

Abstract
The prevalence of both cancer and end-stage renal disease is increasing. In addition, medical advances have meant increased survival rates for both diseases. Many chemotherapeutics are renally excreted, and conversely, renal insufficiency promotes a pro-neoplastic state, including genitourinary and other cancers. Dialysis prolongs life while increasing cancer risk. Proposed oncogenic mechanisms include immune dysfunction, chronic inflammation, changes in gut microbiota and stimulation of the renin–angiotensin system. This review summarizes current concepts in the relationship between cancer and renal insufficiency.
Keywords: cancer, carcinogenesis, end-stage renal disease, malignancy, renal transplant
INTRODUCTION
Both cancer and end-stage renal disease (ESRD) are highly prevalent in the general population. The International Agency for Research on Cancer reported a global incidence for all cancers of 18 million and a 5-year prevalence of >43 million in 2018. Diagnostic and therapeutic advancements in recent decades in the management of cancer patients have led to improved survival, with 26% reduced mortality from cancer in the USA according to the Surveillance, Epidemiology, and End Results (SEER) Cancer Database.
ESRD increases mortality and morbidity through an increased risk of associated cardiovascular disease, diabetes and hypertension. Cancer incidence is reported to be higher in patients with ESRD compared with the general population [1–3], although the mechanisms involved are still not completely understood.
Both cancer and ESRD are more commonly diagnosed in older patients, such that there is overlapping of the two conditions in ageing individuals. Several cancer registry studies have reported [2, 3] a higher incidence of bladder [standardized incidence ratio (SIR) 1.5], kidney (SIR 3.6–4.03), liver, thyroid, head and neck and cervical cancer, as well as a higher incidence of multiple myeloma and non-Hodgkin’s lymphoma. This review summarizes current concepts in the relationship between cancer and kidney disease.
RISK FACTORS
Although the mechanisms are still unknown, specific conditions that have linked ESRD and cancer include the following (Figures 1 and 2):
FIGURE 1.

Mechanisms of renal cell carcinoma; ESRD- end stage renal disease; IL-1b - interleukin 1 beta; IL-6- interleukin 6; TNF-a - tumor necrosis factor alpha; Bcl-2 - B-cell lymphoma 2.
FIGURE 2.

Effects of uremic toxins in ESRD patients; miRNA - microRNA; HD - hemodialysis.
Acquired cystic disease of the kidney has been linked to a higher incidence of both clear cell and papillary renal cell carcinoma (RCC) in chronic kidney disease (CKD) patients [4]. Oya et al. [5] suggested possible activation of the proto-oncogene c-Jun in patients with ESRD, secondary to a chronic cytokine environment. ESRD patients have increased levels of inflammatory cytokines such as interleukin (IL)-1β, IL-6 and tumour necrosis factor (TNF)-α [6] due to oxidative stress in uraemia, leading to c-Jun activation and a higher risk of developing RCC. However, there seems to be a correlation with only low-stage and low-grade RCC. Another possible mechanism involves activation of the hepatocyte growth factor–c-Met pathway, leading to cyst formation and progression towards RCC [7]. Other anti-apoptotic molecules seem to be overexpressed, such as Bcl-2, leading to higher proliferative activity of the cystic epithelium.
Cyclophosphamide (CYC) use in patients with systemic autoimmune diseases can increase the risk of both solid tumours and myeloproliferative disorders [8, 9] due to direct chromosomal damage induced by cytotoxic and induced immunosuppression. Bladder and skin cancers were found to be more prevalent in patients previously treated with CYC compared with controls (P < 0.024). The risk increased at 6 years post-treatment and continued to increase at 20 years [8], suggesting a need for prolonged surveillance in these patients. Moreover, there is a higher risk in male patients and those receiving a higher cumulative dose of CYC.
Viral infections [human papillomavirus (HPV), hepatitis B or C virus (HBV/HCV), Epstein–Barr virus (EBV)] in ESRD patients can increase the risk of cancer. Both haemodialysis (HD) and changes in innate and adaptive immunity [10, 11] are already known to increase the risk of infection in ESRD patients, increasing morbidity and mortality. It has been suggested that ∼20% of HD patients become HBV carriers, whereas the prevalence in the general population is ∼5% [12]. However, only 5% of these patients die from liver disease [13] such as cirrhosis or hepatocellular carcinoma, and other coexisting factors such as co-infection with HCV/human immunodeficiency virus, alcohol abuse and impaired immune response contribute to a more rapid progression of HBV-related disease. According to the Dialysis Outcomes and Practice Patterns Study, the prevalence of HCV is nearly 10% [14], being lower in Western European countries and higher in Middle Eastern countries. Antiviral agents have been successfully used to improve outcomes in HCV patients with CKD, being associated with a significant survival benefit [15]. Both HBV and HCV infections lead to an increased risk of HCC, although the risk is higher with HBV [relative risk (RR) 2.49 versus 1.5] [16, 17]. As ESRD patients are generally immunocompromised due to immunosuppressive therapy used in the prevention of transplant rejection, an increased risk (RR 1.53) [18] of other viral infections such as HPV has been observed. Several studies have reported an increased risk of HPV-related cancers of between 1.8-fold and 8.6-fold [3, 18, 19], suggesting a possible theoretical benefit from HPV vaccination in this population. The prevalence of EBV infections in patients undergoing organ transplantation varies between 16% and 50% [20]. Persistent EBV infection may contribute to Hodgkin’s lymphoma, Burkitt’s lymphoma, nasopharyngeal carcinoma and gastric cancer [21]. Alterations in the nuclear factor-κB (NF-κB) pathway, PI3K (phosphatidylinositol 3-kinase) mutations and CpG methylation of the cell genome have been described in these patients. Moreover, EBV avoids immune surveillance through downregulation of the major histocompatibility complex (MHC) or programmed death-ligand 1 (PD-L1) expression, leading to inactivation of T cells.
Prolonged analgesic use has been linked to a 1.25–1.28 increased risk of kidney cancer in a large meta-analysis that included >8000 RCC patients [22]. One suggested mechanism is the alkylating effect of the accumulation of N-hydroxylated phenacetin metabolites in the kidney [23]. This also leads to stimulation of carcinogenesis in the kidney, ureters and bladder due to prolonged exposure and increased concentration of these substances.
