

Abstract
Rationale:
Steroid-resistant nephrotic syndrome (SRNS) is a special kidney disease. SRNS is characterized by steroid-resistant, clinical variability, and genetic heterogeneity. Patients with SRNS often may eventually need renal transplantation.
Patient concerns:
A 10-month-old Chinese male infant presented with oliguria, renal dysfunction, hypertension, and anemia.
Diagnoses:
Combined with clinical manifestations, laboratory testing and sequencing results, the patient was diagnosed as SRNS.
Interventions:
Combined intravenous methylprednisolone and cefoperazone sulbactam did not improve the patient's condition. Thus, SRNS associated with hereditary nephrotic syndrome was strongly suspected. Genetic testing for hereditary renal disease of the patient revealed 2 novel heterozygous mutations in the Nucleoporin 93 (NUP93) gene, which were predicted pathogenic and harmful by bioinformatic softwares of SIFT, PolyPhen_2 and REVEL.
Outcomes:
As general physical health deterioration and renal dysfunction, the patient died of a severe infection.
Lessons:
The novel NUP93 heterozygous mutations identified in the current study broadened the genetic spectrum of SRNS and further deepened our insight into pathogenic mutations of NUP93 to improve disease diagnosis.
Keywords: Chinese infant, genetics, NUP93, steroid-resistant nephrotic syndrome
1. Introduction
Nephrotic syndrome (NS) is a clinical syndrome caused by increased permeability of the glomerular filtration membrane to plasma proteins, resulting in the loss of large quantities of plasma proteins from the urine. This causes a series of pathophysiologic changes characterized by massive proteinuria, hypoproteinemia, hyperlipidemia, and edema. The annual incidence of NS among children <16 years of age in the United States has been reported to be around 2 to 7 per 100,000. Sixteen of every 100,000 children are diagnosed with NS, making NS one of the commonest renal diseases in children.[1] NS can be classified into steroid-sensitive NS and steroid-resistant NS (SRNS) based on the response of NS patients to corticosteroid therapy.[2] Most children with NS respond well to steroids, but around 10% to 20% are diagnosed with SRNS. SRNS is a renal disease presenting with clinical variability and genetic heterogeneity. A higher frequency of genetic variations has been reported in early-onset SRNS than steroid-sensitive NS. Currently, both treatment and prognosis of this disease depend on the renal pathologic diagnosis. Unfortunately, there are still no effective treatment regimens for SRNS, and approximately 50% of patients who have developed SRNS in childhood exhibit focal segmental glomerulosclerosis (FSGS) and are at high risk of developing end-stage renal disease (ESRD). Renal transplantation may eventually be required.[3]
To date, at least 50 genes have been shown to be associated with SRNS when mutated.[4] It has been reported that approximately 29.5% of families with a member diagnosed with SRNS and <25 years of age are caused by a single gene mutation. Among the mutations, the NPHS1, NPHS2, and WT1 are the most common causes of SRNS.[5] It has previously been reported that among 160 SRNS families, a Nucleoporin 93 (NUP93) mutation was responsible for the pathogenesis of 3 families with hereditary renal disease.[6] Based on high-throughput exon sequencing in >1800 SRNS families, NUP93 variations, including compound heterozygous and missense variants, were identified to cause this disease in 3 families.[7] In addition, a case of SRNS caused by NUP93 a gene mutation was recently reported by Japanese scholars.[23] A case of SRNS caused by a novel compound heterozygous mutation of NUP93, which has not been previously reported, is described herein.
2. Case report
A 7-month-old Chinese male infant with edema and proteinuria received symptomatic treatment at a local hospital. At 8 months of age, he was diagnosed clinically with nephritic NS due to edema, proteinuria, hypoproteinemia, microscopic hematuria, and hypertension, and thus received prednisone at a dose of 2 mg/kg/d for 8 weeks in Thailand. The edema was not relieved and the proteinuria did not resolve. At 10 months of age, the patient was admitted to our hospital due to cough, shortness of breath, oliguria, renal dysfunction, hypertension, and anemia. The infant was the third child of the family and delivered with a birth weight of 2400 g. His family has no history of renal disease. Peripheral blood samples of the patient and his parents were collected for gene sequencing.
