

Abstract
The incorporation of nonacetate starter units during type II polyketide biosynthesis helps diversify natural products. Currently, there are few enzymatic strategies for the incorporation of nonacetate starter units in type II polyketide synthase (PKS) pathways. Here we report the crystal structure of AuaEII, the anthranilate:CoA ligase responsible for the generation of anthraniloyl-CoA, which is used as a starter unit by a type II PKS in aurachin biosynthesis. We present structural and protein sequence comparisons to other aryl:CoA ligases. We also compare the AuaEII crystal structure to a model of a CoA ligase homologue, AuaE, which is present in the same gene cluster. AuaE is predicted to have the same fold as AuaEII, but instead of CoA ligation, AuaE catalyzes acyl transfer of anthranilate from anthraniloyl-CoA to the acyl carrier protein (ACP). Together, this work provides insight into the molecular basis for starter unit selection of anthranilate in type II PKS biosynthesis.
Graphical Abstract
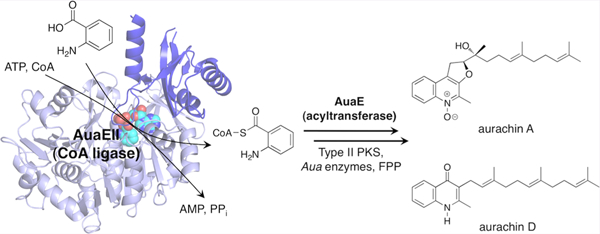
Type II polyketide biosynthesis typically begins by loading an acetyl starter unit onto the terminal thiol of 4′-phosphopantetheine (pPant) linked to an acyl carrier protein (ACP), which is subsequently transferred to the active site cysteine of a ketosynthase (KS) and then undergoes iterative elongation using malonyl-CoA as a building block.1 In some cases, different (nonacetyl) starter units are employed in polyketide biosynthesis, thus yielding more structurally diverse natural products.2 For example, priming KSs use malonyl-CoA to catalyze the extension of an acyl-CoA derived starter unit. The growing polyketide is then transferred to the ketosynthase active site cysteine and further extended by the complex consisting of ketosynthase/chain length factor (KS/CLF).3,4 Nonacetate starter units have been observed in the daunorubicin (propionyl-CoA) and R1128 (propionyl-CoA and isobutyryl-CoA) biosynthetic pathways.4,5 Further, other strategies for diversified starter unit incorporation have been discovered. For example, a bifunctional decarboxylase/acyltransferase (AT) LomC was identified in the lomaiviticin gene cluster and found to decarboxylate methylmalonyl-CoA to form propionyl-CoA, which is then loaded onto the ACP and incorporated into lomaiviticin biosynthesis.6 Additionally, the enterocin pathway harbors the enzyme EncN, which uses ATP to activate and transfer benzoic acid onto the ACP that is then elongated by KS/CLF.7 The incorporation of nonacetate starter units is important for the generation of structural diversity in polyketide biosynthesis. Therefore, there is a need to understand the molecular and mechanistic basis for the enzymatic reactions responsible for nonacetate starter unit selection.
This work extends our current understanding of novel starter unit incorporation by a comprehensive analysis of the crystal structure of the anthranilate:CoA ligase AuaEII, which is involved in anthranilate priming during aurachin biosynthesis. Aurachins are a class of quinoline alkaloids produced by Stigmatella aurantiaca Sg a15 that inhibit the electron transport chain.8,9 A gene cluster responsible for aurachin biosynthesis was previously identified and sequenced.10 Two CoA ligase homologues, AuaEII and AuaE, were found to be responsible for a unique anthranilate priming mechanism that generates and incorporates the anthranilate starter unit into aurachin biosynthesis (Figure 1).11 In vitro experiments revealed that AuaEII is an anthranilate:CoA ligase and AuaE is an anthraniloyl-CoA:ACP acyltransferase.11 Surprisingly, both AuaEII and AuaE are members of the CoA ligase family based on protein sequence analysis, but AuaE only functions as an AT and has no CoA ligase activity. Our current work establishes the structural basis for substrate specificity of AuaEII and provides multiple hypotheses for the acyltransferase mechanism of AuaE.
Figure 1.
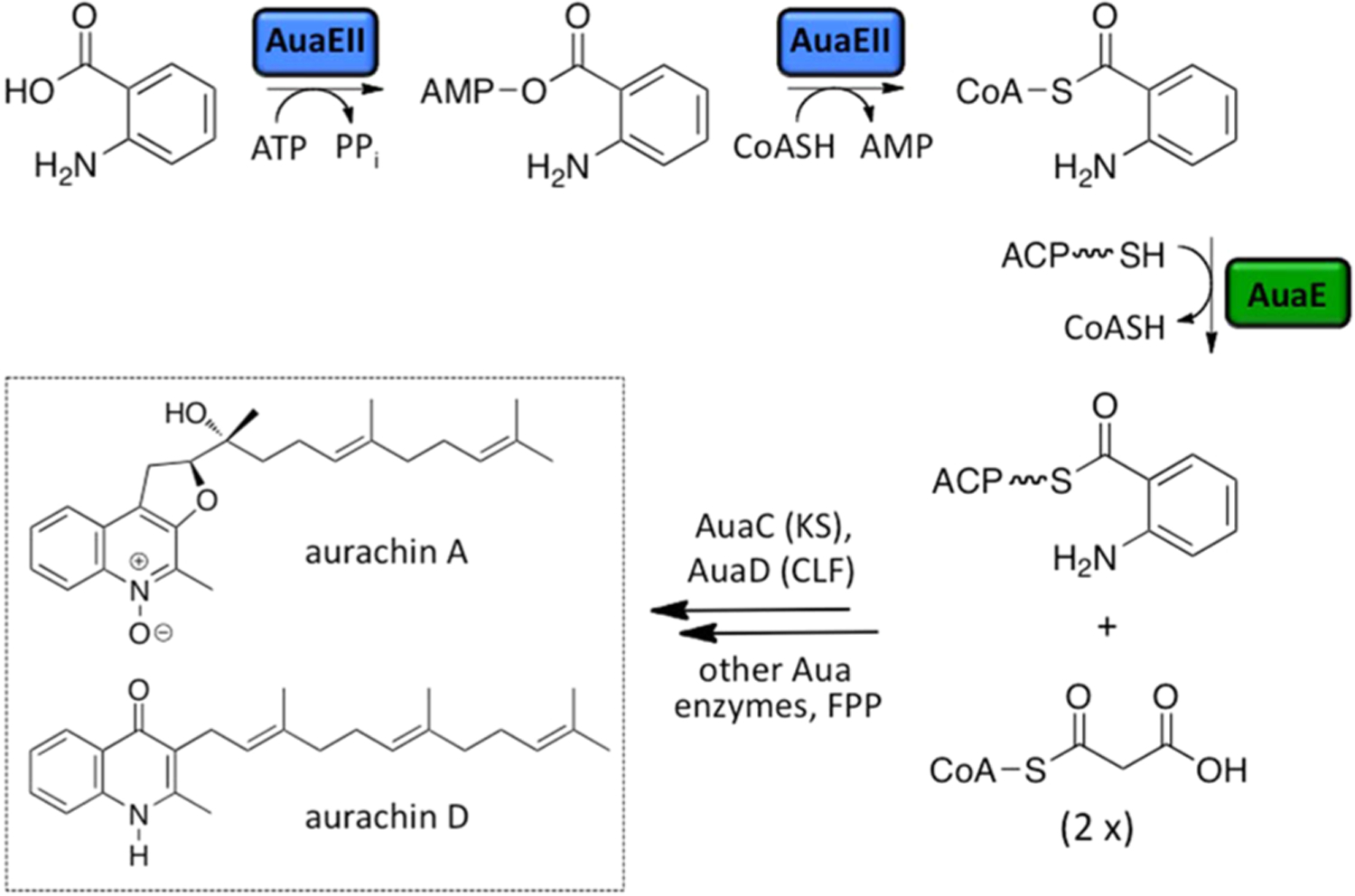
Roles of AuaEII and AuaE in aurachin biosynthesis. Aurachin biosynthesis begins with the activation of anthranilic acid into a CoA ester that is catalyzed by AuaEII. AuaEII first activates anthranilic acid using ATP to form anthraniloyl-AMP, which is susceptible to nucleophilic attack by CoA to yield anthraniloyl-CoA. AuaE uses anthraniloyl-CoA as a substrate and transfers the anthraniloyl group onto ACP (AuaB) that is then elongated by a type II PKS (AuaB, AuaC, and AuaD). Subsequent elongation and cyclization yields a quinoline core, which is modified by farnesyl group. Additional oxidation and rearrangement reactions yield a variety of aurachin natural products (only aurachin A and aurachin D are pictured).
