

Abstract
Intense X-rays available at powerful synchrotron beamlines provide macromolecular crystallographers with an incomparable tool for investigating biological phenomena on an atomic scale. The resulting insights into the mechanism’s underlying biological processes have played an essential role and shaped biomedical sciences during the last 30 years, considered the “golden age” of structural biology. In this review, we analyze selected aspects of the impact of synchrotron radiation on structural biology. Synchrotron beamlines have been used to determine over 70% of all macromolecular structures deposited into the Protein Data Bank (PDB). These structures were deposited by over 13,000 different research groups. Interestingly, despite the impressive advances in synchrotron technologies, the median resolution of macromolecular structures determined using synchrotrons has remained constant throughout the last 30 years, at about 2 Å. Similarly, the median times from the data collection to the deposition and release have not changed significantly. We describe challenges to reproducibility related to recording all relevant data and metadata during the synchrotron experiments, including diffraction images. Finally, we discuss some of the recent opinions suggesting a diminishing importance of X-ray crystallography due to impressive advances in Cryo-EM and theoretical modeling. We believe that synchrotrons of the future will increasingly evolve towards a life science center model, where X-ray crystallography, Cryo-EM, and other experimental and computational resources and knowledge are encompassed within a versatile research facility. The recent response of crystallographers to the COVID-19 pandemic suggests that X-ray crystallography conducted at synchrotron beamlines will continue to play an essential role in structural biology and drug discovery for years to come.
1. Introduction: First uses of synchrotron radiation in structural biology
Synchrotron radiation, the electromagnetic radiation generated by charged particles accelerated in a magnetic field (initially referred to as Magnetobremsstrahlung), was first observed in the research laboratory of the General Electric company in the 1940s. At the time, it was considered a nuisance responsible for energy losses that made the design and use of particle accelerators more difficult [1]. It was soon realized that the powerful X-ray radiation generated as a side-effect of particle accelerators could be beneficial in material science studies and solid-state physics research [2]. The first experiments with X-ray synchrotron radiation were performed at Stanford University, USA, DESY (Deutsche Elektronen-Synchrotron), Germany, and Daresbury Laboratory, UK. A 1977 paper describing a pioneer single-crystal diffraction experiment [3] concluded that “synchrotron radiation is an ideal X-ray source for energy-dispersive diffractometry … especially suited for fast structure identification.” Indeed, it was later demonstrated that energy-dispersive techniques are valuable tools to study time-resolved phenomena like the crystallization of metallic glasses [4].
However, until the 1970s, it was generally doubted that biological samples could withstand the high-intensity X-ray beams generated by synchrotron sources. The 1976 paper “Applications of synchrotron radiation to protein crystallography: Preliminary results” from the Stanford Synchrotron Radiation Project [5] demonstrated that these doubts were unjustified. The paper described experiments in which several protein crystals were irradiated by the X-ray beam produced in the Stanford Linear Accelerator Center (SLAC). Strikingly, the stability of the protein crystals tested in the beam was sufficient to collect good-quality diffraction images. All the tested crystals of different proteins (rubredoxin, azurin, nerve growth factor, and L-glutaminase-asparaginase) suffered only relatively minor radiation damage. Thus, this experiment established the viability of using synchrotron radiation to determine the structure of crystals of biological molecules.
At the time, macromolecular crystallography relied on “home-laboratory sources” or “home sources,” X-ray generators stationed in individual research laboratories or shared within a department or institution. These were typically much weaker than synchrotron sources. In the early years, these generators used a sealed-tube design. Starting in the 1950s, more powerful rotating-anode X-ray generators became commercially produced by companies such as Elliott, Syntex, Rigaku, Enraf-Nonius, and Bruker and became the workhorses of in-house structure determination [6].
Even after synchrotron radiation became available, the wide-spread adoption of synchrotron sources in macromolecular crystallography was not immediate. Until 2000, most of the structures deposited each year to the Protein Data Bank (PDB), a central repository for structural models established in 1971 [7], were determined using data collected on home sources.
It is not straightforward to identify the first structure deposited to the PDB that used synchrotron radiation. Although the PDB deposition format included fields for experimental details, such as the type of detector, radiation source, and software used, the extent to which these fields were used was variable. Many of the early PDB files had upwards of 100 “NULL” (missing) data items. Consequently, even if some of the early deposits used synchrotron radiation, it is impossible to ascertain this based solely on the information in the PDB files. The first entry that filled in the “SYNCHROTRON(Y/N)” field with a “Y” dates from 1989, when the PDB already had more than 400 structures. Diffraction images for this entry, a bovine beta-trypsin (PDB id: 1tld) [8] were collected at DESY in Hamburg. However, even earlier structures used data collected on this synchrotron, e.g., PDB id: 1paz (Z. Dauter, personal communication), but this information was not recorded in the PDB. The determination of who was the first would be an interesting science history project that we may entertain when retired; however, we made a first step toward this project – we found that Max Perutz’s structure of deoxyhaemoglobin (PDB id: 2hhb) used data collected at the LURE facility [9].
In the 30+ years since these first synchrotron structures were determined, numerous synchrotron sources have been built and made available for biological scientists, facilitating hundreds of thousands of data collection attempts for biological molecules, and ushering in the “golden age” of structural biology. As early as 1996, some scientists were claiming that “macromolecular crystallography has benefitted more from the availability of synchrotron radiation than any other single discipline” [10]. The application of synchrotron radiation to structural biology is sometimes cited as the best example of a serendipitous effect of “big science” infrastructure impacting scientific fields far beyond the original application [11].
One aspect of synchrotron radiation that revolutionized structure determination is the ability to change the wavelength of the X-ray beam. Home sources can only generate X-rays that correspond to the emission spectra of the anode, typically copper, molybdenum, or sometimes chromium. Some “dual wavelength” home sources allow the user to switch anode, but most home sources have afixed-wavelength. The ability to tune the wavelength of the radiation permitted the development of anomalous dispersion techniques, which take advantage of differences in the diffraction intensities when the wavelength of the X-rays is close to an absorption edge of an element in the crystal. Anomalous dispersion techniques provided researchers with a third method of solving the “phase problem” of crystallography, supplementing multiple isomorphous replacement and molecular replacement. Nowadays, anomalous techniques and molecular replacement practically solved the “phase problem.”
The impact of synchrotron radiation on structural biology – the subject of this review – is the story of some 50 synchrotrons, the thousands of researchers that used them, and the 120,000 (and counting) biological structures that they have determined. Various aspects of this story have already been presented in multiple accounts from different viewpoints, e.g., [12–14]. In this review, we aim to present our perspective, based on our first-hand experiences and the analysis of data deposited to the PDB and other structural resources.
2. Synchrotron sources revolutionize structural biology with over 120,000 macromolecular structures
2.1. From “parasitic mode” to fourth generation radiation sources
Pioneering research was conducted in particle accelerators equipped with storage rings where particles could circulate for long periods of time at a constant speed. From the point of view of particle physicists, the accelerator operated in a “parasitic mode,” and usage of the radiation by non-physicists was an exception [15]. Synchrotrons of this type, such as the SPEAR storage ring of SSRL (formerly known as SSRP), the original DORIS storage ring at DESY, and the CHESS facility at Cornell University are referred to as first-generation synchrotron sources.
The second-generation synchrotrons were no longer designed as particle accelerators, but rather to serve as “light sources” - sources of intense radiation. One of the earliest facilities of this type was the Synchrotron Radiation Source (SRS) in Daresbury, UK, which started operations in 1981. In the same year, near Berlin, Germany, a synchrotron built by Berliner Elektronenspeicherring-Gesellschaft für Synchrotronstrahlung (BESSY) was inaugurated. In the following years, many other second-generation synchrotron stations were commissioned for radiation studies, including the National Synchrotron Light Source (NSLS) at the Brookhaven National Laboratory, USA, the Photon Factory at KEK Institute in Tsukuba, Japan, and the LURE (Laboratoire pour l’Utilisation du Rayonnement Electromagnétique) in Orsay, France. Some of the older storage rings were retrofitted and operated as second- generation sources, e.g., the DORIS (known as DORIS III since 1993) storage ring at DESY and the SPEAR ring, fully dedicated to SSRL in 1990. Two important detector technologies were introduced at this stage. DESY introduced an automatic image plate scanner constructed in the European Molecular Biology Laboratory by Jules Hendrix, which was later commercialized by MAR Research [16]. A little later, CHESS introduced the first CCD detector, constructed by Sol Gruner, subsequently commercialized by Area Detector System Corporation [17]. Thus, DESY and CHESS and later Photon Factory, NSLS, SSRL and SSRS in Daresbury became leaders in the determination of high resolution and high-quality macromolecular structures.
The third-generation synchrotrons were designed to significantly increase the intensity and stability of radiation using technologies such as undulators and insertion devices. Several such facilities were built in the 1990s. The European Synchrotron Radiation Facility (ESRF) (funded jointly by multiple European countries) was inaugurated in 1994 in Grenoble, France. In the next year, MAX II facility in Lund and a new BESSY II near Berlin were opened. In the USA, the Advanced Light Source (ALS) at the Berkeley National Laboratory and the Advanced Photon Source (APS) at Argonne National Laboratory were opened in the mid-1990s. In Asia, roughly at the same time, National Synchrotron Radiation Research Center (NSRRC) in Taiwan, the Pohang Light Source (PLS) in Korea and the Super Photon ring-8 GeV (SPring8) in Japan were opened to users. By 1997, there were already ten third-generation storage rings in use, complementing over 30 facilities belonging to the first and second generations [18]. Several other third-generation facilities joined the ranks in the first decade of this century, including the SSRLupgraded storage ring at Stanford, the Swiss Light Source (SLS), the Australian Synchrotron, Diamond Light Source in Oxfordshire, UK, and the Shanghai Synchrotron Radiation Facility (SSRF) in China. During the last decade, a new storage ring, PETRA III at DESY, and the most technologically advanced synchrotron, NSLS-II, were opened for users.
Even in the middle of the building boom for the third-generation light sources in the 1990s, synchrotron scientists dreamt of a next generation, which would “exceed the performance of previous sources by one or more orders of magnitude in an important parameter such as brightness, coherence, or shortness of pulse duration” [18]. The first two facilities that satisfied this definition were MAX IV in Lund, Sweden [19], and the new SIRIUS storage ring at the Brazilian Laboratório Nacional de Luz Síncrotron (LNLS) (alongside the existing UVX ring). Many third-generation light sources plan significant upgrades in the coming years, including a “category-jumping” upgrade of APS in Argonne.
All synchrotron facilities/rings reported as a radiation source by at least one macromolecular structure in the PDB, and the corresponding total numbers of structures for these facilities as of September 9, 2020, are listed in Table 1.
Table 1.
