

Abstract
Sepsis is a leading cause of death in intensive care units and survivors develop prolonged immunosuppression and a high incidence of recurrent infections. No definitive therapy exists to treat sepsis and physicians rely on supportive care including antibiotics, intravenous fluids, and vasopressors. With the rising incidence of antibiotic resistant microbes, it is becoming increasingly critical to discover novel therapeutics. Sepsis-induced leukocyte dysfunction and immunosuppression is recognized as an important contributor towards increased morbidity and mortality. Pre-clinical and clinical studies show that specific cell surface inhibitory immune checkpoint receptors and ligands including PD-1, PD-L1, CTLA4, BTLA, TIM3, OX40, and 2B4 play important roles in the pathophysiology of sepsis by mediating a fine balance between host immune competency and immunosuppression. Pre-clinical studies targeting the inhibitory effects of these immune checkpoints have demonstrated reversal of leukocyte dysfunction and improved host resistance of infection. Measurement of immune checkpoint expression on peripheral blood leukocytes may serve as a means of stratifying patients to direct individualized therapy. This review focuses on advances in our understanding of the role of immune checkpoints in the host response to infections, and the potential clinical application of therapeutics targeting the inhibitory immune checkpoint pathways for the management of septic patients.
Keywords: sepsis, immunosuppression, T lymphocyte, myeloid cell, immune checkpoints
Introduction
Sepsis is defined as life-threatening organ dysfunction caused by a dysregulated host response to infection (Sepsis-3 guidelines) (1), and accounts for an annual worldwide case load of 48.9 million patients, leading to 11 million deaths (2). In the United States alone, sepsis is among the costliest hospital conditions and accounts for an annual healthcare burden of ~$24 billion (3). Although recent advances in fluid resuscitation, antibiotic administration, and organ support have decreased in-hospital deaths, septic patients manifest increased long-term morbidity and mortality. Many sepsis survivors suffer from long-term physical and cognitive disabilities and have a higher mortality rate than the general population (4–8). Specifically, sepsis survivors suffer a death rate of 15% in the first year after hospital discharge and a rate of 6–8% per year for the next 5 years.
Sepsis is historically considered a highly inflammatory disease, however clinical trials targeting inflammation have failed and no FDA approved therapy currently exists to treat sepsis (9). Therefore, there is a critical need to advance our understanding of sepsis pathology and identify novel biological targets for the development of new therapeutics. Numerous studies show that sepsis is not only characterized by early severe inflammation but also a concurrent immunosuppressed state that may persist for months after the initial episode of sepsis (10). Importantly, the persistent inflammatory and immunosuppressed states caused by dysfunctional innate and adaptive immune responses drive impaired immunity, multi-organ injury, prolonged hospital length of stay, and death (10–13). Sepsis survivors develop an impaired ability to clear primary infections and manifest an increased incidence of secondary nosocomial infections caused by opportunistic pathogens, indicating significant immunosuppression (14, 15). Studies demonstrate that sepsis causes significant depletion of CD4 and CD8 T cells, which is a major driver of impaired immune responses and reduced host antimicrobial capacity (16–18). Leukocytes harbor specific cell surface checkpoint proteins, which are meant to limit over activation and maintain homeostasis. Immune checkpoint receptors are specific membrane molecules located predominantly, but not exclusively, on T lymphocytes, which recognize complimentary ligands on antigen presenting cells (APCs) such as monocytes, macrophages, and dendritic cells. Major cell surface inhibitory immune checkpoints include programmed cell death receptor-1 (PD-1), programmed cell death receptor ligand-1 (PD-L1), programmed cell death receptor ligand-2 (PD-L2), cytotoxic T lymphocyte antigen-4 (CTLA-4), B and T lymphocyte attenuator (BTLA), lymphocyte activation-gene-3 (LAG-3) and T cell membrane protein-3 (TIM-3) and 2B4. Expression of these inhibitory checkpoint receptors is increased during sepsis, and hypothesized to facilitate sepsis-induced immunosuppression by impairing leukocyte antimicrobial functions (19, 20) (summarized in Table 1). Therapeutics targeting checkpoint inhibitors in preclinical studies and early clinical trials caused improved host resistance to infections and outcomes (18, 19, 25, 62). Certain cell surface immune checkpoints also act to drive co-stimulatory signals such as the interaction between Ox40 receptor on T cells and its cognate ligand Ox40L on APCs, and stimulation of this pathway has recently been shown to improve outcomes in a murine model of sepsis (61). This review will highlight some of the recent advances in our understanding of the role of immune checkpoints during sepsis and novel metabolism focused mechanistic aspects to be considered for their translational application in combination with other immunotherapeutics.
Table 1.
Summary of immune checkpoints cellular distribution and function.
| Immune Checkpoint Receptor | Cells Expressing Receptor | Ligand | Cells Expressing Ligand | Signaling Outcome in Sepsis |
|---|---|---|---|---|
| PD-1 (CD279) | - T cells - B cells - Monocytes - NK cells - Dendritic cells (21) |
PD-L1 | - T cells - B cells - Dendritic cells - Macrophages - Tumor cells - Lung, liver, heart, placenta, kidney and pancreas and other parenchymal cells (21, 22) |
- T cell exhaustion - Decreases myeloid cell function - Intestinal injury - Lung injury (18, 23–29) |
| PD-L2 | - Dendritic cells - Monocytes - Lung, liver, colon, small intestine, and placenta parenchymal cells (22, 30) |
- Deletion of PD-L2 improves bacterial clearance, with no observed survival benefit (30) |
||
| 2B4 (CD244) | - NK cells - T cells (31) |
CD48 | - All hematopoietic cells in humans (32) |
- T cell apoptosis - Increases PD-1 expression (33–35). |
| CTLA-4 (CD152) | - Activated CD4 and CD8 T cells | - CD80 - CD86 |
- Antigen presenting cells (36) |
- T cell apoptosis - Increased mortality (37, 38) |
| HVEM (CD270, TNFRSF14) | - Monocytes - Dendritic cells - Neutrophils - NK cells - Resting T cells - Immature B cells (39, 40) |
-BTLA (CD272) | - T cells - Monocytes - Macrophages - Dendritic cells (39–42) |
- Promotes CD4 T cell death - Inhibits innate leukocyte activation - Reduces bacterial clearance - Promotes organ injury (43) |
| LAG-3 (CD223) | - CD4, CD8, and regulatory T cells - B cells - Dendritic cells |
MHC II | - Antigen presenting cells | - Inhibits T cell responses - Decreases bacterial clearance (44) |
| TIM-3 | - NK cells - Macrophages - Dendritic cells - Mast cells - CD4 and CD8 T cells |
CEACAM1 | - Microvascular endothelia - Epithelial cells - T cells - B cells - Macrophages - Dendritic cells - Tumor cells (45) |
- Decreases inflammation - Decreases T cell apoptosis (46, 47) |
| Galectin 9 | - APCs, T cells, B cells -Liver - Small intestine - Thymus - Kidney - Spleen - Lung - Cardiac and skeletal muscle (48, 49) |
|||
| Ox40 (CD134) | - Activated CD4, CD8, and regulatory T cells - Activated NK cells (50–52) |
Ox40L (CD134L, CD252, glycoprotein 34) | - Macrophages - Dendritic cells - Langerhans cells - Vascular endothelial cells - Bronchial smooth muscle cells - Mast cells - Activated NK cells (53–60) |
- Improves outcomes in murine sepsis (61) |
Ox40 and Ox40L
Along with T-cell receptor ligation, optimal T cell activation requires potent costimulatory signals such as the classical interaction between CD28 on T cells and CD80/86 on APCs (63). Interaction between T cell-expressed Ox40 and Ox40L on APCs is also recognized as a potent costimulatory signal for effector T cells (64). Ox40 (CD134) is a member of the tumor necrosis factor receptor superfamily and it is primarily expressed on activated CD4 and CD8 T lymphocytes (50). Ox40 receptor consists of a single transmembrane domain and a cysteine rich extracellular domain (65). Ox40 is also expressed on regulatory T cells and activated NKT cells (51, 52). The cognate ligand of Ox40 is Ox40L, which is also named as CD134L, CD252 or glycoprotein 34 (gp34) (66). Ox40L was first identified on human T-cell leukemia virus type I (HTLV-I) transformed T cells (53). Expression of Ox40L is not only restricted to classical APCs such as macrophages (54), dendritic cells (55), and Langerhans cells (56), but is also expressed on vascular endothelial cells (57), bronchial smooth muscle cells (58), mast cells (59), and activated natural killer (NK) cells (60).
Ox40 is exclusively expressed on activated CD4 and CD8 T lymphocytes and not on naïve T cells, thereby implicating Ox40-Ox40L interaction to specifically provide costimulatory signals in activated effector T cells (67). Ox40 signals via its cytoplasmic domain which is linked to the signal transduction pathway involving TNFR- associated factors such as TRAF2 and TRAF5 leading to NF-κB activation (Figure 1) (68, 69). Signaling through Ox40 drives clonal expansion of T cells, and also increases the expression of anti-apoptotic proteins such as Bcl-2 and Bcl-xl leading to increased survival of T cells (70, 71). Sepsis functionally impairs and depletes T cells (16–18). Therefore, therapeutics targeting Ox40 pathway hold significant potential to boost T cell function during sepsis.
Figure 1.
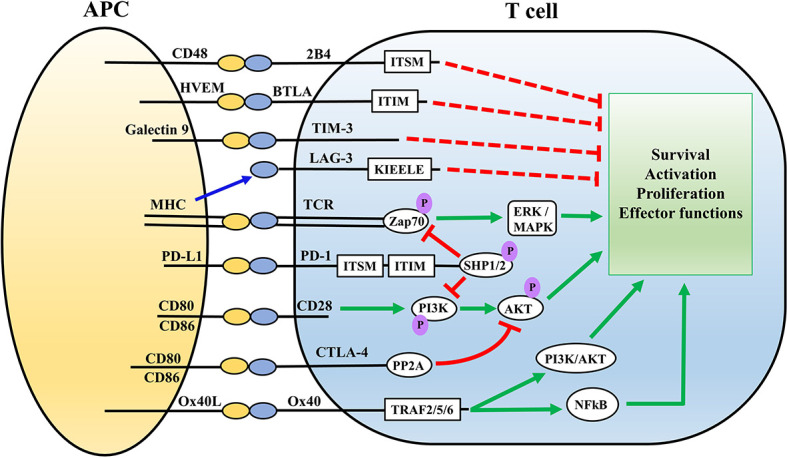
Overview of the major immune cell checkpoints and associated signaling pathways. Antigen presentation via MHC on APCs to the TCR complex on T cells executes activation of T cells via Zap70 and ERK/MAPK signaling pathways. Ligation of CD28 on T cells with CD80/86 on APCs provides co-stimulatory signals. PD-1/PD-L and CTLA-4 signaling impair T cell activation via inhibition of the AKT signaling pathway. PD-1signaling involves SHP mediated inhibition of Zap20 and PI3K/AKT signaling pathway, whereas CTLA-4 directly inhibits the AKT pathway via recruiting PP2A. 2B4, TIM3, BTLA, and LAG-3 also inhibit T cell activation through interaction with their associated ligands on APCs. Ox40-Ox40L serves as a co-stimulatory signal for T cells via PI3K/AKT and NFkB pathways. Red lines represent inhibitory signals while green represents stimulatory signals. ITSM, ITIM, KIEELE represent specific intracellular domains of the immune checkpoints which mediate their intracellular signaling APC, antigen presenting cell; HVEM, herpes virus entry mediator; MHC, Major histocompatibility complex; BTLA, B and T lymphocyte attenuator; TIM3, T cell membrane protein-3; LAG-3, Lymphocyte activation-gene-3; TCR, T cell receptor; PD-1, Programmed death-1; CTLA4, Cytotoxic T lymphocyte antigen-4; ITSM, Immunoreceptor Tyrosine-based Motif; ZAP70, Zeta Chain of T Cell Receptor Associated Protein Kinase 70; PI3K, Phosphoinositide 3 kinase; ITIM, immunoreceptor tyrosine-based inhibition motif; SHP, Src homology region 2 domain-containing phosphatase; PP2A, protein phosphatase 2A; TRAF, TNF receptor associated factor; ERK, Extracellular signal-regulated kinase; MAPK, mitogen activated protein kinase; AKT, protein kinase B.
