuyama T.; Komatsu M.; Shin-ya K.; Omura S.; Cane D. E.; Ikeda H. Terpene Synthases are Widely Distributed in Bacteria. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 857–862. 10.1073/pnas.1422108112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikuni Y.; Ferrin T. E.; Keasling J. D. Designed Divergent Evolution of Enzyme Function. Nature 2006, 440, 1078–1082. 10.1038/nature04607. [DOI] [PubMed] [Google Scholar]
- Kampranis S. C.; Ioannidis D.; Purvis A.; Mahrez W.; Ninga E.; Katerelos N. A.; Anssour S.; Dunwell J. M.; Degenhardt J.; Makris A. M.; Goodenough P. W.; Johnson C. B. Rational Conversion of Substrate and Product Specificity in a Salvia Monoterpene Synthase: Structural Insights into the Evolution of Terpene Synthase Function. Plant Cell 2007, 19, 1994–2005. 10.1105/tpc.106.047779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele C. L.; Crock J.; Bohlmann J.; Croteau R. Sesquiterpene Synthases from Grand Fir (Abies grandis). J. Biol. Chem. 1998, 273, 2078–2089. 10.1074/jbc.273.4.2078. [DOI] [PubMed] [Google Scholar]
- Greenhagen B. T.; O'Maille P. E.; Noel J. P.; Chappell J. Identifying and Manipulating Structural Determinates Linking Catalytic Specificities in Terpene Synthases. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 9826–9831. 10.1073/pnas.0601605103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Maille P. E.; Malone A.; Dellas N.; Andes Hess B.; Smentek L.; Sheehan I.; Greenhagen B. T.; Chappell J.; Manning G.; Noel J. P. Quantitative Exploration of the Catalytic Landscape Separating Divergent Plant Sesquiterpene Synthases. Nat. Chem. Biol. 2008, 4, 617–623. 10.1038/nchembio.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köllner T. G.; Schnee C.; Gershenzon J.; Degenhardt J. The Variability of Sesquiterpenes Emitted from Two Zea mays Cultivars Is Controlled by Allelic Variation of Two Terpene Synthase Genes Encoding Stereoselective Multiple Product Enzymes. Plant Cell 2004, 16, 1115–1131. 10.1105/tpc.019877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leferink N. G. H.; Ranaghan K. E.; Karuppiah V.; Currin A.; Van der Kamp M. W.; Mulholland A. J.; Scrutton N. S. Experiment and Simulation Reveal How Mutations in Functional Plasticity Regions Guide Plant Monoterpene Synthase Product Outcome. ACS Catal. 2018, 8, 3780–3791. 10.1021/acscatal.8b00692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez V.; Touchet S.; Grundy D. J.; Faraldos J. A.; Allemann R. K. Evolutionary and Mechanistic Insights from the Reconstruction of α-Humulene Synthases from a Modern (+)-Germacrene A Synthase. J. Am. Chem. Soc. 2014, 136, 14505–14512. 10.1021/ja5066366. [DOI] [PubMed] [Google Scholar]
- Dixit M.; Weitman M.; Gao J.; Major D. T. Chemical Control in the Battle against Fidelity in Promiscuous Natural Product Biosynthesis: The Case of Trichodiene Synthase. ACS Catal. 2017, 7, 812–818. 10.1021/acscatal.6b02584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F.; An T.; Tang X.; Zi J.; Luo H.-B.; Wu R. Enzyme Promiscuity versus Fidelity in Two Sesquiterpene Cyclases (TEAS versus ATAS). ACS Catal. 2020, 10, 1470–1484. 10.1021/acscatal.9b05051. [DOI] [Google Scholar]
- Raz K.; Driller R.; Dimos N.; Ringel M.; Brü T.; Loll B.; Major D. T. The Impression of a Nonexisting Catalytic Effect: The Role of CotB2 in Guiding the Complex Biosynthesis of Cyclooctat-9-en-7-ol. J. Am. Chem. Soc. 2020, 142, 21562. 10.1021/jacs.0c11348. [DOI] [PubMed] [Google Scholar]