CoA ligases are members of the adenylate-forming enzymes and catalyze the transformation of a carboxylic acid substrate into an acyl-CoA using ATP, Mg2+, and CoA.12 These adenylate-forming enzymes are composed of three classes: type I (the adenylation domains of nonribosomal peptide synthetases (NRPS), acyl- and aryl-CoA synthetases, and oxidoreductases), type II (aminoacyl-tRNA synthetases), and type III (NRPS independent synthetases).12 AuaEII is an aryl:CoA ligase and belongs to the type I ligase class. Other related aryl:CoA ligases include benzoate:CoA ligase, coumarate:CoA ligase, and 4-chlorobenzoate:CoA ligase13–15 (Supporting Information (SI) Table 1). AuaE, due to its functional divergence to serve as an acyltransferase, cannot be classified by any of the above categories.11 How AuaEII and AuaE can have such different functions remains a mystery.
Here, we describe the crystal structure of AuaEII and a comprehensive comparison of AuaE and AuaEII. The AuaEII structure agrees with the previously described structural characteristics of aryl:CoA ligases, thus giving us an excellent opportunity to systematically evaluate the molecular basis of substrate specificity of the CoA ligases. Further, the AuaEII structure represents the first crystal structure of a dedicated anthranilate:CoA ligase. Below, we present the structural analysis of AuaEII in comparison to other aryl:CoA ligase crystal structures and an AuaE homology model. These comparisons reveal the molecular details of substrate specificity of the aryl:CoA ligases and provide insight into this novel type II PKS priming mechanism during aurachin biosynthesis.
RESULTS AND DISCUSSION
The Structure of AuaEII from S. aurantiaca Sg a15.
AuaEII crystallized in the space group P212121 with two AuaEII molecules per asymmetric unit. The AuaEII dimer is composed of two monomers that are related by a 2-fold rotational axis of symmetry. The protein–protein interface between monomers is not extensive and is largely mediated by solvent accessible hydrophilic contacts (Figure 2A). Each monomer contains a large N-terminal and small C-terminal domain, which is in accordance with other structurally characterized CoA ligases.16 The N-terminal domain is responsible for substrate specificity, and the flexible C-terminal domain adopts different conformations, depending on the reaction that the enzyme catalyzes: adenylation or CoA acylation.17 The N-terminal domain has been described as a combination of a β-barrel and a five-layered α–β–α–β–α structure, where two β-sheets are flanked by a series of short α-helices.16 The C-terminal domain sits on top of the N-terminal domain and consists of a core of three antiparallel β-sheets surrounded by three α-helices.18
Figure 2.
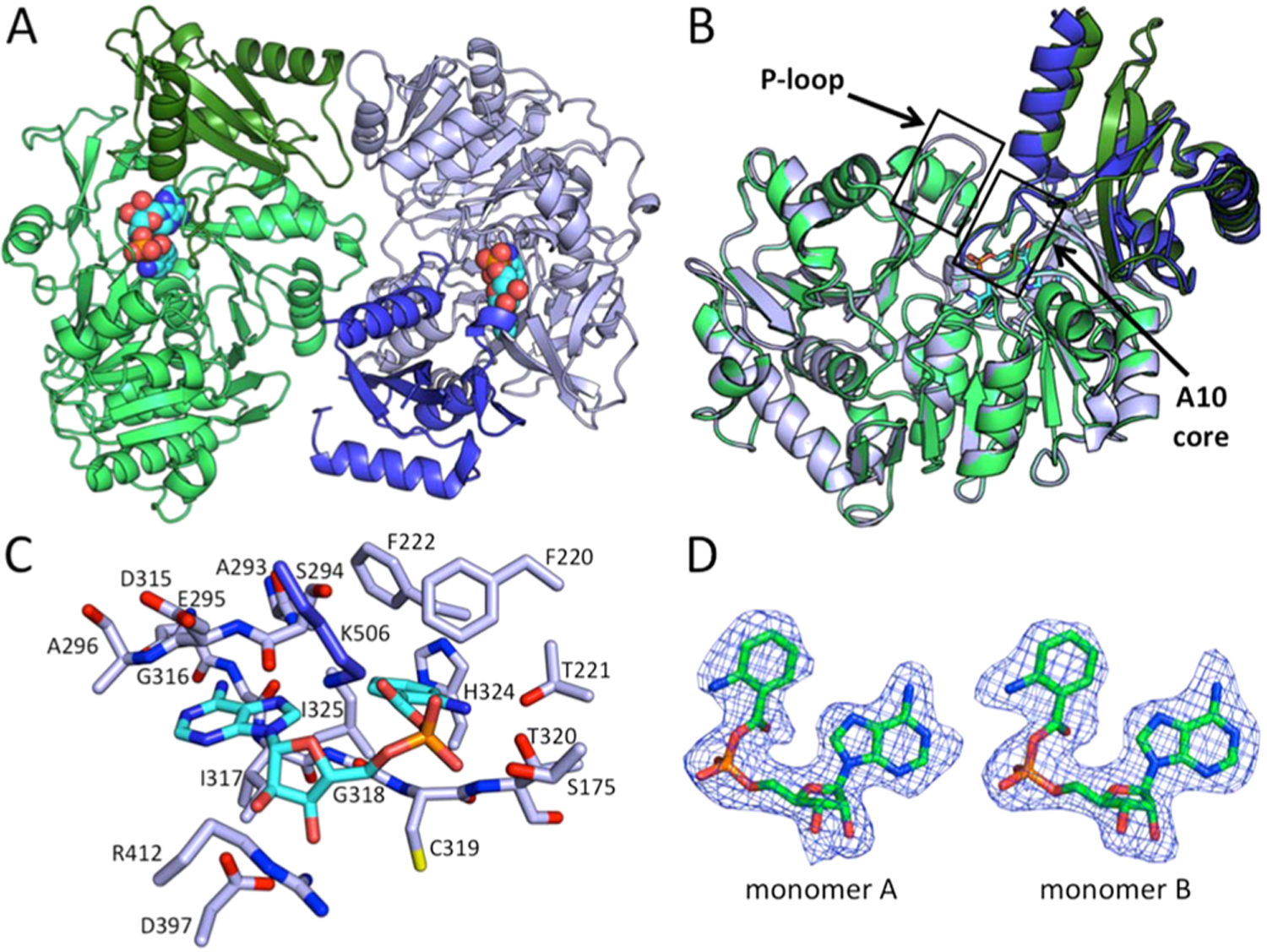
Crystal structure of AuaEII bound to anthraniloyl-AMP. (A) The crystal structure of AuaEII is composed of two monomers per asymmetric unit that are related by noncrystallographic symmetry. The N-terminal domains of monomers A and B are colored light green and light blue, respectively, and the C-terminal domains of monomers A and B are colored deeper green and blue, respectively. Anthraniloyl-AMP is represented in spheres and lies between the N- and C-terminal domains of both monomers. (B) AuaEII monomers A and B overlay well with minor structural differences. Conserved regions of the CoA ligase family (P-loop and A10 core) are labeled. The P-loop of AuaEII monomer A could not be completely modeled in the crystal structure because of weak electron density. (C) The anthraniloyl-AMP intermediate is locked into place by a series of interactions with both the protein side chains, as well as backbone amide nitrogens and carbonyl oxygens. The adenine ring is bound between two regions via the protein backbone. The ribose ring forms hydrogen bonds with Lys506, Arg412, and Asp397. The aryl-binding region is hydrophobic except for Thr221, which forms hydrogen bonds with the 2-amino group of the anthraniloyl moiety. (D) The SA Fo-Fc omit map (contoured at 1 σ) of anthraniloyl-AMP intermediates in each monomer shows well-defined electron density for the ligands.
In AuaEII, the structures of both monomers are highly similar to an overall RMSD of 0.217 Å. The largest structural deviations occur in two α-helices in the C-terminal domain (Figure 2B). The N-terminal domain consists of 12 α-helices and 19 β-strands connected by 28 loops. The core structure of the N-terminal domain contains a series of short central α-helices, which are flanked on both sides by β-sheets. The β-sheets on both sides are further surrounded by α-helices, which define the solvent accessible surface of the N-terminal domain. The N-terminal domain also contains a pseudo-β-barrel structure formed by three antiparallel two-stranded β-sheets, which is situated directly below the C-terminal domain.
The C-terminal domain is structurally distinct from the N-terminal domain. Starting with a short β-hairpin followed three antiparallel β-strands surrounded by three α-helicies, the C-terminal domain also contains a long, highly conserved loop, termed the A10 core, which leads into the final C-terminal α-helix. Anthraniloyl-AMP is located in the active site between the N- and C-terminal domains, and all atoms of both ligands were modeled with well-defined electron density (Figure 2D). In summary, the crystal structure of AuaEII matches well with other members of the aryl:CoA ligase family, and the flexible C-terminal domains of both monomers have elevated B-factors relative to the large N-terminal domains (Figure S3).
The C-Terminal Conformation of AuaEII.