Major synchrotron facilities and the number of structures deposited to the PDB (as of September 9, 2020) reporting collecting X-ray diffraction data on these facilities. Defunct facilities are marked with darker background.
| Short name | Full name | Location | PDB deposits |
|---|---|---|---|
| APS | The Advanced Photon Source | Argonne, USA | 26157 |
| ESRF | European Synchrotron Radiation Facility | Grenoble, France | 15646 |
| Diamond | Diamond Light Source | Oxfordshire, UK | 9810 |
| NSLS | National Synchrotron Light Source | Brookhaven, USA | 8170 |
| ALS | The Advanced Light Source | Berkeley, USA | 8118 |
| SLS | Swiss Light Source at the Paul Scherrer Institute | Villigen, Switzerland | 7227 |
| SSRL | Stanford Synchrotron Radiation Lightsource | Stanford, USA | 7076 |
| SPring-8 | Super Photon ring 8 GeV | Soyo, Japan | 5718 |
| SSRF | Shanghai Synchrotron Radiation Facility | Shanghai, China | 5103 |
| Photon Factory | Photon Factory | Tsukuba, Japan | 4792 |
| BESSY | Berliner Elektronenspeicherring-Gesellschaft für Synchrotronstrahlung m. b. H. | Berlin, Germany | 3375 |
| DESY | Doris/Doris III ring at Deutsches Elektronen-Synchrotron (DESY) | Hamburg, Germany | 3195 |
| Australian | ANSTO’s Australian Synchrotron | Clayton, Australia | 2681 |
| CHESS | Cornell High Energy Synchrotron Source | Ithaca, USA | 1942 |
| Pohang | PAL - Pohang Light Source | Pohang, South Korea | 1879 |
| SOLEIL | Synchrotrone SOLEIL | Saint-Aubin, France | 1665 |
| SRS | Synchrotron Radiation Source | Daresbury, UK | 1566 |
| NSRRC | National Synchrotron Radiation Research Center | Hsinchu, Taiwan | 1469 |
| CLSI | Canadian Light Source Inc. | Saskatoon, Canada | 1403 |
| MAX II | MAX II Laboratory | Lund, Sweden | 1128 |
| PETRA III | Petra III ring at Deutsches Elektronen-Synchrotron | Hamburg, Germany | 924 |
| ELETTRA | Elettra Sincrotrone Trieste | Trieste, Italy | 711 |
| LNLS | Laboratório Nacional de Luz Síncrotron UVX | Campinas, Brazil | 648 |
| ALBA | ALBA Synchrotron | Barcelona, Spain | 529 |
| MAX IV | MAX IV Laboratory | Lund, Sweden | 349 |
| BSRF | Beijing Synchrotron Radiation Facility | Beijing, China | 278 |
| LURE | Laboratoire pour l’Utilisation du Rayonnement Electromagnétique | Gif-sur-Yvette, France | 273 |
| NSLS-II | National Synchrotron Light Source II | Brookhaven, USA | 173 |
| RRCAT INDUS-2 | Raja Ramanna Centre for Advanced Technology, Indus-2 | Indore, India | 124 |
| CAMD | The Center for Advanced Microstructures and Devices | Baton Rouge, USA | 56 |
| AichiSR | Aichi Synchrotron Radiation Center | Seto, Japan | 43 |
| KURCHA TOV SNC | Kurchatov Center for Synchrotron Radiation and Nanotechnology | Moscow, Russia | 39 |
| SAGA-LS | SAGA Light Source Kyushu Synchrotron Light Research Center | Tosu, Japan | 5 |
| LNLS SIRIUS | Laboratório Nacional de Luz Síncrotron SIRIUS | Campinas, Brazil | 2 |
| NSRL | National Synchrotron Radiation Laboratory | Hefei, China | 1 |
| SLRI | Synchrotron Light Research Institute | Nakhon Ratchasima, Thailand | 1 |
In addition, there are a number of synchrotrons that have been serving biomedical scientists and commercial users but have not yet made contributions to the PDB. Sometimes, decommissioned synchrotrons have a second life. This happened to the original BESSY I synchrotron, which upon the construction of its successor, BESSY II, was dismantled, shipped to Jordan and reassembled to serve as a foundation of the Synchrotron-Light for Experimental Science and Applications in the Middle East (SESAME) facility [20]. A twin of the smaller MAX IV storage ring takes the center place at the new SOLARIS synchrotron in Krakow, Poland [21]. This facility, together with SSRL and Diamond, represents an emerging trend of creating joint laboratories for X-ray, Cryo-EM, computational, and functional research.
The traditional synchrotron X-ray radiation sources are complemented by other facilities that are listed in Table 2.
Table 2.
Neutron diffraction facilities and the number of PDB deposits to (as of September, 9, 2020) which have reported collecting diffraction data using these facilities
| Short name | Full name | Country | PDB eposits |
|---|---|---|---|
| FRM II | Forschungs-Neutronenquelle Heinz Maier-Leibnitz | Garching, Germany | 27 |
| ILL | Institut Laue-Langevin | Grenoble, France | 26 |
| ORNL Neutron | Oak Ridge National Laboratory the High Flux Isotope Reactor | Oak Ridge, USA | 23 |
| JRR-3M | Japan Research Reactor No.3 Modified JAEA | Tokai, Japan | 13 |
| LANSCE | The Los Alamos Neutron Science Center | Los Alamos, USA | 10 |
| JPARC MLF | Japan Proton Accelerator Research Complex Materials and Life Science Experimental Facility | Tokai, Japan | 5 |
| ISIS | ISIS Neutron and Muon Source | Oxfordshire, UK | 3 |
In 1975, Wood and Chapline [22,23] suggested using intense, short X-ray pulses to examine biological structures. Nearly 35 years were needed until their idea was implemented in the world’s first high-energy XFEL facility at the Stanford Linear Accelerator Center Linac Coherent Light Source (SLAC LCLS) [24] and the first molecular structures were determined by this method [25] The XFELs, opened a new field in structural biology: the ultra-short pulses allow data collection before radiation damage destroys crystals and allow tracking the course and changes in the structure due to chemical reactions [26]. Currently, five XFEL sources operate and two more are in construction [27] (Table 3).
Table 3.
XFEL facilities and the number of structures deposited to the PDB (as of September, 9, 2020) which have reported collecting data using these facilities.
| Short name | Full name | Location | PDB deposits |
|---|---|---|---|
| SLAC LCLS | Stanford Linear Accelerator Center Linac Coherent Light Source | Stanford, USA | 210 |
| SACLA | SPring-8 Angstrom Compact free electron Laser, | Sayo, Japan | 116 |
| European XFEL | European XFEL | Hamburg, Germany | 15 |
| SwissFEL ARAMIS | Switzerland’s X-ray free-electron laser at the Paul Scherrer Institute ARAMIS | Villingen Switzerland | 15 |
| PAL-XFEL | Pohang Accelerator Laboratory XFEL | Pohang, South Korea | 10 |
Parallel to the development synchrotron facilities, there was also progress in home sources. Generators based on rotating anode are usually high maintenance and have largely been supplanted over the last 20 years by sealed tube X-ray generators with comparable flux but substantially reduced maintenance requirements. Recently, powerful metal-jet generators, in which a solid target of conventional rotating anode generators is replaced by a high-speed jet of liquid metal, have become available [28], but they still have relatively few deposits. In the last five years, home sources have been used to produce roughly 8% of recent X-ray structures.
2.2. A stream of macromolecular structures
The availability of second- and third-generation synchrotron sources has profoundly influenced the output of structural biology. Fig.1 presents the numbers of structures based on either synchrotron or home sources and released by the PDB annually between 1989 and 2019.
Fig. 1.
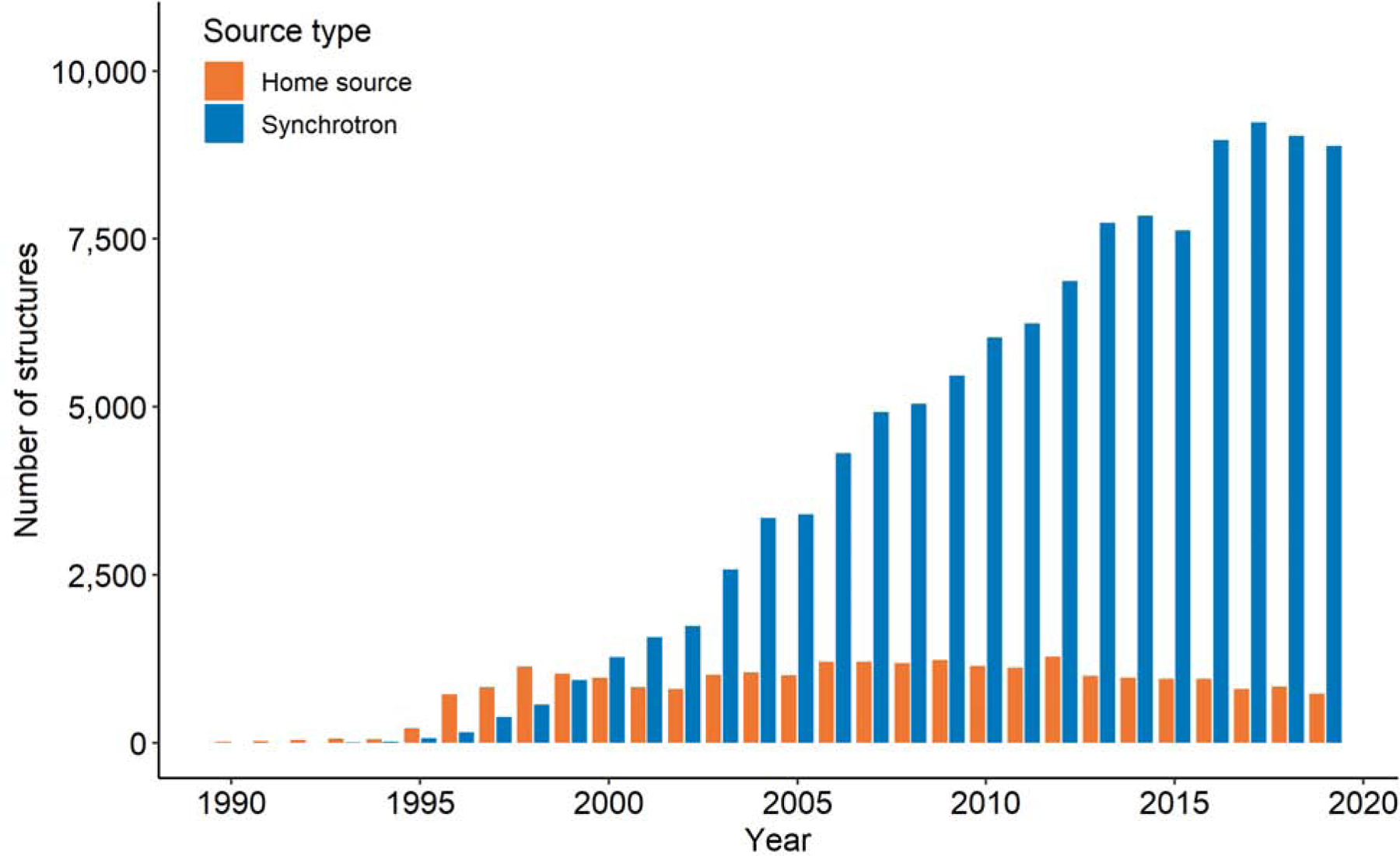
Numbers of macromolecular X-ray structures released annually by the PDB that reported the use of home radiation sources or synchrotron stations. For about 1500 X-ray structures, the radiation source has not been reported to the PDB.
Three different stages can be distinguished:
Years 1989–1999, in which most structures released by the PDB were solved using home sources
Years 2000–2016, characterized by the dominance of synchrotron usage and rapid growth
The period since 2017, characterized by a plateau in the yearly output of X-ray structures
First- and second-generation synchrotrons shaped the first period. In the early 1990s, the most important sources were second-generation synchrotrons: DESY, SRS, LURE in Europe, and CHESS in the USA. The latter part of the decade included contributions from the Photon Factory in Japan and NSLS in the USA.