The physical examination findings of the infant at the time of admission were as follows: body temperature, 36.3°C; heart rate, 126 bpm; respiratory rate, 36/min; blood pressure, 152/103 mm Hg; weight, 9.5 kg; an appearance of anemia; anasarca; abdominal distension; no systemic rashes or papulovesicles; and moist rales auscultated in the lungs bilaterally.
The biochemical indices of the patient are summarized in Table 1. The albumin and globulin levels were decreased, the creatinine, urea, uric acid, lactate dehydrogenase, total cholesterol, triglycerides, and complement C4 levels were increased, and the serum complement C3 level was normal. All the patient's immune-related antibodies were negative, which included antinuclear antibodies, anti-Sm antibodies, antineutrophil cytoplasmic antibodies, etc. the urine analysis revealed 4+ proteinuria, and there were 13 red blood cell/high power field. The hematologic examination revealed hypochromic microcytic anemia (hemoglobin, 69 g/L; mean corpuscular volume, 79.6 fL; mean corpuscular hemoglobin, 23.7 pg; and mean corpuscular hemoglobin concentration, 305 g/L), while the platelet count and the serum C-reactive protein level were normal.
Table 1.
Laboratory examination results of the patient.
| Items | Results | References |
| Albumin | 20.2 g/L | 35 g/L–50 g/L |
| Globulin | 21.9 g/L | 20 g/L–40 g/L |
| Creatinine | 239.75 μmol/L | 27 μmol/L–62 μmol/L |
| Urea nitrogen | 16.42 μmol/L | 1.8 μmol/L–6.4 μmol/L |
| Lactic dehydrogenase | 499 μ/L | 67 μ/L–394 μ/L |
| Total cholesterol | 9.12 mmol/L | 3.12 mmol/L–5.2 mmol/L |
| Triglyceride | 5.4 mmol/L | 0.8 mmol/L–1.8 mmol/L |
| Complement C3 | 0.85 g/L | 0.8 g/L–1.5 g/L |
| Complement C4 | 0.48 g/L | 0.12 g/L–0.4 g/L |
| Urinary protein | 4+ | negative |
| Urine red cell | 13/HPF | 0-3/HPF |
Relevant imaging examinations were obtained. Abdominal color Doppler ultrasonography showed a moderate peritoneal effusion, and a chest X-ray revealed exudation in both lungs and a pleural effusion.
Based on the clinical manifestations and the results of the laboratory testing, hereditary NS was strongly suspected. Therefore, all exons of 506 genes associated with urinary system diseases were analyzed using next generation sequencing. The infant harbored 2 novel heterozygous mutations in NUP93 (NM_014669), and were subsequently confirmed by Sanger sequencing: a paternal missense variant c.1655A>G (p.Tyr552Cys) and a maternal nonsense variant c.1732C>T (p. Arg578∗) (Fig. 1). After searching the mutation records of normal population in 1000 Genome Project, NHLBI Exome Sequencing Project (ESP6500), Exome Aggregation Consortium (ExAC_ALL), and Exome Aggregation Consortium East Asian (ExAC_EAS), a very low frequency of c.1655A>G (p.Tyr552Cys) and no information of the frequency of c.1732C>T (p. Arg578∗) in normal population were found. No related reports have been found in the Human Gene Mutation Database Professional database. The patient was treated with intravenous methylprednisolone (2 mg/kg/d) and cefoperazone-sulbactam (100 mg/kg/d) for 1 week did not improve the patient's condition. The clinical disease progressed rapidly and was accompanied with anemia and renal dysfunction. The patient died of a severe infection.
Figure 1.

Genotype and conservation of the patient's mutations in NUP93: A. Table with Chromosome position, c. change, p. change, exon number, nomal frequency, pathogenic analysis and inheritance pattern in our patient. B. Sanger tracing for the patient, the mother and father for each allele.