Currently, there is great interest in the immunotherapeutic potential of Ox40 as an anticancer therapy (72), however, studies of Ox40 in sepsis are limited. A recent study by Unsinger et al., demonstrated that treatment with an agonistic antibody (Ab) to Ox40 improved T cell function and reduced mortality in a cecal ligation and puncture (CLP) model of murine sepsis (61). CLP-induced sepsis increased expression of Ox40 on splenic CD4 T cells which persisted up to 5 days, with no change on CD8 T cells. Treatment with Ox40 agonistic Ab increased splenic CD4 T cell count, and surprisingly further augmented the CLP-induced increase in splenic myeloid cell (macrophages and monocytes) numbers at day 5 post sepsis. Ox40 Ab improved T lymphocyte function, as measured by increased ability to produce IFNγ, not only in murine cells but also in peripheral blood mononuclear cells derived from sepsis patients, lending significant translational relevance to the study. It is important to note that treatment with Ox40 agonistic antibody was effective even when administered 6 and 48 h after the onset of sepsis, which closely mimics the clinical scenario for the treatment of sepsis patients. In contrast with these findings, a study by Karulf et al., showed that sepsis leads to significant upregulation of Ox40L on circulating monocytes and neutrophils at 24 h after sepsis diagnosis in their patient cohort and the level of monocyte Ox40L was higher in non-survivors (73). Treatment with a blocking antibody against Ox40L or Ox40L knock out reduced inflammation and organ damage leading to improved survival in a murine CLP model of sepsis. The protective effect of blocking Ox40L was predominantly dependent on intact macrophages and independent of T lymphocytes. A major difference between the two contrasting studies is that Unsinger et al. employed a less severe model of sepsis with 50% of septic mice surviving at day 7 post CLP, as opposed to the sepsis model by Karulf et al. which showed 100% mortality within 48 h after CLP (61, 73). Therefore, these findings indicate that there exists a fine balance for maintaining immune homeostasis during sepsis and therapeutics targeting Ox40-Ox40L could be a double-edged sword. Future studies should carefully evaluate their therapeutic potential using diverse models of sepsis in context of myeloid and T cell functions and inflammation.
Programmed Cell Death Receptor-1 (PD-1) and PD-L1/PD-L2
PD-1 receptor, also known as CD279, is a type I transmembrane glycoprotein belonging to the immunoglobulin (Ig) superfamily and it is composed of an extracellular Variable-type (V-type) Ig domain, a transmembrane domain and an intracellular cytoplasmic signaling domain (74–76). PD-1 is expressed on variety of leukocytes including T and B lymphocytes, monocytes, NK cells, and dendritic cells (21). Signaling through PD-1 is executed upon engagement with its cognate ligands including PD-L1 and PD-L2 (77, 78). PD-L1 is expressed on activated leukocytes including T and B lymphocytes, dendritic cells, macrophages and parenchymal cells of lung, liver, heart, placenta kidney, tumor cells, and pancreas (21, 22). PD-L2 has been shown to be expressed on APCs such as dendritic cells and monocytes, along with tissues including lung, liver, colon, small intestine, and placenta (22, 30). The interaction between PD-1 and PD-L1 has been the focus of majority of the immune checkpoint studies during sepsis, while studies implicating PD-L2 are limited.
Overview of PD-1/PD-L Signaling Pathways
The interaction between PD-1 and its ligands generates inhibitory signals to attenuate T cell responses (79). The intracellular signaling mechanisms leading to the T cell inhibitory effect of PD-1 ligation is a topic of great interest and not completely understood. The intracellular cytoplasmic domain of PD-1 is composed of tyrosine-based motifs including immuno-receptor tyrosine-based inhibitory motif (ITIM) and immuno-receptor tyrosine-based switch motif (ITSM) (75). The inhibitory effect of PD-1 proceeds predominantly though ITSM phosphorylation and to a lesser extent ITIM phosphorylation, leading to recruitment of Src homology region 2 domain-containing phosphatase-2 (SHP-2) which drives the downstream signaling pathways (75, 80). Upon PD-1 ligation, SHP-2 dephosphorylates phosphoinositide 3-kinase (PI3K) leading to inhibition of Protein Kinase B (Akt) and extracellular signal-regulated kinase/mitogen-activated protein kinase (ERK/MAPK) signaling pathways (Figure 1) (79, 81). SHP-2 is not the only mediator of PD-1 signaling mediated T cell exhaustion (82), and SHP-1 could compensate for absence of SHP-2 (83, 84). PD-1 mediated signaling pathway targets the primary T cell receptor (TCR) as well as the co-stimulatory CD28 pathways for downregulating T cell effector functions (85, 86). On the other hand, whether PD-L1 can reverse signal is highly debated, because the intracellular signaling domains of PD-L1 remain poorly characterized. Only a limited number of studies focused of PD-L1 harboring tumor cells have demonstrated some PD-L1 mediated effects which are independent of PD-1 (87). For example, PD-L1 has been shown to promote tumor cell proliferation and survival independent of PD-1 (88), acting through specific conserved sequence motifs to shield melanoma cells from IFNγ cytotoxicity through a STAT3/caspase-7-mediated pathway (89). Interestingly, a study by Hartley et al. showed PD-L1 delivers a constitutive inhibitory signal within macrophages, and treatment with anti-PD-L1 antibody increased proliferation, survival, and activation of murine and human macrophages which triggered tumoricidal effects of macrophages (90). Such effects, if any, for PD-L2 are unknown. The inhibitory effects of PD-1:PD-L interaction has been well documented in sepsis-induced T cell exhaustion using PD-1 and PD-L1 blocking antibodies (18, 25, 26), However, specific details about the intracellular signaling pathways executing the PD-1:PD-L interaction driven inhibitory signals are yet to be fully characterized in the context of sepsis.
PD-1 and PD-L Mediated Immunosuppression and Organ Injury During Sepsis
Increased expression of PD-1 on T cells and PD-L1 on APCs causes T cell exhaustion during sepsis which is associated with impaired microbial clearance, multi-organ injury, and increased mortality (18, 19, 23–26). The characteristic features of T cell exhaustion include reduced co-stimulatory receptor expression such as CD28, upregulation of inhibitory immune checkpoints, and metabolic derangements leading to impaired production of effector cytokines (interferon-gamma, and granzyme-B), along with impaired proliferation and increased susceptibility to apoptotic cell death (42). A postmortem study of sepsis patients by Boomer et al. provided critical evidence that sepsis increases PD-1 and reduces CD28 and IL7 receptor expression on T cells, along with increased PD-L1 and PD-L2 expression dendritic cells (23). Increased PD-1 expression on T cells and PD-L1 expression of APCs in septic patient has been shown to be associated with lymphopenia, T cell apoptosis, and higher mortality (23, 91–93). A recent study by Wilson et al. shows upregulation of PD-1 and PD-L1 on both T and B lymphocytes, and that expression of PD-1 was found to be higher in lymphocytes associated with CD27+ memory status (94). Although, the study demonstrated an association between higher lymphocyte PD-L1 expression and secondary nosocomial infections in patients with >7 days of ICU length of stay, the expression of PD-1, PD-L1, or PD-L2 on lymphocytes was not predictive of eventual mortality (94). The findings of this study is in contrast to other studies which show an association between T cell PD-1 expression with sepsis mortality (91, 95). However, it is important to note that study conducted by Wilson et al. provided a snapshot of PD-1 expression at an early single time point within 12h of ICU admission (94), while a time course study by Tomino et al. shows that higher PD-1 expression on day 7 after ICU stay correlates with increased mortality among sepsis patients (95). Fungal Candida infected sepsis patients also demonstrate an increased PD-1 expression on circulating CD4 and CD8 T cells, and decreased CD28 expression (96). Studies using PD-1 knockout mice show improved survival outcomes after sepsis using a neonatal cecal slurry model (97).
In addition to T cells, PD-1/PD-L1 axis also impairs myeloid cell function during sepsis. A study by Patera et al. showed that increased PD-L1 expression on neutrophils and monocytes correlates with a decrease in their phagocytic capacity in septic patients (25). CLP-induced murine sepsis has been shown to increase PD-1 expression on liver Kuppfer cells and deletion of PD-1 improves their phagocytic capacity (98). Increased PD-L1 expression on circulating neutrophils is shown to correlate with increased pro and anti-inflammatory cytokines and higher mortality in a CLP model of sepsis (27). Shao et al. showed that increased PD-L1 expression on circulating monocytes predicts higher 28 day mortality in septic shock patients (99). Increased PD-L1 expression on NK cells within 24 h of ICU admission has recently been shown to correlate with increased Sequential Organ Failure Assessment Score (SOFA) and sepsis severity (100). From a mechanistic standpoint, a recent study by Deng et al. showed that PD-L1 expression on myeloid cells is regulated by a circadian clock gene, BMAL1 (101). Specifically, BMAL1 blocks pyruvate kinase M2 and STAT1 driven upregulation of PD-L1 expression on macrophages, reduces T cell exhaustion and immunosuppression, and protects against sepsis (101). However, detailed cellular signaling mechanisms leading to sepsis-induced upregulation of leukocyte PD-1 and PD-L1, and the downstream effects of PD-1/PD-L1 ligation needs further investigations. In the context of the ongoing COVID-19 pandemic, SARS-CoV-2 has been shown to cause lymphopenia, and impaired functional capacity of T cells and monocytes (102). However, the direct role of immune checkpoints such as PD-1/PD-L1 in mediating immunosuppression during SARS-CoV-2 infection and evaluating it as potential therapeutic target requires further investigation (103).