As a result of new third-generation facilities becoming available to users, for the first time more structures were determined in 2000 using synchrotron sources than with home generators. In 2007, there were four times as many synchrotron deposits as those reporting using home generators. The rapid growth in the number of structures solved using synchrotron sources in that period was in part driven by the Structural Genomics (SG) programs [29]. Out of 15,000 structures that these programs have contributed, over 11,300 (75%) used data collected on synchrotrons. Since 2017, the stream of structures determined using synchrotron sources, as measured by the number of structures released yearly by the PDB has been slowing down (Fig. 1). One of the reasons is the loss of funding for SG projects. Only three major SG centers have been operating in this period; the Structural Genomic Consortium (SGC) [30] and the two centers focused on infectious diseases: the Seattle Structural Center for Infectious Genomics (SSGCID) [31,32] and the Center for Structural Genomics of Infectious Diseases (CSGID) [33]. Another reason for the plateau in the number of structures using data collected on synchrotrons is a shift in focus of some research groups towards Cryo-EM (discussed below in Section 5). Of course, the numbers of structures determined by the pharmaceutical industry, both in house and at synchrotron sources, have not been disclosed and thus are not reflected in the size of the PDB or the statistics in this paper.
Altogether, over 30 synchrotrons and 5 XFEL facilities were used for data collection in structures reported to the PDB, with contributions of individual synchrotrons varying by up to four orders of magnitude (Table 1).
2.3. Synchrotron stations put structure determination within reach of thousands of research groups
The high cost of home X-ray generators limited access to them to better-funded laboratories. In 1989 there were only around 160 research groups worldwide that had contributed structures to the PDB. Within the next decade, more than 700 research groups deposited at least one structure determined using a synchrotron source (with about 300 of these having no documented previous experience in determining structures using home sources) (Fig. 2). The opening of new synchrotron facilities made the process of structure determination available to many research groups, including scientists from developing countries. The funding for synchrotrons and beamlines has been coming from many national and international bodies, funding agencies, charitable organizations, and industry. The estimated cumulative number of different research groups that have determined at least one structure each using a synchrotron source now stands at 13,700 (as of September 9, 2020); only about one-fourth of these group have demonstrated experience in structure determination using a home source.
Fig. 2.
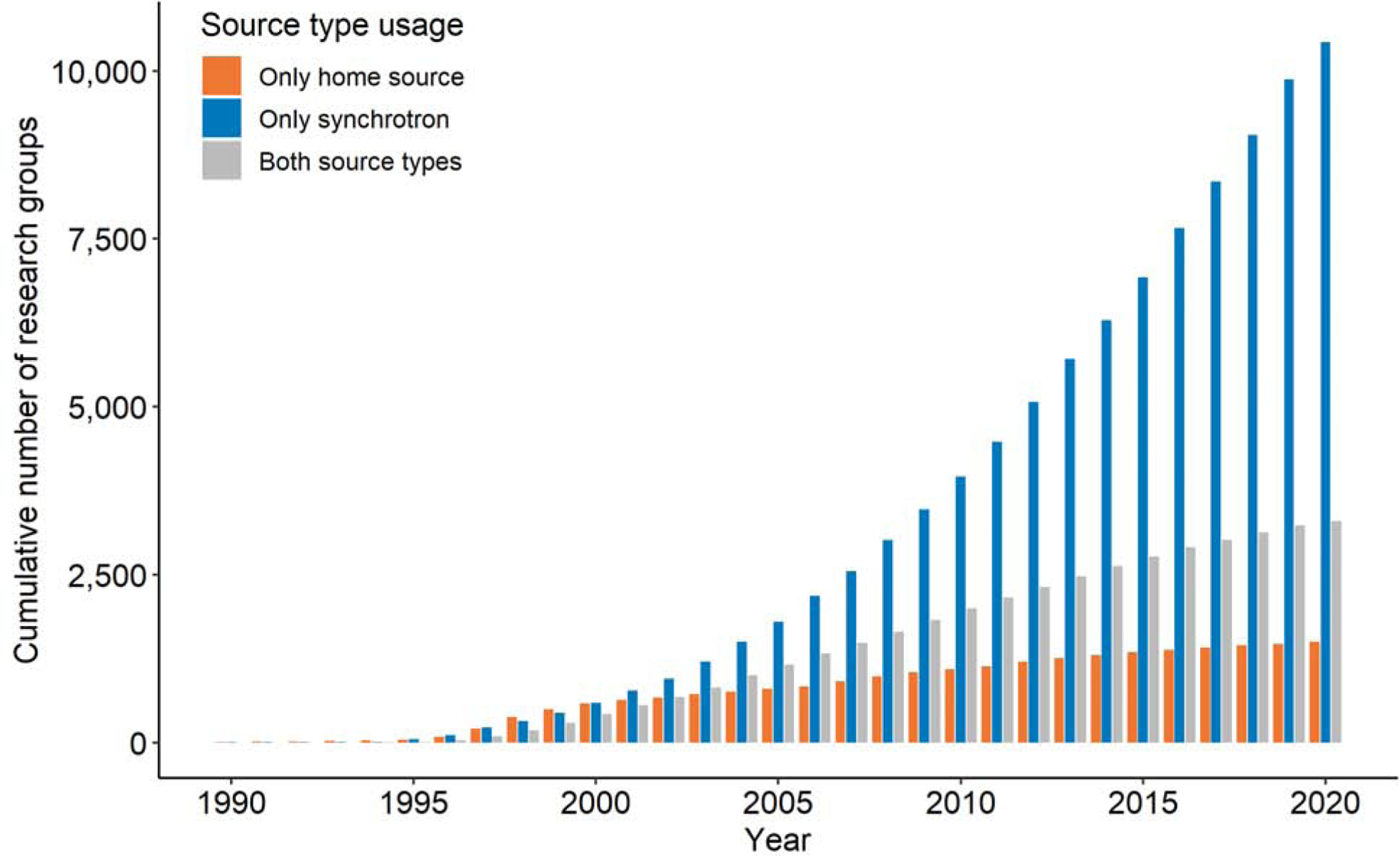
The estimated number of X-ray crystallography research groups having determined at least one structure in the PDB using home sources (orange), synchrotron sources (blue), or using both sources (gray). The number of research groups was estimated based on the last name of the author of the primary citation, or the name of the next to last author if the last one was a Structural Genomics center. In cases where an SG center was the sole author of a primary citation, it was considered as a single research group.
3. From sample to high quality macromolecular structure
3.1. Are we achieving the most from the data collected on synchrotrons?
Even though many synchrotron beamlines are equipped with robots that allow users to collect data remotely and sometimes automatically, it does not mean that this will result in an optimal structure. As Zbyszek Dauter remarked, “it is finally the responsibility of the experimenter, not of the robot, to ensure that the diffraction data are measured optimally. This requires the correct adjustment of a large number of parameters and finding an optimal compromise between several factors.” [34]. The same is also true for the entire process of structure determination. It is possible to gain some insight about how well the data have been collected and then refined, by analyzing quality metrics for the resulting macromolecular structures [35,36].
The quality of macromolecular structures stored in the PDB is a topic that has been repeatedly discussed over the years [30,37–39]. The PDB itself is constantly developing new tools and standards for the assessment of the quality of the structural models deposited in its archives [40]. As a result, there are many complementary ways in which one can measure structure quality. However, most of them correlate with each other [41]; therefore, trends observed using one metric are usually also observed when analyzing other metrics. Here, we analyze trends observed at different synchrotrons using an aggregated measure of overall structure quality (PQ1) that combines five different indicators: Rfree, RSRZ (normalized real-space R-factor) outliers, Ramachandran outliers, rotamer outliers, and Clashscore [39]. Using PQ1, each structure is ranked within the population of all PDB deposits to obtain its final ranking percentile, with the lowest (worst) value at 0% and highest (best) at 100%. The results of this analysis are presented in Fig. 3.
Fig. 3.
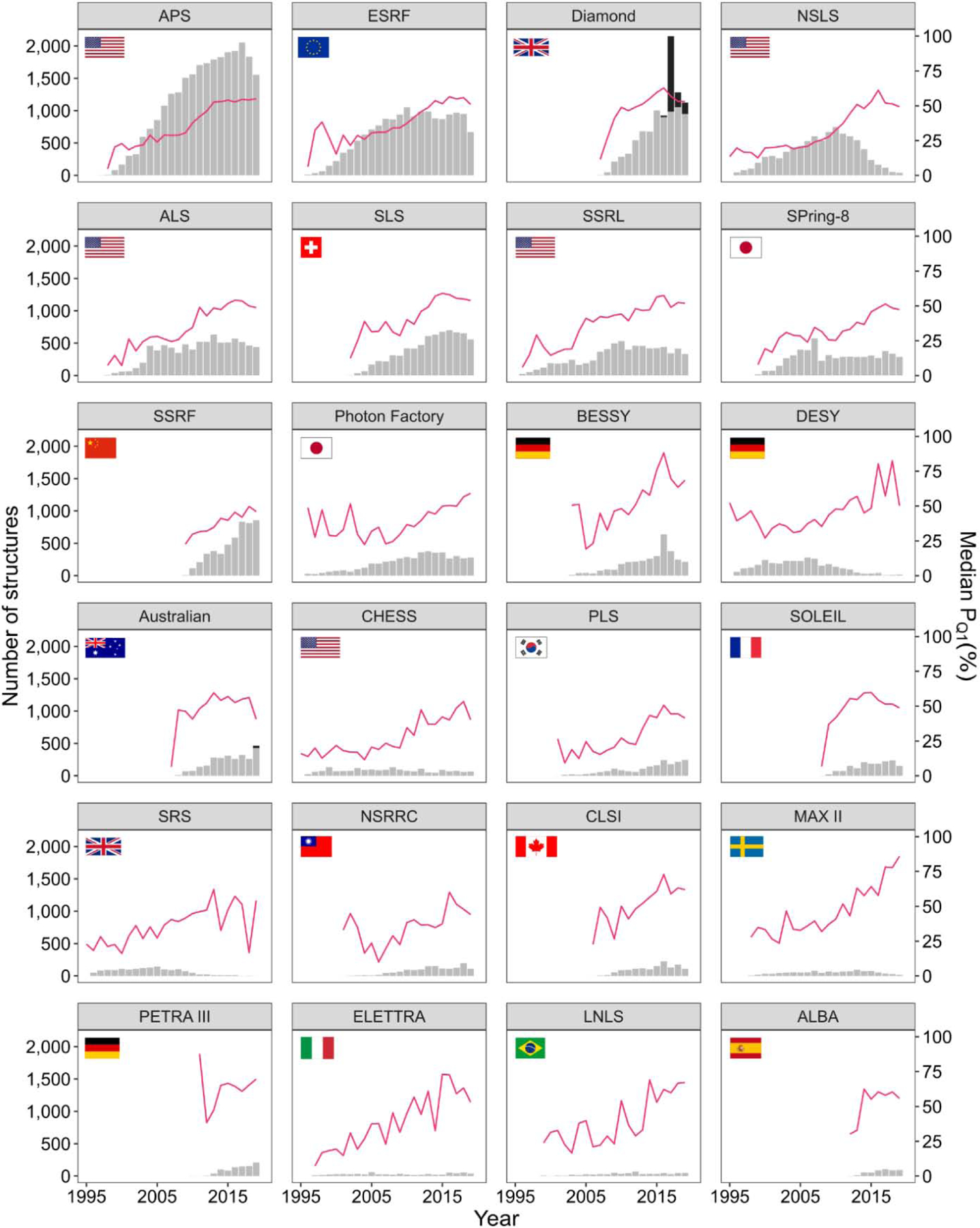
The number of structures (gray bar chart, left y-axis) and median PQ1 overall structure quality (magenta line, right y-axis) for each year (x-axis) at the 24 synchrotrons with the most deposited structures (panels). PanDDA fragment screening deposits represented as black bars.