CoA ligases typically crystallize with the C-terminal domain in conformation 1 or conformation 2. Conformation 1 has the A10 core facing the active site of the N-terminal domain, and this conformation facilitates the adenylation reaction.19 Conformation 2 represents the thioester forming conformation. In conformation 2, the C-terminal domain is rotated ∼140° relative to conformation 1, resulting in the movement of the A10 core motif away from the N-terminal active site.17 In the AuaEII crystal structure, the C-terminal domain is in conformation 1, and the conserved lysine Lys506 of the A10 core interacts with the phosphate of the anthraniloyl-AMP intermediate (Figure 2C). The charge–charge interaction between Lys506 and the anthraniloyl-AMP phosphate may be important for stabilizing conformation 1 of AuaEII and promoting crystallization.
Multiple aryl:CoA ligase crystal structures (benzoate:CoA ligase, 4-chlorobenzoate:CoA ligase, 4-coumarate:CoA ligase, and phenylacetyl:CoA ligase) have been solved with an aryl-AMP intermediate bound in the N-terminal pocket, yet the C-terminal domains of these structures do not all adopt the same conformation.13,18,20,21 It has been hypothesized that in different CoA ligase enzymes, there is an equilibrium between conformations 1 and 2, and this equilibrium may be different for different CoA ligases during crystallization.17 In the AuaEII crystal structure, the C-terminal domains of both monomers have elevated B-factors for the majority of residues except in the A10 core region, where Lys506 interacts with the anthraniloyl-AMP intermediate (Figure S3). In summary, based on our structure, anthraniloyl-AMP is located in the active site of AuaEII, and the C-terminal domain is in conformation 1 with Lys506 of the A10 core interacting with the phosphate of anthraniloyl-AMP.
Anthraniloyl-AMP Interactions in the AuaEII Active Site.
The AuaEII active site, similar to those of other aryl:CoA ligases, lies in a cleft between the N- and C-terminal domains. The anthraniloyl portion of the anthraniloyl-AMP intermediate is bound in an internal pocket of the N-terminal domain, whereas the AMP portion interacts with both the N- and C-terminal domains (Figure 2). The adenine portion of the anthraniloyl-AMP is situated between the backbone amides of Ser294, Glu295, Ala296 from the top, and Ile317 on the bottom and forms multiple hydrogen bonds with other residues. The adenine C6 amine forms hydrogen bonds to the carboxylate of Asp315 and the backbone carbonyl group of Gly316. The N1 of the adenine ring also forms a hydrogen bond to a solvent-accessible water molecule. The ribose portion of the anthraniloyl-AMP intermediate interacts with two residues with opposing charges, Lys506 and Asp397. Lys506 is located on the highly conserved A10 core of the C-terminal domain and forms hydrogen bonds with the 5′ hydroxyl and oxygen in the ribose ring. The Asp397 carboxylate forms tight hydrogen bonds with the 2′ and 3′ hydroxyls of the ribose ring, and Arg412 is also in hydrogen bond distance to the 3′ hydroxyl. The phosphate forms weak hydrogen bonds with the amide hydrogen of Thr320 and the hydroxyl of Ser175. Additionally, three solvent linked water molecules form hydrogen bonds to the phosphate. The anthraniloyl aromatic ring is located under the end of an α-helix where three consecutive residues (Phe220, Thr221, and Phe222) interact with the anthraniloyl ring. First, Phe222 has hydrophobic interactions with the edge of the anthraniloyl group and defines the bottom of the substrate pocket. Second, Thr221 forms a hydrogen bond with the aromatic amine of anthraniloyl that locks the intermediate into the observed orientation. Third, Phe220 has hydrophobic interactions with the top of the anthraniloyl aromatic ring, and the edge of the Phe220 aromatic ring is perpendicular to the carbonyl group of the anthraniloyl ester. The back edge of the anthraniloyl aromatic ring is bound in a pocket defined by His324, Ile325, and Ala293. His324 is engaged in face-on-edge π–π interactions with the anthraniloyl aromatic ring from one side, while the adenine ring of the anthraniloyl-AMP intermediate sits edge to edge with the anthraniloyl aromatic ring. The bottom of the anthraniloyl aromatic ring undergoes π–π stacking with the amide bond between Gly318 and Cys319 (Figure 2C). In summary, the anthraniloyl-AMP intermediate sits between the N-terminal and C-terminal domains and reveals residues in the N-terminal domain that are important for substrate specificity.
A Comparison between the Aryl Binding Regions of AuaEII and AuaE.
The selectivity of aryl:CoA ligases is determined entirely by residues in the aryl binding region of the N-terminal domain. The aryl-binding region is where the carboxylic acid substrate binds. This region contains specific residues that interact with the aryl substituent. To date, many CoA ligase crystal structures have been solved that are bound with an acid substrate or aryl-adenylate intermediate, and these structures reveal residues that are important protein–substrate interactions.13,17,18,21 An in depth comparison of the AuaEII crystal structure to the energy minimized AuaE homology model and six previously determined aryl:CoA ligase crystal structures reveal unprecedented insights into aryl substrate binding (Figure 3; Table 1). The aryl-binding region of AuaEII consists of histidine (His324), isoleucine (Ile325), alanine (Ala293), and phenyalanine (Phe222). In the case of AuaEII, a threonine (Thr221) is located near the aryl binding region and forms a hydrogen bond with the 2-amino group of the anthranilate moiety of the bound anthraniloyl-AMP. Therefore, the aryl binding region of AuaEII is most similar to benzoate CoA-ligases except for the residue Thr221, which is important for anthranilate specificity.
Figure 3.
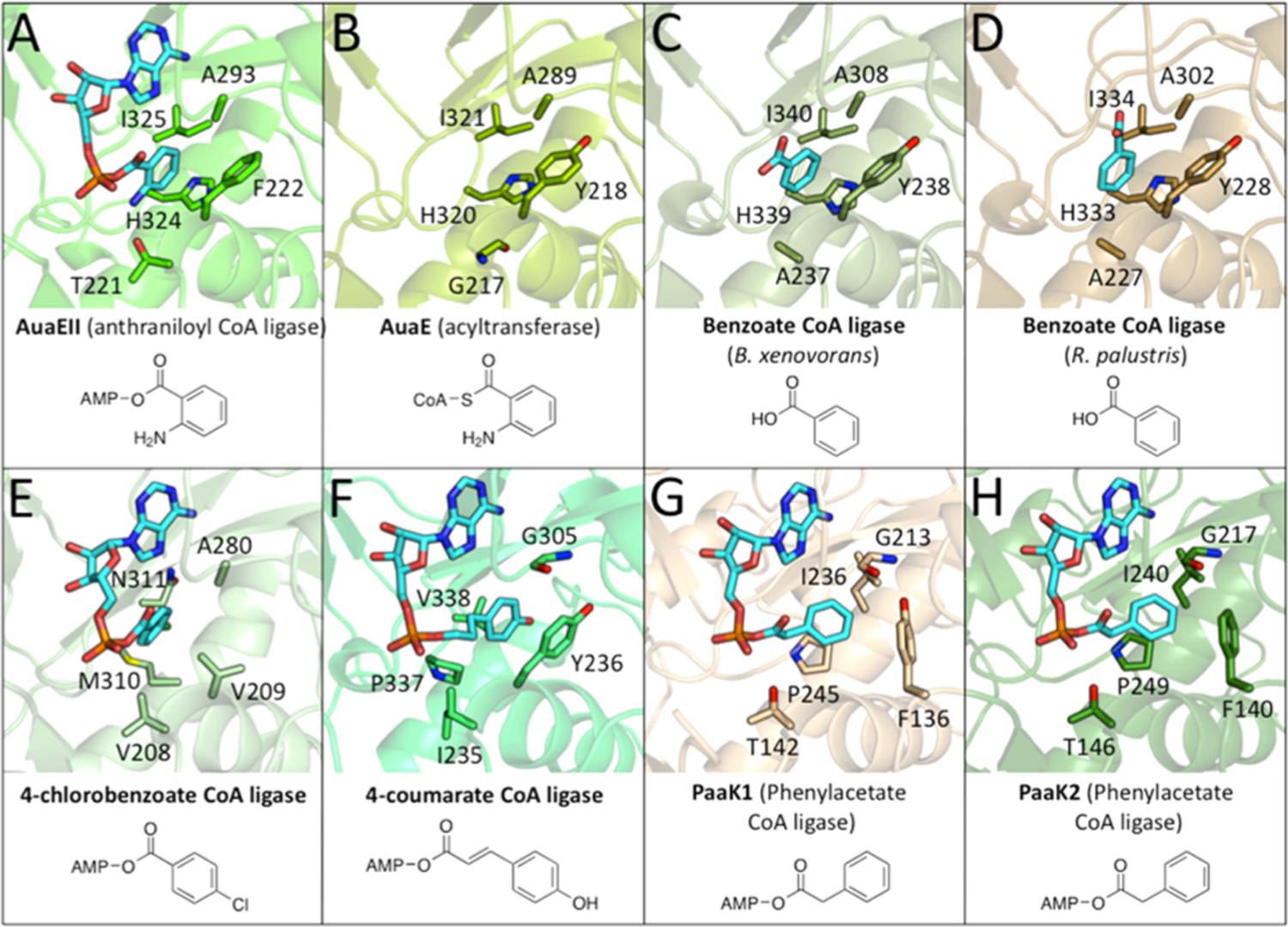
Structural comparison of the active sites of the AuaEII crystal structure and AuaE homology model with other aryl-CoA ligases. (A) AuaEII (anthraniloyl CoA ligase) bound to anthraniloyl-AMP. (B) AuaE homology model (no ligand). (C) Benzoate CoA ligase (B. xenovorans) bound to benzoate. (D) Benzoate CoA ligase (R. palustris) bound to benzoate. (E) 4-chlorobenzoate ligase bound to 4-chlorobenzoyl-AMP. (F) 4-coumarate CoA ligase bound to 4-coumaroyl-AMP. (G) PaaK1 (phenylacetate CoA ligase) bound to phenyacetyl-AMP. (H) PaaK2 (phenylacetate CoA ligase) bound to phenyacetyl-AMP.