- Shishova E. Y.; Yu F.; Miller D. J.; Faraldos J. A.; Zhao Y.; Coates R. M.; Allemann R. K.; Cane D. E.; Christianson D. W. X-ray Crystallographic Studies of Substrate Binding to Aristolochene Synthase Suggest a Metal Ion Binding Sequence for Catalysis. J. Biol. Chem. 2008, 283, 15431–15439. 10.1074/jbc.M800659200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Kamp M. W.; Sirirak J.; Zurek J.; Allemann R. K.; Mulholland A. J. Conformational Change and Ligand Binding in the Aristolochene Synthase Catalytic Cycle. Biochemistry 2013, 52, 8094–8105. 10.1021/bi400898k. [DOI] [PubMed] [Google Scholar]
- Croteau R. Biosynthesis and Catabolism of Monoterpenoids. Chem. Rev. 1987, 87, 929–954. 10.1021/cr00081a004. [DOI] [Google Scholar]
- Cane D. E. Enzymatic Formation of Sesquiterpenes. Chem. Rev. 1990, 90, 1089–1103. 10.1021/cr00105a002. [DOI] [Google Scholar]
- Nakano C.; Kudo F.; Eguchi T.; Ohnishi Y. Genome Mining Reveals Two Novel Bacterial Sesquiterpene Cyclases: (−)-Germacradien-4-ol and (−)-epi-α-Bisabolol Synthases from Streptomyces citricolor. ChemBioChem 2011, 12, 2271–2275. 10.1002/cbic.201100418. [DOI] [PubMed] [Google Scholar]
- Nakano C.; Tezuka T.; Horinouchi S.; Ohnishi Y. Identification of the SGR6065 Gene Product as a Sesquiterpene Cyclase Involved in (+)-Epicubenol Biosynthesis in Streptomyces griseus. J. Antibiot. 2012, 65, 551–558. 10.1038/ja.2012.68. [DOI] [PubMed] [Google Scholar]
- Attia M.; Kim S. U.; Ro D. K. Molecular Cloning and Characterization of (+)-epi-α-Bisabolol Synthase, Catalyzing the First Step in the Biosynthesis of the Natural Sweetener, Hernandulcin, in Lippia dulcis. Arch. Biochem. Biophys. 2012, 527, 37–44. 10.1016/j.abb.2012.07.010. [DOI] [PubMed] [Google Scholar]
- Piechulla B.; Bartelt R.; Brosemann A.; Effmert U.; Bouwmeester H.; Hippauf F.; Brandt W. The α-Terpineol to 1,8-Cineole Cyclization Reaction of Tobacco Terpene Synthases. Plant Physiol. 2016, 172, 2120–2131. 10.1104/pp.16.01378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia M.; O’Brien T. E.; Zhang Y.; Siegel J. B.; Tantillo D. J.; Peters R. J. A Key Residue for the Addition of Water by Sclareol Synthase. ACS Catal. 2018, 8, 3133–3137. 10.1021/acscatal.8b00121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starks C. M.; Back K.; Chappell J.; Noel J. P. Structural Basis for Cyclic Terpene Biosynthesis by Tobacco 5-epi-Aristolochene Synthase. Science 1997, 277, 1815–1820. 10.1126/science.277.5333.1815. [DOI] [PubMed] [Google Scholar]
- Rynkiewicz M. J.; Cane D. E.; Christianson D. W. Structure of Trichodiene Synthase from Fusarium sporotrichioides Provides Mechanistic Inferences on the Terpene Cyclization Cascade. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 13543–13548. 10.1073/pnas.231313098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M.; Chou W. K. W.; Al-Lami N.; Faraldos J. A.; Allemann R. K.; Cane D. E.; Christianson D. W. Probing the Role of Active Site Water in the Sesquiterpene Cyclization Reaction Catalyzed by Aristolochene Synthase. Biochemistry 2016, 55, 2864–2874. 10.1021/acs.biochem.6b00343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittington D. A.; Wise M. L.; Urbansky M.; Coates R. M.; Croteau R. B.; Christianson D. W. Bornyl Diphosphate Synthase: Structure and Strategy for Carbocation Manipulation by a Terpenoid Cyclase. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 15375–15380. 10.1073/pnas.232591099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baer P.; Rabe P.; Fischer K.; Citron C. A.; Klapschinski T. A.; Groll M.; Dickschat J. S. Induced-Fit Mechanism in Class I Terpene Cyclases. Angew. Chem., Int. Ed. 2014, 53, 7652–7656. 10.1002/anie.201403648. [DOI] [PubMed] [Google Scholar]