PD-1/PD-L signaling has also been implicated in sepsis-induced organ injury. In addition to leukocytes, PD-L1 and PD-L2 are also expressed on non-immune cells including liver, lung, kidney, small intestine, colon, tissue endothelial cells (21, 22, 30). A study by Rossi et al. showed that deletion of PD-L1 attenuated the rise in hepatic dysfunction indices including serum bilirubin, alanine aminotransferase (ALT) and aspartate amino transferase (AST) and protected the endothelial permeability barrier, which was associated with improved systemic bacteria clearance and improved survival in a murine model of CLP-induced sepsis (30). Although, deletion of PD-L2 improved systemic bacterial clearance, it did not protect against the rise in hepatic injury makers including serum bilirubin and AST, and offered no survival benefit (30). Using a CLP model of sepsis, Hutchins et al. showed increased PD-1 and PD-L1 on liver Kuppfer cells and sinusoidal endothelial cells respectively, and deletion of PD-L1 protected against sepsis-induced increase in liver vascular leakage, edema, and endothelial cell apoptosis (104). PD-L1 expression was shown to be increased in the lung tissue of postmortem samples from sepsis patients (23). Using a two hit murine model of hemorrhage followed by CLP-induced sepsis, recent studies demonstrate a deleterious role of PD-1/PD-L1 pathway in mediating sepsis-induced lung injury (29, 105, 106). Specifically, hemorrhagic shock and sepsis upregulated PD-L1 expression on lung endothelial and parenchymal cells, and deletion of PD-1 or PD-L1 attenuated sepsis-induced increase in pulmonary endothelial cell permeability and reduced lung injury (29, 106). Intravenous delivery of siRNA targeting PD-L1 expression reduces neutrophil influx into lungs and attenuates sepsis-induced lung injury, indicating targeting PD-L1 as an attractive therapeutic target to protect against sepsis-induced lung injury. Adoptive transfer of regulatory T cells (Tregs) before induction sepsis in mice subjected to hemorrhagic insult demonstrates reduced infiltration of neutrophils and lung injury, an effect which is ablated using adoptive transfer of PD-1 deleted Tregs, indicating that PD-1 expressing Tregs dampen the innate leukocyte mediated exaggerated inflammation and protect against sepsis-induced acute lung injury (105). PD-L1 also plays a critical role in regulating sepsis-induced intestinal injury. Human and murine sepsis studies show that upregulated PD-L1 expression on intestinal tissue causes increased inflammation and intestinal permeability, and deletion of PD-L1 or treatment with anti-PD-L1 antibody attenuates sepsis-induced intestinal injury (27, 28).
Therapeutic Targeting of the PD-1/PD-L1 Pathway
Antibodies blocking PD-1/PD-L1pathway are in clinical use for treating cancer (107), and remain of great interest as a means of treating sepsis. Hotchkiss and colleagues showed that administration of anti-PD-1 antibody at 24 h after CLP-induced sepsis induction, attenuated sepsis-induced T cell dysfunction and improved survival (108). Treatment with anti-PD-1 antibody after sepsis induction has also shown to improve T cell function, myeloid cell MHC-II expression and survival in a two-hit model of CLP-induced sepsis followed by fungal infection with C. albicans (38, 109). Ex vivo treatment with anti-PD-1 antibody reduces apoptosis and improves IFNγ production in circulating CD8 T cells obtained from septic patients (93). Likewise, Patera et al. showed that ex vivo treatment with anti-PD-1 antibody not only improves T cell function but also myeloid cell phagocytic function in peripheral blood leukocytes isolated from septic patients (25). Interestingly, a recent study by Phares et al. shows that treatment with a novel peptide (LD01) which blocks PD-1 signaling improved macrophage phagocytic capacity, T cell function and survival following CLP-induced sepsis (110). Collectively, these experimental studies show that treatment with anti-PD-1 is an attractive therapeutic option as it could be effective even when administered after sepsis induction. In a clinical trial, Hotchkiss and colleagues administered nivolumab, an anti-PD-1 antibody, to 31 septic patients beginning 24 h after sepsis diagnosis (111). Treatment did not result in significant induction of pro-inflammatory cytokine production, but adverse events possibly related to drug administration or immune dysfunction were identified in five subjects. Further large-scale trials are needed to assess the safety and efficacy of anti-PD-1 antibodies in septic patients.
Hotchkiss and colleagues recently conducted a Phase 1 clinical trial of anti-PD-L1 antibody (BMS-936559 from Bristol-Myers Squibb) in sepsis patients (111). Anti-PD-L1 was administered using a dose escalation strategy starting at 24 h after the onset of organ dysfunction to avoid treating during the peak inflammatory phase of sepsis. Importantly, treatment with anti-PD-L1 antibody was well tolerated and did not fuel inflammation in septic patients, and the antibody showed full PD-L1 receptor occupancy at 28 days after 900 mg single dose regimen (111). Previous studies by Patera et al. and Chang et al. demonstrated that ex vivo treatment with anti-PD-L1 antibody reverses sepsis-induced T cell dysfunction and improves phagocytic function of neutrophils and monocytes, in circulating blood cells obtained from septic patients (25, 93). In murine models of CLP-induced sepsis, treatment with anti-PD-L1 antibody reduces T cell apoptosis, enhances bacterial clearance, and reduces organ injury leading to improved survival (112, 113). Delayed treatment with anti-PD-L1 antibody at 24 h after induction of fungal sepsis with C. albicans also preserves T cell function and improves survival (38). Treatment with Compound 8, a novel PD-L1 blocking peptide, halved the mortality rate in a two-hit model of CLP-induced sepsis followed by fungal sepsis with C. albicans (114). Together, these studies suggest that targeting the PD-1/PD-L1 axis would likely be highly beneficial in the treatment of septic patients.
2B4
2B4 (CD244) is predominantly expressed on NK cells and T lymphocytes (31). The 2B4 belongs to the Signaling Lymphocyte Activation Molecule (SLAM) family of immunoregulatory receptors in the Ig superfamily (115). 2B4 receptor is composed of an extracellular Ig-like domains, a transmembrane region, and a cytoplasmic domain consisting of four ITSMs (115, 116). CD48, expressed on APCs, is the known ligand for 2B4 (32). High expression of 2B4 on T cells relays an inhibitory signal and low-moderate 2B4 expression leads to a co-stimulatory signal (116). Using a lymphocytic choriomeningitis virus (LCMV) infection model, Blackburn et al. showed that blocking the 2B4 pathway with CD48 antibody reduced IFNγ production in CD8 T cells expressing intermediate levels of 2B4, whereas increased IFNγ production in CD8 T cells expressing high levels of 2B4 (117). These studies demonstrate that high 2B4 expression leads to functional exhaustion of T cells (117). Interestingly, the signaling pathway upon CD48-2B4 ligation could be bidirectional. A study by Assarsson et al. showed that NK cells can augment T cell proliferation and activation, which involves interactions between 2B4 expressed on NK cells and CD48 expressed on CD8 T cells (118).
A seminal study by Chen al. showed significant upregulation of 2B4 on CD4 and CD8 T cells in human sepsis patients and in a CLP model of murine sepsis within 24 h of sepsis induction (33). Using conditional knock out mice, these studies revealed that deletion of 2B4 expressed specifically on CD4 T cells offers survival advantage following sepsis (33). Follow-up studies from the same research group showed 2B4 specifically mediated the loss of memory T cells, which was attenuated by 2B4 deletion leading to improved survival after sepsis (34). 2B4+ T cells from septic patients showed increased caspase-3/7+ apoptotic T cells, and 2B4 deletion reduced caspase-3/7+ apoptotic T cells in a murine CLP model of sepsis, thereby implicating 2B4 as an important mediator of sepsis-induced T cell apoptosis (34). Using a two-hit model of CLP sepsis followed by murine Epstein-Barr virus infection (gHV), a recent study by Xie et al. shows that deletion of 2B4 protects against sepsis-induced loss of antigen specific CD8 T cells and attenuates the increased viral load following sepsis (35). This study noted an important observation that CD8 T cells from wild type mice exhibited increased PD-1 expression, which was attenuated in 2B4 knockout mice, implying that 2B4 could modulate PD-1 expression during sepsis. These results suggest that blocking 2B4 pathway could serve to limit expression of multiple co-inhibitory receptors causing T cell exhaustion and open a new area of investigation for future studies. Therefore, 2B4 signaling pathway represents an attractive therapeutic target to reverse sepsis-induced T cell exhaustion and merits further investigation.
Cytotoxic T Lymphocyte Antigen-4 (CTLA-4)
CTLA-4 (CD152) is a dimeric cell surface glycoprotein, which binds to CD80 and CD86 cell surface receptors on APCs (36). CTLA-4 is a known negative regulator of T cell effector functions and strongly counteracts CD28-mediated T cell co-simulation (42). CTLA-4 regulates T cell function through recruitment of cell intrinsic serine/threonine Protein Phosphatase 2A (PP2A), which inhibits CD3/CD28-mediated upregulation of cellular glucose metabolism via blocking of AKT phosphorylation (Figure 1) (81, 119). CTLA-4 signaling reduces cell surface expression of CD80 and CD86 on APCs via promoting their endocytosis, thereby limiting the availability of these co-stimulatory receptors for CD28 (119). CTLA-4 engagement reduces IL-2 receptor expression and IL-2 generation leading to cell cycle arrest, and reduced proliferation and activation of T cells (121, 122). Therefore, CTLA-4 signaling pathway downregulates T cell function via both cell intrinsic and extrinsic pathways, and represents an important immune checkpoint regulating T cell effector functions.
Limited studies have evaluated the role of CTLA-4 pathway during sepsis. Boomer et al. showed a time-dependent progressive increase in CTLA-4 expression on T cells in septic patients (123). Studies by Inoue et al. demonstrated that increased CTLA-4 expression on CD4 and CD8 T cells within 24 h of sepsis induction was associated with increased T cell depletion via apoptosis (37). Treatment with anti-CTLA4 antibody attenuates T cell apoptosis and improves survival in CLP induced sepsis (using a dose of 50 µg per mouse) and in a two-hit model of CLP induced sepsis followed by fungal infection with C. albicans (using a dose of 50 µg per mouse) (37, 38). A higher dose of anti-CTLA-4 antibody (200 µg) worsened survival outcomes, indicating that excess CTL-4 inhibition could fuel increased inflammation via uncontrolled T cell activation (37). Recent studies have also explored the correlation between single nucleotide polymorphisms (SNP) within the CTLA-4 gene and sepsis severity in humans. The functional SNPs such as rs231775, rs733618, and rs3087243 in the CTLA-4 gene have been shown to be associated with autoimmune diseases such as rheumatoid arthritis (124), Hashimoto thyroiditis (124, 125), and autoimmune diabetes (126, 127). These CTLA-4 SNPs are associated with higher T cell activation and proliferation capacity owing to a reduced CTLA-4 expression (128). Therefore, it is possible that the presence of specific CTLA-4 SNPs could be beneficial during sepsis in regard to alleviation of CTLA-4 mediated T cell exhaustion. Interestingly, a recent study by Mewes et al. reported a significantly lower 28- and 90-day mortality risk among septic Caucasian patients with rs231775 GG homozygous SNP in CTLA-4 gene (129). A follow-up study from the same group further demonstrated that CTLA-4 rs3087243 G allele carriers showed lower 28- and 90-day mortality risk (130). These demonstrate that specific SNPs in CTLA-4 could serve as prognostic predictors in septic patients. However, the significance of these specific CTLA-4 SNPs in regulating T cell function during sepsis remains to be explored, and hold significant potential for future research in personalized medicine approach.