Table 1.
N-Terminal Domain of 6 Aryl CoA Ligases and the AuaE Homology Model Aligned to the N-Terminal Domain of AuaEII for a Comparison of the Aryl Binding Region in Order to Gain Insight into Substrate Specificity of These Aryl:CoA Ligasesa
| enzyme | PDB ID | compound present crystal structure | RMSD (Å) compared to AuaEII | % identity to AuaEII | aryl binding region | 2-amino binding region | |||
|---|---|---|---|---|---|---|---|---|---|
| AuaEII (anthranilate:CoA ligase) | 4WV3 | anthraniloyl-AMP | 100.00 | H324 | I325 | A293 | F222 | T221 | |
| AuaE (acyltransferase) | 0.785 | 28.40 | H320 | I321 | A289 | Y218 | G217 | ||
| benzoate:CoA ligase (B. xenovorans) | 2V7B | benzoate | 0.773 | 40.00 | H339 | I340 | A308 | Y238 | A237 |
| benzoate:CoA ligase (R. palustris) | 4EAT | benzoate | 0.795 | 40.82 | H333 | I334 | A302 | Y228 | A227 |
| 4-chlorobenzoate:CoA ligase | 3CW8 | 4-chlorobenzoyl-AMP | 1.862 | 21.21 | M310 | N311 | A280 | V209 | V208 |
| 4-coumarate:CoA ligase | 3NI2 | 4-coumaroyl-AMP | 1.542 | 24.51 | P337 | V338 | G305 | Y236 | I235 |
| PaaK1 (phenylacetate:CoA ligase) | 2Y4N | phenylacetyl-AMP | 2.332 | 18.08 | P245 | I236 | G213 | Y136 | T142 |
| PaaK2 (phenylacetate:CoA ligase) | 2Y4O | phenylacetyl-AMP | 2.514 | 18.22 | P249 | I240 | G217 | F140 | T146 |
The overall % identity, residues in the aryl binding region, and 2-amino binding region are listed for comparison with AuaEII and AuaE.
AuaE has an overall low sequence identity of 28.4% with AuaEII. The aryl binding site of the AuaE homology model consists of histidine (His320), isoleucine (Ile321), alanine (Ala289), and tyrosine (Tyr218). The aryl binding region of AuaE is very similar to that of AuaEII. The only difference is that AuaE has a tyrosine (Tyr218) in place of phenylalanine (Phe222) of AuaEII. Moreover, AuaE has a glycine (Gly217) at the position corresponding to Thr221 of AuaEII. Thr221 of AuaEII is responsible for forming a hydrogen bond with the anthraniloyl 2-amino group. Therefore, it is surprising that the AuaE model would have glycine at this position, because the aryl binding region of both enzymes is proposed to process the 2-amino bearing anthraniloyl moiety. The small size of glycine would not exclude anthraniloyl-CoA binding, but it would lack a hydrogen bond donating/accepting side chain present in threonine. It is possible that the anthraniloyl group of AuaE’s substrate, anthraniloyl-CoA, binds in a different orientation. Alternatively, the cavity of the aryl-binding region alone may be sufficient for productive binding of the aromatic substrate, and additional hydrogen bonding to the 2-amino moiety is not necessary.
The Observation of Low Sequence Homology among Aryl:CoA Ligases.
Among aryl:CoA ligases with very similar substrates (benzoate, 4-chlorobenzoate, 4-coumarate, phenylacetate, and anthranilate), the overall sequence identity can be very low (Table 1; Figure S1). For example, the substrate differences of AuaEII and 4-chlorobenzoate-CoA ligase are minor (2-amino vs 4-chloro substituent), yet the two enzymes only share 21.2% of sequence identity. CoA ligases are very common in primary metabolism, and diversification of substrate specificity is easily achievable by a small number of mutations in the N-terminal substrate-binding region. Therefore, it is surprising that low sequence identity is observed for different CoA ligases acting on similar substrates. This is especially intriguing in the case of AuaE and AuaEII, because AuaE only has an overall sequence identity of 28.4% with AuaEII, despite both being present in the S. aurantiaca Sg a15 genome and their joint roles in the same biosynthetic pathway. These observations suggest that AuaE did not arise from duplication of AuaEII (or vice versa). The situation is quite different in the active site. Even though the overall sequence identity is only 28.4% between AuaE and AuaEII, residues in the aryl binding region that directly interact with the substrates (based on the AuaEII crystal structure and AuaE homology model) are highly conserved (His324/His320, Ile325/Ile321, Ala293/Ala289, Phe222/Tyr218, for AuaEII/AuaE, respectively). AuaE and AuaEII are most closely related to benzoate CoA ligases (Table 1). Therefore, both enzymes could have independently evolved from different benzoate (or other aryl) CoA ligases.
Rationale for Low Overall Sequence Conservation of Aryl:CoA Ligases.
Aryl:CoA ligases are often employed by bacteria for detoxification of environmental contaminants such as 4-chlorobenzoate and benzoate.22–25 The driving force for activation of aromatic acids as CoA thioesters may have directed the evolution of CoA ligases, resulting in a group of aryl:CoA ligases that share conserved active site residues but low overall sequence identity (such as AuaEII and benzoate:- CoA ligases). When analyzing the aryl:CoA ligase crystal structures, it is clear that the aryl binding regions of AuaEII, AuaE, and both benzoate-CoA ligases are similar. However, the aryl binding region is different from that of 4-chlorobenzoate-CoA ligase, despite the structural similarities between benzoate and 4-chlorobenzoate. The aryl binding regions of 4-coumarate:CoA ligase and both phenylacetyl:CoA ligases are also distinct from those of AuaEII, AuaE, the benzoate:CoA ligases, and 4-chlorbenzoate:CoA ligase. Nevertheless, several features are still retained, such as an aromatic amino acid at the position corresponding to F220 of AuaEII (Figure 3; Figure S1). In summary, our analysis concludes that the need for activation of aromatic acids as CoA thioesters, especially in bacteria, has led to the emergence of a wide variety of aryl:CoA ligases often sharing only low overall sequence identity but with highly conserved residues in the aryl binding region.
Overall Motif Differences of AuaE and Aryl:CoA Ligases.
Previously, we completed an in depth sequence analysis of AuaE and identified two regions, the A3 (also known as the P-loop) and A10 core, which vary greatly in sequence when compared to other CoA ligases.11 The A3 region is usually responsible for binding and positioning the β- and γ-phosphates of ATP during the adenylation reaction, whereas the A10 region is mobile and contains a conserved lysine residue that stabilizes the negative charge formed on the α-phosphate during the adenylation reaction. The A10 region then undergoes a conformational change that allows CoA to enter the active site and complete the CoA ligation.17,26–30 In contrast, the function of AuaE is to catalyze acyltransfer from anthraniloyl-CoA to ACP, which does not involve ATP binding or adenylation. For this reason, in AuaE, high conservation of the P-loop and A10 regions is not critical for its function. In AuaEII, these regions are conserved. However, the P-loop could not be completely modeled in both AuaEII monomers, and only monomer B has well-defined electron density for this region. The B-factors of the P-loop region in AuaEII are elevated (Figure S3), and this has also been observed in other CoA ligase crystal structures in which ATP is not bound, suggesting high flexibility of the P-loop in the absence of ATP. In summary, AuaE lacks conservation of the important A10 and P-loop regions of aryl:CoA ligases.
Absence of Conserved C-terminal Residues in AuaE.
The N-terminal domain of AuaE shares a high degree of sequence identity to other CoA ligases. However, the C-terminal domain is truncated and lacks the highly conserved A10 core region, which is important for the ligase activity (Figures S1 and S2). A BLAST search followed by sequence alignment of the AuaE C-terminal domain revealed that ∼20 residues are missing when compared to the C-terminal domain of other CoA ligases. The A10 core is comprised of a highly conserved loop that leads into a C-terminal α-helix in AuaEII. The conserved loop contains a lysine residue, which has been shown to stabilize the negative charge of the phosphate pentahedral intermediate formed when the acyl-adenylate is generated.26,27 AuaE does not catalyze a typical CoA ligase reaction; therefore, it is likely that the highly conserved A10 core is missing because it is not crucial for the enzyme activity. A second BLAST search using only the AuaE C-terminal domain as a query sequence showed that the most similar proteins are CoA ligases and, when aligned, all the sequences have a full length C-terminal domain (Figure S2). This result suggests that AuaE is a unique member of the CoA ligase family where the loss of the A10 core has occurred at the cost of losing the CoA ligase function. However, AuaE somehow gained the AT activity.