As shown in Fig. 3, the overall quality of structures has generally improved over the years for all synchrotrons. This trend is correlated with the stronger X-ray sources, better data collection protocols, better detectors and beamline equipment, and better data reduction and refinement software that allows to determine structures even from poor-quality crystals. There has been enormous progress in crystallization methodology and technology [42,43]; however, getting high quality crystals is still extremely difficult for challenging projects, like membrane proteins. Better standards for structure quality were not only the result of better validation tools. They were rather enforced by scientific journals which started requesting validation reports from the PDB to accompany manuscripts reporting new structures, as well as various “at-large” groups that are detecting, correcting, and helping to re-deposit non-optimal structures [44]. There are two approaches to structure optimization: a) an across-the-board automatic optimization using the latest refinement software [45–47] and b) optimization done by groups that are interested in the structures related to a particular disease, a particular biomedical issue, or a particular protein family [48,49].
As of now, the Advanced Photon Source has produced the largest cumulative number of structures and has also been the most productive synchrotron in recent years. The Diamond Light Source doubled its released structures in 2017 and then halved it again the next year. This peak in deposits reflects 1167 Pan-Dataset Density Analysis (PanDDA) fragment screening deposits (black bars on the bar chart in Fig. 3) in 2017. MAX II, currently replaced by MAX IV, seems to be the synchrotron with the best median structure quality in recent years.
3.2. From the data collection to PDB deposition and peer reviewed publication
For structures released by the PDB in 2019, the median time from the date of data collection to the date of deposition release was about two years. This time span was decreasing between 1995 and 2005, but afterwards it started increasing again. The time between deposition and release is typically much shorter but has shown a similar trend (Fig. 4).
Fig. 4.
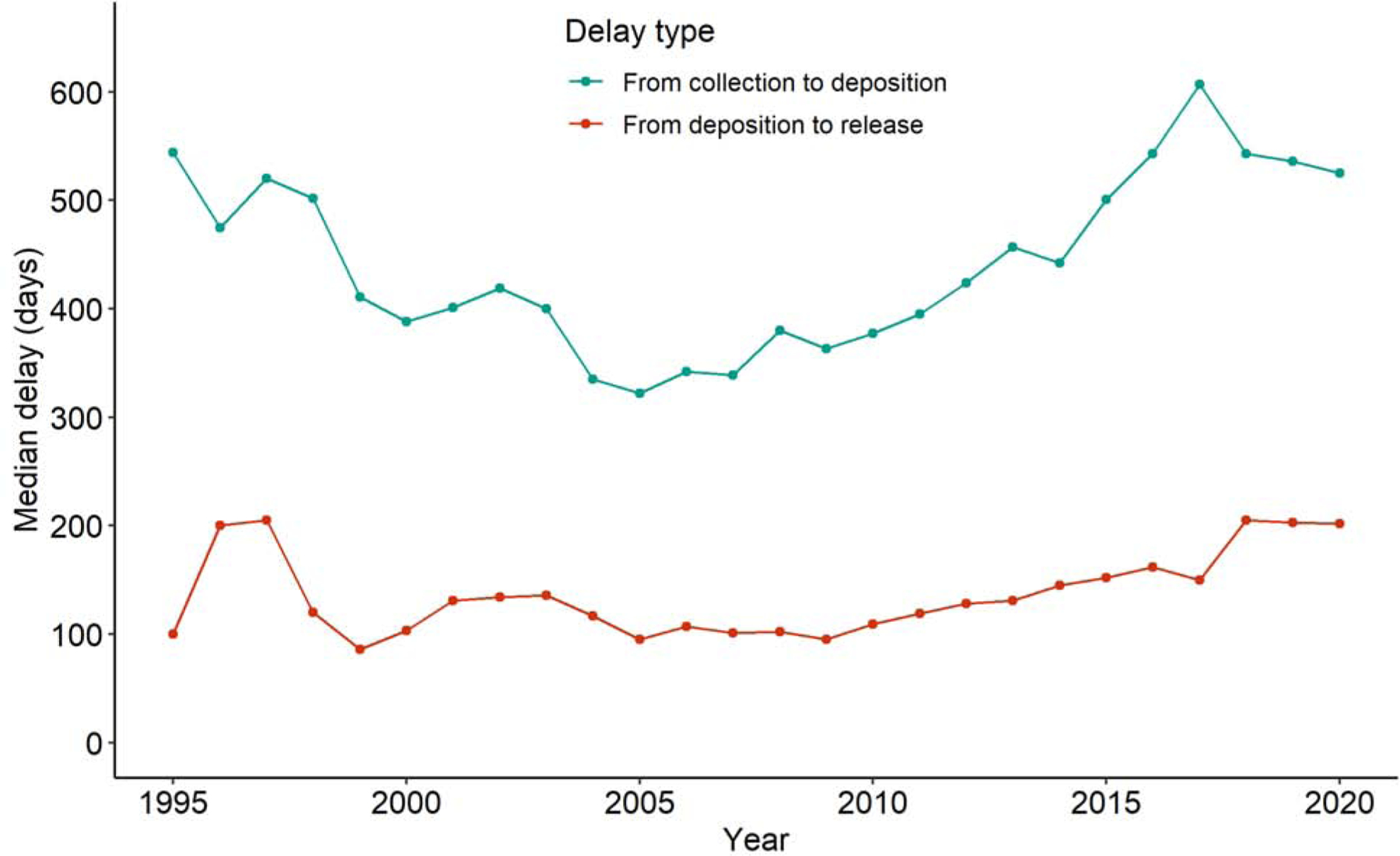
Median time (in days) from data collection to deposition of a structure in the PDB (teal) and from deposition to release (red) for structures reporting using a synchrotron and released in a given year. Deposits that were replacements for structures obsoleted by the PDB and structures which clearly had erroneous data collection dates (e.g. after deposition or before the date the synchrotron started operating) were excluded from the calculation.
3.3. Higher resolution, better quality, and reproducibility of structures are necessary for novel drug discovery. Can we remove bottlenecks?
Despite the tremendous progress in increasing the brightness of synchrotron sources and the sensitivity of detectors, the median resolution of X-ray structures remained roughly the same (Fig. 5). The increased brightness of modern synchrotrons does allow smaller crystals and has undoubtedly decreased the time necessary to collect a dataset. However, the achievable resolution is are primarily limited by the quality (long-range order) of the crystalline sample. The technologies of X-ray detectors have kept pace with improvements in beam brightness, progressing from film to image plates, CCD detectors, and now Pixel Array Detectors (PADs). These advancements may have contributed to the progress in structure quality seen in Fig. 3. At the same time, the improvement of sample preparation, detectors, and software has dramatically increased the achievable resolution of Cryo-EM structures (Fig. 5), which led to a substantial increase in the use of this technique.
Fig. 5.
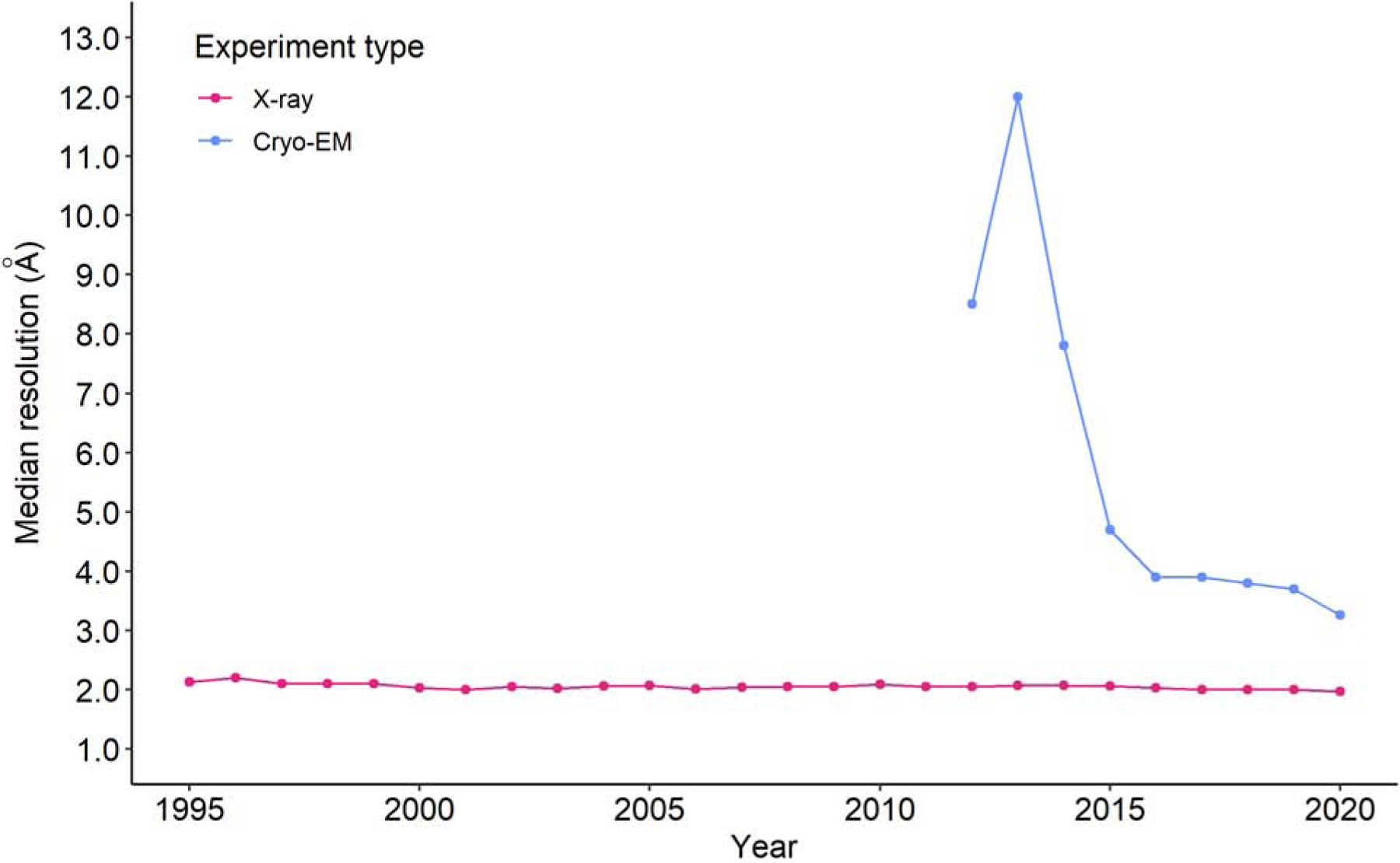
Comparison of median resolutions for macromolecular structures in the PDB determined by X-ray crystallography and electron microscopy as a function of the year of release. Median resolution for a given experimental method has been plotted only for years with at least 30 released structures with a defined resolution.
There are a number of issues related to data collection that have not been fully addressed at synchrotron facilities, and in our opinion, correcting these issues could substantially improve structure quality and indirectly lead to shortening the time between structure determination, PDB deposition, release, and peer reviewed publication.
One of these issues is the lack of standards and enforcement mechanism for the metadata. During the last thirty years, there were many meetings and many discussions about the format of diffraction images and, in particular, about what information should be included in the image header. There was always a general agreement that the “header” (metadata embedded in the image files) should not only have information that would allow any program to reduce (integrate, scale, and merge) diffraction data but should also include all information about the diffraction experiment. The information should be harvested in an automatic or semi-automatic way and should contain all information necessary for a PDB deposit. In principle, these seem to be simple and modest requirements for science of the XXI century, but in practice, there are hundreds of frame formats, some of which are very difficult to handle. An outside observer could easily suspect that scientific programmers proliferate frame formats to provide themselves with job security. One company is known for changing frame formats for almost any new instrument. As a result, authors of data reduction programs may face a situation where two essentially identical instruments have different frame formats. A slight change in the format, e.g., a change of the overload table, may not be noticed by users but can lead to inferior structures.