- Grundy D. J.; Chen M.; González V.; Leoni S.; Miller D. J.; Christianson D. W.; Allemann R. K. Mechanism of Germacradien-4-ol Synthase-Controlled Water Capture. Biochemistry 2016, 55, 2112–2121. 10.1021/acs.biochem.6b00115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loizzi M.; González V.; Miller D. J.; Allemann R. K. Nucleophilic Water Capture or Proton Loss: Single Amino Acid Switch Converts δ-Cadinene Synthase into Germacradien-4-ol Synthase. ChemBioChem 2018, 19, 100–105. 10.1002/cbic.201700531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinde S. S.; Navale G. R.; Said M. S.; Thulasiram H. V. Stereoselective Quenching of Cedryl Carbocation in Epicedrol Bio-synthesis. Tetrahedron Lett. 2016, 57, 1161–1164. 10.1016/j.tetlet.2016.01.109. [DOI] [Google Scholar]
- Cane D. E.; Ke N. Epicubenol Synthase. Origin of the Oxygen Atom of a Bacterial Sesquiterpene Alcohol. Bioorg. Med. Chem. Lett. 2000, 10, 105–107. 10.1016/S0960-894X(99)00650-2. [DOI] [PubMed] [Google Scholar]
- Baer P.; Rabe P.; Citron C. A.; De Oliveira Mann C. C.; Kaufmann N.; Groll M.; Dickschat J. S. Hedycaryol Synthase in Complex with Nerolidol Reveals Terpene Cyclase Mechanism. ChemBioChem 2014, 15, 213–216. 10.1002/cbic.201300708. [DOI] [PubMed] [Google Scholar]
- Leferink N. G. H.; Ranaghan K. E.; Battye J.; Johannissen L. O.; Hay S.; Van der Kamp M. W.; Mulholland A. J.; Scrutton N. S. Taming the Reactivity of Monoterpene Synthases to Guide Regioselective Product Hydroxylation. ChemBioChem 2020, 21, 985. 10.1002/cbic.201900672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driller R.; Janke S.; Fuchs M.; Warner E.; Mhashal A. R.; Major D. T.; Christmann M.; Brück T.; Loll B. Towards a Comprehensive Understanding of the Structural Dynamics of a Bacterial Diterpene Synthase During Catalysis. Nat. Commun. 2018, 9, 3971. 10.1038/s41467-018-06325-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J.; He X.; Cane D. E. Geosmin Biosynthesis. Streptomyces coelicolor Germacradienol/Germacrene D Synthase Converts Farnesyl Diphosphate to Geosmin. J. Am. Chem. Soc. 2006, 128, 8128–8129. 10.1021/ja062669x. [DOI] [PubMed] [Google Scholar]
- Jiang J.; He X.; Cane D. E. Biosynthesis of the Earthy Odorant Geosmin by a Bifunctional Streptomyces coelicolor Enzyme. Nat. Chem. Biol. 2007, 3, 711–715. 10.1038/nchembio.2007.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cane D. E.; Watt R. M. Expression and Mechanistic Analysis of a Germacradienol Synthase from Streptomyces coelicolor Implicated in Geosmin Biosynthesis. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 1547–1551. 10.1073/pnas.0337625100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X.; Cane D. E. Mechanism and Stereochemistry of the Germacradienol/Germacrene D Synthase of Streptomyces coelicolor A3(2). J. Am. Chem. Soc. 2004, 126, 2678–2679. 10.1021/ja039929k. [DOI] [PubMed] [Google Scholar]