B and T Lymphocyte Attenuator (BTLA) and Herpes Virus Entry Mediator (HVEM)
BTLA (CD272) is a type I transmembrane glycoprotein, and its cytoplasmic domain is comprised of distinct intracellular motifs including growth factor receptor-bound protein-2 (Grb-2), ITIM and ITSM (131). BTLA exhibits bidirectional regulation, as the ITIM motif could deliver inhibitory signals via SHP1 and SHP2 mediated inhibition of cell activation, and stimulatory signals via Grb-2 motif which recognizes Grb-2 protein leading to activation of the PI3K pathway (132, 133). BTLA is expressed on T cells and innate immune cells including monocytes, macrophages, and dendritic cells (41, 42). BTLA interacts with HVEM (CD270 or TNFRSF14), a type I transmembrane receptor belonging to the tumor necrosis factor receptor superfamily, and is known to also interact with multiple additional ligands such as CD160, LIGHT, and LTα (39, 134). Montgomery et al. discovered HVEM as a required receptor for cellular entry of herpes simplex virus-1 (135). HVEM has diverse cell type distribution on APCs such as monocytes, dendritic cells, neutrophils, and NK cells, and it is also expressed on resting T cells and immature B cells (39, 40). Engagement of BTLA with its receptor HVEM predominantly inhibits TCR signaling mediated effector functions including T cell activation and proliferation (133).
Recent studies have implicated a role for BTLA as an important immune checkpoint molecule during sepsis-induced immunosuppression. In some of the first evidence supporting this role, Boomer et al. demonstrated the increased presence of HVEM in postmortem lungs of septic patients as compared to non-septic controls (23). Shubin et al. showed that increased BTLA expression on CD4 T cells is predictive of susceptibility to secondary nosocomial infections among sepsis patients and ablation of BTLA protects against sepsis-induced CD4 T cell depletion in a murine model of CLP-induced sepsis (43). In contrast to these findings, Spec et al. observed no difference between leukocyte BTLA expression in septic patients infected by fungal Candida albicans infection (96), which could point to a difference in the role of BTLA depending on the etiology of sepsis. Shubin and colleagues also demonstrated that sepsis leads to increased infiltration of high BTLA and HVEM expressing innate immune cells including macrophages, inflammatory monocytes, dendritic cells, and neutrophils at the site of infection in the peritoneum (136). Using BTLA knockout mice, this study also showed enhanced activation of innate leukocytes, reduced bacterial burden, and attenuation of organ injury leading to improved survival (136). Therefore, targeting BTLA could serve to activate both innate and adaptive immunity during sepsis. Higher plasma levels of soluble BTLA (sBTLA) greater than 21 ng/ml, were predictive of five-fold increase in mortality among sepsis patients, implying sBTLA as a prognostic marker during sepsis (137). However, the functional significance of increased sBTLA during sepsis is unknown. Treatment with anti-BTLA antibody in a two-hit murine model of hemorrhage followed by sepsis lead to increased inflammation, organ injury and mortality (138). Therefore, future studies employing anti-BTLA antibody should be carefully evaluated in a dose and time response manner along with its effects on T cell and myeloid functions.
Lymphocyte Activation-Gene-3 (LAG-3) and T Cell Membrane Protein-3 (TIM-3)
LAG-3 and TIM-3 are the lesser investigated immune checkpoints during sepsis as compared to those discussed above. LAG-3 (CD223) is expressed on T cells and belongs to the Ig superfamily. It binds to major histocompatibility class II (MHC II) on APCs and competes for binding of the MHC molecules to TCR on T cells, thereby inhibiting T cell immune responses (139). LAG-3 signaling is mediated via its intracellular KIEELE motif, which causes cell cycle arrest in T cells and suppresses T cell proliferation (140). TIM-3 is a member of the Ig superfamily and it interacts with CEACAM1 (carcinoembryonic antigen-related cell adhesion molecule 1) or Galectin 9 (42), both of which are expressed on a variety of immune cells and tissues (45, 48, 49). TIM-3/Galectin 9 interaction has been shown to cause apoptosis in CD4 T cells (141).
Using a CLP model of murine sepsis, a recent study by Lou et al. showed increased LAG-3 expression not only on CD4 and CD8 T cells, but also on B cells, regulatory T cells, and dendritic cells, and treatment with anti-LAG-3 antibody improved T cell function, bacterial clearance and survival after sepsis (44). Importantly, anti-LAG-3 antibody was administered in a therapeutic manner at 3 h after sepsis induction, which increases the clinical relevance. A recent study by Niu et al. shows T cells co-expressing LAG-3 and PD-1 synergistically inhibit CD4 and CD8 T cells, and this correlated with increased length of hospital stay and higher mortality (142). Interestingly, this study showed that LAG-3 was only significantly elevated at day 5 post sepsis, as compared to PD-1 which was elevated early at 12 h after sepsis, indicating an important role for LAG-3 immunosuppression during progression of sepsis. Studies by Boomer et al. showed increased expression of LAG-3 and TIM-3 on CD4 T cells, with a relatively higher level of LAG-3 on CD8 T cells as compared to TIM-3 (122). Immune checkpoints are critical in maintaining immune tolerance and homeostasis to limit excess inflammation. Studies by Yang et al. demonstrated a direct correlation between reduced TIM-3 mRNA expression in peripheral blood mononuclear cells and increased pro-inflammatory status, and blocking TIM-3 signaling with anti-TIM-3 antibody worsened outcomes in murine model of CLP-induced sepsis (47). In line with these findings, the same research group demonstrated that soluble TIM-3 immunoglobulin which blocks TIM-3 signaling exacerbates inflammation and T cell apoptosis during sepsis, which was attenuated in mice overexpressing TIM-3 (46). Studies by Ren et al. show increased TIM-3 expression on monocytes in sepsis patients, but no change in severe sepsis or septic shock patients (143). Furthermore, soluble TIM-3 levels correlated with mortality in the septic shock group alone (143). Spec et al. showed no difference in TIM-3 expression on CD4 and CD8 T cell in sepsis patients affected by C. albicans infection, implying that fungal etiology of sepsis may not involve TIM-3 (95). It is important to note that studies showing increased inflammation with anti-TIM-3 administered it in a prophylactic manner at least 24 h before sepsis induction (46, 47). Based on all these findings, it is possible that TIM-3 serves to limit excess inflammation during sepsis and strategies to prophylactically block TIM-3 might accentuate inflammation during sepsis. Future studies should carefully evaluate a dose and time dependent response of anti-TIM-3 antibody as a therapeutic during sepsis, and test whether blocking TIM-3 signaling during the acute inflammatory phase after sepsis induction is a viable strategy.
Immune Checkpoint Inhibitors as a Therapeutic Strategy to Protect Against Nosocomial Infections
Although judicious fluid resuscitation and antibiotic strategies have decreased short-term death among sepsis patients, sepsis survivors manifest increased morbidity and mortality from secondary nosocomial infections. Sepsis-induced immunosuppression increases susceptibility to secondary life-threatening infections caused by microbes such as Pseudomonas, Candida, Acinetobacter, and Enterococcus (15). Septic patients also demonstrate increased reactivation of latent viruses including cytomegalovirus and herpes simplex (144, 145). As discussed above, increased expression of immune checkpoints including PD-1, PD-L1, 2B4, and BTLA play a major role in sepsis-induced immunosuppression. Targeting immune checkpoints could be harnessed to reinvigorate host immune responses and protect critically ill patients against secondary hospital acquired infections. Blocking PD-1/PD-L signaling pathways is protective in two-hit models of CLP-induced bacterial sepsis followed by fungal C. albicans infection (38, 108, 113). Double LAG-3 and PD-1 positive T cells show significant dysfunction and LAG-3 is upregulated later in the course of sepsis progression in patients (142). 2B4 is implicated in inhibiting the T cell specific responses, thereby compromising host immune responses (34, 35). Apart from sepsis, critically ill patients from other etiologies could also be a target population which could benefit from targeting immune checkpoints. Patients with acute liver failure demonstrate reduced proliferation capacity and increased expression of CTLA-4 on circulating CD4 T cells, and ex vivo treatment with anti-CTLA-4 antibody restores the proliferative response (146). Studies by Shubin et al. demonstrated increased risk of acquiring nosocomial infections among non-septic critically ill patients with greater than 80% BTLA expression on CD4 T cells (43). Therefore, this evidence brings to light an important observation that therapeutics targeting immune checkpoints could be prime candidates to limit the risk of developing secondary infections among septic patients, and this merits detailed future investigations using clinically relevant models of sepsis.
Can Therapeutics Targeting Immune Checkpoints Augment Trained Immunity-Mediated Protection Against Infections?
Trained immunity refers to a long-term functional reprogramming of innate leukocytes, evoked by a prior exposure to microbial ligands or a non-lethal infectious insult, which leads to an amplified anti-microbial response to a broad range of secondary infections (147). Trained immunity induced by microbial ligands such as Toll-like receptor 4 agonists (TLR4), including Monophosphoryl lipid-A (MPLA) and fungal ligand β-glucan, potently protects against life-threatening pathogens such as Pseudomonas aeruginosa, Staphylococcus aureus, and Mycobacterium tuberculosis (148–151). TLR4 agonist-induced trained immunity involves metabolic reprogramming of macrophages including mitochondrial biogenesis and augmentation of glycolysis and mitochondrial oxidative phosphorylation (149, 152).
The general consensus holds that trained immunity is predominantly mediated via innate immune cells and independent of the adaptive immunity (147, 149). As discussed above, immune checkpoints, specifically the PD-1/PD-L pathway also affects innate leukocyte responses during sepsis (20). Studies by Huang et al. showed increased PD-1 expression on macrophages at the site of infection correlated with their dysfunction and while deletion of PD-1 protected against sepsis lethality (153). PD-L1 delivers a constitutive inhibitory signal within macrophages, and treatment with anti-PD-L1 antibody activates both murine and human macrophages (89). Stimulation of TLR4 receptor by lipopolysaccharide and MPLA has been shown to induce PD-L1 expression on innate leukocytes including macrophages and dendritic cells (154, 155). Metabolic reprogramming is the hallmark of trained immunity, and interaction between immune checkpoint pathways and cellular metabolism in innate leukocytes during sepsis is not well characterized. A recent study by Deng et al. shows that pyruvate kinase M2 (glycolytic enzyme) and STAT1 signaling drive upregulation of PD-L1 expression on macrophages (100). Hypoxia-inducible factor-1α (HIF-1α) signaling has also been shown to upregulate PD-L1 expression in endotoxin tolerant monocytes isolated from septic patients, and ex vivo co-culture experiments using knockdown of HIF-1α or PD-L1 from septic monocytes significantly improved T cell proliferation capacity (156). Interestingly, induction of trained immunity via ligands such as β-glucan and MPLA require HIF-1α mediated increase in glycolysis (157, 158). Therefore, it is possible that stimulation with ligands which induce trained immunity such as MPLA could also cause increased PD-L1 expression, and the therapeutic efficacy of these ligands could be further boosted by blocking specific immune checkpoint pathways such as PD-1/PD-L. Immune checkpoint blockers could thus serve as adjunct therapeutics in combination with clinically applicable microbial ligands such as MPLA. However, the role of immune checkpoints in regulating leukocyte metabolic reprogramming and generation of microbial ligand-induced trained immunity, and the metabolic effects of blocking immune checkpoint pathways during sepsis are currently unknown. Apart from innate leukocytes, signaling through immune checkpoints including PD-1 and CTLA-4 predominantly impair the uptake of glucose and subsequent glucose metabolism via inhibiting glycolysis, leading to impaired T cell effector responses (80, 159). PD-1 signaling impairs T cell functions by inhibiting PI3K and Akt signaling, which diverts cellular metabolism away from glycolysis to fatty acid oxidation (159). CTLA-4 also blocks the Akt pathway and glycolysis, although through a different mechanism involving PP2A (80, 118). Therefore, the T cell protective effects of therapeutics blocking immune checkpoints such as PD-1 and CTLA-4 pathways during sepsis could also involve metabolic reprogramming and re-invigoration of the glycolytic pathway, which will be an interesting topic for future studies.