Conserved Aspartic Acid of the Hinge Region.
The 140° rotation of the C-terminal domain between adenylation and thioester forming reactions has been well studied in 4-chlorobenzoate:CoA ligase, which revealed that a conserved aspartic acid in the hinge region between the N- and C-terminal domains is crucial for the conformational change.28 In the AuaE homology model, the aspartic acid (Asp410) is located in the hinge region at the same position as the conserved aspartic acid of 4-chlorobenzoate:CoA ligase (Figure S4). Because of the missing A10 motif of AuaE, the influence of this conformational change on the acyltransferase activity of AuaE is unclear. However, because of the presence of the conserved aspartic acid within the hinge region, AuaE may undergo conformational changes during catalysis. Furthermore, in conformation 2 (thioester forming conformation), the aspartic acid of the hinge region forms a salt bridge with a nearby arginine, which stabilizes conformation 2. AuaE also has arginine (Arg408) at this position, further supporting the possibility of a conformational change mechanism, or the possibility that conformation 2 may be important for the acyltransferase activity of AuaE. AuaEII has the conserved aspartic acid/arginine pair in the hinge region as expected for a functional CoA ligase. However, the two residues are not engaged in a charge–charge interaction, because the C-terminal domain of AuaEII is in conformation 1 (Figure S4). In summary, AuaE contains a conserved aspartic acid/arginine pair, which has been shown to stabilize conformation 2 in aryl-CoA ligases. Therefore, conformation 2 may be important for the acyltransferase activity of AuaE.
Possible Mechanisms of AuaE Acyltransferase Activity.
CoA ligases have a CoA binding site, which allows the pPant of CoA to extend into the N- and C-terminal domain interface and react with the acyl-adenylate intermediate. In vitro, it has been demonstrated that the substrate for AuaE is anthraniloyl-CoA and the product is anthraniloyl-ACP; however, the exact mechanism of this reaction remains unknown. There are two likely possibilities for how AuaE catalyzes the acyl transfer from anthraniloyl-CoA to holo-ACP: (1) A classic AT-like mechanism or (2) AuaE acts simply as a reaction surface.
The classic AT mechanism is used by enzymes such as malonyl-CoA:ACP transacylase (MCAT) that has an active site serine, with a multistep mechanism starting from an acyl-CoA as the substrate. The acyl group is trans-esterified onto an active site serine to form an acyl-enzyme intermediate, which then reacts with holo-ACP to yield the acyl-ACP product. Other ATs have also been identified that use cysteine as the active site residue to produce the covalent acyl-enzyme intermediate.31 Enzymes with a CoA ligase fold, such as AuaE, are able to produce and bind acyl-CoAs; therefore, AuaE may bind anthraniloyl-CoA and undergo trans-esterification to form a stable enzyme intermediate on a serine or cysteine. Once the AuaE enzyme acyl-intermediate is formed, holo-ACP can form a complex with AuaE and allow the pPant to react with the acyl-intermediate and complete the AT reaction. The classic AT mechanism is an attractive hypothesis, because there are multiple serine and cysteine residues in the C-terminal domain, which may be the site of acylation. In the AuaE homology model, Cys428, Ser431, Ser484, Cys485, and Ser487 are all located on the active site side of the C-terminal domain while in the thioester forming conformation 2. Ser493, Ser495, and Ser497 are all located in the truncated A10 region and would be close to the active site, if AuaE adopts conformation 1 during catalysis (Figure 4). Both conformations could potentially facilitate acyl transfer. Additionally, in conformation 2, a series of positively charged residues are located on the C-terminal domain facing the aryl-binding region, and these residues form a positive patch that could bind the ACP (Figure 5). Molecular dynamics simulations for AuaE modeled in conformation 1 and conformation 2 show that both conformations are stable; therefore, one or both conformations may be important for catalysis (Figure S5).
Figure 4.
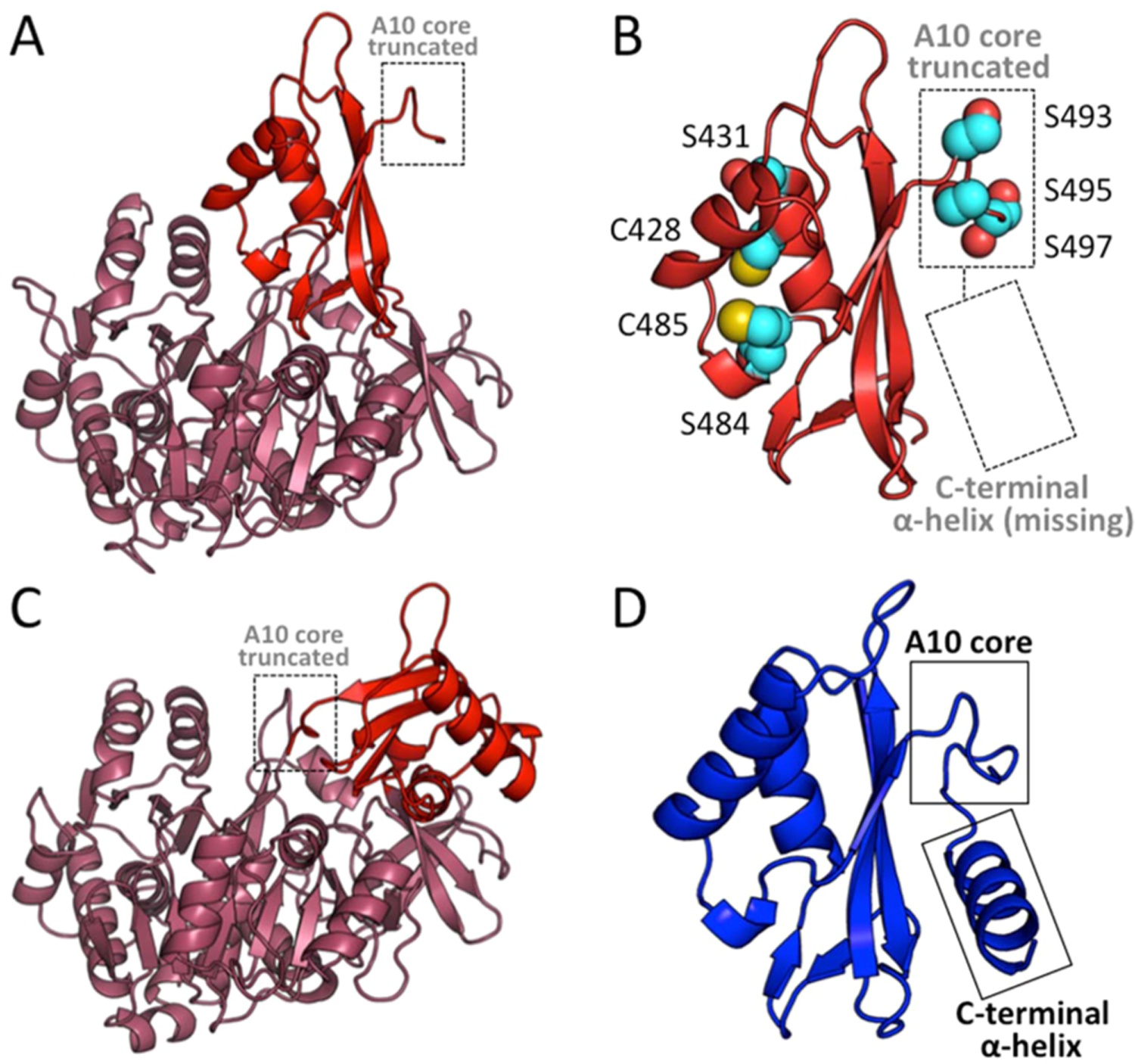
AuaE homology model in both conformation 1 and conformation 2, and a comparison of the C-terminal domains of AuaEII and AuaE. (A) Cartoon representation of an AuaE homology model generated using the structure of benzoate:CoA ligase (PDB ID: 4EAT) with the C-terminal domain (colored in red) in conformation 1 (adenylate forming conformation). The truncated A10 core is labeled and located away from the N-terminal (colored in raspberry) active site. (B) Close up view of the C-terminal domain of the AuaE homology model in conformation 1, showing the truncated A10 core and missing C-terminal helix. Residues (serines and cysteines) that may be involved in acyltransfer are represented in spheres. (C) Cartoon representation of the AuaE homology model generated using the structure of benzoate:CoA ligase (PDB ID: 2V7B) with the C-terminal domain (colored in red) in conformation 2 (thioester forming conformation). (D) The C-terminal domain of AuaEII displaying the conserved A10 core and C-terminal helix.
Figure 5.