The study was approved by the Ethics Committee at the Kunming Children's Hospital, Yunnan, China (2016-03-022-K01). All of the patients provided written informed consent for publication of this case report and accompanying images.
3. Discussion
NS is a clinical syndrome caused by damage to the glomerular filtration barrier. NS is a common glomerular disease in children, the incidence and responsiveness to steroids of which varies with ethnicity.[8] In our case, the infant was diagnosed with SRNS based on the clinical manifestations and results of laboratory testing.
A growing body of literature has shown that 29.8% of SRNS cases are caused by single gene mutations in various genes, such as NPHS2, NPSH1,[9]WT1, LAMB2, and PLCE1.[10,11] In our patient, the genetic analysis revealed a compound heterozygous variations in NUP93, one is a novel paternal missense variant in exon 14, c.1655A>G (p.Tyr552Cys), with a frequency of 0.00006 in normal population in the gnomAD_genome database (http://gnomad.broadinstitute.org/). This site has not been reported before. The effect of this mutation on the protein function was predicted pathogenic and harmful by bioinformatic softwares of SIFT, PolyPhen_2, and REVEL. Another is a maternal nonsense variant in exon 15, c.1732C>T (p. Arg578∗), which was rated to be likely pathogenic according to the American College of Medical Genetics and Genomics guidelines. No information of the frequency of this site in normal population was found. The 2 sites of NUP93 (NM_014669): c.1655A>G (p.Tyr552Cys)/c.1732C>T (p. Arg578∗) are highly conserved sequence across multiple species (Fig. 2). When modeled by SWISS-MODEL, one key benzene ring structure was found to be absent in the three-dimensional structural model of the Nuclear Pore Complex Protein NUP93 (Fig. 3). A mutation in the NUP93 is one of the very rare causes of hereditary SRNS.[12,13]
Figure 2.

Conservative analysis of amino acid 552 and amino acid 578 of NUP93.
Figure 3.
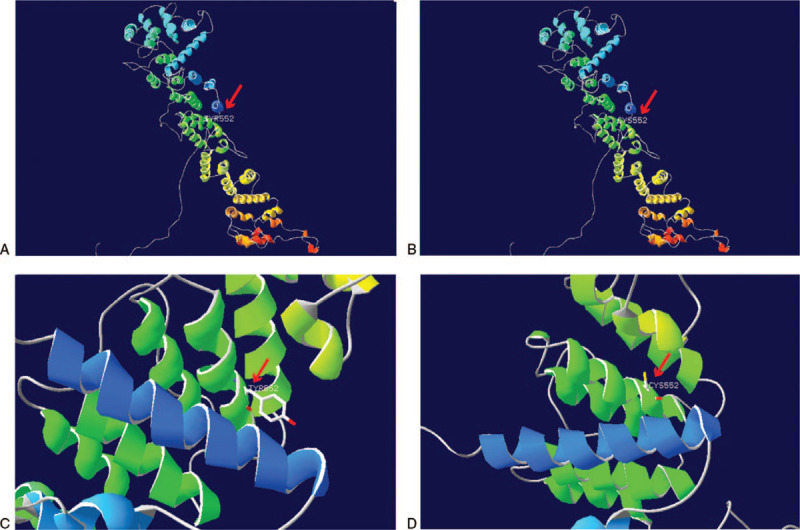
Structural modeling of the wild type and mutant NUP93 mutation (Y552C): The structure modeling is based on the Electron Microscopy determined coordinates of the composite structure of the inner ring of the human nuclear pore complex using SWISS-MODEL (SMTL ID: 5ijn.1). A and C: A structural model of the wild NPC protein NUP93 with 100% sequence identity is demonstrated. B and D: A structural alteration of the mutant NPC protein NUP93 with 99.88% sequence identity is shown. The locations of the NUP93 mutation (Y552C) mutations (red arrows) are indicated. The benzene ring structure (red arrows) was disrupted when tyrosine was replaced by cysteine as a result of c. 1655A>G mutation.