Concluding Remarks for Translational Application of Therapeutics Targeting Immune Checkpoints
Therapeutics targeting immune checkpoints hold significant potential to reverse sepsis-induced immunosuppression and preserve host immunity against primary and secondary infections. Future studies should also evaluate a combinatorial therapeutic approach using immune checkpoint blockade in conjunction with other therapeutics such as interleukin-7, which augments T cell functions, as well as microbial ligands such as MPLA which induce trained immunity mediated augmentation of innate immune functions. A recent study by Hotchkiss and colleagues demonstrated the safety of PD-L1 antibody in septic patients (110). However, cancer clinical trials have documented a broad spectrum of serious adverse events associated with immune checkpoint blockade such as liver injury, thyroiditis, colitis, pneumonitis, thrombocytopenia, vasculitis and others (160), and in a murine model of CLP sepsis, anti-CTLA-4 antibody worsened survival at higher doses (37). Clinical use of such therapeutics will need to be tailored to individual patients based on immunophenotypic analysis of circulating immune cells. The concurrent existence of proinflammatory and immunosuppressed states in sepsis calls for a careful consideration of the translational application of immune checkpoint targeted therapeutics. Nonetheless, many studies reviewed here show therapeutic potential for this class of drugs in a disease currently managed only with source control and supportive care.
Author Contributions
NKP and ERS conceptualized the manuscript. NKP, ERS, MM, and TKP contributed to writing the initial draft of the manuscript. MM drafted the table. MM and TKP drafted the figure. ERS, JKB, and AH critically revised the manuscript for important intellectual concepts. NKP supervised the drafting and performed final editing. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by National Institutes of Health (NIH) grants R01 GM119197 and R01 AI151210 to ERS; T32 GM108554-05, Shock Society Faculty Research Award and Vanderbilt Faculty Scholars Award to NKP, R01 GM12171 to JKB, K08 GM123345 to AH, and T32 GM007347-41 to MM.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- 1. Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA (2016) 315(8):801–10. 10.1001/jama.2016.0287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rudd KE, Johnson SC, Agesa KM, Shackelford KA, Tsoi D, Kievlan DR, et al. Global, regional, and national sepsis incidence and mortality, 1990-2017: analysis for the Global Burden of Disease Study. Lancet (2020) 395(10219):200–11. 10.1016/S0140-6736(19)32989-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Torio CM, Moore BJ. National Inpatient Hospital Costs: The Most Expensive Conditions by Payer, 2013. (2016). Available at: https://wwwhcup-usahrqgov/reports/statbriefs/sb204-Most-Expensive-Hospital-Conditionsjsp. [PubMed]
- 4. Iwashyna TJ, Ely EW, Smith DM, Langa KM. Long-term cognitive impairment and functional disability among survivors of severe sepsis. JAMA (2010) 304(16):1787–94. 10.1001/jama.2010.1553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mayr FB, Yende S, Angus DC. Epidemiology of severe sepsis. Virulence (2014) 5(1):4–11. 10.4161/viru.27372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pandharipande PP, Girard TD, Ely EW. Long-term cognitive impairment after critical illness. N Engl J Med (2014) 370(2):185–6. 10.1056/NEJMc1313886 [DOI] [PubMed] [Google Scholar]
- 7. Angus DC, Opal S. Immunosuppression and Secondary Infection in Sepsis: Part, Not All, of the Story. JAMA (2016) 315(14):1457–9. 10.1001/jama.2016.2762 [DOI] [PubMed] [Google Scholar]
- 8. Prescott HC, Osterholzer JJ, Langa KM, Angus DC, Iwashyna TJ. Late mortality after sepsis: propensity matched cohort study. BMJ (2016) 353:i2375. 10.1136/bmj.i2375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nedeva C, Menassa J, Puthalakath H. Sepsis: Inflammation Is a Necessary Evil. Front Cell Dev Biol (2019) 7:108. 10.3389/fcell.2019.00108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Delano MJ, Ward PA. Sepsis-induced immune dysfunction: can immune therapies reduce mortality? J Clin Invest (2016) 126(1):23–31. 10.1172/JCI82224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tang BM, Huang SJ, McLean AS. Genome-wide transcription profiling of human sepsis: a systematic review. Crit Care (2010) 14(6):R237. 10.1186/cc9392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Boomer JS, Green JM, Hotchkiss RS. The changing immune system in sepsis: is individualized immuno-modulatory therapy the answer? Virulence (2014) 5(1):45–56. 10.4161/viru.26516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Patil NK, Bohannon JK, Sherwood ER. Immunotherapy: A promising approach to reverse sepsis-induced immunosuppression. Pharmacol Res (2016) 111:688–702. 10.1016/j.phrs.2016.07.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Monneret G, Venet F, Kullberg BJ, Netea MG. ICU-acquired immunosuppression and the risk for secondary fungal infections. Med Mycol (2011) 49 Suppl 1:S17–23. 10.3109/13693786.2010.509744 [DOI] [PubMed] [Google Scholar]
- 15. Otto GP, Sossdorf M, Claus RA, Rodel J, Menge K, Reinhart K, et al. The late phase of sepsis is characterized by an increased microbiological burden and death rate. Crit Care (2011) 15(4):R183. 10.1186/cc10332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hotchkiss RS, Tinsley KW, Swanson PE, Schmieg RE, Jr., Hui JJ, Chang KC, et al. Sepsis-induced apoptosis causes progressive profound depletion of B and CD4+ T lymphocytes in humans. J Immunol (2001) 166(11):6952–63. 10.4049/jimmunol.166.11.6952 [DOI] [PubMed] [Google Scholar]
- 17. Unsinger J, McDonough JS, Shultz LD, Ferguson TA, Hotchkiss RS. Sepsis-induced human lymphocyte apoptosis and cytokine production in “humanized”. Mice J Leukoc Biol (2009) 86(2):219–27. 10.1189/jlb.1008615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hotchkiss RS, Monneret G, Payen D. Immunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approach. Lancet Infect Dis (2013) 13(3):260–8. 10.1016/S1473-3099(13)70001-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Patil NK, Guo Y, Luan L, Sherwood ER. Targeting Immune Cell Checkpoints during Sepsis. Int J Mol Sci (2017) 18(11):2413. 10.3390/ijms18112413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fallon EA, Biron-Girard BM, Chung CS, Lomas-Neira J, Heffernan DS, Monaghan SF, et al. A novel role for coinhibitory receptors/checkpoint proteins in the immunopathology of sepsis. J Leukoc Biol (2018). 103:1151–64 10.1002/JLB.2MIR0917-377R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol (2008) 26:677–704. 10.1146/annurev.immunol.26.021607.090331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Collins M, Ling V, Carreno BM. The B7 family of immune-regulatory ligands. Genome Biol (2005) 6(6):223. 10.1186/gb-2005-6-6-223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Boomer JS, To K, Chang KC, Takasu O, Osborne DF, Walton AH, et al. Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA (2011) 306(23):2594–605. 10.1001/jama.2011.1829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McNab FW, Berry MP, Graham CM, Bloch SA, Oni T, Wilkinson KA, et al. Programmed death ligand 1 is over-expressed by neutrophils in the blood of patients with active tuberculosis. Eur J Immunol (2011) 41(7):1941–7. 10.1002/eji.201141421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Patera AC, Drewry AM, Chang K, Beiter ER, Osborne D, Hotchkiss RS. Frontline Science: Defects in immune function in patients with sepsis are associated with PD-1 or PD-L1 expression and can be restored by antibodies targeting PD-1 or PD-L1. J Leukoc Biol (2016) 100(6):1239–54. 10.1189/jlb.4HI0616-255R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Patil NK, Luan L, Bohannon JK, Hernandez A, Guo Y, Sherwood ER. Frontline Science: Anti-PD-L1 protects against infection with common bacterial pathogens after burn injury. J Leukoc Biol (2018) 103(1):23–33. 10.1002/JLB.5HI0917-360R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Huang X, Chen Y, Chung CS, Yuan Z, Monaghan SF, Wang F, et al. Identification of B7-H1 as a novel mediator of the innate immune/proinflammatory response as well as a possible myeloid cell prognostic biomarker in sepsis. J Immunol (2014) 192(3):1091–9. 10.4049/jimmunol.1302252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wu Y, Chung CS, Chen Y, Monaghan SF, Patel S, Huang X, et al. A Novel Role for Programmed Cell Death Receptor Ligand-1 (PD-L1) in Sepsis-Induced Intestinal Dysfunction. Mol Med (2016) 22:830–40. 10.2119/molmed.2016.00150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xu S, Yang Q, Bai J, Tao T, Tang L, Chen Y, et al. Blockade of endothelial, but not epithelial, cell expression of PD-L1 following severe shock attenuates the development of indirect acute lung injury in mice. Am J Physiol Lung Cell Mol Physiol (2020) 318(4):L801–12. 10.1152/ajplung.00108.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rossi AL, Le M, Chung CS, Chen Y, Fallon EA, Matoso A, et al. A novel role for programmed cell death receptor ligand 2 in sepsis-induced hepatic dysfunction. Am J Physiol Gastrointest Liver Physiol (2019) 316(1):G106–14. 10.1152/ajpgi.00204.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Garni-Wagner BA, Purohit A, Mathew PA, Bennett M, Kumar V. A novel function-associated molecule related to non-MHC-restricted cytotoxicity mediated by activated natural killer cells and T cells. J Immunol (1993) 151(1):60–70. [PubMed] [Google Scholar]
- 32. McArdel SL, Terhorst C, Sharpe AH. Roles of CD48 in regulating immunity and tolerance. Clin Immunol (2016) 164:10–20. 10.1016/j.clim.2016.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen CW, Mittal R, Klingensmith NJ, Burd EM, Terhorst C, Martin GS, et al. Cutting Edge: 2B4-Mediated Coinhibition of CD4+ T Cells Underlies Mortality in Experimental Sepsis. J Immunol (2017) 199(6):1961–6. 10.4049/jimmunol.1700375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xie J, Chen CW, Sun Y, Laurie SJ, Zhang W, Otani S, et al. Increased attrition of memory T cells during sepsis requires 2B4. JCI Insight (2019) 4(9). 10.1172/jci.insight.126030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Xie J, Crepeau RL, Chen CW, Zhang W, Otani S, Coopersmith CM, et al. Sepsis erodes CD8(+) memory T cell-protective immunity against an EBV homolog in a 2B4-dependent manner. J Leukoc Biol (2019) 105(3):565–75. 10.1002/JLB.4A0718-292R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Walker LS, Sansom DM. The emerging role of CTLA4 as a cell-extrinsic regulator of T cell responses. Nat Rev Immunol (2011) 11(12):852–63. 10.1038/nri3108 [DOI] [PubMed] [Google Scholar]