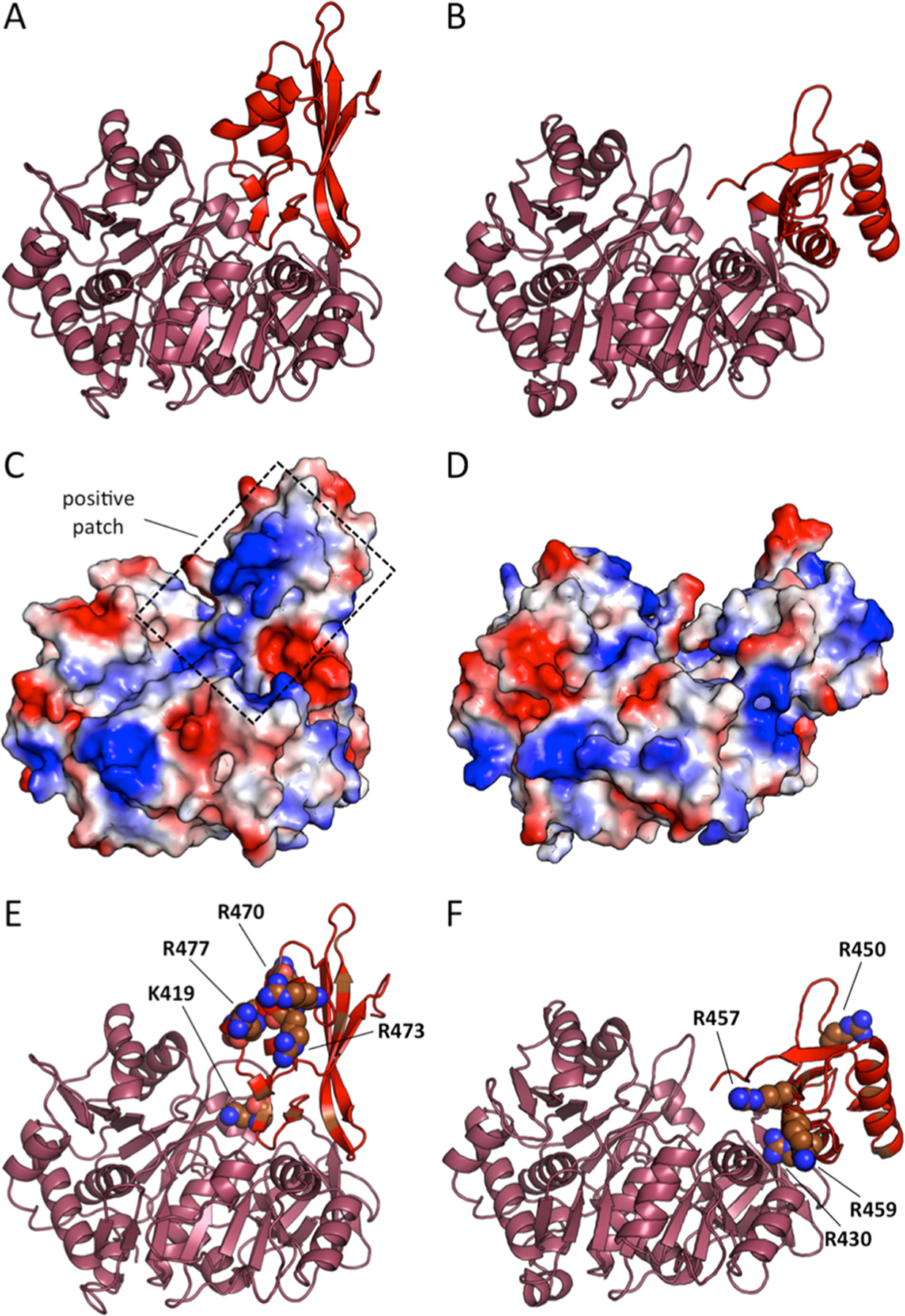
AuaE homology models in conformations 1 and 2 with electrostatic surface analysis. (A) AuaE model of conformation 2. (B) AuaE model of conformation 1. (C) AuaE conformation 2 electrostatic surface*. (D) AuaE conformation 1 electrostatic surface*. (E) Positively charged residues facing the N-terminal of AuaE in conformation 2. (F) Positively charged residues facing the N-terminal of AuaE in conformation 1. *Red represents negative and blue is positive surface charge.
Alternatively, AuaE may simply act as a surface to bring anthraniloyl-CoA and holo-ACP together. The thioester carbonyl of anthraniloyl-CoA could be bound in an oxyanion hole to stabilize the negative charge formed during ACP acylation. In this scenario, anthraniloyl-CoA would be stabilized in a favorable conformation by AuaE for acyl transfer. Once anthraniloyl-CoA is bound to AuaE, the positive surface of the AuaE C-terminal domain can dock with the negative surface of ACP to facilitate acyl transfer (Figure 5). Mutagenesis studies and functional assays are needed to test this hypothesis; however, structural analysis of the AuaE homology model suggests that AuaE has the potential to act as a reaction surface to catalyze acyl transfer.
Conclusions.
We have solved the crystal structure the CoA ligase AuaEII that is responsible for the generation of anthraniloyl-CoA from anthranilic acid. AuaEII activity is essential for the production of the aurachin class of natural products. It is part of a novel starter unit activation strategy to incorporate anthranilic acid into the type II polyketide biosynthesis. The crystal structure of AuaEII bound to anthraniloyl-AMP reveals specific residues involved in substrate selectivity and adds to a growing body of work about this diverse enzyme class. AuaE is a novel enzyme that is predicted to have the same overall fold as a CoA ligase, except that the C-terminal domain is truncated. Instead of possessing CoA ligase activity, AuaE serves as an AT. We generated multiple homology models of AuaE and used a combination of molecular dynamics, structural comparisons, and sequence alignments to visualize the potential function of AuaE as AT instead of a CoA ligase. Additional structural studies of AuaE are needed to fully understand the uncommon AT activity of AuaE. Taken together, these results represent the first crystal structure of an anthraniloyl-CoA ligase, which paves the foundation for understanding the priming mechanism of a novel starter unit in type II polyketide biosynthesis.
METHODS
Protein Expression and Purification.
A pET-29b (+) (Novagen) derived DNA plasmid encoding a C-terminal noncleavable His-tagged AuaEII (AuaEII/pET29) was transformed into E. coli BL21(DE3) competent cells and plated on LB-agar plates containing kanamycin (50 µg/mL). The plates were incubated overnight at 37 °C. Positive transformants were transferred to a 5 mL starter culture of Luria–Bertani (LB) broth containing kanamycin (50 µg/mL) and grown overnight at 37 °C with shaking, which was then used to inoculate 2 L of LB with kanamycin (50 µg/mL) and grown at 37 °C until the A600 reached 0.4–0.6. The cells were then cooled to 18 °C, and 0.1 mM IPTG was added to induce protein expression. After 12–18 h of incubation at 18 °C, the cells were harvested by centrifugation at 5000 rpm for 15 min. The cell pellets were flash-frozen in liquid nitrogen and stored at −80 °C. The frozen cell pellets were thawed on ice and resuspended in lysis buffer (50 mM Tris pH 8.0, 300 mM NaCl, 10% glycerol, and 10 mM imidazole). The cell suspension was lysed using sonication (8 × 30 s cycles), and the cell debris was removed by centrifugation at 14 000 rpm for 45 min. The clarified lysate was incubated with 5 mL of Ni-IMAC resin (BioRad) at 4 C for 1 h. The resin was poured into a fritted column, and the flow through fraction was collected. The resin was washed with 100 mL of lysis buffer, then eluted with lysis buffer plus increasing amounts of imidazole (20–500 mM). The elutions were analyzed using SDS-PAGE, and elutions containing the protein of interest were pooled and dialyzed overnight into a storage buffer (50 mM Tris 8.0, 300 mM NaCl, 10% glycerol, and 2 mM DTT). The dialyzed protein was concentrated to 8–10 mg mL−1 by centrifugal filtration using a 30 kDa MWCO concentrator (Millipore), then flash frozen in liquid nitrogen and stored at −80 °C for further use. The theoretical molecular weight of AuaEII of 58 477 Da was confirmed by MALDI-TOF MS (data not shown).
In Vitro Production of Anthraniloyl-AMP and AuaEII Crystallization.
AuaEII was thawed on ice and buffer exchanged into a crystallization buffer (25 mM Tris 8.0 and 2 mM DTT) using a PD-10 column (GE Healthcare). In order to produce diffracting crystals, AuaEII was incubated with different combinations of anthranilic acid, ATP, AMP, CoA, and MgCl2 before cocrystallization screening. The most promising cocrystallization condition was found by incubating AuaEII (95 µM) with anthranilic acid (2 mM), ATP (5 mM), and MgCl2 (2 mM) at 37 °C for 1 h to form the anthraniloyl-AMP intermediate. The reaction was then filtered with a 0.22 µM spin filter and screened for crystallization. For crystallization optimization, 1.7 µL of the incubated AuaEII reaction mixture was mixed with 1.7 µL of well solution (8% PEG 8000, 0.1 M magnesium acetate, and 0.1 sodium acetate, pH 4.5) and allowed to equilibrate over 500 µL of well solution using the sitting drop vapor diffusion method. Small rod-shaped crystals grew over 1–3 days. Crystals also formed when AuaEII was incubated with AMP + anthranilic acid and incubated on ice for 45 min prior to crystallization, but the optimized crystals from this cocrystallization did not diffract beyond ∼10 Å. Despite many different cocrystallization strategies, we were unable to obtain any diffraction-quality crystals of AuaE.