The situation at some synchrotron floors is similar. Two synchrotron stations that are close to each other sometimes use two slightly different frame formats. The differences are usually small and can be easily accommodated by data reduction programs, but scientists do not always know or remember which beam station their data were collected on. This shows that almost 200 years after Napoleon’s death, his statement that one poor general (format) is better than two excellent ones is still very true, but clearly not understood by detector vendors and facilities personnel.
Another problem is insufficient knowledge or control of some experimental parameters. When researchers are physically present at the beamline, they can monitor parameters that may be inaccessible when collecting data remotely. For example, the temperature at the position of the crystal requires mounting a thermocouple at the position of the crystal. Researchers who collect remotely must assume that the nominal cryo system temperature is correct, but this is not always the case. Slight positional misalignment or an ice buildup in the nozzle can dramatically affect the temperature. Regardless of whether an experimenter is at the beamline, the temperature of data collection seems to be one parameter that is frequently not recorded correctly. An analysis of measurement temperatures reported to the PDB shows that temperatures submitted to the PDB are frequently inaccurate (Fig 6). There are several maxima seen in Fig. 6 that can be explained: 77K, i.e., boiling liquid nitrogen temperature, 100K – the usual temperature, as measured in the nozzle of the cryo-stream, and the vicinity of 295K – the room temperature. However, many other values occurring in the plot appear to have no justification at all. Even reported values of 100K may not correspond to actual conditions as the difference between temperature in a nozzle and temperature of the sample is usually between 5K and 10K as measured in CLSI (Pawel Grochulski, personal communication). What is somewhat shocking is that there are hundreds of papers discussing radiation decay, but many of these studies do not even attempt to measure the temperature in the sample they describe. Recording (and adjusting) real-time crystal temperatures in the beam may offer valuable insights as to how to handle radiation decay. Some synchrotrons, including CLSI and SSRL (Aina Cohen, personal communication), are planning to provide information about sample temperature in the metadata.
Fig. 6.
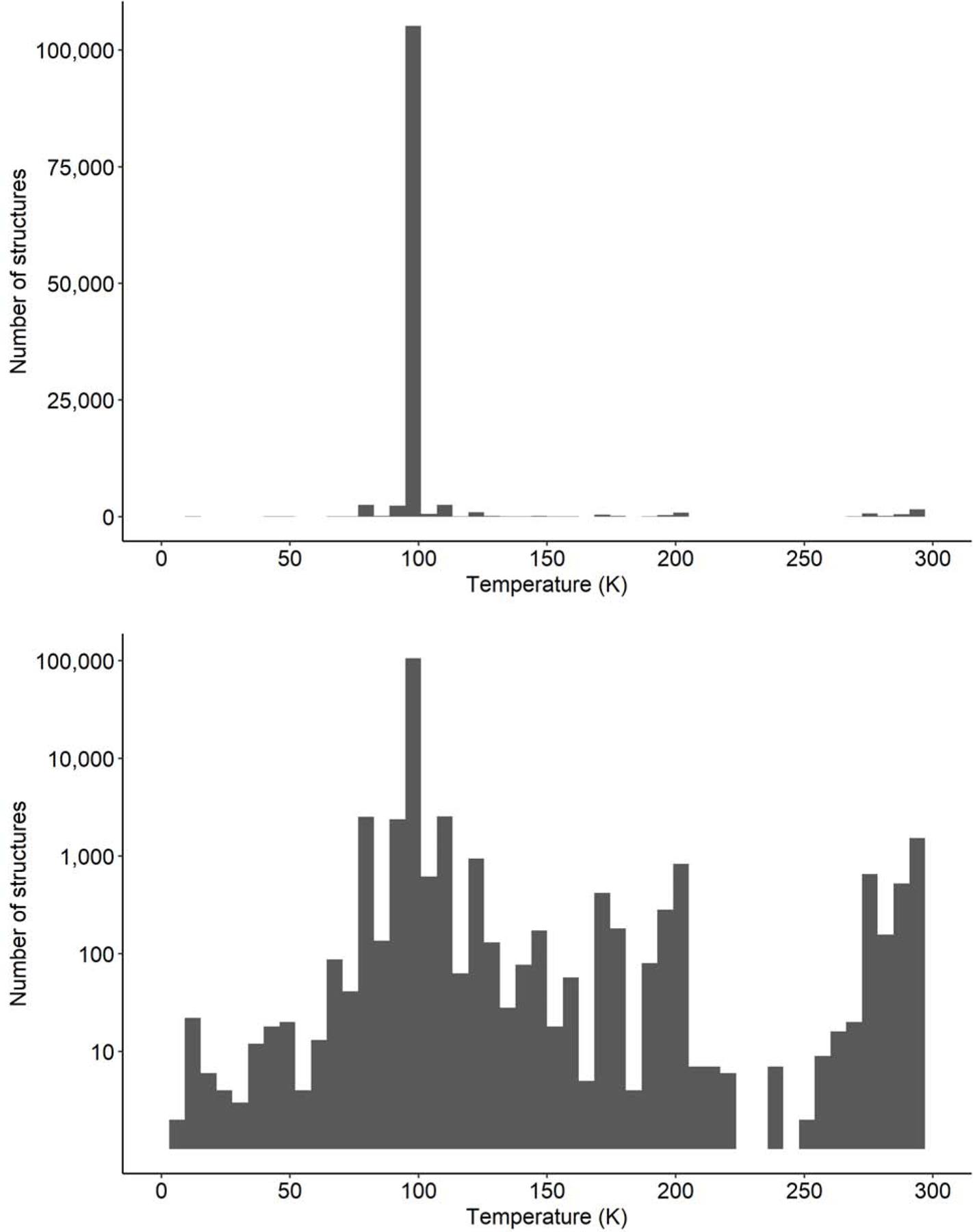
Distribution of the reported temperature used for X-ray diffraction experiments of macromolecular crystals on synchrotron beamlines, as submitted to the PDB: (top) structure counts presented on a linear scale, (bottom) structure counts presented on a log-scale.
There are other side effects of remote data collection. Most groups that perform synchrotron experiments do not have easy access to X-ray data collection systems in the vicinity of their laboratories. Therefore, crystals may be shipped to synchrotrons without checking their quality and optimization of cryo-cooling conditions. The presence of ice rings or other artifacts may affect data reduction as they may overlap with diffraction spots (Fig. 7). One can argue that the effect of ice rings can be eliminated by outlier rejection during scaling, but these types of rejections affect absorption and radiation decay corrections that are based on measured redundancy. Severe artifacts can also affect data completeness. The various pathologies that one can observe on diffraction patterns affect the final quality of electron density maps.
Fig. 7.

An example of a diffraction image showing spots related to the presence of ice.
The spots that are generated by ice or by other contamination of the sample are detrimental for scaling and for structure quality. In the case of structure determination by SAD (single-wavelength anomalous diffraction), various contamination can make structure determination difficult or even impossible.
Remote data collection has become very popular as shipping crystals to the synchrotron is perceived as less time-consuming, much less expensive, and more convenient. However, one should note that expenses related to data collection, including travel, are a very small fraction of the entire cost and effort of structure determination. Moreover, a remote experiment does not have the advantage of “immersion” into the experiment for a few days, which results in better training of younger scientists and better knowledge of experimental details. Being present at the beamline can also speed up the progress of a project because researchers are able to react to feedback immediately. If the first crystals tested indicate that the crystals’ cryoprotectant is not correct, then freezing other crystals using a different procedure could help.
Being able to respond to suboptimal cryo-cooling conditions or instantaneously use feedback from ligand screening while at the synchrotron is one of the primary advantages of going to the synchrotron over remote data collection. The added responsiveness can substantially decrease the entire time a project takes. Of course, responding to feedback from the first few crystals requires either proteins that crystallize extremely fast or traveling to the synchrotron with crystallization plates. Cutting-edge science is not always the most convenient and inexpensive path.
3.4. What is possible and what is beneficial for a particular diffraction experiment?
Technological progress removed many limitations that affected many experiments performed five or ten years ago. The large-size detectors allow collection of high-resolution data without optimizing the sample-detector distance or offsetting the detector center from the position of the direct beam (by shift or 2theta swing). Similarly, the computer speed and storage capacity do not pose a limit from a practical point of view. However, the ability to engage a human brain is still the most serious limitation [34]. The slow speed of processing is often caused by placing the data on NAS (network attached storage) with a suboptimal network configuration. One of the authors sped up processing 100 times at one beamline by properly configuring the network, which unfortunately occurred after the beamline had already spent $100,000 for a super-powerful computer with the aim to improve the speed of processing.
Large detectors with incredibly fast readout times, fast computers/networks, and virtually unlimited storage allowed for the implementation of sophisticated experiments in a reasonable amount of time. However, even the most sophisticated experiment is not necessarily beneficial for a particular project. When one collects data for a SAD experiment, it is important to measure diffraction spots very accurately to 2.5–2.0 Å resolution. In such experiments, one should avoid overloads but, at the same time, collect data that will have reasonable statistics. The strategy for final refinement should be slightly different: one should collect data to the highest resolution possible in order to obtain a high-resolution map. Both experiments can be easily performed, but sometimes experimenters use averaged simple one size fits all protocol to collect data regardless of the experiment goal.
PAD detectors have very short readout times. Moreover, they are readout noise free, which allows for shutterless data collection and eliminates synchronization errors between shutter and spindle axis movement. However, this error is not completely eliminated as read-out time, although very fast, is not infinitely small. Unfortunately, users have a tendency to move the spindle axis quite fast, which leads to a relatively low exposure time for each individual frame. As a result, experimenters frequently collect data with the oscillation range 0.05 degree, which results in 3,600 frames for 180 degrees data. The extreme that the authors encountered was 0.01 degree, resulting in 18,000 frames. There is no proof that an ultra-small angular range is beneficial for data quality. The analysis of data collected at 0.05 degree for an average quality crystal with mosaicity 0.34–0.36 degree gave a somewhat surprising result. We reprocessed these data to simulate different angular widths by summing the frames to effectively create oscillation ranges of 0.25 and 0.5 degrees. When we compare original 0.05 angle (too small) with 0.25 (about right) and 0.5 (too large), we obtained very similar Rmerge, number of rejections, and resolution. The ‘narrow angular range’ protocol may work slightly better for very well-diffracting crystals like lysozyme, but real-life diffraction patterns are much worse than lysozyme, thaumatin or myoglobin diffraction that some experimenters love to use to support their theories. Moreover, data reduction of huge, weak datasets is much more difficult and time consuming.
The errors most relevant to drug discovery are the incorrect identification and modeling of metals and other ligands in protein-ligand complexes. The identification of metals was greatly improved by Check My Metal (CMM) server [50,51] that so far has been used to identify metals in over 80,000 uploaded macromolecular structures submitted by over 4900 scientists. However, a simple experimental technique that can identify the metal beyond any doubt by measuring the anomalous signal above and below absorption edge appears to be seldom used nowadays [52]. The same approach can be used for cases when the ligand contains a metal.
3.5. Is the primary experimental data preserved yet?
X-ray crystallography experiments at synchrotron beamlines generate massive amounts of data that, so far, are not necessarily preserved at synchrotron facilities or in the experimenters’ home laboratories. The International Union of Crystallography (IUCr) recommended that diffraction data used for structure solution should be uploaded to publicly accessible resources when ‘important’ or novel experiments are performed [53]. Several repositories for diffraction images, including the Integrated Resource for Reproducibility in Macromolecular Crystallography [35,54] and SBGRID [55], have been created; however, all these resources account only for a small percentage of diffraction data used in the determination of structures in the PDB, even in recent years. In particular, only a handful of datasets for SARS-CoV-2 structures can be located in these repositories. Our requests for primary data for these structures, during our ongoing work on the resource for validation of structural models related to Covid-19 [56,57], often went unanswered. Very often, the re-examination of structures would benefit from reprocessing the primary diffraction data [48,49].