Balkan endemic nephropathy accompanied by urothelial carcinoma after exposure to aristolochic acid (AA) has been described in southeastern European populations [24, 25]. The prevalence of the disease varies between 0.5 and 4.4 in those with a genetic predisposition after exposure to plants of the genera Aristolochia and Asarum. Besides nephrotoxicity, AA has been linked to upper urinary tract cancer. Its carcinogenic potential has been indicated by the overexpression of p53 in these patients, suggesting a role for p53 gene mutations and the presence of AA DNA adducts in the kidney [26, 27].
Genetic factors. Inherited genetic mutations lead to 10% of all cancers, thus a detailed assessment of the family cancer history may indicate a hereditary cancer syndrome. RCC can be linked to a VHL gene abnormality in 1 of 36 000 individuals [28] who are at risk of developing multiple renal cysts and multicentric and bilateral clear cell carcinoma of the kidney. Other genetic alterations, such as the BRCA1-associated protein-1 mutation responsible for encoding a nuclear deubiquitinase, polybromo-1 (PBRM1) gene mutation, inactivation of histone-modifying genes or alterations of the ubiquitin-mediated proteolysis pathway have been described in RCC [29–32]. The Cancer Genome Atlas Research Network documented MET germline mutations in patients with type 1 papillary RCC [33] and fumarate hydratase germline mutations in type 2 papillary RCC.
Ultraviolet light exposure has been linked to a 7.43% cumulative incidence of non-melanoma skin cancer [squamous cell carcinoma (SCC)] in organ transplant recipients [34] under immunosuppressive treatment. Induction of oxidative stress and DNA damage has been documented in patients on azathioprine, and a lower incidence of SCC has been observed with mammalian target of rapamycin–based immunosuppressants.
Glomerulonephritis (GN) in a study by Ryu et al. [35] has been associated with an increased risk of malignancy (colon cancer, lung cancer, multiple myeloma and Kaposi’s sarcoma) via mechanisms that are not fully understood. The observed:expected cancer ratio varied between 0.8 and 28.57, being highest with amyloidosis. Traditionally GN has been considered as a paraneoplastic syndrome associated with cancer diagnosis, however, the use of immunosuppressive drugs interfering with the immune system may explain the higher observed cancer risk [36].
Retrospective studies [2, 3] suggest a higher risk of cancer of the bladder (SIR 1.5) and kidney or renal pelvis (SIR 3.6–4.3) among ESRD patients. In a retrospective cohort study of Taiwanese dialysis patients [37], the highest cancer risk was in the younger population, unlike in the previously mentioned studies. A meta-analysis of prospective studies [38] suggests a correlation between renal function decline and a trend towards an increased risk of urinary tract cancers {hazard ratio [HR] 2.34 [95% confidence interval (CI) 1.31–4.18]; P = 0.06}.
MECHANISMS INVOLVED IN CARCINOGENESIS
The management of cancer is often complicated by coexisting ESRD, and there is interest in early detection of this overlap population whose survival is doubly negatively impacted (Figures 2 and 3).
FIGURE 3.
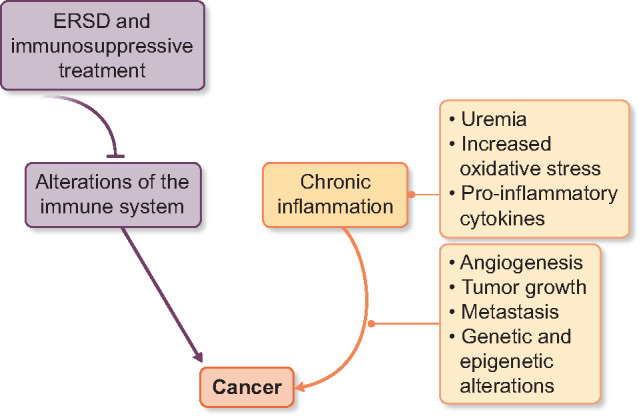
Chronic inflammation and cancer; ESRD - end stage renal disease.
Studies reporting an increased cancer incidence in ESRD patients [39–41] offer valuable epidemiologic information regarding cancer risk in this specific population. These observational studies show an increased incidence of colon and lung cancers after transplantation, as well as virus-associated cancers, possibly as a result of immunosuppressive drugs; endocrine-related cancers such as breast, prostate and ovarian cancers have a similar rate; several cancers related to kidney dysfunction are more frequently diagnosed, such as kidney and urinary tract cancers, myeloma and lymphoma; geographic differences in viral patterns may influence cancer risk in some areas; and differences in cancer screening strategies may also impact cancer diagnosis and influence reported outcomes in these studies.
Immune dysfunction in ESRD
Both innate and adaptive immunity play a major role in immune surveillance, with different specificity of recognition and speed of response. The innate immune response is less specific in target recognition but compensates in the speed of the response. On the other side, the adaptive immune response has a higher target specificity but offers a more delayed, but durable, response. Cancer cells can elude the immune system by blocking the ‘immune checkpoint molecules’, such as cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), programme cell death protein 1 (PD-1) and PD-L1 expression by tumour cells, which represents an important mechanism of tumour immune evasion. Modulating the immune response with checkpoint inhibitors has been recognized as a standard of care for many types of cancers in the last decade.
In ESRD patients, changes in the immune system have been observed in the context of both the condition itself and the immunosuppressive treatment. In the context of decreased renal clearance, increased oxidative stress and volume overload, hypercytokinaemia has been observed [42, 43]. Moreover, uraemia has also been associated with immunosuppression, as demonstrated by changes in immunocompetent cells. In terms of innate immunity changes, an increased production of pro-inflammatory cytokines has been described, such as IL-1β, IL-6 and TNF-α, as a consequence of decreased renal function [43]. Chronic inflammation has also been observed, as neutrophils are more prone to apoptosis, as a direct effect of the uraemic milieu [44].