NUP93 is a widely expressed gene located on chromosome 16q13 and includes 22 exons and 21 introns that encodes a highly conserved nucleoporin. Thirty different nucleoporins (NUPs) will form a nuclear pore complex (NPC), which is a macromolecular (approximately 50 MDa) protein assembly embedded in the nuclear membrane.[14] NPC, composed of multiple copies of NUPs,[15] mediates bidirectional nucleo-cytoplasmic transport of proteins, RNA, and ribonucleoprotein particles. There are 2 major supporting modules in the NUP that constitute a symmetric structure spanning from the inner nuclear membrane to the outer nuclear membrane, thus forming a central pathway.[16] These supporting modules coordinate structures of the NUP93 complex by interacting directly or indirectly with other NUPs (NUP53, NUP155, NUP188, and NUP205) at the C-terminal α-helical domain of NUP93, and this complex regulates the permeability barrier of pores.[17] Thus, NUP93 plays a crucial role in organizing nuclear pores in NPC; the aberrant NUP93 can disrupt the NUP assembly, and in turn induce dysfunction of nucleo-cytoplasmic transport.[18,19] In addition, a recent study suggested that mutation of NUP93 inhibits the proliferation of podocytes and possibly cause FSGS,[20] but the underlying molecular mechanism has not been established.
NUP93 mutation-related SRNS has been reported in 11 separate SRNS families (Table 2), with different ethnicities, ages of onset, and different mutation sites and patterns. Our patient and 10 previously reported cases all presented with SRNS. Eight out of the 10 reported cases was subjected to renal biopsy and their renal pathology was all determined as FSGS. Of the 10 reported cases, 3 had extra-renal manifestations and 7 had not. Their average ages of the disease onset and of developing ESRD were 3.5 years and 5.4 years, respectively. Of these 10 children, 8 were heterozygous mutations and 2 were homozygous mutations, and their mutation sites were distributed in exons 11 to 14, 16 to 19, 7, and 21. In contrast, the age of the disease onset in our child was smaller than that of the previously reported cases, with an age of only 0.6 years, and he was only 0.8 years old when he developing ESRD. In addition, the extra-renal symptoms were absent in our patient. Importantly, he was found to harbor NUP93 mutations in exons 14 and 15, constituting heterozygous mutation.
Table 2.
Description of genetic diagnosis of NUP93 gene mutations in 11 different families with SRNS, gene locus detected with variations, and some clinical features.
| Family/individual | Nucleotide change | Amino acid change | Zygosity | Exon | Gender | Ancestry | Onset/ ESRD year | Proteinuria | Haematuria | Edema | Hypertension | Extrarenal manifestation/DS | Renal biopsy | Therapy/response | RTX | Ref |
| No.1 | c.1162 C>T /c.1772G>T | p.A388T /p.G591V | Het | Ex11/16 | Female | Serbian | 6 yr/6 years | Y | Y | Y | UK | N | FSGS | RTX /HK | Y | [20] |