4. Rapid responses to biomedical threats - SARS-CoV-2 related structures
Synchrotron sources have played a crucial role in the immediate reaction of structural biology to the current COVID-19 pandemic [58]. Within five days after scientists isolated the coronavirus responsible for the outbreak in Wuhan, crystals of the main protease were grown and used for data collection at SSRF. A dataset collected at SSRF on January 12, 2020, was used to solve the structure of the main protease (PDB id: 6lu7) [59]. The second data collection of a COVID-19 protein happened on February 1, 2020, at BESSY. These data were used to determine the structure of a complex arising in the reaction of the main protease with several drug candidates (PDB id: 6y2g, 6y2f, 6y2e) [60].
By the end of February, twenty structures related to SARS-COV-2 were deposited to the PDB. Two of these used “home sources” (PDB id: 6lvn, 6lxt); the others were determined using synchrotron sources; nine in SSRF, six at APS, and three at BESSY.
As of September 23, 2020, 406 SARS-CoV-2 structures had been deposited in the PDB, with three-quarters of them (309) being determined by X-ray crystallography, and the rest – except for a single NMR structure – by Cryo-EM (Fig. 8). Synchrotron sources contributed 289 deposits, one structure was done at the SLAC LLS XFEL facility, and 18 structures used home generators. PanDDA structures collected at the Diamond Light Source accounted for 115 of the synchrotron structures. Excluding the PanDDA deposits, the synchrotrons which contributed the most structures of the virus were APS (74), SSRF (41), and Diamond (12).
Fig. 8.
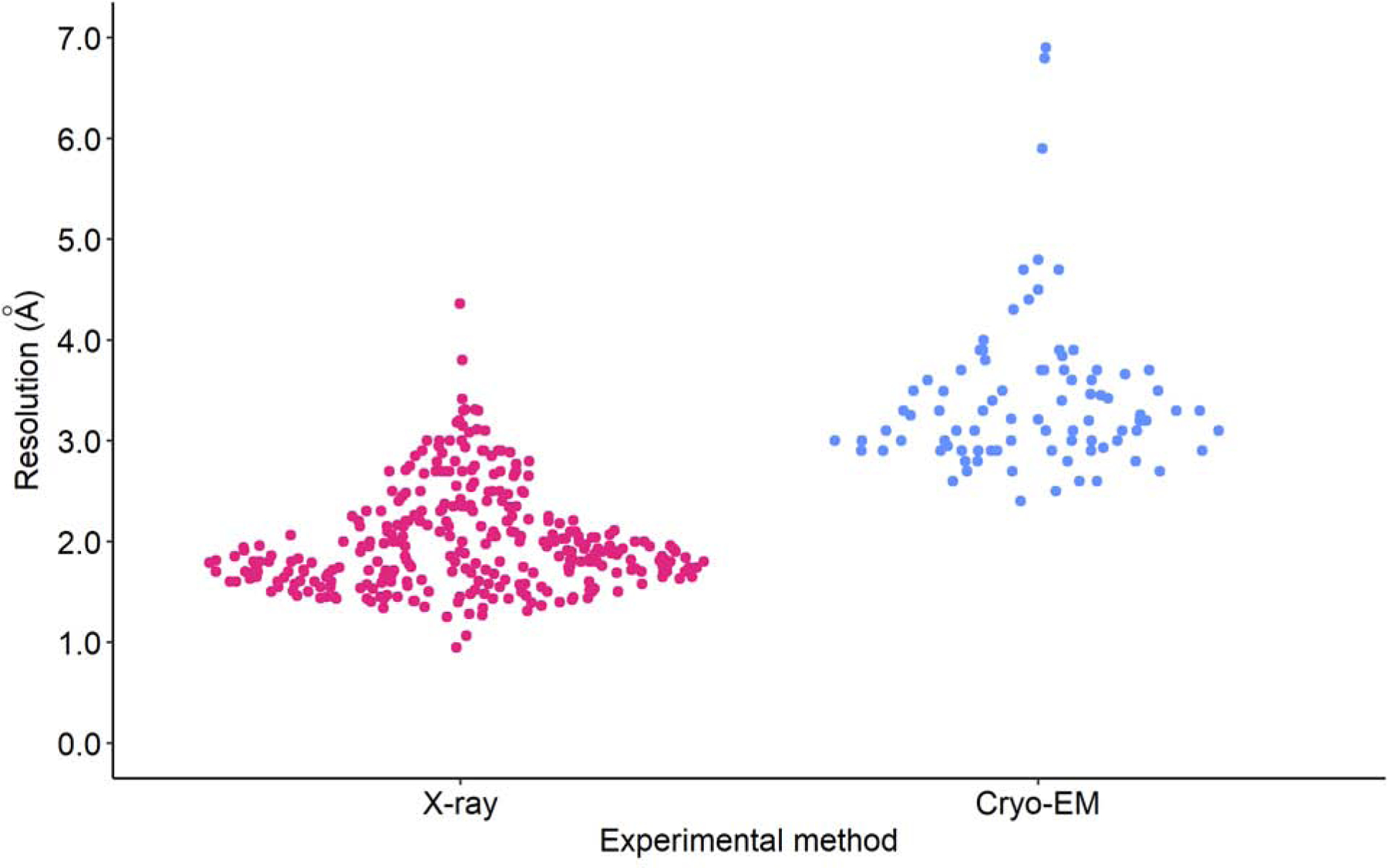
The swarm plot of resolutions of SARS-CoV-2 structures determined using X-ray crystallography and Cryo-EM (excluding PanDDA structures).
5. Where are we heading next
Synchrotrons have made a remarkable contribution to many areas of science, most importantly, to life sciences, drug discovery, physics, chemistry, and material science. Due to these multifaceted applications with evolving technologies and emerging new applications, it is safe to assume that we will soon have more fourth-generation synchrotrons that will serve various areas of science for a long time. Competent and well-working synchrotron beamlines will be critical, especially for life sciences and drug discovery.
There has also been a great progress in ab initio modelling [61], leading to suggestions that it will diminish the role of experimental structure determination. We would argue that it is just the opposite – more robust ab initio modelling will make crystallography more powerful by generating templates for molecular replacement and suggesting ligands for ligand-protein screening. An accurate modelling of ligand binding has been an elusive target for many years, thus making crystallography a go-to method for determining the chemical details of ligand binding. The CASP-14 results presented by a DeepMind team may lead to a revolution in structure determination by MR when the ALPHAFOLD2 server for structure prediction becomes publically available.
We are aware that some scientists believe that the next biomedical “revolution will not be crystallized” and that Cryo-EM will make the use of synchrotrons for the determination of macromolecular structures less relevant [62,63]. However, we do not share their opinion. We believe that more synchrotrons will be transferred into life science centers that will allow their users to perform an array of experiments using X-ray, Cryo-EM, mass spectrometry, NMR, functional studies, etc. The integrative approach, i.e. resource employing scientists that are experts in their respective fields will make such a center an invaluable resource for science of the XXI century.
Cryo-EM is definitely better suited for large proteins (over 1000 kDa) and is maturing at an incredible speed, both in terms of improving resolution and lowering the limit of protein size. The use of Cryo-EM has been rapidly increasing in recent years (Fig. 9). On the other hand, X-ray crystallography is already quite mature and may need new stimuli to make a quantum leap to produce more and higher quality structures. It is impossible to overestimate the role of new integrated resource centers with synchrotrons as a central component. However, synchrotron beamlines should not only purchase the newest multi-million detectors, but also address the issues important for biomedical sciences that are listed in this paper. We hope that this paper will provide a small contribution toward this goal.
Fig. 9.
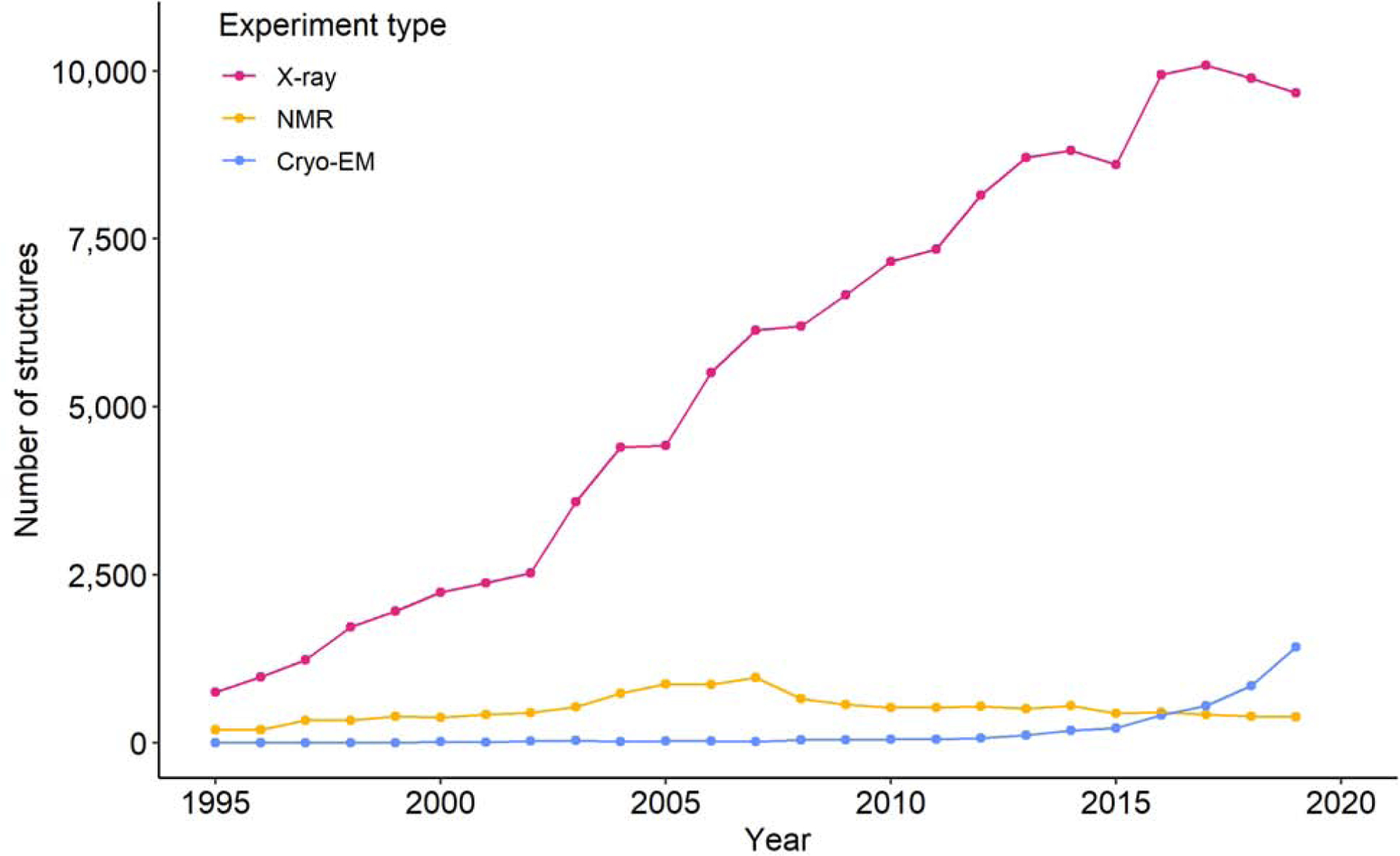
Trends in the growth in the yearly number of X-ray, NMR and Electron Microscopy structures deposited in the PDB between 1995 and 2019 (according to the date of release). Line is for eye-guide only.