A reduced immunogenic response has been observed in ESRD patients undergoing vaccination [45], as a consequence of the uraemic milieu [46]. Only 50–75% of HD patients who receive the usual HBV vaccine develop protective antibody levels, compared with 90% in the general population. This has also been observed when HD patients were vaccinated against influenza virus, Clostridium tetani and Corynebacterium diphtheriae [45]. The most important adaptive immune system representatives are B cells and T helper (Th) lymphocytes. Th lymphocytes activate macrophages and neutrophils and also play a role in promoting humoral immunity. In HD patients, there is an altered Th1:Th2 ratio in favour of Th1 due to increased Th1 differentiation induced by the production of IL-2 [47, 48]. Another change in the immune system in HD patients is B cell lymphopenia due to increased apoptosis [49].
Such an imbalance has been correlated with the development of SCC in post-transplant patients [50, 51]. Due to impairment of dendritic cells, natural killer cells and effector T cells, combined with an increased expression of CD57hi immunosenescent CD8+ T cells (cells with impaired cytotoxic activity), the immune surveillance in HD patients is altered. A possible role of γσ T cells in SCC carcinogenesis has been suggested by Rei et al. [52], who observed a direct association between cancer incidence and the number of γσ T cells. The authors suggested patient stratification according to the CD8+CD57hi phenotype when assessing cancer risk in post-transplant patients.
Chronic inflammation in ESRD/inflammation and oxidative stress
Various transcriptional and epigenetic changes can contribute to the progression of renal damage through oxidative stress, inflammation and uraemic toxins, as identified in genome-wide association studies.
HD may cause acquired immunodeficiency over time [53]. The dialysis membrane used for the procedure and the bacterial products present in the dialysis water may abnormally activate a series of immune cells, such as monocytes. This phenomenon has been mostly observed with cellulose membranes, compared with non-cellulose membranes, and was dependent on interaction with the dialysis membrane. The authors also observed an increased expression of adhesion molecules such as CD18, CD49, CD54 and the lipopolysaccharide ligand CD14. Moreover, HD induces the expression of complement factors and endotoxins, further contributing to cellular activation, with several consequences such as cytokine secretion and induced apoptosis of cells coming into contact with the membrane. As back-filtration and back-diffusion of contaminated dialysate have been linked to cytokine production during the process, the use of ultrapure dialysis fluid may decrease IL-6 and C-reactive protein (CRP) levels, thus reducing the inflammatory process [54, 55]. Compared with HD, peritoneal dialysis (PD) seems to induce a modest inflammatory response [56], although the prevalence of chronic inflammation is similar in both [57]. This is probably due to the fact that PD relies on the use of a physiological membrane, not an artificial one.
Genetic factors are involved in the development of chronic inflammation, which explains variations in the prevalence and consequences of inflammation in various patient groups. Studies of patients of Asian descent showed a lower predisposition towards chronic inflammation, along with better survival outcome [58, 59]. Szalai et al. [60] identified a single-nucleotide polymorphism (SNP) in the CRP gene that caused increased CRP levels in Caucasians compared with Asian populations. Decreased IL-10 production associated with a SNP can also lead to a more active inflammatory response [61], while a SNP in the LGALS2 gene that encodes galectin-2 was found to have a regulatory role in inflammation [62]. Although genetic polymorphism suggests differences in inflammatory response, it has not been established yet if this translates into differences in inflammatory response during dialysis treatment in both HD and PD.
There is growing evidence that suggests increased levels of oxidative stress in CKD patients, especially in later stages, where there seems to be an inverse correlation between the two [63], promoting inflammation. Moreover, HD can decrease antioxidant levels during the procedure, favouring the accumulation of various oxidative products [64]. Therefore it has been speculated that supplementation of various antioxidants (vitamins E and C) may reduce oxidative stress in these patients, although interventional studies have been inconclusive [65]. Increased oxidative damage to DNA, such as single- or double-strand breakage, base modifications or DNA cross-linking, may increase the mutational load and play an important role in carcinogenesis [66, 67]. Activation of the Ras signalling pathway, as well as c-Myc overexpression, can increase genomic instability and promote DNA damage [68, 69].
During carcinogenesis, the chronic inflammatory milieu can influence the immune response and induce angiogenesis, tumour growth and metastasis [70]. Further genomic instability and accumulation of mutations lead to cancer due to oncogene activation and tumour suppressor gene inactivation. Several pathways are altered in most solid tumours, some being driven by inflammatory conditions, leading to a malignant phenotype [71].
Accumulation of genetic and epigenetic alterations induced by either environmental factors or chronic inflammation causes aberrant DNA methylation [72], and oncogenesis can be further stimulated by various carcinogenic exposures [73]. An epigenome-wide association study associated DNA methylation level in specific cytosine–phosphate–guanine sites with a reduction in estimated glomerular filtration rate, incidence of CKD and renal fibrosis in patients from the Atherosclerosis Risk in Communities and Framingham Heart Studies [74]. Moreover, increased levels of DNA methylation in certain genes, such as connective tissue growth factor, methylenetetrahydrofolate reductase (MTHFR) and insulin like growth factor binding protein 1 (IGFBP1), have been observed in ESRD patients [75–77]. Other mechanisms involved in the development of CKD include histone crotonylation and acetylation that can contribute to the inflammatory process [78] or higher levels of pro-inflammatory microRNA (miRNA) such as miR-34 [79]. Of note, miRNA and DNA methylation have been studied as biomarkers of CKD progression and these may become treatment targets in the future.
Gut microbiota and inflammation
Gut microbiota are influenced by and maintain equilibrium via nutrition, metabolism and the immune system [80, 81]. Microbiota evolve with the host over time, with variations in genome, diet and lifestyle factors [82]. The Human Microbiome Project Consortium used next-generation sequencing and metagenomics in order to characterize the diversity of the human microbiome, mapping the microbial signature of normal individuals and highlighting the important role in various physiological functions and its implications in various diseases [83, 84]. As gut microbiota represent a dynamic system, it has been suggested that alterations in commensal flora can contribute to the pathogenesis of various conditions, including CKD and cancer. Although several studies have focused on describing the microbiome composition and its impact on human health, there is a need for further research corroborated by results from larger clinical trials [85].