- Harris G. G.; Lombardi P. M.; Pemberton T. A.; Matsui T.; Weiss T. M.; Cole K. E.; Köksal M.; Murphy F. V. IV; Sangeetha Vedula L.; Chou W. K.; Cane D. E.; Christianson D. W. Structural Studies of Geosmin Synthase, a Bifunctional Sesquiterpene Synthase with αα Domain Architecture That Catalyzes a Unique Cyclization–Fragmentation Reaction Sequence. Biochemistry 2015, 54, 7142–7155. 10.1021/acs.biochem.5b01143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M.; Al-Lami N.; Janvier M.; D’Antonio E. L.; Faraldos J. A.; Cane D. E.; Allemann R. K.; Christianson D. W. Mechanistic Insights from the Binding of Substrate and Carbocation Intermediate Analogues to Aristolochene Synthase. Biochemistry 2013, 52, 5441–5453. 10.1021/bi400691v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Rijn J. P. M.; Escorcia A. M.; Thiel W. QM/MM Study of the Taxadiene Synthase Mechanism. J. Comput. Chem. 2019, 40, 1902–1910. 10.1002/jcc.25846. [DOI] [PubMed] [Google Scholar]
- Faraldos J. A.; Antonczak A. K.; González V.; Fullerton R.; Tippmann E. M.; Allemann R. K. Probing Eudesmane Cation-π Interactions in Catalysis by Aristolochene Synthase with Non-canonical Amino Acids. J. Am. Chem. Soc. 2011, 133, 13906–13909. 10.1021/ja205927u. [DOI] [PubMed] [Google Scholar]
- Srividya N.; Davis E. M.; Croteau R. B.; Lange B. M. Functional Analysis of (4S)-Limonene Synthase Mutants Reveals Determinants of Catalytic Outcome in a Model Monoterpene Syn-thase. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 3332–3337. 10.1073/pnas.1501203112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikuni Y.; Martin V. J. J.; Ferrin T. E.; Keasling J. D. Engineering Cotton (+)-δ-Cadinene Synthase to an Altered Function: Germacrene D-4-ol Synthase. Chem. Biol. 2006, 13, 91–98. 10.1016/j.chembiol.2005.10.016. [DOI] [PubMed] [Google Scholar]
- McMurry J. E.; Bosch G. K. Synthesis of Bicyclogermacrene and Lepidozene. Tetrahedron Lett. 1985, 26, 2167–2170. 10.1016/S0040-4039(00)98952-4. [DOI] [Google Scholar]
- Hardt I. H.; Rieck A.; König W. A.; Muhle H. Isolepidozene, A Diastereomer of Bicyclogermacrene, in Some Liverworts. Phytochemistry 1995, 40, 605–606. 10.1016/0031-9422(95)00324-Z. [DOI] [Google Scholar]
- Helgaker T.; Watson M.; Handy N. C. Analytical Calculation of Nuclear Magnetic Resonance Indirect Spin-Spin Coupling Constants at the Generalized Gradient Approximation and Hybrid Levels of Density-Functional Theory. J. Chem. Phys. 2000, 113, 9402–9409. 10.1063/1.1321296. [DOI] [Google Scholar]
- Deng W.; Cheeseman J. R.; Frisch M. J. Calculation of Nuclear Spin-Spin Coupling Constants of Molecules with First and Second Row Atoms in Study of Basis Set Dependence. J. Chem. Theory Comput. 2006, 2, 1028–1037. 10.1021/ct600110u. [DOI] [PubMed] [Google Scholar]
- Chai J.-D.; Head-Gordon M. Long-range Corrected Hybrid Density Functionals with Damped Atom-Atom Dispersion Corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. 10.1039/b810189b. [DOI] [PubMed] [Google Scholar]
- Krishnan R.; Binkley J. S.; Seeger R.; Pople J. A. Self-consistent Molecular Orbital Methods. XX. A Basis Set for Correlated Wave Functions. J. Chem. Phys. 1980, 72, 650–654. 10.1063/1.438955. [DOI] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam J. M.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas Ö.; Foresman J. B.; Ortiz J. V.; Cioslowski J.. Gaussian09; Gaussian, Inc.: Wallingford, CT, 2009. [Google Scholar]
- Jiang J.; Cane D. E. Geosmin Biosynthesis. Mechanism of the Fragmentation-Rearrangement in the Conversion of Germacradienol to Geosmin. J. Am. Chem. Soc. 2008, 130, 428–429. 10.1021/ja077792i. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.