However, even as it stands now, crystallography is a critical tool for drug discovery. The evaluations of ligand-protein interaction and removal of uncertainty of ligand identifications requires at least 2.5 Å resolution and so far, only about 100 of Cryo-EM structures report reconstruction resolution 2.5 Å or better. Thus, crystallography remains better suited to determine precise atomic coordinates of macromolecules under a few hundred kDa in size. Also, ligand screening can be done much faster and more reliably with crystallography (if well-diffracting crystals can be reproducibly obtained). Moreover, high-resolution crystal structures play a very important role in drug lead optimization because they can provide very accurate information about the determinants of compound binding, which is crucial for further compound optimization. In addition, the challenges of sample preparation for electron microscopy should not be underestimated: many proteins are reluctant to produce high resolution Cryo-EM structures. Instead of being direct competitors, the two techniques, crystallography and electron microscopy, are often complementary [64,65]. One of the best examples of the importance of crystallographic beamlines for rapid response to emerging threats is the COVID-19 pandemic: the vast majority of structures that contribute to virus understanding and its interactions with the host were determined by X-ray crystallography.
Acknowledgments
We thank Alex Wlodawer, Zbyszek Dauter, Aina Cohen, Chloe Estrada and Chad Brautigam for their critical reading and discussions of the manuscript. This work was supported by the National Institute of General Medical Sciences grants R01-GM132595, R01-GM117080, R01-GM118619 and federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, United States Department of Health and Human Services under contract HHSN272201200026C. DB acknowledges the support of the Polish National Agency for Academic Exchange under Grant No. PPN/BEK/2018/1/00058/U/00001. We also thank Aina Cohen, Andrew Thompson, Pawel Grochulski, Marian Szebenyi, Manfred Weiss and Sarah Lee for providing valuable information about their facilities.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Bibliography
- [1].Blewett JP, Synchrotron radiation — 1873 to 1947, Nucl. Instruments Methods Phys. Res. Sect. A Accel. Spectrometers, Detect. Assoc. Equip 266 (1988) 1–9. 10.1016/0168-9002(88)90349-X. [DOI] [Google Scholar]
- [2].Winick H, Bienenstock A, Synchrotron Radiation Research, Annu. Rev. Nucl. Part. Sci 28 (1978) 33–113. 10.1146/annurev.ns.28.120178.000341. [DOI] [Google Scholar]
- [3].Buras B, Olsen JS, Gerward L, Will G, Hinze E, X-ray energy-dispersive diffractometry using synchrotron radiation, J. Appl. Crystallogr 10 (1977) 431–438. 10.1107/s0021889877013910. [DOI] [Google Scholar]
- [4].Minor W, Schönfeld B, Lebech B, Buras B, Dmowski W, Crystallization of Fe-Si-B metallic glasses studied by X-ray synchrotron radiation, J. Mater. Sci 22 (1987) 4144–4152. 10.1007/BF01133371. [DOI] [Google Scholar]
- [5].Phillips JC, Wlodawer A, Yevitz MM, Hodgson KO, Applications of synchrotron radiation to protein crystallography: preliminary results., Proc. Natl. Acad. Sci. U. S. A 73 (1976) 128–32. 10.1073/pnas.73.1.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Skarzynski T, Collecting data in the home laboratory: evolution of X-ray sources, detectors and working practices., Acta Crystallogr. D. Biol. Crystallogr 69 (2013) 1283–8. 10.1107/S0907444913013619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Berman HM, Battistuz T, Bhat TN, Bluhm WF, Bourne PE, Burkhardt K, Feng Z, Gilliland GL, Iype L, Jain S, Fagan P, Marvin J, Padilla D, Ravichandran V, Schneider B, Thanki N, Weissig H, Westbrook JD, Zardecki C, The protein data bank, Acta Crystallogr. Sect. D Biol. Crystallogr 58 (2002) 899–907. 10.1107/S0907444902003451. [DOI] [PubMed] [Google Scholar]
- [8].Bartunik HD, Summers LJ, Bartsch HH, Crystal structure of bovine beta-trypsin at 1.5 A resolution in a crystal form with low molecular packing density. Active site geometry, ion pairs and solvent structure., J. Mol. Biol 210 (1989) 813–28. 10.1016/0022-2836(89)90110-1. [DOI] [PubMed] [Google Scholar]
- [9].Fermi G, Perutz MF, Shaanan B, Fourme R, The crystal structure of human deoxyhaemoglobin at 1.74 Å resolution, J. Mol. Biol 175 (1984) 159–174. 10.1016/0022-2836(84)90472-8. [DOI] [PubMed] [Google Scholar]
- [10].Ealick SE, Editorial, Synchrotron Radiat. News. 9 (1996) 2–3. 10.1080/08940889608602910. [DOI] [Google Scholar]
- [11].Wareham JD, Pujol Priego L, Nordberg M, Garcia Tello P, Systematising Serendipity for Big Science Infrastructures: the ATTRACT Project, SSRN Electron. J (2019). 10.2139/ssrn.3355674. [DOI]
- [12].Dauter Z, Jaskolski M, Wlodawer A, Impact of synchrotron radiation on macromolecular crystallography: A personal view, J. Synchrotron Radiat 17 (2010) 433–444. 10.1107/S0909049510011611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Helliwell JR, The evolution of synchrotron radiation and the growth of its importance in crystallography, Crystallogr. Rev 18 (2012) 33–93. 10.1080/0889311X.2011.631919. [DOI] [Google Scholar]
- [14].Helliwell JR, Mitchell EP, Synchrotron radiation macromolecular crystallography: science and spin-offs, IUCrJ 2 (2015) 283–291. 10.1107/S205225251402795X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Thompson A, Attwood D, Gullikson E, Howells M, Kim K-J, Kirz J, Kortright J, Lindau I, Liu Y, Pianetta P, Robinson A, Scofield J, Underwood J, Williams G, Winick H, Center for X-Ray Optics and Advanced Light Source X-Rray Data Booklet, 2009.
- [16].Hendrix J, The EMBL-Hamburg imaging plate scanner (abstract), Rev. Sci. Instrum 63 (1992) 641–641. 10.1063/1.1142677. [DOI] [Google Scholar]
- [17].Walter RL, Thiel DJ, Barna SL, Tate MW, Wall ME, Eikenberry EF, Gruner SM, Ealick SE, High-resolution macromolecular structure determination using CCD detectors and synchrotron radiation., Structure 3 (1995) 835–44. 10.1016/s0969-2126(01)00218-0. [DOI] [PubMed] [Google Scholar]
- [18].Winick H, Synchrotron Radiation Sources – Present Capabilities and Future Directions, J. Synchrotron Radiat 5 (1998) 168–175. 10.1107/S0909049597018761. [DOI] [PubMed] [Google Scholar]
- [19].Castelvecchi D, Next-generation X-ray source fires up, Nature 525 (2015) 15–16. 10.1038/nature.2015.18253. [DOI] [PubMed] [Google Scholar]
- [20].Voss G-A, Rabedeau T, Raither S, Schopper H, Weihreter E, Winick H, SESAME: an extended spectral range synchrotron radiation facility in the Middle East based on an upgrade to BESSY I, Nucl. Instruments Methods Phys. Res. Sect. A Accel. Spectrometers, Detect. Assoc. Equip 467–468 (2001) 55–58. 10.1016/S0168-9002(01)00215-7. [DOI] [Google Scholar]
- [21].Wawrzyniak AI, Marendziak A, Kisiel A, Borowiec P, Nietubyć R, Wiechecki J, Karaś K, Szamota-Leandersson K, Zając M, Bocchetta CJ, Stankiewicz MJ, Solaris a new class of low energy and high brightness light source, Nucl. Instruments Methods Phys. Res. Sect. B Beam Interact. with Mater. Atoms (2017). 10.1016/j.nimb.2016.12.046. [DOI]
- [22].Wood L, Chapline G, Towards gamma-ray lasers, Nature 252 (1974) 447–450. 10.1038/252447a0. [DOI] [Google Scholar]
- [23].Chapline G, Wood L, X-ray lasers, Phys. Today 28 (1975) 40–48. 10.1063/1.3069004. [DOI] [Google Scholar]
- [24].Emma P, Akre R, Arthur J, Bionta R, Bostedt C, Bozek J, Brachmann A, Bucksbaum P, Coffee R, Decker F-J, Ding Y, Dowell D, Edstrom S, Fisher A, Frisch J, Gilevich S, Hastings J, Hays G, Hering P, Huang Z, Iverson R, Loos H, Messerschmidt M, Miahnahri A, Moeller S, Nuhn H-D, Pile G, Ratner D, Rzepiela J, Schultz D, Smith T, Stefan P, Tompkins H, Turner J, Welch J, White W, Wu J, Yocky G, Galayda J, First lasing and operation of an ångstrom-wavelength free-electron laser, Nat. Photonics 4 (2010) 641–647. 10.1038/nphoton.2010.176. [DOI] [Google Scholar]
- [25].Chapman HN, Fromme P, Barty A, White TA, Kirian RA, Aquila A, Hunter MS, Schulz J, DePonte DP, Weierstall U, Doak RB, Maia FRNC, Martin AV, Schlichting I, Lomb L, Coppola N, Shoeman RL, Epp SW, Hartmann R, Rolles D, Rudenko A, Foucar L, Kimmel N, Weidenspointner G, Holl P, Liang M, Barthelmess M, Caleman C, Boutet S, Bogan MJ, Krzywinski J, Bostedt C, Bajt S, Gumprecht L, Rudek B, Erk B, Schmidt C, Hömke A, Reich C, Pietschner D, Strüder L, Hauser G, Gorke H, Ullrich J, Herrmann S, Schaller G, Schopper F, Soltau H, Kühnel K-U, Messerschmidt M, Bozek JD, Hau-Riege SP, Frank M, Hampton CY, Sierra RG, Starodub D, Williams GJ, Hajdu J, Timneanu N, Seibert MM, Andreasson J, Rocker A, Jönsson O, Svenda M, Stern S, Nass K, Andritschke R, Schröter C-D, Krasniqi F, Bott M, Schmidt KE, Wang X, Grotjohann I, Holton JM, Barends TRM, Neutze R, Marchesini S, Fromme R, Schorb S, Rupp D, Adolph M, Gorkhover T, Andersson I, Hirsemann H, Potdevin G, Graafsma H, Nilsson B, Spence JCH, Femtosecond X-ray protein nanocrystallography., Nature 470 (2011) 73–7. 10.1038/nature09750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Fukuda Y, Tse KM, Nakane T, Nakatsu T, Suzuki M, Sugahara M, Inoue S, Masuda T, Yumoto F, Matsugaki N, Nango E, Tono K, Joti Y, Kameshima T, Song C, Hatsui T, Yabashi M, Nureki O, Murphy MEPP, Inoue T, Iwata S, Mizohata E, Iwatae S, Mizohata E, Redox-coupled proton transfer mechanism in nitrite reductase revealed by femtosecond crystallography., Proc. Natl. Acad. Sci. U. S. A 113 (2016) 2928–33. 10.1073/pnas.1517770113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Liu H, Lee W, The XFEL Protein Crystallography: Developments and Perspectives., Int. J. Mol. Sci 20 (2019). 10.3390/ijms20143421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].METALJET - Single Crystal X-ray Diffraction, X-ray Source - Sources - Single Crystal X-ray Diffraction | Bruker, (n.d). https://www.bruker.com/products/x-ray-diffraction-and-elemental-analysis/single-crystal-x-ray-diffraction/sc-xrd-components/sc-xrd-components/overview/sc-xrd-components/sources/metaljet.html (accessed September 26, 2020).