In CKD patients, the uraemic milieu influences the intestinal barrier, promoting chronic inflammation and translocation of bacteria and endotoxins across the intestinal wall. In turn, there is a decreased population of beneficial bacteria that produce short-chain fatty acids and an increase in uraemic toxin–producing bacteria [86], generating bound uraemic solutes produced by fermentation in the large intestine [87]. The continuous gut production of endotoxin (constituents of the outer membrane of Gram-negative bacteria) triggers an immune response through the activation of macrophages and endothelial cells, contributing to chronic inflammation in CKD patients [88, 89].
In ESRD patients, there is a bidirectional relationship between the microbiota and the uraemic milieu, as the uraemic milieu has a negative impact on the microbiota on the one hand and, on the other hand, alterations in the microbiota lead to uraemic toxin production [90]. As a consequence, dietary interventions in CKD patients seem to provide benefits in reducing uraemic toxin generation by both the microbiota and the disease itself.
The relationship between gut microbiota and cancer has been intensely studied and a link has been described with gastrointestinal tumours through either oncogene activation or tumour suppressor gene inactivation. A ‘gut–brain axis’ has been described, suggesting a crosstalk between the enteroendocrine system and the central nervous system, the hypothalamic–pituitary–adrenal axis, the autonomic nervous system and the enteric system [91]. The microbiome can modulate host metabolism, the immune system, xenobiotics and drug metabolism [92–94], being involved in both carcinogenesis through dysbiosis and inflammation and the modulation of treatment response in cancer patients treated with immunotherapy. Treating dysbiosis with probiotics or faecal microbiota transplantation has proven beneficial in preclinical studies, with some authors recommending integrating probiotics alongside anticancer therapy in order to modulate treatment response [95, 96].
Given the increased risk of gastrointestinal tumours in ESRD patients, one possible mechanism could be long-term alterations in gut microbiota, promoting carcinogenesis in this population. However, this association has not been confirmed yet in preclinical or clinical studies and further research is needed, especially in the context of possible modulation of dysbiosis with dietary interventions.
Intrarenal renin–angiotensin system (RAS) activation as a hallmark of cancer
The RAS regulates haemodynamics [97] via renal sodium transport under both physiological and pathological conditions. However, preclinical studies have suggested that RAS is also involved in various other functions such as cell cycle regulation and angiogenesis inhibition [98]. In ESRD patients, higher intrarenal angiotensin II levels and angiotensin II receptor type 1 (AT1R) expression have been observed, secondary to RAS activation, contributing to renal damage such as tubulointerstitial fibrosis [99].
The extravascular impact of RAS may be oncogenic. Several acquired properties of cancer cells, as defined in 2000 by Hanahan and Weinberg [100], include angiogenesis, cell proliferation, metastasis and alteration of metabolism and the microenvironment. The effects of the RAS on cancer cells seem to favour the described hallmarks. However, it is still unclear if RAS alterations are secondary to cancer development or if they play a role in carcinogenesis.
RAS signalling induces receptor-mediated cell proliferation and differentiation in vascular smooth muscle cells and breast, pituitary, adrenocortical and endometrial cells [101, 102]. One of the possible mechanisms by which angiotensin peptides are involved in cellular growth is through mitogen-activated protein kinase kinase (MEK) and PI3K signalling [103], which are important signalling pathways overexpressed in various cancers. The receptor-mediated signalling of the RAS may be dependent on both the overexpression and the type of receptor, as AT1R is mainly responsible for cell proliferation and angiogenesis, whereas angiotensin II receptor type 2 (AT2R) has inhibitory effects, thus stimulating apoptosis [103].
Arrieta et al. [104] analysed the expression of AT1R and AT2R in high-grade astrocytoma, correlating receptor expression with the mitotic index and vascular density, suggesting a co-dependent relationship between the two receptors. Inhibition of AT1R may result in AT2R overexpression and activation of cell death through apoptosis.
RAS activation through AT1R may favour cancer cell survival through activation of the PI3K–Akt pathway, suppression of the caspase pathway and production of anti-apoptotic Bcl proteins [105]. It is involved in reshaping the tumour microenvironment through pro-inflammatory and pro-angiogenic signalling, along with generation of reactive oxygen species involved in DNA damage.
The RAS in tumour invasion and metastasis has been suggested by two trials that included gastric cancer [106] and renal cell cancer patients [107], where AT1R overexpression could have an impact on tumour behaviour, suggesting a possible link between RAS and tumour capacity for metastasis. Keizman et al. [107] retrospectively analysed the possible effects of adding an angiotensin-converting enzyme (ACE) inhibitor to sunitinib. The combination significantly improved patient outcome, with a 7-month progression-free survival benefit (HR 0.537; P = 0.0055), suggesting that adding an ACE inhibitor may decrease the expression of vascular endothelial growth factor and inhibit the growth factor receptor through a secondary mechanism.
Genetic determinants have been associated with an increased risk of cancer. In a population-based study, van der Knaap et al. [108] suggested that carriers of the high-activity DD genotype have an increased risk of breast cancer compared with those with the low-activity II/ID genotype [HR 1.47 (95% CI 1.05–2.04)]. Moreover, DD carriers who were exposed to long-term high-dose medication were at lower risk of cancer [HR 0.28 (95% CI 0.10–0.79)] and short-term high-dose patients had a higher risk of colorectal cancer progression in the II/ID stratum [HR 3.83 (95% CI 1.67–8.79)].
CONCLUSIONS
Mechanisms related to immune dysfunction and inflammation have been suggested to be involved in promoting cancer in ESRD patients. Current data focus on the role the microbiota and uraemic milieu play in modulating inflammation in CKD patients and on understanding their role in cancer initiation. In the ‘-omics’ era, identification of predictive biomarkers can better stratify patients at risk and incorporate better surveillance or preventive strategies for the CKD patient.
Onconephrology has already been recognized as an emerging field, due to the importance of a multidisciplinary approach of ESRD patients with cancer. Complex management strategies for these patients have influenced research strategies that have led to a better understanding of the underlying mechanisms of cancer in these patients. However, there is still an unmet need in the field, as the mechanisms involved in cancer initiation and progression in this particular setting are still not fully understood. The focus of future research will likely include preventive strategies that can lead to improved cancer screening and treatment in patients with CKD.