- 37. Inoue S, Bo L, Bian J, Unsinger J, Chang K, Hotchkiss RS. Dose-dependent effect of anti-CTLA-4 on survival in sepsis. Shock (2011) 36(1):38–44. 10.1097/SHK.0b013e3182168cce [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chang KC, Burnham CA, Compton SM, Rasche DP, Mazuski RJ, McDonough JS, et al. Blockade of the negative co-stimulatory molecules PD-1 and CTLA-4 improves survival in primary and secondary fungal sepsis. Crit Care (2013) 17(3):R85. 10.1186/cc12711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. del Rio ML, Lucas CL, Buhler L, Rayat G, Rodriguez-Barbosa JI. HVEM/LIGHT/BTLA/CD160 cosignaling pathways as targets for immune regulation. J Leukoc Biol (2010) 87(2):223–35. 10.1189/jlb.0809590 [DOI] [PubMed] [Google Scholar]
- 40. Steinberg MW, Cheung TC, Ware CF. The signaling networks of the herpesvirus entry mediator (TNFRSF14) in immune regulation. Immunol Rev (2011) 244(1):169–87. 10.1111/j.1600-065X.2011.01064.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Murphy TL, Murphy KM. Slow down and survive: Enigmatic immunoregulation by BTLA and HVEM. Annu Rev Immunol (2010) 28:389–411. 10.1146/annurev-immunol-030409-101202 [DOI] [PubMed] [Google Scholar]
- 42. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol (2015) 15(8):486–99. 10.1038/nri3862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shubin NJ, Monaghan SF, Heffernan DS, Chung CS, Ayala A. B and T lymphocyte attenuator expression on CD4+ T-cells associates with sepsis and subsequent infections in ICU patients. Crit Care (2013) 17(6):R276. 10.1186/cc13131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lou JS, Wang JF, Fei MM, Zhang Y, Wang J, Guo Y, et al. Targeting Lymphocyte Activation Gene 3 to Reverse T-Lymphocyte Dysfunction and Improve Survival in Murine Polymicrobial Sepsis. J Infect Dis (2020) 222(6):1051–61. 10.1093/infdis/jiaa191 [DOI] [PubMed] [Google Scholar]
- 45. Khairnar V, Duhan V, Patil AM, Zhou F, Bhat H, Thoens C, et al. CEACAM1 promotes CD8+ T cell responses and improves control of a chronic viral infection. Nat Commun (2018) 9(1):2561. 10.1038/s41467-018-04832-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhao Z, Jiang X, Kang C, Xiao Y, Hou C, Yu J, et al. Blockade of the T cell immunoglobulin and mucin domain protein 3 pathway exacerbates sepsis-induced immune deviation and immunosuppression. Clin Exp Immunol (2014) 178(2):279–91. 10.1111/cei.12401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yang X, Jiang X, Chen G, Xiao Y, Geng S, Kang C, et al. T cell Ig mucin-3 promotes homeostasis of sepsis by negatively regulating the TLR response. J Immunol (2013) 190(5):2068–79. 10.4049/jimmunol.1202661 [DOI] [PubMed] [Google Scholar]
- 48. Matsumoto R, Matsumoto H, Seki M, Hata M, Asano Y, Kanegasaki S, et al. Human ecalectin, a variant of human galectin-9, is a novel eosinophil chemoattractant produced by T lymphocytes. J Biol Chem (1998) 273(27):16976–84. 10.1074/jbc.273.27.16976 [DOI] [PubMed] [Google Scholar]
- 49. Wada J, Kanwar YS. Identification and characterization of galectin-9, a novel beta-galactoside-binding mammalian lectin. J Biol Chem (1997) 272(9):6078–86. 10.1074/jbc.272.9.6078 [DOI] [PubMed] [Google Scholar]
- 50. Baum PR, Gayle RB, Ramsdell F, Srinivasan S, Sorensen RA, Watson ML, et al. Molecular characterization of murine and human OX40/OX40 ligand systems: identification of a human OX40 ligand as the HTLV-1-regulated protein gp34. EMBO J (1994) 13(17):3992–4001. 10.1002/j.1460-2075.1994.tb06715.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Takeda I, Ine S, Killeen N, Ndhlovu LC, Murata K, Satomi S, et al. Distinct roles for the OX40-OX40 ligand interaction in regulatory and nonregulatory T cells. J Immunol (2004) 172(6):3580–9. 10.4049/jimmunol.172.6.3580 [DOI] [PubMed] [Google Scholar]
- 52. Zaini J, Andarini S, Tahara M, Saijo Y, Ishii N, Kawakami K, et al. OX40 ligand expressed by DCs costimulates NKT and CD4+ Th cell antitumor immunity in mice. J Clin Invest (2007) 117(11):3330–8. 10.1172/JCI32693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tanaka Y, Inoi T, Tozawa H, Yamamoto N, Hinuma Y. A glycoprotein antigen detected with new monoclonal antibodies on the surface of human lymphocytes infected with human T-cell leukemia virus type-I (HTLV-I). Int J Cancer (1985) 36(5):549–55. 10.1002/ijc.2910360506 [DOI] [PubMed] [Google Scholar]
- 54. Weinberg AD, Wegmann KW, Funatake C, Whitham RH. Blocking OX-40/OX-40 ligand interaction in vitro and in vivo leads to decreased T cell function and amelioration of experimental allergic encephalomyelitis. J Immunol (1999) 162(3):1818–26. [PubMed] [Google Scholar]
- 55. Ohshima Y, Tanaka Y, Tozawa H, Takahashi Y, Maliszewski C, Delespesse G. Expression and function of OX40 ligand on human dendritic cells. J Immunol (1997) 159(8):3838–48. [PubMed] [Google Scholar]
- 56. Sato T, Ishii N, Murata K, Kikuchi K, Nakagawa S, Ndhlovu LC, et al. Consequences of OX40-OX40 ligand interactions in langerhans cell function: enhanced contact hypersensitivity responses in OX40L-transgenic mice. Eur J Immunol (2002) 32(11):3326–35. [DOI] [PubMed] [Google Scholar]
- 57. Imura A, Hori T, Imada K, Ishikawa T, Tanaka Y, Maeda M, et al. The human OX40/gp34 system directly mediates adhesion of activated T cells to vascular endothelial cells. J Exp Med (1996) 183(5):2185–95. 10.1084/jem.183.5.2185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Krimmer DI, Loseli M, Hughes JM, Oliver BG, Moir LM, Hunt NH, et al. CD40 and OX40 ligand are differentially regulated on asthmatic airway smooth muscle. Allergy (2009) 64(7):1074–82. 10.1111/j.1398-9995.2009.01959.x [DOI] [PubMed] [Google Scholar]
- 59. Nakae S, Suto H, Iikura M, Kakurai M, Sedgwick JD, Tsai M, et al. Mast cells enhance T cell activation: importance of mast cell costimulatory molecules and secreted TNF. J Immunol (2006) 176(4):2238–48. 10.4049/jimmunol.176.4.2238 [DOI] [PubMed] [Google Scholar]
- 60. Zingoni A, Sornasse T, Cocks BG, Tanaka Y, Santoni A, Lanier LL. Cross-talk between activated human NK cells and CD4+ T cells via OX40-OX40 ligand interactions. J Immunol (2004) 173(6):3716–24. 10.4049/jimmunol.173.6.3716 [DOI] [PubMed] [Google Scholar]
- 61. Unsinger J, Walton AH, Blood T, Tenney DJ, Quigley M, Drewry AM, et al. OX40 agonistic antibody reverses immune suppression and improves survival in sepsis. J Leukoc Biol (2020). 10.1002/JLB.5HI0720-043R [DOI] [PMC free article] [PubMed]
- 62. Rudick CP, Cornell DL, Agrawal DK. Single versus combined immunoregulatory approach using PD-1 and CTLA-4 modulators in controlling sepsis. Expert Rev Clin Immunol (2017) 13(9):907–19. 10.1080/1744666X.2017.1357469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lenschow DJ, Walunas TL, Bluestone JA. CD28/B7 system of T cell costimulation. Annu Rev Immunol (1996) 14:233–58. 10.1146/annurev.immunol.14.1.233 [DOI] [PubMed] [Google Scholar]
- 64. Sugamura K, Ishii N, Weinberg AD. Therapeutic targeting of the effector T-cell co-stimulatory molecule OX40. Nat Rev Immunol (2004) 4(6):420–31. 10.1038/nri1371 [DOI] [PubMed] [Google Scholar]
- 65. Mallett S, Fossum S, Barclay AN. Characterization of the MRC OX40 antigen of activated CD4 positive T lymphocytes–a molecule related to nerve growth factor receptor. EMBO J (1990) 9(4):1063–8. 10.1002/j.1460-2075.1990.tb08211.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Webb GJ, Hirschfield GM, Lane PJ. OX40, OX40L and Autoimmunity: a Comprehensive Review. Clin Rev Allergy Immunol (2016) 50(3):312–32. 10.1007/s12016-015-8498-3 [DOI] [PubMed] [Google Scholar]
- 67. al-Shamkhani A, Birkeland ML, Puklavec M, Brown MH, James W, Barclay AN. OX40 is differentially expressed on activated rat and mouse T cells and is the sole receptor for the OX40 ligand. Eur J Immunol (1996) 26(8):1695–9. 10.1002/eji.1830260805 [DOI] [PubMed] [Google Scholar]
- 68. Arch RH, Thompson CB. 4-1BB and Ox40 are members of a tumor necrosis factor (TNF)-nerve growth factor receptor subfamily that bind TNF receptor-associated factors and activate nuclear factor kappaB. Mol Cell Biol (1998) 18(1):558–65. 10.1128/mcb.18.1.558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kawamata S, Hori T, Imura A, Takaori-Kondo A, Uchiyama T. Activation of OX40 signal transduction pathways leads to tumor necrosis factor receptor-associated factor (TRAF) 2- and TRAF5-mediated NF-kappaB activation. J Biol Chem (1998) 273(10):5808–14. 10.1074/jbc.273.10.5808 [DOI] [PubMed] [Google Scholar]
- 70. Rogers PR, Song J, Gramaglia I, Killeen N, Croft M. OX40 promotes Bcl-xL and Bcl-2 expression and is essential for long-term survival of CD4 T cells. Immunity (2001) 15(3):445–55. 10.1016/s1074-7613(01)00191-1 [DOI] [PubMed] [Google Scholar]
- 71. Song J, So T, Cheng M, Tang X, Croft M. Sustained survivin expression from OX40 costimulatory signals drives T cell clonal expansion. Immunity (2005) 22(5):621–31. 10.1016/j.immuni.2005.03.012 [DOI] [PubMed] [Google Scholar]
- 72. Linch SN, McNamara MJ, Redmond WL. OX40 Agonists and Combination Immunotherapy: Putting the Pedal to the Metal. Front Oncol (2015) 5:34. 10.3389/fonc.2015.00034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Karulf M, Kelly A, Weinberg AD, Gold JA. OX40 ligand regulates inflammation and mortality in the innate immune response to sepsis. J Immunol (2010) 185(8):4856–62. 10.4049/jimmunol.1000404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J (1992) 11(11):3887–95. 10.1002/j.1460-2075.1992.tb05481.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Okazaki T, Maeda A, Nishimura H, Kurosaki T, Honjo T. PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc Natl Acad Sci U S A (2001) 98(24):13866–71. 10.1073/pnas.231486598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Zhang X, Schwartz JC, Guo X, Bhatia S, Cao E, Lorenz M, et al. Structural and functional analysis of the costimulatory receptor programmed death-1. Immunity (2004) 20(3):337–47. 10.1016/S1074-7613(04)00051-2 [DOI] [PubMed] [Google Scholar]
- 77. Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med (1999) 5(12):1365–9. 10.1038/70932 [DOI] [PubMed] [Google Scholar]
- 78. Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol (2001) 2(3):261–8. 10.1038/85330 [DOI] [PubMed] [Google Scholar]