- [29].Grabowski M, Niedzialkowska E, Zimmerman MD, Minor W, The impact of structural genomics: the first quindecennial., J. Struct. Funct. Genomics 17 (2016) 1–16. 10.1007/s10969-016-9201-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Brown EN, Ramaswamy S, Quality of protein crystal structures., Acta Crystallogr. D. Biol. Crystallogr 63 (2007) 941–50. 10.1107/S0907444907033847. [DOI] [PubMed] [Google Scholar]
- [31].Stacy R, Begley DW, Phan I, Staker BL, Van Voorhis WC, Varani G, Buchko GW, Stewart LJ, Myler PJ, Structural genomics of infectious disease drug targets: the SSGCID., Acta Crystallogr. Sect. F. Struct. Biol. Cryst. Commun 67 (2011) 979–84. 10.1107/S1744309111029204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Myler PJ, Stacy R, Stewart L, Staker BL, Van Voorhis WC, Varani G, Buchko GW, The Seattle Structural Genomics Center for Infectious Disease (SSGCID)., Infect. Disord. Drug Targets 9 (2009) 493–506. 10.2174/187152609789105687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Anderson WF, Structural genomics and drug discovery for infectious diseases., Infect. Disord. Drug Targets 9 (2009) 507–17. 10.2174/187152609789105713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Dauter Z, Carrying out an optimal experiment., Acta Crystallogr. D. Biol. Crystallogr 66 (2010) 389–92. 10.1107/S0907444909038578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Grabowski M, Langner KM, Cymborowski M, Porebski PJ, Sroka P, Zheng H, Cooper DR, Zimmerman MD, Elsliger M-A, Burley SK, Minor W, A public database of macromolecular diffraction experiments, Acta Crystallogr. Sect. D Struct. Biol 72 (2016) 1181–1193. 10.1107/S2059798316014716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Shabalin I, Porebski P, Minor W, Refining the macromolecular model - achieving the best agreement with the data from X-ray diffraction experiment., Crystallogr. Rev 24 (2018) 236–262. 10.1080/0889311X.2018.1521805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Read RJ, Kleywegt GJ, Case-controlled structure validation., Acta Crystallogr. D. Biol. Crystallogr 65 (2009) 140–7. 10.1107/S0907444908041085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Pozharski E, Weichenberger CX, Rupp B, Techniques, tools and best practices for ligand electron-density analysis and results from their application to deposited crystal structures., Acta Crystallogr. D. Biol. Crystallogr 69 (2013) 150–67. 10.1107/S0907444912044423. [DOI] [PubMed] [Google Scholar]
- [39].Brzezinski D, Dauter Z, Minor W, Jaskolski M, On the evolution of the quality of macromolecular models in the PDB., FEBS J 287 (2020) 2685–2698. 10.1111/febs.15314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Gore S, Sanz García E, Hendrickx PMS, Gutmanas A, Westbrook JD, Yang H, Feng Z, Baskaran K, Berrisford JM, Hudson BP, Ikegawa Y, Kobayashi N, Lawson CL, Mading S, Mak L, Mukhopadhyay A, Oldfield TJ, Patwardhan A, Peisach E, Sahni G, Sekharan MR, Sen S, Shao C, Smart OS, Ulrich EL, Yamashita R, Quesada M, Young JY, Nakamura H, Markley JL, Berman HM, Burley SK, Velankar S, Kleywegt GJ, Validation of Structures in the Protein Data Bank., Structure 25 (2017) 1916–1927. 10.1016/j.str.2017.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Shao C, Yang H, Westbrook JD, Young JY, Zardecki C, Burley SK, Multivariate Analyses of Quality Metrics for Crystal Structures in the PDB Archive., Structure 25 (2017) 458–468. 10.1016/j.str.2017.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Derewenda ZS, Vekilov PG, Entropy and surface engineering in protein crystallization, in: Acta Crystallogr. Sect. D Biol. Crystallogr, 2006. 10.1107/S0907444905035237. [DOI] [PubMed]
- [43].Luft JR, Newman J, Snell EH, Crystallization screening: The influence of history on current practice, Acta Crystallogr. Sect. FStructural Biol. Commun (2014). 10.1107/S2053230X1401262X. [DOI] [PMC free article] [PubMed]
- [44].Wlodawer A, Dauter Z, Porebski PJ, Minor W, Stanfield R, Jaskolski M, Pozharski E, Weichenberger CX, Rupp B, Detect, correct, retract: How to manage incorrect structural models, FEBS J 285 (2018) 444–466. 10.1111/febs.14320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Joosten RP, Salzemann J, Bloch V, Stockinger H, Berglund A-C, Blanchet C, Bongcam-Rudloff E, Combet C, Da Costa AL, Deleage G, Diarena M, Fabbretti R, Fettahi G, Flegel V, Gisel A, Kasam V, Kervinen T, Korpelainen E, Mattila K, Pagni M, Reichstadt M, Breton V, Tickle IJ, Vriend G, PDB_REDO: automated re-refinement of X-ray structure models in the PDB, J. Appl. Crystallogr 42 (2009) 376–384. 10.1107/S0021889809008784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Joosten RP, Long F, Murshudov GN, Perrakis A, The PDB_REDO server for macromolecular structure model optimization, IUCrJ 1 (2014) 213–220. 10.1107/S2052252514009324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Joosten RP, Joosten K, Murshudov GN, Perrakis A, PDB_REDO: constructive validation, more than just looking for errors., Acta Crystallogr. D. Biol. Crystallogr 68 (2012) 484–96. 10.1107/S0907444911054515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Raczynska JE, Shabalin IG, Minor W, Wlodawer A, Jaskolski M, A close look onto structural models and primary ligands of metallo-β-lactamases., Drug Resist. Updat 40 (2018) 1–12. 10.1016/j.drup.2018.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Shabalin I, Dauter Z, Jaskolski M, Minor W, Wlodawer A, Crystallography and chemistry should always go together: a cautionary tale of protein complexes with cisplatin and carboplatin., Acta Crystallogr. D. Biol. Crystallogr 71 (2015) 1965–79. 10.1107/S139900471500629X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Zheng H, Cooper DR, Porebski PJ, Shabalin IG, Handing KB, Minor W, CheckMyMetal: a macromolecular metal-binding validation tool., Acta Crystallogr. Sect. D, Struct. Biol 73 (2017) 223–233. 10.1107/S2059798317001061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Zheng H, Chordia MD, Cooper DR, Chruszcz M, Müller P, Sheldrick GM, Minor W, Validation of metal-binding sites in macromolecular structures with the CheckMyMetal web server, Nat. Protoc 9 (2014) 156–170. 10.1038/nprot.2013.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Handing KB, Niedzialkowska E, Shabalin IG, Kuhn ML, Zheng H, Minor W, Characterizing metal-binding sites in proteins with X-ray crystallography, Nat. Protoc (2018). 10.1038/nprot.2018.018. [DOI] [PMC free article] [PubMed]
- [53].Helliwell JR, Minor W, Weiss MS, Garman EF, Read RJ, Newman J, van Raaij MJ, Hajdu J, Baker EN, Findable Accessible Interoperable Re-usable (FAIR) diffraction data are coming to protein crystallography., IUCrJ 6 (2019) 341–343. 10.1107/S2052252519005918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Grabowski M, Cymborowski M, Porebski PJ, Osinski T, Shabalin IG, Cooper DR, Minor W, The Integrated Resource for Reproducibility in Macromolecular Crystallography: Experiences of the first four years, Struct. Dyn 6 (2019) 064301 10.1063/1.5128672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Meyer PA, Socias S, Key J, Ransey E, Tjon EC, Buschiazzo A, Lei M, Botka C, Withrow J, Neau D, Rajashankar K, Anderson KS, Baxter RH, Blacklow SC, Boggon TJ, Bonvin AMJJ, Borek D, Brett TJ, Caflisch A, Chang C-I, Chazin WJ, Corbett KD, Cosgrove MS, Crosson S, Dhe-Paganon S, Di Cera E, Drennan CL, Eck MJ, Eichman BF, Fan QR, Ferré-D’Amaré AR, Christopher Fromme J, Garcia KC, Gaudet R, Gong P, Harrison SC, Heldwein EE, Jia Z, Keenan RJ, Kruse AC, Kvansakul M, McLellan JS, Modis Y, Nam Y, Otwinowski Z, Pai EF, Pereira PJB, Petosa C, Raman CS, Rapoport TA, Roll-Mecak A, Rosen MK, Rudenko G, Schlessinger J, Schwartz TU, Shamoo Y, Sondermann H, Tao YJ, Tolia NH, Tsodikov OV, Westover KD, Wu H, Foster I, Fraser JS, Maia FRNC, Gonen T, Kirchhausen T, Diederichs K, Crosas M, Sliz P, Data publication with the structural biology data grid supports live analysis, Nat. Commun 7 (2016) 10882 10.1038/ncomms10882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Wlodawer A, Dauter Z, Shabalin IG, Gilski M, Brzezinski D, Kowiel M, Minor W, Rupp B, Jaskolski M, Ligand-centered assessment of SARS-CoV-2 drug target models in the Protein Data Bank., FEBS J 26 (2020) 32–39. 10.1111/febs.15366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Brzezinski D, Kowiel M, Cooper DR, Cymborowski M, Grabowski M, Wlodawer A, Dauter Z, Shabalin IG, Gilski M, Rupp B, Jaskolski M, Minor W, Covid-19.bioreproducibility.org: A web resource for SARS-CoV-2-related structural models., Protein Sci (2020) pro.3959 10.1002/pro.3959. [DOI] [PMC free article] [PubMed]
- [58].Baker EN, Visualizing an unseen enemy; mobilizing structural biology to counter COVID-19, IUCrJ 7 (2020) 366–367. 10.1107/S2052252520004571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Jin Z, Du X, Xu Y, Deng Y, Liu M, Zhao Y, Zhang B, Li X, Zhang L, Peng C, Duan Y, Yu J, Wang L, Yang K, Liu F, Jiang R, Yang X, You T, Liu X, Yang X, Bai F, Liu H, Liu X, Guddat LW, Xu W, Xiao G, Qin C, Shi Z, Jiang H, Rao Z, Yang H, Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors., Nature 582 (2020) 289–293. 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- [60].Zhang L, Lin D, Sun X, Curth U, Drosten C, Sauerhering L, Becker S, Rox K, Hilgenfeld R, Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors., Science 368 (2020) 409–412. 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Abbass J, Nebel J-C, Rosetta and the journey to predict proteins’ structures, 20 years on, Curr. Bioinform 15 (2020). 10.2174/1574893615999200504103643. [DOI] [Google Scholar]
- [62].Ramakrishnan V, The ribosome emerges from a black box., Cell 159 (2014) 979–984. 10.1016/j.cell.2014.10.052. [DOI] [PubMed] [Google Scholar]
- [63].Callaway E, The revolution will not be crystallized: a new method sweeps through structural biology, Nature 525 (2015) 172–174. 10.1038/525172a. [DOI] [PubMed] [Google Scholar]
- [64].Wang HW, Wang JW, How cryo-electron microscopy and X-ray crystallography complement each other, Protein Sci 26 (2017) 32–39. 10.1002/pro.3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Shoemaker SC, Ando N, X-rays in the Cryo-Electron Microscopy Era: Structural Biology’s Dynamic Future., Biochemistry 57 (2018) 277–285. 10.1021/acs.biochem.7b01031. [DOI] [PMC free article] [PubMed] [Google Scholar]