| No.2 | c.1326delG /c.1772G>T | p.L442A /p.G591V | Het | Ex12/16 | Female | German | 3 yr/3 yr | Y | Y | Y | UK | N | FSGS | CSA/PR | Y | [20] |
| No.3 | c.1537+1G>A /c.1772G>T | del exon13/p.G591V | Het | Ex13/16 | Female | German | 3 yr/4 yr | Y | Y | Y | UK | MG-syndrome | FSGS | SRNS | Y | [20] |
| No.4 | c.1772G>T | p.G591V | Hom | Ex16 | Male | Turkish | 3 yr/11 yr | Y | Y | Y | UK | N | FSGS | SRNS | N | [20] |
| No.5 | c.1886A>G | p.T629C | Hom | Ex17 | Male | Turkish | 1 yr/ 1 yr | Y | Y | Y | UK | N | FSGS | SRNS | Y | [20] |
| No.6 | c.1772G>T /c.1298delA | p.G591V/ p.D433A | Het | Ex16/12 | Female | Czsk | 3 yr/ 3.5 yr | Y | UK | Y | UK | N | FSGS | ESRD | Y | [21] |
| No.7 | c.1772G>T/c.1916 T>C | p.G591V/ p.L639P | Het | Ex16/18 | Male | Czsk | 1.8 yr/ 2.9 yr | Y | UK | Y | UK | N | FSGS | CSA/ESRD | Y | [21] |
| No.8 | c.2084T>C/c.2267T>C | p.L695S/ p.L756S | Het | Ex19/21 | Male | White | 6.1 yr/ 12 yr | Y | UK | UK | UK | UK | UK | SRNS | UK | [22] |
| No.9 | c.1573C>T/c.1886A>G | p.R525W/ p.Y629C | Het | Ex14/17 | Female | Japanese | 4 yr/ 6 yr | Y | Y | Y | Y | RA | FSGS | SRNS | Y | [23] |
| No.10 | c.575A>G/c. 1605C>G | p.T192C/ p.Tyr535Ter | Het | Ex7/14 | Female | American | 5 yr/ 5 yr | Y | Y | Y | UK | CM, AF | UK | SRNS | Y | [13] |
| New report | c.1655A>G/c.1732C>T | p.T192C/ p.Tyr535Ter | Het | Ex14/15 | Male | Chinese | 0.6 yr/ 0.8 yr | Y | Y | Y | Y | N | UK | SRNS | N | [∗] |
AF = autistic features, CM = cardiomyopathy, CSA = cyclosporin A, CZSK = Czech and Slovak, DS = dysmorphic-syndrome, ESRD = end stage renal disease, Ex = Exon, HK = hyperechogenic kidneys, MG = Marcus-Gunn, N = No, PR = partial response, RA = rheumatoid arthritis, Ref = reference, RTX = renal transplantation, UK = unknown, Y = Yes.
The present case.
Taken together, the novel mutation of the NUP93 detected in this patient might cause SRNS, but this mutation resulted in the functional alteration of the corresponding protein has only been validated in protein function prediction and 3D protein structure models prediction. In clinical practice, gene sequencing should be performed for infantile-onset SRNS to determine whether the disease is congenital, thereby accurate and adequate treatment methods can be used to improve the outcome of this disease.
Author contributions
Conceptualization: Bo Zhao, Li Li.
Formal analysis: Ji-Yu Chen, Jing-Jing Cui.
Methodology: Ya-Bin Liao, Yan-Fang Li.
Resources: Yan-Fang Li, Xue-Mei Jiang.
Software: Ji-Yu Chen.
Supervision: Xue-Mei Jiang, Xin Bi, Mi-Feng Yang.
Validation: Xin Bi, Mi-Feng Yang.
Writing – original draft: Bo Zhao, Ji-Yu Chen.
Writing – review & editing: Ya-Bin Liao, Li Li, Jing-Jing Cui, Ji-Yu Chen.
Glossary
Abbreviations: ESRD = end-stage renal disease, FSGS = focal segmental glomerulosclerosis, NPC = nuclear pore complex, NS = nephrotic syndrome, NUP93 = Nucleoporin 93, NUPs = nucleoporins, SRNS = steroid-resistant nephrotic syndrome, SSNS = steroid-sensitive nephrotic syndrome.