- 79. Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med (2000) 192(7):1027–34. 10.1084/jem.192.7.1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, Saito T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med (2012) 209(6):1201–17. 10.1084/jem.20112741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol (2005) 25(21):9543–53. 10.1128/MCB.25.21.9543-9553.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Rota G, Niogret C, Dang AT, Barros CR, Fonta NP, Alfei F, et al. Shp-2 Is Dispensable for Establishing T Cell Exhaustion and for PD-1 Signaling In Vivo. Cell Rep (2018) 23(1):39–49. 10.1016/j.celrep.2018.03.026 [DOI] [PubMed] [Google Scholar]
- 83. Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol (2004) 173(2):945–54. 10.4049/jimmunol.173.2.945 [DOI] [PubMed] [Google Scholar]
- 84. Celis-Gutierrez J, Blattmann P, Zhai Y, Jarmuzynski N, Ruminski K, Gregoire C, et al. Quantitative Interactomics in Primary T Cells Provides a Rationale for Concomitant PD-1 and BTLA Coinhibitor Blockade in Cancer Immunotherapy. Cell Rep (2019) 27(11):3315–30.e3317. 10.1016/j.celrep.2019.05.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Adler G, Steeg C, Pfeffer K, Murphy TL, Murphy KM, Langhorne J, et al. B and T lymphocyte attenuator restricts the protective immune response against experimental malaria. J Immunol (2011) 187(10):5310–9. 10.4049/jimmunol.1101456 [DOI] [PubMed] [Google Scholar]
- 86. Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science (2017) 355(6332):1428–33. 10.1126/science.aaf1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Lecis D, Sangaletti S, Colombo MP, Chiodoni C. Immune Checkpoint Ligand Reverse Signaling: Looking Back to Go Forward in Cancer Therapy. Cancers (Basel) (2019) 11(5). 10.3390/cancers11050624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Clark CA, Gupta HB, Sareddy G, Pandeswara S, Lao S, Yuan B, et al. Tumor-Intrinsic PD-L1 Signals Regulate Cell Growth, Pathogenesis, and Autophagy in Ovarian Cancer and Melanoma. Cancer Res (2016) 76(23):6964–74. 10.1158/0008-5472.CAN-16-0258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Gato-Canas M, Zuazo M, Arasanz H, Ibanez-Vea M, Lorenzo L, Fernandez-Hinojal G, et al. PDL1 Signals through Conserved Sequence Motifs to Overcome Interferon-Mediated Cytotoxicity. Cell Rep (2017) 20(8):1818–29. 10.1016/j.celrep.2017.07.075 [DOI] [PubMed] [Google Scholar]
- 90. Hartley GP, Chow L, Ammons DT, Wheat WH, Dow SW. Programmed Cell Death Ligand 1 (PD-L1) Signaling Regulates Macrophage Proliferation and Activation. Cancer Immunol Res (2018) 6(10):1260–73. 10.1158/2326-6066.CIR-17-0537 [DOI] [PubMed] [Google Scholar]
- 91. Guignant C, Lepape A, Huang X, Kherouf H, Denis L, Poitevin F, et al. Programmed death-1 levels correlate with increased mortality, nosocomial infection and immune dysfunctions in septic shock patients. Crit Care (2011) 15(2):R99. 10.1186/cc10112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Zhang Y, Li J, Lou J, Zhou Y, Bo L, Zhu J, et al. Upregulation of programmed death-1 on T cells and programmed death ligand-1 on monocytes in septic shock patients. Crit Care (2011) 15(1):R70. 10.1186/cc10059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Chang K, Svabek C, Vazquez-Guillamet C, Sato B, Rasche D, Wilson S, et al. Targeting the programmed cell death 1: programmed cell death ligand 1 pathway reverses T cell exhaustion in patients with sepsis. Crit Care (2014) 18(1):R3. 10.1186/cc13176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Wilson JK, Zhao Y, Singer M, Spencer J, Shankar-Hari M. Lymphocyte subset expression and serum concentrations of PD-1/PD-L1 in sepsis - pilot study. Crit Care (2018) 22(1):95. 10.1186/s13054-018-2020-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Tomino A, Tsuda M, Aoki R, Kajita Y, Hashiba M, Terajima T, et al. Increased PD-1 Expression and Altered T Cell Repertoire Diversity Predict Mortality in Patients with Septic Shock: A Preliminary Study. PLoS One (2017) 12(1):e0169653. 10.1371/journal.pone.0169653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Spec A, Shindo Y, Burnham CA, Wilson S, Ablordeppey EA, Beiter ER, et al. T cells from patients with Candida sepsis display a suppressive immunophenotype. Crit Care (2016) 20:15. 10.1186/s13054-016-1182-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Young WA, Fallon EA, Heffernan DS, Efron PA, Cioffi WG, Ayala A. Improved survival after induction of sepsis by cecal slurry in PD-1 knockout murine neonates. Surgery (2017) 161(5):1387–93. 10.1016/j.surg.2016.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Wang F, Huang X, Chung CS, Chen Y, Hutchins NA, Ayala A. Contribution of programmed cell death receptor (PD)-1 to Kupffer cell dysfunction in murine polymicrobial sepsis. Am J Physiol Gastrointest Liver Physiol (2016) 311(2):G237–245. 10.1152/ajpgi.00371.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Shao R, Fang Y, Yu H, Zhao L, Jiang Z, Li CS. Monocyte programmed death ligand-1 expression after 3-4 days of sepsis is associated with risk stratification and mortality in septic patients: a prospective cohort study. Crit Care (2016) 20(1):124. 10.1186/s13054-016-1301-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Jiang W, Li X, Wen M, Liu X, Wang K, Wang Q, et al. Increased percentage of PD-L1(+) natural killer cells predicts poor prognosis in sepsis patients: a prospective observational cohort study. Crit Care (2020) 24(1):617. 10.1186/s13054-020-03329-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Deng W, Zhu S, Zeng L, Liu J, Kang R, Yang M, et al. The Circadian Clock Controls Immune Checkpoint Pathway in Sepsis. Cell Rep (2018) 24(2):366–78. 10.1016/j.celrep.2018.06.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Remy KE, Mazer M, Striker DA, Ellebedy AH, Walton AH, Unsinger J, et al. Severe immunosuppression and not a cytokine storm characterizes COVID-19 infections. JCI Insight (2020) 5(17). 10.1172/jci.insight.140329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Sullivan RJ, Johnson DB, Rini BI, Neilan TG, Lovly CM, Moslehi JJ, et al. COVID-19 and immune checkpoint inhibitors: initial considerations. J Immunother Cancer (2020) 8(1). 10.1136/jitc-2020-000933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Hutchins NA, Wang F, Wang Y, Chung CS, Ayala A. Kupffer cells potentiate liver sinusoidal endothelial cell injury in sepsis by ligating programmed cell death ligand-1. J Leukoc Biol (2013) 94(5):963–70. 10.1189/jlb.0113051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Tang L, Bai J, Chung CS, Lomas-Neira J, Chen Y, Huang X, et al. Active players in resolution of shock/sepsis induced indirect lung injury: immunomodulatory effects of Tregs and PD-1. J Leukoc Biol (2014) 96(5):809–20. 10.1189/jlb.4MA1213-647RR [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Lomas-Neira J, Monaghan SF, Huang X, Fallon EA, Chung CS, Ayala A. Novel Role for PD-1:PD-L1 as Mediator of Pulmonary Vascular Endothelial Cell Functions in Pathogenesis of Indirect ARDS in Mice. Front Immunol (2018) 9:3030. 10.3389/fimmu.2018.03030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Chen L, Han X. Anti-PD-1/PD-L1 therapy of human cancer: past, present, and future. J Clin Invest (2015) 125(9):3384–91. 10.1172/JCI80011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Brahmamdam P, Inoue S, Unsinger J, Chang KC, McDunn JE, Hotchkiss RS. Delayed administration of anti-PD-1 antibody reverses immune dysfunction and improves survival during sepsis. J Leukoc Biol (2010) 88(2):233–40. 10.1189/jlb.0110037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Shindo Y, Unsinger J, Burnham CA, Green JM, Hotchkiss RS. Interleukin-7 and anti-programmed cell death 1 antibody have differing effects to reverse sepsis-induced immunosuppression. Shock (2015) 43(4):334–43. 10.1097/SHK.0000000000000317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Phares TW, Kotraiah V, Chung CS, Unsinger J, Mazer M, Remy KE, et al. A Peptide-Based Checkpoint Immunomodulator Alleviates Immune Dysfunction in Murine Polymicrobial Sepsis. Shock (2020). 10.1097/SHK.0000000000001682 [DOI] [PMC free article] [PubMed]
- 111. Hotchkiss RS, Colston E, Yende S, Crouser ED, Martin GS, Albertson T, et al. Immune checkpoint inhibition in sepsis: a Phase 1b randomized study to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics of nivolumab. Intensive Care Med (2019) 45(10):1360–71. 10.1007/s00134-019-05704-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Zhang Y, Zhou Y, Lou J, Li J, Bo L, Zhu K, et al. PD-L1 blockade improves survival in experimental sepsis by inhibiting lymphocyte apoptosis and reversing monocyte dysfunction. Crit Care (2010) 14(6):R220. 10.1186/cc9354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Zhu W, Bao R, Fan X, Tao T, Zhu J, Wang J, et al. PD-L1 blockade attenuated sepsis-induced liver injury in a mouse cecal ligation and puncture model. Mediators Inflamm (2013) 2013:361501. 10.1155/2013/361501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Shindo Y, McDonough JS, Chang KC, Ramachandra M, Sasikumar PG, Hotchkiss RS. Anti-PD-L1 peptide improves survival in sepsis. J Surg Res (2017) 208:33–9. 10.1016/j.jss.2016.08.099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Mathew PA, Garni-Wagner BA, Land K, Takashima A, Stoneman E, Bennett M, et al. Cloning and characterization of the 2B4 gene encoding a molecule associated with non-MHC-restricted killing mediated by activated natural killer cells and T cells. J Immunol (1993) 151(10):5328–37. [PubMed] [Google Scholar]
- 116. Chlewicki LK, Velikovsky CA, Balakrishnan V, Mariuzza RA, Kumar V. Molecular basis of the dual functions of 2B4 (CD244). J Immunol (2008) 180(12):8159–67. 10.4049/jimmunol.180.12.8159 [DOI] [PubMed] [Google Scholar]
- 117. Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol (2009) 10(1):29–37. 10.1038/ni.1679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Assarsson E, Kambayashi T, Schatzle JD, Cramer SO, von Bonin A, Jensen PE, et al. NK cells stimulate proliferation of T and NK cells through 2B4/CD48 interactions. J Immunol (2004) 173(1):174–80. 10.4049/jimmunol.173.1.174 [DOI] [PubMed] [Google Scholar]
- 119. Chuang E, Fisher TS, Morgan RW, Robbins MD, Duerr JM, Vander Heiden MG, et al. The CD28 and CTLA-4 receptors associate with the serine/threonine phosphatase PP2A. Immunity (2000) 13(3):313–22. 10.1016/s1074-7613(00)00031-5 [DOI] [PubMed] [Google Scholar]