CONFLICT OF INTEREST STATEMENT
None declared.
REFERENCES
- 1. Wong G, Hayen A, Chapman JR. et al. Association of CKD and cancer risk in older people. J Am Soc Nephrol 2009; 20: 1341–1350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Butler AM, Olshan AF, Kshirsagar AV. et al. Cancer incidence among US Medicare ESRD patients receiving hemodialysis, 1996–2009. Am J Kidney Dis 2015; 65: 763–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Maisonneuve P, Agodoa L, Gellert R. et al. Cancer in patients on dialysis for end-stage renal disease: an international collaborative study. Lancet 1999; 354: 93–99 [DOI] [PubMed] [Google Scholar]
- 4. Sun Y, Argani P, Tickoo S. et al. Acquired cystic disease-associated renal cell carcinoma (ACKD-RCC)-like cysts. Am J Surg Pathol 2018; 42: 1396–1401 [DOI] [PubMed] [Google Scholar]
- 5. Oya M, Mikami S, Mizuno R. et al. c-Jun activation in acquired cystic kidney disease and renal cell carcinoma. J Urol 2005; 174: 726–730 [DOI] [PubMed] [Google Scholar]
- 6. Herbelin A, Ureña P, Nguyen A. et al. Elevated circulating levels of interleukin-6 in patients with chronic renal failure. Kidney Int 1991; 39: 954–960 [DOI] [PubMed] [Google Scholar]
- 7. Konda R, Sato H, Hatafuku F. et al. Expression of hepatocyte growth factor and its receptor c-Met in acquired renal cystic disease associated with renal cell carcinoma. J Urol 2004; 171: 2166–2170 [DOI] [PubMed] [Google Scholar]
- 8. Radis CD, Kahl LE, Baker GL. et al. Effects of cyclophosphamide on the development of malignancy and on long-term survival of patients with rheumatoid arthritis a 20-year followup study. Arthritis Rheum 1995; 38: 1120–1127 [DOI] [PubMed] [Google Scholar]
- 9. Bernatsky S. Hematologic malignant neoplasms after drug exposure in rheumatoid arthritis. Arch Intern Med 2008; 168: 378–381 [DOI] [PubMed] [Google Scholar]
- 10. Lewis SL, Van Epps DE, Chenoweth DE. et al. Alterations in chemotactic factor-induced responses of neutrophils and monocytes from chronic dialysis patients. Clin Nephrol 1988; 30: 63–72 [PubMed] [Google Scholar]
- 11. Eleftheriadis T, Papazisis K, Kortsaris A. et al. Impaired T cell proliferation and zeta chain phosphorylation after stimulation with Staphylococcal enterotoxin-B in hemodialysis patients. Nephron Clin Pract 2004; 96: c15–c20 [DOI] [PubMed] [Google Scholar]
- 12. Burdick RA, Bragg-Gresham JL, Woods JD. et al. Patterns of hepatitis B prevalence and seroconversion in hemodialysis units from three continents: the DOPPS. Kidney Int 2003; 63: 2222–2229 [DOI] [PubMed] [Google Scholar]
- 13. Harnett JD, Parfrey PS, Kennedy M. et al. The long-term outcome of hepatitis B infection in hemodialysis patients. Am J Kidney Dis 1988; 11: 210–213 [DOI] [PubMed] [Google Scholar]
- 14. Jadoul M, Bieber BA, Martin P. et al. Prevalence, incidence, and risk factors for hepatitis C virus infection in hemodialysis patients. Kidney Int 2019; 95: 939–947 [DOI] [PubMed] [Google Scholar]
- 15. Söderholm J, Millbourn C, Büsch K. et al. Higher risk of renal disease in chronic hepatitis C patients: antiviral therapy survival benefit in patients on hemodialysis. J Hepatol 2018; 68: 904–911 [DOI] [PubMed] [Google Scholar]
- 16. Fabrizi F, Martin P, Dixit V. et al. HBsAg seropositive status and survival after renal transplantation: meta-analysis of observational studies. Am J Transplant 2005; 5: 2913–2921 [DOI] [PubMed] [Google Scholar]
- 17. Fabrizi F, Martin P, Dixit V. et al. Effect of hepatitis C virus infection on mortality in dialysis. Aliment Pharmacol Ther 2004; 20: 1271–1277 [DOI] [PubMed] [Google Scholar]
- 18. Skov Dalgaard L, Fassel U, Østergaard LJ. et al. Risk of human papillomavirus-related cancers among kidney transplant recipients and patients receiving chronic dialysis: an observational cohort study. BMC Nephrol 2013; 14: 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Birkeland SA, Storm HH, Lamm LU. et al. Cancer risk after renal transplantation in the Nordic countries, 1964–1986. Int J Cancer 1995; 60: 183–189 [DOI] [PubMed] [Google Scholar]
- 20. Morton M, Coupes B, Roberts SA. et al. Epstein–Barr virus infection in adult renal transplant recipients. Am J Transplant 2014; 14: 1619–1629 [DOI] [PubMed] [Google Scholar]
- 21. Farrell PJ. Epstein-Barr virus and cancer. Annu Rev Pathol Mech Dis 2019; 14: 29–53 [DOI] [PubMed] [Google Scholar]
- 22. Choueiri TK, Je Y, Cho E. et al. Analgesic use and the risk of kidney cancer: a meta-analysis of epidemiologic studies. Int J Cancer 2014; 134: 384–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McCredie M, Stewart JH, Carter JJ. et al. Phenacetin and papillary necrosis: independent risk factors for renal pelvic cancer. Kidney Int 1986; 30: 81–84 [DOI] [PubMed] [Google Scholar]
- 24. Čeović S, Hrabar A, Šarić M.. Epidemiology of Balkan endemic nephropathy. Food Chem Toxicol 1992; 30: 183–188 [DOI] [PubMed] [Google Scholar]
- 25. Stefanovic V, Toncheva D, Atanasova S. et al. Etiology of Balkan endemic nephropathy and associated urothelial cancer. Am J Nephrol 2006; 26: 1–11 [DOI] [PubMed] [Google Scholar]
- 26. Cosyns J-P, Jadoul M, Squifflet J-P. et al. Urothelial malignancy in nephropathy due to Chinese herbs. Lancet 1994; 344: 188. [DOI] [PubMed] [Google Scholar]
- 27. Lord GM, Cook T, Arit VM. et al. Urothelial malignant disease and Chinese herbal nephropathy. Lancet 2001; 358: 1515–1516 [DOI] [PubMed] [Google Scholar]