References
- [1].Eddy Allison A, SYMONS Jordan M. Nephrotic syndrome in childhood. Lancet 2003;362:629–39. [DOI] [PubMed] [Google Scholar]
- [2].Lennon R, Watson L, Webb NJ. Nephrotic syndrome in children. Paediatr Child Health 2012;20:36–42. [Google Scholar]
- [3].Trautmann A, Bodria M, Ozaltin F, et al. Spectrum of steroid-resistant and congenital nephrotic syndrome in children: the PodoNet registry cohort. Clin J Am Soc Nephrol 2015;10:592–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Karp AM, Gbadegesin RA. Genetics of childhood steroid-sensitive nephrotic syndrome. Pediatr Nephrol 2017;32:1481–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Sadowski CE, Lovric S, Ashraf S, et al. A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome. J Am Soc Nephrol 2015;26:1279–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hildebrandt F, Heeringa SF, Rüschendorf F, et al. A systematic approach to mapping recessive disease genes in individuals from outbred populations. PLoS Genet 2009;5:e1000353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Halbritter J, Porath JD, Diaz KA, et al. GPN Study Group. Identification of 99 novel mutations in a worldwide cohort of 1,056 patients with a nephronophthisis-related ciliopathy. Hum Genet 2013;132:865–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Chanchlani R, Parekh RS. Ethnic differences in childhood nephrotic syndrome. Front Pediatr 2016;4:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Zhuo L, Huang L, Yang Z, et al. A comprehensive analysis of NPHS1 gene mutations in patients with sporadic focal segmental glomerulosclerosis. BMC Med Genet 2019;20:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sharief SN, Hefni NA, Alzahrani WA, et al. Genetics of congenital and infantile nephrotic syndrome. World J Pediatr 2019;15:198–203. [DOI] [PubMed] [Google Scholar]
- [11].Li GM, Cao Q, Shen Q, et al. Gene mutation analysis in 12 Chinese children with congenital nephrotic syndrome. BMC Nephrol 2018;19:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Seeman T, Vondrak K. First report of recurrent nephrotic syndrome after kidney transplantation in a patient with NUP93 gene mutations: a case report. Transplant Proc 2018;50:3954–6. [DOI] [PubMed] [Google Scholar]
- [13].Sandokji I, Marquez J, Ji W, et al. Identification of novel mutations and phenotype in the steroid resistant nephrotic syndrome gene NUP93: a case report. BMC Nephrol 2019;20:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Lim RYH, Fahrenkrog B. The nuclear pore complex up close. Curr Opin Cell Biol 2006;18:342–7. [DOI] [PubMed] [Google Scholar]
- [15].Nofrini V, Giacomo DD, Meccuci C. Nucleoporin genes in human diseases. Eur J Hum Genet 2016;24:1388–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].von Appen A, Kosinski J, Sparks L, et al. In situ structural analysis of the human nuclear pore complex. Nature 2015;526:140–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Magistris PD, Tatarek-Nossol M, Dewor M, et al. A self-inhibitory interaction within Nup155 and membrane binding are required for nuclear pore complex formation. J Cell Sci 2018;131: [DOI] [PubMed] [Google Scholar]
- [18].Alber F, Dokudovskaya S, Veenhoff LM, et al. Determining the architectures of macromolecular assemblies. Nature 2007;450:683–94. [DOI] [PubMed] [Google Scholar]
- [19].Bui KH, von Appen A, DiGuilio AL, et al. Integrated structural analysis of the human nuclear pore complex scaffold. Cell 2013;155:1233–43. [DOI] [PubMed] [Google Scholar]
- [20].Braun DA, Sadowski CE, Kohl S, et al. Mutations in nuclear pore genes NUP93, NUP205 and XPO5 cause steroid-resistant nephrotic syndrome. Nat Genet 2016;48:457–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Bezdicka M, Stolbova S, Seeman T, et al. Genetic diagnosis of steroid-resistant nephrotic syndrome in a longitudinal collection of Czech and Slovak patients: a high proportion of causative variants in NUP93. Pediatr Nephrol 2018;33:1347–63. [DOI] [PubMed] [Google Scholar]
- [22].Bierzynska A, McCarthy HJ, Soderquest K, et al. Genomic and clinical profiling of a national nephrotic syndrome cohort advocates a precision medicine approach to disease management. Kidney Int 2017;91:937–47. [DOI] [PubMed] [Google Scholar]
- [23].Hashimoto T, Harita Y, Takizawa K, et al. In vivo expression of NUP93 and its alteration by NUP93 mutations causing focal segmental glomerulosclerosis. Kidney Int Rep 2019;4:1312–22. [DOI] [PMC free article] [PubMed] [Google Scholar]