X-ray Data Collection, Processing, and Refinement.
Crystals of AuaEII were flash frozen directly in liquid nitrogen before data collection. X-ray diffraction data using monochromatic X-rays (0.9537 Å) were collected for an AuaEII crystal to a resolution of 2.6 Å at Stanford Synchrotron Radiation Laboratory (SSRL) on beamline 12–2. The data were indexed, integrated, and scaled using HKL2000.32 AuaEII crystallized as one dimer per asymmetric unit in the space group P212121, and initial phases were determined using molecular replacement (PHENIX Phaser-MR) with the structure of benzoate CoA ligase (PDB ID: 4EAT) as a search model.33 A preliminary model was built (PHENIX AutoBuild), and then this model was used for iterative rounds of manual model building (COOT) and refinement (PHENIX Refine).34–36 Well-defined electron density for anthraniloyl-AMP was observed in each AuaEII monomer after multiple rounds of refinement. The anthraniloyl-AMP ligand was drawn in Chemdraw and then converted to a PDB using the NCI SMILES converter server (http://cactus.nci.nih.gov/translate/), and ligand restraints were generated using PHENIX Elbow.37 Anthraniloyl-AMP was placed in each monomer manually using COOT. Water molecules were added with PHENIX Refine. Refinement was continued until Rfree and Rwork reached values of 22.4% and 17.4%, respectively. Data collection and refinement statistics can be found in Table 2. The following residues could not be modeled confidently in the AuaEII crystal structure due to missing or weak electron density: Pro141, Gln142, Glu143, Gly144, Tyr145, and Thr179 of monomer A and Ser451, Arg452, and Leu435 of monomer B.
Table 2.
AuaEII Crystallographic Data Collection and Refinement Statistics
| AuaEIIa | |
|---|---|
| beamline | SSRL Beamline 12–2 |
| PDB ID | 4WV3 |
| crystallization | 0.1 M sodium acetate pH 4.5, 0.1 M magnesium acetate, 8% PEG 8000 |
| crystallographic data | |
| wavelength (Å) | 0.9537 |
| space group | P212121 |
| cell dimensions (a, b, c) (Å) | 50.054, 132.219, 160.118 |
| α=β=γ=90° | |
| resolution (Å) | 50.00–2.60 |
| no. of observations | 215077 |
| no. of unique observations | 33543 |
| completeness % | 99.7 (97.4) |
| I/σ(I) | 15.1 (5.1) |
| Rmerge % | 11.2 (32.2) |
| redundancy | 6.4 |
| refinement | |
| resolution (Å) | 46.81–2.60 (2.69–2.60) |
| no. of protein atoms | 8062 |
| no. of ligand atoms | 64 |
| no. of water atoms | 488 |
| Rfree % | 0.224 (0.267) |
| Rcrys % | 0.174 (0.203) |
| geometry | |
| RMS bonds (Å) | 0.017 |
| RMS angles (deg) | 0.99 |
| Ramachandran favored (%) | 98 |
| Ramachandran allowed (%) | 1.8 |
| Ramachandran disallowed (%) | 0.2 |
| average B-factors (Å2) | |
| protein | 21.90 |
| water | 22.30 |
| ligands | 15.00 |
Numbers in parentheses denote the highest resolution shell.
AuaE Homology Model Generation, Molecular Dynamics, and Energy Minimization.
HHPred was used to generate two homology models of AuaE using benzoate:CoA ligases (PDB IDs: 2V7B and 4EAT) as templates with two different C-terminal conformations, described below as M1 and M2, respectively.38,39 The C-terminal domains of CoA ligases have been observed in multiple conformations as determined by the crystal structures of several enzymes.19 Even when two different enzymes are bound to a similar intermediate (such as an acid substrate), the C-terminal domains could be in different conformations. The template for the first AuaE homology model, benzoate:CoA ligase from Rhodopseudomonas palustris (PDB ID: 4EAT), has its C-terminal domain in the thioester conformation (conformation 2), with the A10 core rotated away from the active site. The template for the second AuaE homology model, benzoate:CoA ligase (from Burkholderia xenovorans LB400) (PDB ID: 2V7B), has its C-terminal domain in the adenylation conformation (conformation 1), with the A10 core rotated toward the active site. Because of the AuaE C-terminal truncation, it is difficult to predict the exact conformation that the C-terminal domain will adopt during catalysis; therefore, we have chosen to generate two models, which represent the two most common conformations observed in the crystal structures of CoA ligases.19
Molecular dynamics on models M1 and M2 was carried out using Amber14 and AmberTools15.40 The Amber14 Stony Brook force field (ff14SB) was used to model both protein models.41 LEaP was used to add hydrogens and neutralize the systems by adding four Na+ ions to each system, followed by explicit solvation in a 12-Å water buffer TIP3P truncated octahedron box. The fully solvated M1 system contained 51 436 atoms, and the fully solvated M2 system contained 46 978 atoms. Periodic boundry conditions were used along with PME to model the long-range electrostatics and a real-space nonbonded interaction cutoff of 10 Å. Minimization using SANDER was performed in two stages to remove steric clashes present in the homology models. The initial stage was carried out over 5000 steps for the solvent and ions with the homology model restrained by a force constant of 500 kcal/mol/Å2, followed by a second stage carried out over 5000 steps for the entire system. A short 100 ps simulation with weak restraints (force constant of 10 kcal/mol/Å2 on the homology model) was used to heat up the system to a temperature of 300 K using a Langevin temperature equilibration scheme (ntb = 2, ntp = 1, ntt = 3, gamma_ln = 1.0). Periodic boundary conditions were used, along with a nonbonded interaction cutoff of 10 Å. The systems were equilibrated using a short 100 ps equilibration with constant pressure (ntb = 2, ntp = 1, ntt = 3, gamma_ln = 1.0). For the simulation, hydrogen atoms were constrained using the SHAKE algorithm, allowing for a 2 fs time step. Refinement production using PMEMD was was run using NPT (ntb = 2, ntp = 1, ntt = 3, gamma_ln = 1.0) over 100 ns (50 000 000 timesteps). Simulation speeds of 22.9–24.9 ns/day were observed. A total of 1000 frames were collected, with the average structure being generated for the final 200 frames using cpptraj and methods described by Nurisso et al.42,43 cpptraj was used to calculate backbone trace RMSD (Figure S5) using the script provided by Nurisso et al. and plotted using a custom in-house Python script. The average structures for each model were minimized using SANDER and the same method described above to obtain the final homology models.
AuaEII and AuaE Sequence Alignment and Structural Comparison.
Proteins similar to AuaEII and AuaE were identified using BLAST, searching both nonredundant protein sequences and also Protein Data Bank (PDB) proteins. Sequence alignments were conducted using ClustalW,44 and graphics were generated using Espript (http://espript.ibcp.fr).45
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health grant GM100305.
Footnotes
ASSOCIATED CONTENT
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschem-bio.5b00500.
Five figures and one table (PDF)
Accession Codes
The atomic coordinates and structure factors have been deposited with the Protein Data Bank (PDB ID: 4WV3).
The authors declare no competing financial interest.