- 120. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science (2011) 332(6029):600–3. 10.1126/science.1202947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, et al. CTLA-4 can function as a negative regulator of T cell activation. Immunity (1994) 1(5):405–13. 10.1016/1074-7613(94)90071-X [DOI] [PubMed] [Google Scholar]
- 122. Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med (1995) 182(2):459–65. 10.1084/jem.182.2.459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Boomer JS, Shuherk-Shaffer J, Hotchkiss RS, Green JM. A prospective analysis of lymphocyte phenotype and function over the course of acute sepsis. Crit Care (2012) 16(3):R112. 10.1186/cc11404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Benhatchi K, Jochmanova I, Habalova V, Wagnerova H, Lazurova I. CTLA4 exon1 A49G polymorphism in Slovak patients with rheumatoid arthritis and Hashimoto thyroiditis-results and the review of the literature. Clin Rheumatol (2011) 30(10):1319–24. 10.1007/s10067-011-1752-z [DOI] [PubMed] [Google Scholar]
- 125. Zaletel K, Krhin B, Gaberscek S, Bicek A, Pajic T, Hojker S. Association of CT60 cytotoxic T lymphocyte antigen-4 gene polymorphism with thyroid autoantibody production in patients with Hashimoto’s and postpartum thyroiditis. Clin Exp Immunol (2010) 161(1):41–7. 10.1111/j.1365-2249.2010.04113.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Marron MP, Raffel LJ, Garchon HJ, Jacob CO, Serrano-Rios M, Martinez Larrad MT, et al. Insulin-dependent diabetes mellitus (IDDM) is associated with CTLA4 polymorphisms in multiple ethnic groups. Hum Mol Genet (1997) 6(8):1275–82. 10.1093/hmg/6.8.1275 [DOI] [PubMed] [Google Scholar]
- 127. Douroudis K, Prans E, Kisand K, Nemvalts V, Uibo R. Cytotoxic T-lymphocyte antigen 4 gene polymorphisms are associated with latent autoimmune diabetes in adults. Clin Chim Acta (2009) 403(1-2):226–8. 10.1016/j.cca.2009.03.036 [DOI] [PubMed] [Google Scholar]
- 128. Ligers A, Teleshova N, Masterman T, Huang WX, Hillert J. CTLA-4 gene expression is influenced by promoter and exon 1 polymorphisms. Genes Immun (2001) 2(3):145–52. 10.1038/sj.gene.6363752 [DOI] [PubMed] [Google Scholar]
- 129. Mewes C, Buttner B, Hinz J, Alpert A, Popov AF, Ghadimi M, et al. The CTLA-4 rs231775 GG genotype is associated with favorable 90-day survival in Caucasian patients with sepsis. Sci Rep (2018) 8(1):15140. 10.1038/s41598-018-33246-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Mewes C, Buttner B, Hinz J, Alpert A, Popov AF, Ghadimi M, et al. CTLA-4 Genetic Variants Predict Survival in Patients with Sepsis. J Clin Med (2019) 8(1). 10.3390/jcm8010070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Murphy KM, Nelson CA, Sedy JR. Balancing co-stimulation and inhibition with BTLA and HVEM. Nat Rev Immunol (2006) 6(9):671–81. 10.1038/nri1917 [DOI] [PubMed] [Google Scholar]
- 132. Gavrieli M, Murphy KM. Association of Grb-2 and PI3K p85 with phosphotyrosile peptides derived from BTLA. Biochem Biophys Res Commun (2006) 345(4):1440–5. 10.1016/j.bbrc.2006.05.036 [DOI] [PubMed] [Google Scholar]
- 133. Yu X, Zheng Y, Mao R, Su Z, Zhang J. BTLA/HVEM Signaling: Milestones in Research and Role in Chronic Hepatitis B Virus Infection. Front Immunol (2019) 10:617. 10.3389/fimmu.2019.00617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Rodriguez-Barbosa JI, Schneider P, Weigert A, Lee KM, Kim TJ, Perez-Simon JA, et al. HVEM, a cosignaling molecular switch, and its interactions with BTLA, CD160 and LIGHT. Cell Mol Immunol (2019) 16(7):679–82. 10.1038/s41423-019-0241-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Montgomery RI, Warner MS, Lum BJ, Spear PG. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell (1996) 87(3):427–36. 10.1016/s0092-8674(00)81363-x [DOI] [PubMed] [Google Scholar]
- 136. Shubin NJ, Chung CS, Heffernan DS, Irwin LR, Monaghan SF, Ayala A. BTLA expression contributes to septic morbidity and mortality by inducing innate inflammatory cell dysfunction. J Leukoc Biol (2012) 92(3):593–603. 10.1189/jlb.1211641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Lange A, Sunden-Cullberg J, Magnuson A, Hultgren O. Soluble B and T Lymphocyte Attenuator Correlates to Disease Severity in Sepsis and High Levels Are Associated with an Increased Risk of Mortality. PLoS One (2017) 12(1):e0169176. 10.1371/journal.pone.0169176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Cheng T, Bai J, Chung CS, Chen Y, Biron BM, Ayala A. Enhanced Innate Inflammation Induced by Anti-BTLA Antibody in Dual Insult Model of Hemorrhagic Shock/Sepsis. Shock (2016) 45(1):40–9. 10.1097/SHK.0000000000000479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Goldberg MV, Drake CG. LAG-3 in Cancer Immunotherapy. Curr Top Microbiol Immunol (2011) 344:269–78. 10.1007/82_2010_114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Workman CJ, Cauley LS, Kim IJ, Blackman MA, Woodland DL, Vignali DA. Lymphocyte activation gene-3 (CD223) regulates the size of the expanding T cell population following antigen activation in vivo. J Immunol (2004) 172(9):5450–5. 10.4049/jimmunol.172.9.5450 [DOI] [PubMed] [Google Scholar]
- 141. Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ, et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol (2005) 6(12):1245–52. 10.1038/ni1271 [DOI] [PubMed] [Google Scholar]
- 142. Niu B, Zhou F, Su Y, Wang L, Xu Y, Yi Z, et al. Different Expression Characteristics of LAG3 and PD-1 in Sepsis and Their Synergistic Effect on T Cell Exhaustion: A New Strategy for Immune Checkpoint Blockade. Front Immunol (2019) 10:1888. 10.3389/fimmu.2019.01888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Ren F, Li J, Jiang X, Xiao K, Zhang D, Zhao Z, et al. Plasma soluble Tim-3 emerges as an inhibitor in sepsis: sepsis contrary to membrane Tim-3 on monocytes. Tissue Antigens (2015) 86(5):325–32. 10.1111/tan.12653 [DOI] [PubMed] [Google Scholar]
- 144. Luyt CE, Kaiser L. Virus detection in patients with severe pneumonia: still more questions than answers? Am J Respir Crit Care Med (2012) 186(4):301–2. 10.1164/rccm.201206-1119ED [DOI] [PubMed] [Google Scholar]
- 145. Walton AH, Muenzer JT, Rasche D, Boomer JS, Sato B, Brownstein BH, et al. Reactivation of multiple viruses in patients with sepsis. PLoS One (2014) 9(2):e98819. 10.1371/journal.pone.0098819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Khamri W, Abeles RD, Hou TZ, Anderson AE, El-Masry A, Triantafyllou E, et al. Increased Expression of Cytotoxic T-Lymphocyte-Associated Protein 4 by T Cells, Induced by B7 in Sera, Reduces Adaptive Immunity in Patients With Acute Liver Failure. Gastroenterology (2017) 153(1):263–76. e268. 10.1053/j.gastro.2017.03.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Netea MG, Dominguez-Andres J, Barreiro LB, Chavakis T, Divangahi M, Fuchs E, et al. Defining trained immunity and its role in health and disease. Nat Rev Immunol (2020) 20(6):375–88. 10.1038/s41577-020-0285-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Bohannon JK, Luan L, Hernandez A, Afzal A, Guo Y, Patil NK, et al. Role of G-CSF in monophosphoryl lipid A-mediated augmentation of neutrophil functions after burn injury. J Leukoc Biol (2016) 99(4):629–40. 10.1189/jlb.4A0815-362R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Fensterheim BA, Young JD, Luan L, Kleinbard RR, Stothers CL, Patil NK, et al. The TLR4 Agonist Monophosphoryl Lipid A Drives Broad Resistance to Infection via Dynamic Reprogramming of Macrophage Metabolism. J Immunol (2018) 200(11):3777–89. 10.4049/jimmunol.1800085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Hernandez A, Luan L, Stothers CL, Patil NK, Fults JB, Fensterheim BA, et al. Phosphorylated Hexa-Acyl Disaccharides Augment Host Resistance Against Common Nosocomial Pathogens. Crit Care Med (2019) 47(11):e930–8. 10.1097/CCM.0000000000003967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Moorlag S, Khan N, Novakovic B, Kaufmann E, Jansen T, van Crevel R, et al. beta-Glucan Induces Protective Trained Immunity against Mycobacterium tuberculosis Infection: A Key Role for IL-1. Cell Rep (2020) 31(7):107634. 10.1016/j.celrep.2020.107634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Hernandez A, Patil NK, Stothers CL, Luan L, McBride MA, Owen AM, et al. Immunobiology and application of toll-like receptor 4 agonists to augment host resistance to infection. Pharmacol Res (2019) 150:104502. 10.1016/j.phrs.2019.104502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Huang X, Venet F, Wang YL, Lepape A, Yuan Z, Chen Y, et al. PD-1 expression by macrophages plays a pathologic role in altering microbial clearance and the innate inflammatory response to sepsis. Proc Natl Acad Sci U S A (2009) 106(15):6303–8. 10.1073/pnas.0809422106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Allam JP, Peng WM, Appel T, Wenghoefer M, Niederhagen B, Bieber T, et al. Toll-like receptor 4 ligation enforces tolerogenic properties of oral mucosal Langerhans cells. J Allergy Clin Immunol (2008) 121(2):368–74.e361. 10.1016/j.jaci.2007.09.045 [DOI] [PubMed] [Google Scholar]
- 155. Rodriguez-Garcia M, Porichis F, de Jong OG, Levi K, Diefenbach TJ, Lifson JD, et al. Expression of PD-L1 and PD-L2 on human macrophages is up-regulated by HIV-1 and differentially modulated by IL-10. J Leukoc Biol (2011) 89(4):507–15. 10.1189/jlb.0610327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Avendano-Ortiz J, Maroun-Eid C, Martin-Quiros A, Toledano V, Cubillos-Zapata C, Gomez-Campelo P, et al. PD-L1 Overexpression During Endotoxin Tolerance Impairs the Adaptive Immune Response in Septic Patients via HIF1alpha. J Infect Dis (2018) 217(3):393–404. 10.1093/infdis/jix279 [DOI] [PubMed] [Google Scholar]
- 157. Cheng SC, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, et al. mTOR- and HIF-1alpha-mediated aerobic glycolysis as metabolic basis for trained immunity. Science (2014) 345(6204):1250684. 10.1126/science.1250684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Fensterheim BA, Guo Y, Sherwood ER, Bohannon JK. The Cytokine Response to Lipopolysaccharide Does Not Predict the Host Response to Infection. J Immunol (2017) 198(8):3264–73. 10.4049/jimmunol.1602106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun (2015) 6:6692. 10.1038/ncomms7692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Martins F, Sofiya L, Sykiotis GP, Lamine F, Maillard M, Fraga M, et al. Adverse effects of immune-checkpoint inhibitors: epidemiology, management and surveillance. Nat Rev Clin Oncol (2019) 16(9):563–80. 10.1038/s41571-019-0218-0 [DOI] [PubMed] [Google Scholar]