- 28. Lonser RR, Glenn GM, Walther M. et al. von Hippel–Lindau disease. Lancet 2003; 361: 2059–2067 [DOI] [PubMed] [Google Scholar]
- 29. Peña-Llopis S, Vega-Rubín-de-Celis S, Liao A. et al. BAP1 loss defines a new class of renal cell carcinoma. Nat Genet 2012; 44: 751–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Varela I, Tarpey P, Raine K. et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature 2011; 469: 539–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dalgliesh GL, Furge K, Greenman C. et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature 2010; 463: 360–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Guo G, Gui Y, Gao S. et al. Frequent mutations of genes encoding ubiquitin-mediated proteolysis pathway components in clear cell renal cell carcinoma. Nat Genet 2012; 44: 17–19 [DOI] [PubMed] [Google Scholar]
- 33.Linehan WM, Spellman PT, Ricketts CJ et al. Comprehensive molecular characterization of papillary renal-cell carcinoma. N Engl J Med 2016; 374: 135–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ulrich C, Stockfleth E.. Azathioprine, UV light, and skin cancer in organ transplant patients—do we have an answer? Nephrol Dial Transplant 2007; 22: 1027–1029 [DOI] [PubMed] [Google Scholar]
- 35. Ryu J, Ryu H, Kim S. et al. Comparison of cancer prevalence between patients with glomerulonephritis and the general population at the time of kidney biopsy. PLoS One 2019; 14: e0224024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pani A, Porta C, Cosmai L. et al. Glomerular diseases and cancer: evaluation of underlying malignancy. J Nephrol 2016; 29: 143–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lin HF, Li YH, Wang CH. et al. Increased risk of cancer in chronic dialysis patients: a population-based cohort study in Taiwan. Nephrol Dial Transplant 2012; 27: 1585–1590 [DOI] [PubMed] [Google Scholar]
- 38. Wong G, Staplin N, Emberson J. et al. Chronic kidney disease and the risk of cancer: an individual patient data meta-analysis of 32,057 participants from six prospective studies. BMC Cancer 2016; 16: 488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vajdic CM, McDonald SP, McCredie MRE. et al. Cancer incidence before and after kidney transplantation. JAMA 2006; 296: 2823–2831 [DOI] [PubMed] [Google Scholar]
- 40. Stewart JH, Vajdic CM, van Leeuwen MT. et al. The pattern of excess cancer in dialysis and transplantation. Nephrol Dial Transplant 2009; 24: 3225–3231 [DOI] [PubMed] [Google Scholar]
- 41. Yanik EL, Clarke CA, Snyder JJ. et al. Variation in cancer incidence among end-stage renal disease patients during kidney function and non-function intervals. J Am Soc Nephrol 2016; 27: 1495–1504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kimmel PL, Phillips TM, Simmens SJ. et al. Immunologic function and survival in hemodialysis patients. Kidney Int 1998; 54: 236–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Stenvinkel P, Ketteler M, Johnson RJ. et al. IL-10, IL-6, and TNF-α: central factors in the altered cytokine network of uremia—the good, the bad, and the ugly. Kidney Int 2005; 67: 1216–1233 [DOI] [PubMed] [Google Scholar]
- 44. Cendoroglo M, Jaber BL, Balakrishnan VS. et al. Neutrophil apoptosis and dysfunction in uremia. J Am Soc Nephrol 1999; 10: 93–100 [DOI] [PubMed] [Google Scholar]
- 45. Eleftheriadis T, Antoniadi G, Liakopoulos V. et al. Disturbances of acquired immunity in hemodialysis patients. Semin Dial 2007; 20: 440–451 [DOI] [PubMed] [Google Scholar]
- 46. Meuer SC, Hauer M, Kurz P. et al. Selective blockade of the antigen receptor-mediated pathway of T cell activation in patients with impaired primary immune responses. J Clin Invest 1987; 80: 743–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ando M, Shibuya A, Yasuda M. et al. Impairment of innate cellular response to in vitro stimuli in patients on continuous ambulatory peritoneal dialysis. Nephrol Dial Transplant 2005; 20: 2497–2503 [DOI] [PubMed] [Google Scholar]
- 48. Sester U, Sester M, Hauk M. et al. T-cell activation follows Th1 rather than Th2 pattern in haemodialysis patients. Nephrol Dial Transplant 2000; 15: 1217–1223 [DOI] [PubMed] [Google Scholar]
- 49. Fernandez-Fresnedo G, Ramos MA, González-Pardo MC. et al. B lymphopenia in uremia is related to an accelerated in vitro apoptosis and dysregulation of Bcl-2. Nephrol Dial Transplant 2000; 15: 502–510 [DOI] [PubMed] [Google Scholar]
- 50. Bottomley M, Harden P, Wood K.. CD81 immunosenescence predicts post-transplant cutaneous squamous carcinoma in high-risk patients. J Am Soc Nephrol 2016; 27: 1505–1515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Carroll RP, Segundo DS, Hollowood K. et al. Immune phenotype predicts risk for posttransplantation squamous cell carcinoma. J Am Soc Nephrol 2010; 21: 713–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rei M, Pennington DJ, Silva-Santos B.. The emerging protumor role of γδ T lymphocytes: implications for cancer immunotherapy. Cancer Res 2015; 75: 798–802 [DOI] [PubMed] [Google Scholar]
- 53. Carracedo J, Ramírez R, Madueño JA et al. Cell apoptosis and hemodialysis-induced inflammation. Kidney Int Suppl 2002; 80: 89–93 [DOI] [PubMed] [Google Scholar]
- 54. Panichi V. Cytokine production in haemodiafiltration: a multicentre study. Nephrol Dial Transplant 1998; 13: 1737–1744 [DOI] [PubMed] [Google Scholar]