REFERENCES
- (1).Das A, and Khosla C (2009) Biosynthesis of aromatic polyketides in bacteria. Acc. Chem. Res 42, 631–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Moore BS, and Hertweck C (2002) Biosynthesis and attachment of novel bacterial polyketide synthase starter units. Nat. Prod. Rep 19, 70–99. [DOI] [PubMed] [Google Scholar]
- (3).Das A, and Khosla C (2009) In vivo and in vitro analysis of the hedamycin polyketide synthase. Chem. Biol 16, 1197–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Meadows ES, and Khosla C (2001) In vitro reconstitution and analysis of the chain initiating enzymes of the R1128 polyketide synthase. Biochemistry 40, 14855–14861. [DOI] [PubMed] [Google Scholar]
- (5).Bao W, Sheldon PJ, Wendt-Pienkowski E, and Hutchinson CR (1999) The Streptomyces peucetius dpsC gene determines the choice of starter unit in biosynthesis of the daunorubicin polyketide. J. Bacteriol 181, 4690–4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Waldman AJ, and Balskus EP (2014) Lomaiviticin biosynthesis employs a new strategy for starter unit generation. Org. Lett 16, 640–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Kalaitzis JA, Cheng Q, Thomas PM, Kelleher NL, and Moore BS (2009) In vitro biosynthesis of unnatural enterocin and wailupemycin polyketides. J. Nat. Prod 72, 469–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Friedrich T, van Heek P, Leif H, Ohnishi T, Forche E, Kunze B, Jansen R, Trowitzsch-Kienast W, Hofle G, Reichenbach H, et al. (1994) Two binding sites of inhibitors in NADH: ubiquinone oxidoreductase (complex I). Relationship of one site with the ubiquinone-binding site of bacterial glucose:ubiquinone oxidoreductase. Eur. J. Biochem 219, 691–698. [DOI] [PubMed] [Google Scholar]
- (9).Meunier B, Madgwick SA, Reil E, Oettmeier W, and Rich PR (1995) New inhibitors of the quinol oxidation sites of bacterial cytochromes bo and bd. Biochemistry 34, 1076–1083. [DOI] [PubMed] [Google Scholar]
- (10).Sandmann A, Dickschat J, Jenke-Kodama H, Kunze B, Dittmann E, and Muller R (2007) A Type II polyketide synthase from the gram-negative Bacterium Stigmatella aurantiaca is involved in Aurachin alkaloid biosynthesis. Angew. Chem., Int. Ed 46, 2712–2716. [DOI] [PubMed] [Google Scholar]
- (11).Pistorius D, Li Y, Mann S, and Muller R (2011) Unprecedented anthranilate priming involving two enzymes of the acyl adenylating superfamily in aurachin biosynthesis. J. Am. Chem. Soc 133, 12362–12365. [DOI] [PubMed] [Google Scholar]
- (12).Schmelz S, and Naismith JH (2009) Adenylate-forming enzymes. Curr. Opin. Struct. Biol 19, 666–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Bains J, and Boulanger MJ (2007) Biochemical and structural characterization of the paralogous benzoate CoA ligases from Burkholderia xenovorans LB400: defining the entry point into the novel benzoate oxidation (box) pathway. J. Mol. Biol 373, 965–977. [DOI] [PubMed] [Google Scholar]
- (14).Hu Y, Gai Y, Yin L, Wang X, Feng C, Feng L, Li D, Jiang XN, and Wang DC (2010) Crystal structures of a Populus tomentosa 4-coumarate:CoA ligase shed light on its enzymatic mechanisms. Plant Cell 22, 3093–3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Coschigano PW, Haggblom MM, and Young LY (1994) Metabolism of both 4-chlorobenzoate and toluene under denitrifying conditions by a constructed bacterial strain. Appl. Environ. Microbiol 60, 989–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Conti E, Franks NP, and Brick P (1996) Crystal structure of firefly luciferase throws light on a superfamily of adenylate-forming enzymes. Structure 4, 287–298. [DOI] [PubMed] [Google Scholar]
- (17).Reger AS, Wu R, Dunaway-Mariano D, and Gulick AM (2008) Structural characterization of a 140 degrees domain movement in the two-step reaction catalyzed by 4-chlorobenzoate:CoA ligase. Biochemistry 47, 8016–8025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Gulick AM, Lu X, and Dunaway-Mariano D (2004) Crystal structure of 4-chlorobenzoate:CoA ligase/synthetase in the unliganded and aryl substrate-bound states. Biochemistry 43, 8670–8679. [DOI] [PubMed] [Google Scholar]
- (19).Gulick AM (2009) Conformational dynamics in the Acyl-CoA synthetases, adenylation domains of non-ribosomal peptide synthetases, and firefly luciferase. ACS Chem. Biol 4, 811–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Wu R, Reger AS, Cao J, Gulick AM, and Dunaway-Mariano D (2007) Rational redesign of the 4-chlorobenzoate binding site of 4-chlorobenzoate: coenzyme a ligase for expanded substrate range. Biochemistry 46, 14487–14499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Law A, and Boulanger MJ (2011) Defining a structural and kinetic rationale for paralogous copies of phenylacetate-CoA ligases from the cystic fibrosis pathogen Burkholderia cenocepacia J2315. J. Biol. Chem 286, 15577–15585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Goris J, De Vos P, Caballero-Mellado J, Park J, Falsen E, Quensen JF 3rd, Tiedje JM, and Vandamme P (2004) Classification of the biphenyl- and polychlorinated biphenyl-degrading strain LB400T and relatives as Burkholderia xenovorans sp. nov. Int. J. Syst. Evol. Microbiol 54, 1677–1681. [DOI] [PubMed] [Google Scholar]
- (23).Denef VJ, Klappenbach JA, Patrauchan MA, Florizone C, Rodrigues JL, Tsoi TV, Verstraete W, Eltis LD, and Tiedje JM (2006) Genetic and genomic insights into the role of benzoate-catabolic pathway redundancy in Burkholderia xenovorans LB400. Appl. Environ. Microbiol 72, 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Chang KH, Liang PH, Beck W, Scholten JD, and Dunaway-Mariano D (1992) Isolation and characterization of the three polypeptide components of 4-chlorobenzoate dehalogenase from Pseudomonas sp. strain CBS-3. Biochemistry 31, 5605–5610. [DOI] [PubMed] [Google Scholar]
- (25).Dunaway-Mariano D, and Babbitt PC (1994) On the origins and functions of the enzymes of the 4-chlorobenzoate to 4-hydroxybenzoate converting pathway. Biodegradation 5, 259–276. [DOI] [PubMed] [Google Scholar]
- (26).Horswill AR, and Escalante-Semerena JC (2002) Characterization of the propionyl-CoA synthetase (PrpE) enzyme of Salmonella enterica: residue Lys592 is required for propionyl-AMP synthesis. Biochemistry 41, 2379–2387. [DOI] [PubMed] [Google Scholar]
- (27).Branchini BR, Murtiashaw MH, Magyar RA, and Anderson SM (2000) The role of lysine 529, a conserved residue of the acyl-adenylate-forming enzyme superfamily, in firefly luciferase. Biochemistry 39, 5433–5440. [DOI] [PubMed] [Google Scholar]
- (28).Wu R, Reger AS, Lu X, Gulick AM, and Dunaway-Mariano D (2009) The mechanism of domain alternation in the acyl-adenylate forming ligase superfamily member 4-chlorobenzoate: coenzyme A ligase. Biochemistry 48, 4115–4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Osman KT, Du L, He Y, and Luo Y (2009) Crystal structure of Bacillus cereus D-alanyl carrier protein ligase (DltA) in complex with ATP. J. Mol. Biol 388, 345–355. [DOI] [PubMed] [Google Scholar]
- (30).Kochan G, Pilka ES, von Delft F, Oppermann U, and Yue WW (2009) Structural snapshots for the conformation-dependent catalysis by human medium-chain acyl-coenzyme A synthetase ACSM2A. J. Mol. Biol 388, 997–1008. [DOI] [PubMed] [Google Scholar]
- (31).Bretschneider T, Zocher G, Unger M, Scherlach K, Stehle T, and Hertweck C (2012) A ketosynthase homolog uses malonyl units to form esters in cervimycin biosynthesis. Nat. Chem. Biol 8, 154–161. [DOI] [PubMed] [Google Scholar]
- (32).Otwinowski Z, and Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276, 307–326. [DOI] [PubMed] [Google Scholar]
- (33).Mccoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007) Phaser crystallographic software. J. Appl. Crystallogr 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Terwilliger TC, Grosse-Kunstleve RW, Afonine PV, Moriarty NW, Zwart PH, Hung LW, Read RJ, and Adams PD (2008) Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr., Sect. D: Biol. Crystallogr 64, 61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, and Adams PD (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr., Sect. D: Biol. Crystallogr 68, 352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Emsley P, and Cowtan K (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- (37).Moriarty NW, Grosse-Kunstleve RW, and Adams PD (2009) electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr., Sect. D: Biol. Crystallogr 65, 1074–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Soding J, Biegert A, and Lupas AN (2005) The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res 33, W244–W248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Hildebrand A, Remmert M, Biegert A, and Soding J (2009) Fast and accurate automatic structure prediction with HHpred. Proteins: Struct., Funct., Genet 77 (Suppl 9), 128–132. [DOI] [PubMed] [Google Scholar]
- (40).Case DA, Berryman JT, Betz RM, Cerutti DS, Cheatham TE III, Darden TA, Duke RE, Giese TJ, Gohlke H, Goetz AW, et al. (2015) AMBER 2015, Univerity of California, San Francisco. [Google Scholar]
- (41).Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, and Simmerling C (2006) Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins: Struct., Funct., Genet 65, 712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Nurisso A, Daina A, and Walker RC (2011) A practical introduction to molecular dynamics simulations: applications to homology modeling. Methods Mol. Biol 857, 137–173. [DOI] [PubMed] [Google Scholar]
- (43).Roe DR, and Cheatham TE III. (2013) PTRAJ and CCPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput 9, 3084–3095. [DOI] [PubMed] [Google Scholar]
- (44).Chenna R, Sugawara H, Koike T, Lopez R, Gibson TJ, Higgins DG, and Thompson JD (2003) Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res 31, 3497–3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Robert X, and Gouet P (2014) Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res 42, W320–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.