- 55. Schiffl H, Lang SM, Stratakis D. et al. Effects of ultrapure dialysis fluid on nutritional status and inflammatory parameters. Nephrol Dial Transplant 2001; 16: 1863–1869 [DOI] [PubMed] [Google Scholar]
- 56. Haubitz M, Brunkhorst R, Wrenger E. et al. Chronic induction of C-reactive protein by hemodialysis, but not by peritoneal dialysis therapy. Perit Dial Int 1996; 16: 158–162 [PubMed] [Google Scholar]
- 57. Chung SH, Heimburger O, Stenvinkel P. et al. Association between residual renal function, inflammation and patient survival in new peritoneal dialysis patients. Nephrol Dial Transplant 2003; 18: 590–597 [DOI] [PubMed] [Google Scholar]
- 58. Xia B, Crusius JB, Wu J. et al. CTLA4 gene polymorphisms in Dutch and Chinese patients with inflammatory bowel disease. Scand J Gastroenterol 2002; 37: 1296–1300 [DOI] [PubMed] [Google Scholar]
- 59. Wong JS, Port FK, Hulbert-Shearon TE. et al. Survival advantage in Asian American endstage renal disease patients. Kidney Int 1999; 55: 2515–2523 [DOI] [PubMed] [Google Scholar]
- 60. Szalai AJ, McCrory MA, Cooper GS. et al. Association between baseline levels of C-reactive protein (CRP) and a dinucleotide repeat polymorphism in the intron of the CRP gene. Genes Immun 2002; 3: 14–19 [DOI] [PubMed] [Google Scholar]
- 61. Girndt M, Kaul H, Sester U. et al. Anti-inflammatory interleukin-10 genotype protects dialysis patients from cardiovascular events. Kidney Int 2002; 62: 949–955 [DOI] [PubMed] [Google Scholar]
- 62. Ozaki K, Inoue K, Sato H. et al. Functional variation in LGALS2 confers risk of myocardial infarction and regulates lymphotoxin-α secretion in vitro. Nature 2004; 429: 72–75 [DOI] [PubMed] [Google Scholar]
- 63. Yilmaz MI, Saglam M, Caglar K. et al. The determinants of endothelial dysfunction in CKD: oxidative stress and asymmetric dimethylarginine. Am J Kidney Dis 2006; 47: 42–50 [DOI] [PubMed] [Google Scholar]
- 64. Loughrey CM, Young IS, Lightbody JH. et al. Oxidative stress in haemodialysis. QJM 1994; 87: 679–683 [PubMed] [Google Scholar]
- 65. Antoniadi G, Eleftheriadis T, Liakopoulos V. et al. Effect of one-year oral α-tocopherol administration on the antioxidant defense system in hemodialysis patients. Therapher Dial 2008; 12: 237–242 [DOI] [PubMed] [Google Scholar]
- 66. Klaunig JE, Kamendulis LM.. The role of oxidative stress in carcinogenesis. Annu Rev Pharmacol Toxicol 2004; 44: 239–267 [DOI] [PubMed] [Google Scholar]
- 67. Bhattacharyya A, Chattopadhyay R, Mitra S. et al. Oxidative stress: an essential factor in the pathogenesis of gastrointestinal mucosal diseases. Physiol Rev 2014; 94: 329–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Felsher DW, Bishop JM.. Transient excess of MYC activity can elicit genomic instability and tumorigenesis. Proc Natl Acad Sci USA 1999; 96: 3940–3944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Irani K, Xia Y, Zweier JL. et al. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science 1997; 275: 1649–1652 [DOI] [PubMed] [Google Scholar]
- 70. Fox P, Hudson M, Brown C. et al. Markers of systemic inflammation predict survival in patients with advanced renal cell cancer. Br J Cancer 2013; 109: 147–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Mantovani A, Garlanda C, Allavena P.. Molecular pathways and targets in cancer-related inflammation. Ann Med 2010; 42: 161–170 [DOI] [PubMed] [Google Scholar]
- 72. Niwa T, Tsukamoto T, Toyoda T. et al. Inflammatory processes triggered by Helicobacter pylori infection cause aberrant DNA methylation in gastric epithelial cells. Cancer Res 2010; 70: 1430–1440 [DOI] [PubMed] [Google Scholar]
- 73. Yamashita S, Kishino T, Takahashi T. et al. Genetic and epigenetic alterations in normal tissues have differential impacts on cancer risk among tissues. Proc Natl Acad Sci USA 2018; 115: 1328–1333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Chu AY, Tin A, Schlosser P. et al. Epigenome-wide association studies identify DNA methylation associated with kidney function. Nat Commun 2017; 8: 1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zhang HAO, Cai XU, Yi BIN. et al. Correlation of CTGF gene promoter methylation with CTGF expression in type 2 diabetes mellitus with or without nephropathy. Mol Med Rep 2014; 9: 2138–2144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Ghattas M, El-Shaarawy F, Mesbah N. et al. DNA methylation status of the methylenetetrahydrofolate reductase gene promoter in peripheral blood of end-stage renal disease patients. Mol Biol Rep 2014; 41: 683–688 [DOI] [PubMed] [Google Scholar]
- 77. Gu T, Falhammar H, Gu HF. et al. Epigenetic analyses of the insulin-like growth factor binding protein 1 gene in type 1 diabetes and diabetic nephropathy. Clin Epigenet 2014; 6: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Miao F, Gonzalo IG, Lanting L. et al. In vivo chromatin remodeling events leading to inflammatory gene transcription under diabetic conditions. J Biol Chem 2004; 279: 18091–18097 [DOI] [PubMed] [Google Scholar]
- 79. Amrouche L, Desbuissons G, Rabant M. et al. MicroRNA-146a in human and experimental ischemic AKI: CXCL8-dependent mechanism of action. J Am Soc Nephrol 2017; 28: 479–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Hooper LV, Gordon JI.. Commensal host–bacterial relationships in the gut. Science 2001; 292: 1115–1118 [DOI] [PubMed] [Google Scholar]
- 81. Bäckhed F, Ley RE, Sonnenburg JL. et al. Host–bacterial mutualism in the human intestine. Science 2005; 307: 1915–1920 [DOI] [PubMed] [Google Scholar]
- 82. Nicholson JK, Holmes E, Kinross J. et al. Host–gut microbiota metabolic interactions. Science 2012; 336: 1262–1267 [DOI] [PubMed] [Google Scholar]
- 83. Human Microbiome Project Consortium. A framework for human microbiome research. Nature 2012; 486: 215–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Hattori M. Advanced technologies for the human gut microbiome analysis. Jpn J Clin Immun 2014; 37: 412–422 [DOI] [PubMed] [Google Scholar]
- 85. Rothschild D, Weissbrod O, Barkan E. et al. Environment dominates over host genetics in shaping human gut microbiota. Nature 2018; 555: 210–215 [DOI] [PubMed] [Google Scholar]
- 86. Lau WL, Savoj J, Nakata MB. et al. Altered microbiome in chronic kidney disease: systemic effects of gut-derived uremic toxins. Clin Sci 2018; 132: 509–522 [DOI] [PubMed] [Google Scholar]
- 87. Meijers BKI, Evenepoel P.. The gut–kidney axis: indoxyl sulfate, p-cresyl sulfate and CKD progression. Nephrol Dial Transplant 2011; 26: 759–761 [DOI] [PubMed] [Google Scholar]
- 88. McIntyre CW, Harrison LEA, Eldehni MT. et al. Circulating endotoxemia: a novel factor in systemic in flammation and cardiovascular disease in chronic kidney disease. Clin J Am Soc Nephrol 2011; 6: 133–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Feroze U, Kalantar-Zadeh K, Sterling KA. et al. Examining associations of circulating endotoxin with nutritional status, inflammation, and mortality in hemodialysis patients. J Ren Nutr 2012; 22: 317–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Evenepoel P, Poesen R, Meijers B.. The gut–kidney axis. Pediatr Nephrol 2017; 32: 2005–2014 [DOI] [PubMed] [Google Scholar]
- 91. Carabotti M, Scirocco A, Maselli MA. et al. The gut–brain axis: interactions between enteric microbiota, central and enteric nervous systems. Ann Gastroenterol 2015; 28: 203–209 [PMC free article] [PubMed] [Google Scholar]
- 92. Gensollen T, Iyer SS, Kasper DL. et al. How colonization by microbiota in early life shapes the immune system. Science 2016; 352: 539–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Bultman SJ. Emerging roles of the microbiome in cancer. Carcinogenesis 2014; 35: 249–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Schmidt TSB, Raes J, Bork P.. The human gut microbiome: from association to modulation. Cell 2018; 172: 1198–1215 [DOI] [PubMed] [Google Scholar]
- 95. Larkin J, Hodi FS, Wolchok JD.. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 2015; 373: 1270–1271 [DOI] [PubMed] [Google Scholar]
- 96. Kakihana K, Fujioka Y, Suda W. et al. Fecal microbiota transplantation for patients with steroid-resistant acute graft-versus-host disease of the gut. Blood 2016; 128: 2083–2088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Kobori H, Urushihara M.. Augmented intrarenal and urinary angiotensinogen in hypertension and chronic kidney disease. Pflugers Arch 2013; 465: 3–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Vincent F, Bonnin P, Clemessy M. et al. Angiotensinogen delays angiogenesis and tumour growth of hepatocarcinoma in transgenic mice. Cancer Res 2009; 69: 2853–2860 [DOI] [PubMed] [Google Scholar]
- 99. Ohashi N, Isobe S, Ishigaki S. et al. Intrarenal renin–angiotensin system activity is augmented after initiaion of dialysis. Hypertens Res 2017; 40: 364–370 [DOI] [PubMed] [Google Scholar]
- 100. Hanahan D, Weinberg RA.. The hallmarks of cancer. Cell 2000; 100: 57–70 [DOI] [PubMed] [Google Scholar]
- 101. Dolley-Hitze T, Jouan F, Martin B. et al. Angiotensin-2 receptors (AT1-R and AT2-R), new prognostic factors for renal clear-cell carcinoma? Br J Cancer 2010; 103: 1698–1705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Lewandowska U, Lachowicz-Ochędalska A, Domińska K.. Angiotensin II ASA factor modulating protein tyrosine kinase activity in two breast cancer lines – MCF-7 and MDA-MB-231. Endokrynol Pol 2011; 62: 151–158 [PubMed] [Google Scholar]
- 103. Ager EI, Neo J, Christophi C.. The renin–angiotensin system and malignancy. Carcinogenesis 2008; 29: 1675–1684 [DOI] [PubMed] [Google Scholar]
- 104. Arrieta O, Pineda-Olvera B, Guevara-Salazar P. et al. Expression of AT1 and AT2 angiotensin receptors in astrocytomas is associated with poor prognosis. Br J Cancer 2008; 99: 160–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. George AJ, Thomas WG, Hannan RD.. The renin–angiotensin system and cancer: old dog, new tricks. Nat Rev Cancer 2010; 10: 745–759 [DOI] [PubMed] [Google Scholar]
- 106. Rocken C, Rohl F-W, Diebler E. et al. The angiotensin II/angiotensin II receptor system correlates with nodal spread in intestinal type gastric cancer. Cancer Epidemiol Biomarkers Prev 2007; 16: 1206–1212 [DOI] [PubMed] [Google Scholar]
- 107. Keizman D, Huang P, Eisenberger MA. et al. Angiotensin system inhibitors and outcome of sunitinib treatment in patients with metastatic renal cell carcinoma: a retrospective examination. Eur J Cancer 2011; 47: 1955–1961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. van der Knaap R, Siemes C, Coebergh J-WW. et al. Renin–angiotensin system inhibitors, angiotensin I-converting enzyme gene insertion/deletion polymorphism, and cancer: the Rotterdam Study. Cancer 2008; 112: 748–757 [DOI] [PubMed] [Google Scholar]